
Method Article
Мышиная модель перехода Streptococcus pneumoniae от колонизатора к патогену при вирусной коинфекции повторяет возрастное заболевание
* Эти авторы внесли равный вклад
В этой статье
Резюме
В этой статье описана новая мышиная модель перехода пневмококка от бессимптомного колонизатора к болезнетворному патогену во время вирусной инфекции. Эта модель может быть легко адаптирована для изучения полимикробных взаимодействий и взаимодействий хозяина с патогеном на разных этапах прогрессирования заболевания и у разных хозяев.
Аннотация
Streptococcus pneumoniae (пневмококк) является бессимптомным колонизатором носоглотки у большинства людей, но может прогрессировать до легочного и системного патогена при инфекции вирусом гриппа А (IAV). Пожилой возраст повышает восприимчивость хозяина к вторичной пневмококковой пневмонии и связан с ухудшением исходов заболевания. Факторы-хозяева, управляющие этими процессами, четко не определены, отчасти из-за отсутствия животных моделей, которые воспроизводят переход от бессимптомной колонизации к тяжелому клиническому заболеванию.
В этой статье описывается новая модель мыши, которая воссоздает переход пневмококков от бессимптомного носительства к заболеванию при вирусной инфекции. В этой модели мышей сначала интраназально инокулируют выращенные в биопленке пневмококки для установления бессимптомного носительства, а затем инфекцию IAV как носоглотки, так и легких. Это приводит к распространению бактерий в легкие, воспалению легких и очевидным признакам болезни, которые могут прогрессировать до летального исхода. Степень заболевания зависит от бактериального штамма и факторов хозяина.
Важно отметить, что эта модель воспроизводит восприимчивость к старению, потому что по сравнению с молодыми мышами старые мыши проявляют более тяжелое клиническое заболевание и чаще умирают от болезней. Разделяя носительство и заболевание на отдельные этапы и предоставляя возможность анализировать генетические варианты как патогена, так и хозяина, эта модель коинфекции S. pneumoniae/IAV позволяет детально изучить взаимодействие важного патобионта с хозяином на разных этапах прогрессирования заболевания. Эта модель также может служить важным инструментом для выявления потенциальных терапевтических мишеней против вторичной пневмококковой пневмонии у восприимчивых хозяев.
Введение
Streptococcus pneumoniae (пневмококк) - это грамположительные бактерии, которые бессимптомно обитают в носоглотке большинства здоровых людей 1,2. Под влиянием факторов, которые полностью не определены, пневмококки могут переходить от доброкачественных колонизаторов носоглотки к патогенам, которые распространяются на другие органы, что приводит к серьезным инфекциям, включая средний отит, пневмонию и бактериемию3. Проявления пневмококковой инфекции частично зависят от штаммоспецифических различий, включая серотип, который основан на составе капсульных полисахаридов. На сегодняшний день охарактеризовано более 100 серотипов, и некоторые из них связаны с более инвазивными инфекциями 4,5. Ряд других факторов увеличивает риск пневмококковой инфекции. Одним из таких факторов является вирусная инфекция, при которой риск пневмококковой пневмонии увеличивается в 100 раз на 6,7 IAV. Исторически сложилось так, что S. pneumoniae является одной из наиболее частых причин вторичной бактериальной пневмонии после гриппа и ассоциируется с худшими исходами8. Еще одним важным фактором риска является пожилой возраст. Фактически, S. pneumoniae является основной причиной внебольничной бактериальной пневмонии у пожилых людей старше 65 лет 9,10. Пожилые люди составляют большинство (>75%) смертей от пневмонии и гриппа, что указывает на то, что два фактора риска - старение и инфекция IAV - синергетически ухудшают восприимчивость к заболеванию11,12,13,14. Однако механизмы, с помощью которых вирусная инфекция вызывает переход пневмококков от бессимптомного колонизатора к инвазивному патогену, и то, как это формируется факторами хозяина, остаются плохо определенными. Во многом это связано с отсутствием модели мелких животных, которая повторяет переход от бессимптомной пневмококковой колонизации к критическому клиническому заболеванию.
Исследования коинфекции классически моделировались на мышах, инокулированных пневмококками непосредственно в легкие через 7 дней после заражения гриппом15,16. Это воспроизводит восприимчивость к вторичной бактериальной пневмонии и идеально подходит для изучения того, как противовирусные иммунные реакции ослабляют антибактериальную защиту17. Однако лонгитюдные исследования на людях показали, что пневмококковое носительство в носоглотке, где бактерии могут образовывать бессимптомные биопленки18, равномерно связано с инвазивными заболеваниями19,20. Бактериальные изоляты от инфекций среднего уха, легких и крови генетически идентичны тем, которые обнаруживаются в носоглотке20. Таким образом, для изучения перехода от бессимптомного носительства к инвазивному заболеванию после инфекции IAV была создана модель, в которой мышам интраназально вводили пневмококки, выращенные в биопленке, с последующей инфекцией IAV носоглотки21,22. Вирусная инфекция верхних дыхательных путей привела к изменениям в среде хозяина, что привело к рассеиванию пневмококков из биопленок и их распространению в нижние дыхательные пути21. Эти дисперсные бактерии повышали экспрессию факторов вирулентности, важных для инфекции, превращая их из колонизаторов в патогены21. Эти наблюдения подчеркивают сложное взаимодействие между вирусом, хозяином и бактериями и демонстрируют, что изменения в организме хозяина, вызванные вирусной инфекцией, оказывают непосредственное влияние на поведение пневмококковой инфекции, что, в свою очередь, изменяет течение бактериальной инфекции. Тем не менее, эта модель не повторяет тяжелые признаки болезни, наблюдаемые у людей, вероятно, потому, что вирус ограничен носовой полостью, а системное воздействие вирусной инфекции на иммунитет хозяина и повреждение легких не повторяется.
Недавно мы создали модель, которая включает в себя сложное взаимодействие между хозяином и патогенами, но также более точно имитирует тяжесть заболевания, наблюдаемую у людей23. В этой модели мышей сначала интраназально интестируют пневмококками, выращенными в биопленке, чтобы установить бессимптомное носительство, а затем инфекцию IAV как носоглотки, так и легких. Это привело к распространению бактерий в легкие, воспалению легких и заболеванию, которое прогрессировало до летального исхода у части молодых мышей23. Это предыдущее исследование продемонстрировало, что как вирусная, так и бактериальная инфекция изменяет защиту хозяина: вирусная инфекция способствовала распространению бактерий, а предшествующая бактериальная колонизация ухудшала способность хозяина контролировать уровень IAVв легких 23. Изучение иммунного ответа показало, что инфекция IAV снижает антибактериальную активность нейтрофилов, в то время как бактериальная колонизация притупляет ответ интерферона I типа, критически важный для противовирусной защиты23. Важно отметить, что эта модель воспроизводила восприимчивость к старению. По сравнению с молодыми мышами, старые мыши проявляли признаки заболевания раньше, проявляли более тяжелое клиническое заболевание и чаще поддавались инфекции23. Работа, представленная в этой рукописи, показывает, что степень заболевания также зависит от бактериального штамма, поскольку инвазивные пневмококковые штаммы демонстрируют более эффективное распространение при инфекции IAV, проявляют более явные признаки воспаления легких и приводят к ускоренным темпам заболевания по сравнению с неинвазивными штаммами. Таким образом, эта модель коинфекции S. pneumoniae/IAV позволяет детально изучить как патогенные факторы, так и факторы хозяина и хорошо подходит для изучения иммунных реакций на полимикробные инфекции на разных фазах прогрессирования заболевания.
протокол
Все исследования на животных проводились в соответствии с рекомендациями, изложенными в Руководстве по уходу и использованию лабораторных животных. Все процедуры были одобрены Комитетом по уходу за животными и их использованию Университета в Буффало.
1. Подготовка химически определенных сред (CDM)
- Подготовьте подвои следующим образом:
- Растворите соединения смеси I, перечисленные в таблице 1 , в 100 мл сверхчистой воды при перемешивании. Хранить в аликвотах 200 мкл при -20 °C.
- Растворите соединения смеси II, перечисленные в таблице 1, в 20 мл 0,1 М NaOH при перемешивании. Хранить в аликвотах 100 мкл при -20 °C.
- Растворите соединения смеси III, перечисленные в таблице 1, в 1 мл сверхчистой воды при перемешивании. Хранить в аликвотах 10 мкл при температуре 4 °C.
- Растворите соединение смеси IV, указанное в таблице 1, в 1 мл сверхчистой воды при перемешивании. Хранить в аликвотах 10 мкл при -20 °C.
- Соединения, перечисленные в таблице 2 , растворяют первоначально в 15 мл сверхчистой воды при перемешивании. Отрегулируйте рН до 7,0 с помощью нескольких капель 0,1 М NaOH и отрегулируйте конечный объем до 20 мл, используя сверхчистую воду. Хранить в аликвотах по 1 мл при температуре -20 °C.
- Растворите соединения, перечисленные в таблице 3 , в 90 мл сверхчистой воды на горячей плите при температуре 50 ° C при перемешивании. Отрегулируйте рН до 7,0 с помощью 0,1 М NaOH, а затем отрегулируйте конечный объем до 100 мл с помощью сверхчистой воды. Хранить в аликвотах объемом 5 мл при температуре -20 °C.
- Каждый раз готовьте закваску свежей, растворяя соединения, указанные в таблице 4 , в 70 мл сверхчистой воды при перемешивании.
- К свежему стартовому бульону добавьте следующие запасы смеси по порядку: 200 мкл смеси I (таблица 1), 80 мкл смеси II (таблица 1), 10 мкл смеси III (таблица 1), 10 мкл смеси IV (таблица 2), 1 мл витаминной смеси (таблица 3) и 5 мл аминокислоты (таблица 4).
- После того, как запасы будут добавлены, отрегулируйте окончательный объем до 100 мл, добавив 30 мл сверхчистой воды в стакан.
- Дополнить МЧР соединениями из таблицы 4. После тщательного перемешивания процедить-стерилизовать и хранить при температуре 4 °C не более 2 недель.
2. Выращивание биопленки S. pneumoniae
- Приготовьте среду RP-10, смешав 445 мл RPMI 1640 с 50 мл инактивированной теплом эмбриональной бычьей сыворотки (FBS) и 5 мл пенициллина/стрептомицина в концентрации 10 000 ЕД/мл и 10 000 мкг/мл соответственно.
- Вырастите клеточную линию мукоэпидермоидной карциномы NCI-H292 (H292). Добавьте клетки из одного купленного флакона в 5 мл среды RP-10 в колбу, обработанную тканевой культурой T-25. Инкубировать при 37 °C / 5% CO2 в течение 3-5 дней до достижения 100% слияния.
- Проверьте клетки под световым микроскопом, используя 10-кратное увеличение, чтобы оценить слияние.
ПРИМЕЧАНИЕ: Когда все ячейки находятся в контакте с другими клетками и между ними нет промежутков, достигается желаемое 100% слияние. - Промойте клетки 2 раза в 5 мл PBS комнатной температуры. Убедитесь, что буфер не содержит кальция, чтобы избежать хелатирования ЭДТА на следующем этапе.
- Добавьте 1 мл трипсина-ЭДТА в колбу и инкубируйте при 37 ° C / 5% CO2 в течение 5-10 мин, пока клетки не отделятся. Нейтрализуйте 4 мл среды RP-10. Аккуратно перемешайте, пипеткой вверх и вниз, и переложите в коническую пробирку объемом 50 мл.
- Добавьте 500 мкл клеточной суспензии на лунку в 24-луночную пластину, обработанную тканевой культурой. Из сливающейся колбы Т-25 ожидайте 2 × 10 6-4 × 106 клеток/мл.
- На следующий день проверьте клетки под световым микроскопом, чтобы убедиться, что они сливаются, как показано на шаге 2.3. Если их нет, то инкубируют дольше.
- После того, как клетки H292 на 100% сливаются в 24-луночную пластину, осторожно промойте клетки 3 раза 1 мл PBS комнатной температуры, чтобы убедиться, что не осталось среды, содержащей антибиотики или мусор.
- После промывки клеток добавьте 250 мкл / лунку 4% параформальдегида, чтобы зафиксировать клетки. Инкубировать в течение 1 часа на льду или в течение ночи при температуре 4 ° C.
- В ночь перед фиксацией клеток нанесите интересующий штамм S. pneumoniae на пластины с кровяным агаром и инкубируйте в течение ночи при 37 ° C / 5% CO2.
ПРИМЕЧАНИЕ: Представленные здесь данные относятся к следующим штаммам S. pneumoniae, полученным в результате совместного обмена: серотип 19F изолят среднего отита EF3030 24, классический серотип 2 штамм Avery D3925 и бактериемический изолят серотипа 4 TIGR426. Штаммы также доступны из общедоступных коллекций, упомянутых в таблице материалов. - Приготовьте CDM плюс оксиразу (0,15 ЕД / мл), добавив 100 мкл оксиразы (30 ЕД / мл) к 20 мл CDM.
ПРИМЕЧАНИЕ: Оксиразу используют для устранения кислорода, чтобы обеспечить эффективный рост S. pneumoniae в жидкой культуре27. - Инокулируйте бактерии с тарелки в свежий CDM + оксиразу, смывая бактерии с тарелки, добавив 1 мл CDM + оксиразы и осторожно подняв бактериальные колонии, используя сторону наконечника пипетки объемом 1 мл, стараясь не соскребать агар. В качестве альтернативы используйте инокуляционную петлю для подъема бактерий и инокуляции их в пробирку, содержащую 1 мл оксиразы CDM +.
- Разбавьте бактерии в CDM + оксиразе до начального OD600 0,05.
- Выращивайте бактерии в неплотно закрытой конической пробирке объемом 50 мл, стоящей при 37 ° C / 5% CO 2 до тех пор, пока не будет достигнут OD600 0,2 (это займет от2 до 5 часов). Проверяйте OD600 каждый час, чтобы убедиться, что OD не превышает 0,2.
- Как только OD достигнет 0,2, встряхните бактериальную культуральную трубку. Засейте 0,5 мл бактерий на фиксированные клетки H292 и добавьте еще 0,5 мл среды CDM + оксиразы на лунку. Добавьте 1 мл CDM + оксиразы в контрольные лунки без бактерий. Инкубируйте пластину в течение 48 ч при 34 °C/5%CO2.
ПРИМЕЧАНИЕ: Рост при 34 ° C используется для более точной имитации более низкой температуры в носоглотке21. - Каждые 12 ч после первоначального посева осторожно удаляйте 0,5 мл среды и пополняйте 0,5 мл свежего CDM + оксиразы. Будьте осторожны, чтобы не нарушить образующуюся биопленку. Проверьте нижнюю часть пластины на наличие биопленки и обратите внимание на увеличение помутнения с течением времени из-за роста биопленки. Чтобы контролировать загрязнение, проверьте колодцы на отсутствие бактерий, чтобы убедиться, что контрольные колодцы остаются чистыми.
- Через 48 часов после инокуляции удалите надосадочную жидкость и очень осторожно промойте 2 раза 1 мл PBS. Ресуспендировать в 1 мл свежего CDM и энергично пипеткой вверх и вниз, чтобы поднять биопленку. Для каждого бактериального штамма объедините бактерии из всех лунок в коническую пробирку объемом 50 мл. Хорошо перемешайте, осторожно наклонив плотно закрытую трубку вверх и вниз несколько раз.
- В коническую пробирку объемом 50 мл добавьте 40% глицерина в CDM в равных объемах для получения бактериальной суспензии с конечной концентрацией 20% глицерина. Аликвотировать 1 мл в микроцентрифужные пробирки, заморозить на сухом льду и сохранить при -80 °C.
- Перед использованием перечислите бактерии, разморозив одну аликвоту на льду, вращая пробирку при 1,700 × г в течение 5 мин, удалив надосадочную жидкость, ресуспендировав гранулу в 1 мл PBS и нанеся последовательные разведения на пластины28 с кровяным агаром.
- Выращивайте агаровые пластины в течение ночи при 37 ° C / 5% CO2 и подсчитывайте колонии в соответствующих разведениях, чтобы получить концентрацию бактерий в колониеобразующих единицах (КОЕ) / мл.
ПРИМЕЧАНИЕ: Рекомендуется подсчитывать бактерии в запасах не менее чем через сутки после замораживания или позже, так как в течение первых 24 часов наблюдается падение жизнеспособности бактерий. Хранящиеся замороженные аликвоты можно использовать для последующего заражения мышей в течение максимум 2 месяцев.
3. Интраназальная инокуляция мышей S. pneumoniae, выращенным в биопленке
- Приобретайте мышей и используйте в нужном возрасте.
ПРИМЕЧАНИЕ: Мыши в возрасте 3-4 месяцев предпочтительнее для моделирования молодых хозяев, а мыши в возрасте 21-24 месяцев могут быть использованы для моделирования пожилых людей >65 летв возрасте 29 лет. Данные, представленные здесь, относятся к самцам мышей C57BL / 6. - Разморозьте выращенные в биопленке бактериальные аликвоты на льду и вращайте при 1,700 × г в течение 5 минут. Осторожно удалите и выбросьте надосадочную жидкость, не разрушая гранулы, промойте бактерии, ресуспендировав гранулу в 1 мл PBS, и снова отжимайте при 1,700 × г в течение 5 минут. Удалите надосадочную жидкость и ресуспендируйте гранулы в объеме, необходимом для достижения желаемой концентрации (стремитесь к 5 × 106 КОЕ / 10 мкл для интраназальной инокуляции). Подтвердите количество введенных бактерий, нанеся приготовленный инокулят на пластины с кровяным агаром, как показано на шаге 2.19.
- Инокулируйте мышей интраназально 5 × 106 КОЕ путем пипетки 5 мкл разбавленного инокулята в каждый нарис. Обязательно крепко держите мышей, стабилизируя голову, пока объем не будет вдохнут (обычно в течение нескольких секунд после пипетки громкости в ноздри). Выполняйте этот шаг при отсутствии анестезии, чтобы предотвратить легочную аспирацию инокулята.
4. Вирусная инфекция, вызванная вирусом гриппа А (IAV)
- Через 48 ч после интраназальной инокуляции S. pneumoniae разморозьте интересующий штамм IAV на льду.
ПРИМЕЧАНИЕ: Представленные здесь данные относятся к адаптированному к мышам штамму вируса гриппа А A/PR/8/34 H1N1, который был получен в результате совместного обмена30. - Как только вирус оттает, разбавьте вирус в PBS до желаемой концентрации; стремиться к 20 бляшкообразующим единицам (БОЕ)/50 мкл для интратрахеальной инфекции и 200 БОЕ/10 мкл для интраназальной инфекции. Для групп с имитационным заражением и группами, содержащими только бактерии, используйте PBS для инокуляции мышей.
- Поместите офтальмологическую смазку на глаза мышей перед анестезией. Обезболивают мышей 5% изофлураном и подтверждают анестезию крепким щипком пальца ноги.
- После того, как животное будет обезболено, удалите его из камеры изофлурана и немедленно заразите анестезированных мышей 50 мкл (20 БОЕ) IAV интратрахеально, используя тупой пинцет, чтобы вытащить язык изо рта и пипетировать объем жидкости вниз по трахее.
- Поместите мышей в отдельную клетку и наблюдайте за ними до полного выздоровления (они способны поддерживать лежачее положение грудины [способны лежать вертикально на груди]).
- После выздоровления немедленно интраназально инокулируют мышей 10 мкл (200 БОЕ) IAV, используя метод инокуляции на этапе 3.3.
- Домашние мыши, перенесшие одиночную или двойную бактериальную и вирусную инфекцию с одной и той же группой инфекции, отделяют их от других групп.
5. Наблюдение за мышами на наличие симптомов заболевания
- Наблюдайте за мышами ежедневно в течение не менее 10 дней и слепо оценивайте признаки болезни следующим образом:
- Оценка для похудения выглядит следующим образом: 0 = 5% или меньше; 1 = 5%-10%; 2 = 10%-15%; 3 = 20% и более. Усыпляйте мышей с помощью ингаляции CO2 , когда показатель потери веса равен 3.
- Оценка активности выглядит следующим образом: 0 = нормальная/активная; 1 = движущийся, но слегка уменьшенный; 2 = уменьшение; 3 = сильно ослабленный/вялый (двигается только при прикосновении), 4 = кома/неподвижен. Усыпляйте мышей, когда оценка активности будет равна 3.
- Оцените осанку следующим образом: 0 = отсутствие догадки (нормально); 1 = слегка сгорбленная осанка; 2 = сильная догадка. Усыпляйте мышей, когда оценка осанки будет равна 2.
- Оценка глаз выглядит следующим образом: 0 = норма; 1 = выступающий; 1 = затонувший; 1 = закрытый; 1 = разряд. Это может быть комбинация. Сложите итоги для окончательной оценки глаз.
- Оценка дыхания выглядит следующим образом: 0 = нормальное дыхание; 1 = нерегулярный или измененный (более высокая/меньшая ставка); 2 = труд (преувеличенное усилие или вздох). Усыпляйте мышей, когда оценка дыхания будет равна 2.
- Основываясь на приведенных выше критериях, сложите индивидуальные баллы для общего клинического балла от здоровых (0) до крайне больных (15). Считайте, что любая мышь, набравшая общий балл выше 2, больна. Гуманно усыпьте всех мышей, показавших общий балл выше 9 или указанные баллы по каждому критерию, и отметьте их на кривой выживаемости.
6. Обработка инфицированных тканей для бактериального перебора
- Через 48 часов после заражения IAV усыпьте мышей.
- Поместите мышь в положение лежа на спине. Используя 70% этанол, распылите на грудь и живот мыши, чтобы очистить шерсть. Используя щипцы, зажмите мех и кожу посередине мыши и отрежьте шерсть ножницами для рассечения 4,5 дюйма, чтобы обнажить область от печени до груди.
- Забор крови
- С помощью диссекционных ножниц аккуратно врежьте в брюшную полость, чтобы обнажить печень. С помощью щипцов обнажите печеночную воротную вену в верхней части печени возле диафрагмы. Разрежьте воротную вену печени с помощью ножниц для рассечения. Как только кровь начнет скапливаться в брюшной полости, соберите 10 мкл крови с помощью микропипетки и поместите в 90 мкл раствора антикоагулянта (50 мМ раствора ЭДТА в PBS) в микроцентрифужную пробирку для покрытия бактериальной нагрузки.
- Используйте микропипетку P-1000 для сбора остальной крови, поместите ее в пробирку для сбора крови и центрифугу при 7 600 × г в течение 2 минут, чтобы собрать сыворотку. Сохраняют сыворотки в микроцентрифужных пробирках при температуре -80 °C для последующего анализа любого желаемого цитокина или метаболита.
- Сбор легких
- Используя ножницы для рассечения, сделайте надрез по бокам обнаженной грудной клетки и осторожно потяните ребра вверх к голове мыши, чтобы обнажить сердце. Вставьте иглу 25 G, прикрепленную к шприцу объемом 10 мл, предварительно заполненному PBS, в правый желудочек и начните медленно перфузию. Ищите отбеливание легких как показатель успешной перфузии. Медленно промывайте, чтобы не повредить легочную ткань.
- Поднимите сердце щипцами и сделайте разрез, чтобы отделить легкие и сердце. После отделения соберите щипцами все доли легкого и промойте в посуде со стерильным PBS, чтобы удалить остатки крови. В чашке Петри нарежьте легкие небольшими кусочками и хорошо перемешайте. Удалите половину легочной смеси для определения бактериального КОЕ или вирусного БОЕ и поместите его в пробирку объемом 15 мл с круглым дном, предварительно заполненную 0,5 мл PBS для гомогенизации.
ПРИМЕЧАНИЕ: Важно не брать разные доли одного и того же легкого для разных оценок. Вместо этого все доли должны быть измельчены, хорошо перемешаны вместе и одинаково проанализированы для разных оценок. - Удалите другую половину легкого для проточной цитометрии (раздел 7 ниже) и поместите ее в 24-луночную пластину, не обработанную тканевой культурой, с каждой лункой, предварительно заполненной 0,5 мл RP-10. Оставить при комнатной температуре до обработки.
- Коллекция носоглотки
- На шее используйте ножницы для рассечения, чтобы срезать мех, а затем отрежьте мышцу и обнаживите трахею.
ПРИМЕЧАНИЕ: Трахея представляет собой трубчатую структуру, расположенную под мышцей. - Поместите небольшие щипцы под трахею на расстоянии 1 см от челюсти мыши, чтобы стабилизировать ее. С помощью ножниц для рассечения аккуратно сделайте разрез 0,1 см на передней части трахеи, избегая полного разрезания трахеи.
- Подготовьте шприц объемом 1 мл, наполненный 0,5 мл PBS, с трубкой 0,58 мм, прикрепленной к игле 25 G. Соберите средство для промывания носа, вставив трубку в трахею, идущую вверх к носоглотке. Как только сопротивление почувствуется, попадая в носовую полость, поместите микроцентрифужную трубку в нос и медленно промойте PBS через трахею, чтобы собрать носовой лаваж.
- Поместите мышь в положение лежа. Опрыскайте голову мыши этанолом. Используйте ножницы для рассечения, чтобы разрезать мех, и мистовую подушечку, чтобы обнажить головную кость мыши.
- С помощью ножниц для рассечения сделайте разрез на 1 см по бокам нижней челюсти и между глазами. С помощью щипцов медленно оттяните лицевые кости от тела, чтобы обнажить носовую полость.
- Используйте щипцы, чтобы аккуратно удалить ткань носа и поместить ее в пробирку с круглым дном, предварительно заполненную 0,5 мл PBS для гомогенизации.
- На шее используйте ножницы для рассечения, чтобы срезать мех, а затем отрежьте мышцу и обнаживите трахею.
- Чтобы гомогенизировать собранную ткань, сначала очистите зонд гомогенизатора, поместив в него 70% этанол и включив гомогенизатор на 60% мощности на 30 с. Повторите шаг в стерильной воде в течение 10 с. Гомогенизируйте каждую ткань в течение 1 мин. Очистите зонд-гомогенизатор в стерильной воде между каждым образцом и в свежей пробирке с 70% этанолом между каждым органом и группой образцов.
- Перечисление численности бактерий
- После того, как все органы были извлечены и гомогенизированы, пластинчатые серийные разведения на пластинах с кровяным агаром. Чтобы рассчитать общее количество КОЕ, используйте 10 мкл для пластины и запишите окончательный объем в мл для каждого образца. Планшет образцов носоглотки на пластинах с кровяным агаром с добавлением 3 мкг / мл гентамицина для отбора роста S. pneumoniae при одновременном ингибировании роста других микроорганизмов, которые колонизируют эту ткань. Инкубировать в течение ночи при 37 °C/5% CO2.
- Чтобы перечислить бактериальный КОЕ для легких и носоглотки, сначала подсчитайте колонии на пластинах кровяного агара. Затем используйте уравнение (1) и уравнение (2), чтобы рассчитать количество на мл и общее число.
Количество на мл = количество колоний × коэффициент разбавления × 100 (1)
Общее число = количество на мл × общий объем на образец (2)
ПРИМЕЧАНИЕ: В уравнении (1) 100 используется для умножения, так как 10 мкл покрыто, что представляет собой 100-кратное разбавление 1 мл. Общий объем на образец в уравнении (2) взят из шага 6.7.1, что приводит к пределу обнаружения 100 на орган. - Чтобы перечислить бактериальные КОЕ для бактериемии, сначала подсчитайте колонии на пластинах с кровяным агаром. Затем используйте уравнение (3), чтобы определить количество на мл крови.
Количество на мл крови = количество колоний × коэффициент разбавления × 100 × 10 (3)
ПРИМЕЧАНИЕ: В уравнении (3) 100 используется в качестве 10 мкл, что представляет собой 100-кратное разведение 1 мл, а 10 указывает на разбавление крови в антикоагулянте 1:10. Это приводит к пределу обнаружения 1,000 / мл.
7. Обработка образцов легких для проточной цитометрии
- Подготовьте необходимые носители следующим образом:
- Подготовьте RP-10, как описано в шаге 2.1.
- Приготовьте буфер для пищеварения, смешав RP-10 с 2 мг/мл коллагеназы и 30 мкл/мл ДНКазы I.
- Приготовьте буфер для лизиса, растворив 8,29 г NH4Cl, 1 г NaHCO3 и 0,038 г ЭДТА в 1 лH2O.
- Приготовьте 10-кратный буфер FACS, смешав 450 мл HBSS с 50 мл инактивированного теплом FBS и 5 г азида натрия.
- Приготовьте 1x буфер FACS, разбавив 50 мл 10x буфера FACS в 450 мл HBSS.
- Возьмите образцы легких из шага 6.4.3 и поместите в 24-луночную пластину. Добавьте 500 мкл буфера для разложения в каждую лунку. Инкубировать от 45 мин до 1 ч при 37 °C/5%CO2.
- Заполните конические пробирки объемом 50 мл для каждого образца 5 мл RP-10. Когда инкубация закончится, поместите фильтр 100 мкм в верхнюю часть конической пробирки объемом 50 мл и смочите ее 1 мл RP-10.
- С помощью микропипетки Р-1000 переместите переваренные легкие и поместите их на фильтр. Используйте поршень шприца объемом 3 мл, чтобы размять орган. Каждый раз ополаскивайте 2 раза 1 мл RP-10.
- Отжимают образцы при 4 °C и 327 × г в течение 5 мин. Аспирируют надосадочную жидкость и ресуспендируют гранулу в 1 мл лизисного буфера. Оставьте на 3 минуты, чтобы дать возможность лизису эритроцитов. Нейтрализуйте 5 мл RP-10.
- Отжимают образцы при 4 °C и 327 × г в течение 5 мин. Аспирируют надосадочную жидкость, ресуспендируют гранулу в 1 мл RP-10 и берут 10 мкл для подсчета образцов.
- Отжимают образцы при 4 °C и 327 × г в течение 5 мин. Аспирируют надосадочную жидкость и ресуспендируют гранулу в RP-10 в дозе 2 × 10 6-4 × 106 клеток/мл. Добавьте 60 мкл каждого образца в 96-луночную пластину для окрашивания для желаемых типовклеток 23, перечисленных на шаге 7.9, в таблице 5 и в таблице 6.
- Отжимайте пластину при температуре 4 °C и 327 × г в течение 5 мин.
- Тем временем приготовьте основные смеси антител, флуоресцентные минус один (FMO) и контрольные элементы с одним окрашиванием с желаемыми антителами. Для окрашивания полиморфноядерных лейкоцитов (ПМН), макрофагов, моноцитов, дендритных клеток и Т-клеток используют антитела и конечные разведения, перечисленные в таблице 5 и таблице 6. Используйте общий объем смеси антител 100 мкл на лунку. Следуйте разведениям, перечисленным в таблицах, для определения соответствующего объема основной смеси и требуемых отдельных антител.
- Когда отжим завершен (этап 7.8), сцедите надосадочную жидкость, ресуспендируйте гранулы в 100 мкл смесей антител, FMO или контрольных средств с одним пятном и инкубируйте на льду в течение 30 минут в темноте.
- Промойте ячейки 2 раза, добавив 150 мкл буфера FACS в лунки и вращая пластину при 4 ° C и 327 × г в течение 5 мин.
- Когда отжим будет завершен, сцедите надосадочную жидкость, ресуспендируйте гранулы в 100 мкл фиксирующего буфера и инкубируйте на льду в течение 20 минут.
- Промойте ячейки 2 раза, добавив 150 мкл буфера FACS в лунки и вращая пластину при 4 ° C и 327 × г в течение 5 мин.
- Подготовьте маркированные пробирки FACS с 200 мкл буфера FACS. Ресуспендируют гранулы в 150 мкл буфера FACS. Индивидуально фильтруйте каждый образец в соответствующую пробирку FACS с помощью фильтра 100 мкм. Хранить на льду или при температуре 4 °C и защищать от света до тех пор, пока не будет готов к анализу.
- Проанализируйте клетки с помощью проточного цитометра.
8. Анализ бляшек для перечисления IAV
- Подготовьте необходимые носители следующим образом:
- Готовят инфекционную среду, растворяя 2,5 г бычьего сывороточного альбумина (BSA) в 40 мл DMEM при перемешивании при 37 °C в течение 10-20 мин до растворения. Фильтруйте-стерилизуйте в 460 мл DMEM.
- Приготовьте 2,4% микрокристаллической целлюлозы, растворив 1,2 мг микрокристаллической целлюлозы в 50 мл H2O. Автоклав в жидком режиме и храните при комнатной температуре.
- Приготовьте 5% BSA DMEM, растворив 2,5 г BSA в 40 мл DMEM при перемешивании при 37 ° C в течение 10-20 минут. Добавьте оставшиеся 10 мл DMEM для конечного объема 50 мл. Фильтровать-стерилизовать и хранить при температуре 4 °C.
- Приготовьте 2x MEM / 0,5% BSA, смешав 1 мл 5% BSA DMEM с 9 мл 2x MEM.
- Приготовьте наплавочную среду с низкой вязкостью, смешав соотношение 1:1 2,4% микрокристаллической целлюлозы и 2x MEM/0,5% BSA с 1 мг/мл TPCK (ингибитор химотрипсина) трипсина.
- Приготовьте EMEM/10% FBS, смешав 450 мл минимально необходимой среды Eagle's Minimum Essential Medium (EMEM) с 50 мл инактивированного теплом FBS.
- Вырастите клеточную линию собачьей почки Мадина-Дарби (MDCK). Добавьте клетки из одного купленного флакона в 5 мл EMEM/10% FBS в колбу, обработанную тканевой культурой T-25. Инкубировать в течение 3-5 дней при 37 ° C / 5% CO2 до тех пор, пока клетки не достигнут 100% слияния. Проверьте наличие слияния, как показано на шаге 2.3.
- Удалите и выбросьте питательную среду и промойте 2 раза 5 мл PBS комнатной температуры. Добавьте 1 мл трипсина-ЭДТА в колбу и инкубируйте при 37 ° C / 5% CO2 в течение 10-15 мин, пока клетки не отделятся. После подъема нейтрализуйте 4 мл EMEM/10% FBS, чтобы получить клеточную суспензию в дозе 2 × 105 клеток/мл.
- Засейте клетки MDCK в 12-луночную пластину, обработанную культурой тканей, добавив 1 мл ресуспендированных клеток в лунку (при 2 × 105 клеток / лунка) за 1 день до начала анализа бляшек.
ПРИМЕЧАНИЕ: Перед использованием убедитесь, что ячейки достигают 100% слияния, и инкубируйте дольше, если это необходимо для достижения слияния. - Для использования в качестве стандартов сделайте 10-кратные серийные разведения (106-10 1) запаса IAV (известного титра) в инфекционной среде, указанной на этапе 8.1.1. Сделайте 1,2 мл каждого разведения для тестирования в трех экземплярах.
- Орган размораживается на льду. Раскрутите на настольной центрифуге 2000 × г и соберите прозрачную надосадочную жидкость.
- Повторите шаг 8.5, но с надосадочной жидкостью из образцов на шаге 8.6.
- Аспирируйте среду из клеток и промойте 2 раза 1 мл PBS, чтобы удалить все FBS.
- Добавьте 300 мкл каждого стандартного разбавления или образца серийного разбавления осторожно вдоль стенки каждой лунки, начиная с самого высокого разбавления до самого низкого, и сделайте это в трех экземплярах.
- Поместите планшеты в инкубатор при температуре 37 °C/5%CO2, встряхивая пластину каждые 10 минут в течение 50 минут. Обязательно поместите их в инкубатор и не складывайте их друг на друга.
- Через 50 мин промойте клетки 2 раза 1 мл PBS.
- Добавьте 2 мл наплавляемой среды с низкой вязкостью в каждую скважину, за исключением лунок с самым низким разбавлением и без вирусов; К ним добавьте инфекционную среду и трипсин.
- Поместите планшет обратно в инкубатор при 37 ° C / 5% CO 2 на2-4 дня, чтобы получить бляшки, которые можно визуализировать невооруженным глазом.
- Вымойте пластины, быстро добавив 2 мл PBS в каждую лунку сбоку, и осторожно встряхните, чтобы суспендировать отстоявшуюся среду с низкой вязкостью.
- Слейте весь объем жидкости в лунку, осторожно пипетировав среду.
- Повторите промывку еще раз, добавив по 2 мл PBS в каждую лунку, а затем слейте весь объем жидкости путем осторожного пипетирования.
- Чтобы зафиксировать бляшки, добавьте 500 мкл 4% параформальдегида в каждую лунку, встряхните и оставьте на 30 минут.
- Медленно промойте сбоку 1 мл PBS; Затем аккуратно слейте жидкость.
- Добавьте 500 мкл 1% кристаллического фиолетового цвета (разбавленного водой) в каждую лунку, чтобы покрыть монослой ячейки. Инкубировать 5 мин.
- Промыть 1 мл водопроводной воды. Обязательно слейте всю жидкость из лунки путем осторожного пипетирования. Положите тарелку вверх дном на подушечку для подгузника, чтобы она высохла в течение ночи.
- Подсчитайте таблички визуально и сохраните изображения на любом доступном тепловизоре.
Результаты
Выращенные в биопленке S. pneumoniae (рис. 1A) использовали для заражения мышей (рис. 1B) с использованием небольшого инокулята объемом 10 мкл, вводимого интраназально неанестезированным мышам. Этот инокулят небольшого объема приводит к постоянному пневмококковому носительству, ограниченному носоглоткой (рис. 2A, группы + sp), избегая при этом системного распространения (рис. 2B, C, + sp группы). Через два дня после интраназальной инокуляции мыши были инфицированы адаптированным к мышам вирусом гриппа H1N1 A A/PR/8/34 (IAV)22,30, доставленным как интраназально, так и интратрахеально для достижения последовательной доставки определенных количеств в носоглотку и легкие23.
Здесь модель была использована для сравнения течения заболевания после вирусной инфекции у мышей, интраназально зараженных различными штаммами S. pneumoniae, включая TIGR4 и D39, которые являются инвазивными штаммами, которые приводят к пневмонии, прогрессирующей до бактериемии, и EF3030, который является штаммом среднего отита 21,24,25,26,31. Проявление заболевания у мышей с коинфекцией S. pneumoniae/IAV зависело от бактериального штамма (рис. 2). Хотя не было существенной разницы в количестве бактерий в носоглотке (рис. 2A) ни у одного из штаммов, S. pneumoniae TIGR4 и D39, но не EF3030, распространились в легкие через 48 ч после инфекции IAV (рис. 2B). Сорок процентов мышей, интраназально инфицированных S. pneumoniae TIGR4, показали бактериальную диссеминацию в легкие, и из них половина из них стала бактериемической (рис.2C), что согласуется с предыдущими результатами 23.
Мыши, интраназально инфицированные S. pneumoniae D39, показали более эффективное распространение, поскольку распространение в легкие наблюдалось у 100% коинфицированных мышей (рис. 2B). Подобно S. pneumoniae TIGR4, половина из них испытала бактериемию (рис. 2C). При отслеживании общей выживаемости, независимо от бактериального штамма, скорость выживаемости коинфицированных мышей была значительно ниже, чем у мышей, которым по отдельности подвергались воздействию только S. pneumoniae для всех тестируемых штаммов (рис. 2D). По сравнению с контрольными мышами, которым подвергался только IAV, мыши, интраназально инфицированные S. pneumoniae TIGR4 и D39, но не EF3030, показали ускоренные показатели заболевания. Ко 2-му дню после заражения IAV 30% (D39) и 20% (TIGR4) мышей погибли, в то время как контрольные группы, принимавшие только IAV, не начали поддаваться до 5-го дня после заражения (рис. 2D). Мыши, коинфицированные S. pneumoniae EF3030 и IAV, имели отсроченные симптомы, более похожие на контрольную группу, содержащую только IAV (рис. 2D). Эти результаты показывают, что модель коинфекции приводит к заболеванию у молодых здоровых мышей, которое зависит от бактериального штамма, что делает ее идеальной для изучения бактериальных факторов, необходимых на каждом этапе прогрессирования заболевания.
Эта модель была использована для оценки присутствия различных иммунных клеток в легких (типы клеток и стратегия стробирования на рисунке 3) после инфекции IAV у мышей, интраназально инокулированных различными штаммами S. pneumoniae. Бактериальные штаммы D39 и TIGR4, которые рассеялись в легких после инфекции IAV, вызвали значительное увеличение притока воспалительных иммунных клеток из кровообращения по сравнению с исходным уровнем (неинфицированных), таких как нейтрофилы (PMN) и моноциты, в то время как EF3030 этого не произошло (рис. 4A-C). Одна только инфекция IAV вызвала значительное увеличение по сравнению с исходным уровнем притока иммунных клеток, важных для защиты хозяина от вирусной инфекции, таких как NK-клетки и гамма-дельта-Т-клетки (рис. 4A-C). Эти противовирусные реакции были значительно притуплены у мышей, интраназально инфицированных S. pneumoniae до вирусного заражения (рис. 4A-C). Это согласуется с предыдущими исследованиями, оценивающими цитокиновые реакции, которые показали, что носительство S. pneumoniae притупляет выработку интерферонов I типа и ухудшает способность хозяина контролировать нагрузки IAV в легких23. Эти результаты показывают, что модель коинфекции может быть использована для изучения того, как изменяются иммунные реакции при моно- и полимикробных инфекциях.
Эта модель также использовалась для оценки влияния старения на течение заболевания после инфекции IAV у мышей, интраназально инфицированных S. pneumoniae TIGR4. У одиночноинфицированных мышей титры вируса не варьировались между молодыми и старыми когортами (рис. 5А)23. Как и в предыдущих исследованиях23, у старых мышей наблюдались более ранние и значительно более серьезные признаки заболевания по сравнению с их молодыми собратьями, о чем свидетельствуют более высокие клинические показатели (рис. 5B). В соответствии с симптомами заболевания, старые мыши, привитые S. pneumoniae , начали умирать быстрее в течение 24 часов после заражения IAV, и все они умерли от болезни, тогда как молодые контрольные группы пережили инфекцию со значительно более высокой (33%) скоростью (рис. 5C). Эти результаты показывают, что модель коинфекции может быть использована для выявления более тяжелого заболевания у уязвимых хозяев, что делает ее идеальной для изучения факторов хозяина, которые придают устойчивость или восприимчивость к коинфекции.

Рисунок 1: Хронология коинфекции и обработки органов для оценки притока иммунных клеток и нагрузки патогенов . (A) Streptococcus pneumoniae выращивают в биопленках. (B) Мышей инокулируют интраназально 5 × 106 КОЕ указанного штамма S. pneumoniae , выращенного в биопленке, для установления носоглоткового носительства или оставляют без лечения. Через сорок восемь часов мышей либо имитируют лечение PBS, либо получают 200 БОЕ вируса гриппа А PR8 интраназально и 20 БОЕ интратрахеально. Мыши контролируются с течением времени для оценки клинических заболеваний и выживаемости. (C) Через 48 часов после инфекции IAV оценивают бактериальный КОЕ или вирусный БОЕ в различных органах или приток иммунных клеток в легких. Сокращения: КОЕ = колониеобразующие единицы; БОЕ = бляшкообразующие единицы; IAV = вирус гриппа A PR8; ИТ = интратрахеально; NP = носоглотка. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 2: Двойная интраназальная/интратрахеальная инфекция IAV у мышей, инокулированных S. pneumoniae, приводит к распространению бактерий и заболеванию, которое зависит от бактериального штамма. Молодые (10-12 недель) мыши-самцы C57BL/6 (B6) были инфицированы, как показано на рисунке 1. Количество бактерий в носоглотке (А), легких (В) и крови (С) определяли через 48 часов после инфекции IAV. (В,В) Проценты обозначают долю мышей, которые показали распространение. (D) Выживаемость контролировалась в течение 10 дней после заражения IAV. Показаны объединенные данные из (A, B) n = 5, (C) n = 11 и (D) n = 6 мышей в группе. Каждый круг соответствует одной мыши, а пунктирные линии обозначают предел обнаружения. (A-C)*, указывает на достоверную разницу (p < 0,05) между указанными группами, определяемую по критерию Крускала-Уоллиса. (D) * указывает на достоверную разницу (p < 0,05) между мышами +sp и Co-inf на бактериальный штамм, что определено с помощью логарифмического теста (Мантел-Кокс). Сокращения: +sp = мыши, инфицированные интраназально бактериями только с использованием указанного штамма; Co-inf = мыши, инфицированные бактериями, которые были инфицированы IAV; IAV = мыши, получившие вирус гриппа А; КОЕ = колониеобразующие единицы. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 3: Стратегия стробирования иммунных клеток. Легкие были собраны, а приток иммунных клеток определялся с помощью проточной цитометрии. Показана репрезентативная стратегия стробирования различных типов клеток. (A) CD45+, живые одиночные клетки были закрыты и процентное содержание (B) PMN (Ly6G+, CD11b+), макрофагов (Ly6G-, Ly6C-, F480+) и моноцитов (Ly6G-, Ly6C+), (C) DC (Ly6G-, CD11c+) и NK-клеток (NK1.1+, CD3-), (D) TCR-γΔ и CD8 (CD8+, TCRβ+) и CD4 (CD4+, TCRβ+ ) Были определены Т-клетки. Сокращения: SSC-A = площадь пика бокового рассеяния; FSC-A = площадь пика прямого рассеяния; FSC-H = высота пика прямого рассеяния; SSC-W = ширина пика бокового рассеяния; L/D = живой/мертвый; FMO = флуоресцентный минус один; NK = природный убийца; ПМН = полиморфноядерный лейкоцит; DC = дендритная клетка; TCR = рецептор Т-клеток. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
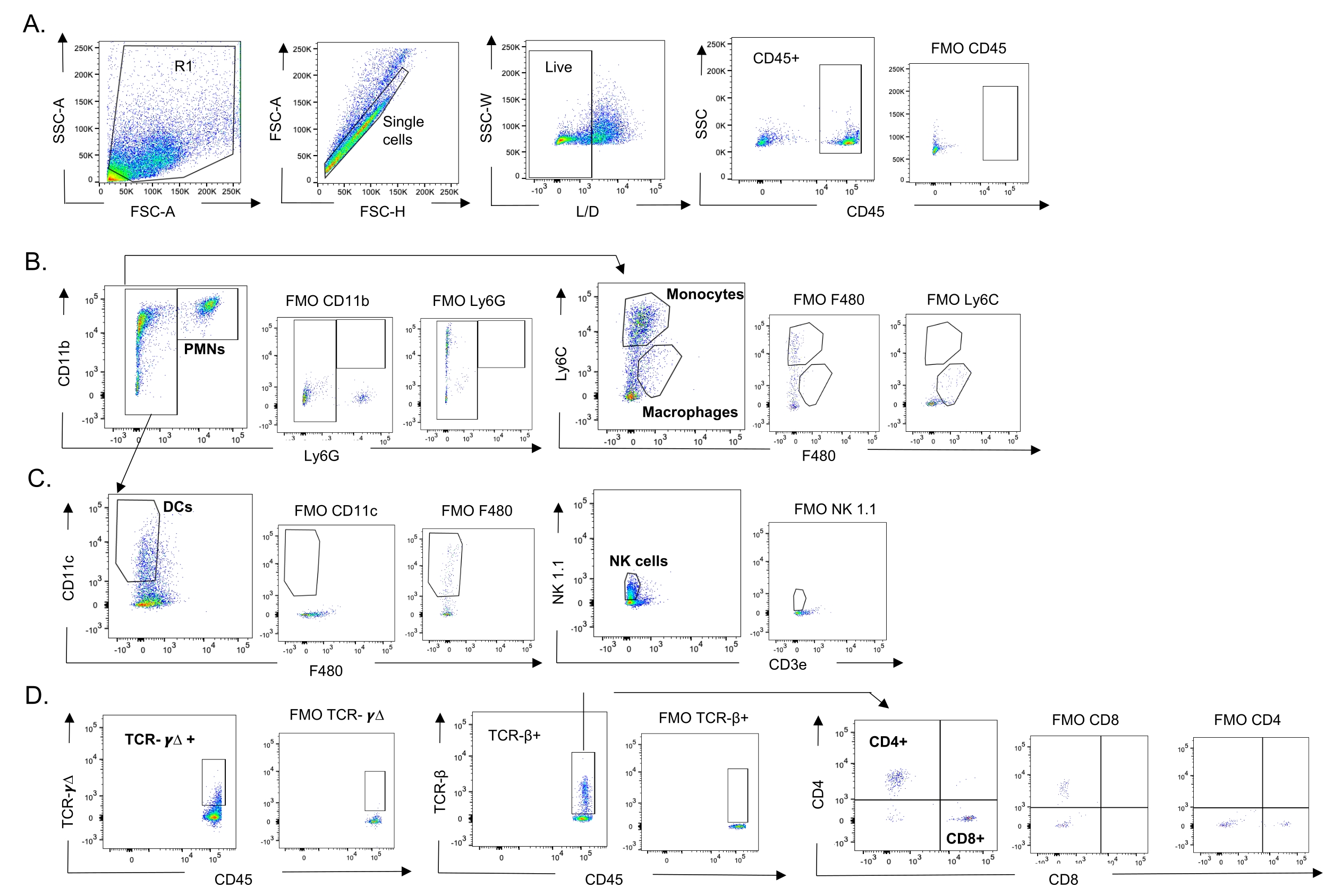{kind=link}

Рисунок 4: Легочные иммунные реакции зависят от штамма бактерий. Молодые (10-12 недель) самцы мышей C57BL/6 были либо неинфицированными, либо однократно инокулированы указанным штаммом Streptococcus pneumoniae (+sp), либо индивидуально заражены IAV (IAV), либо коинфицированы S. pneumoniae и IAV (Co-inf). Через сорок восемь часов после заражения IAV (см. экспериментальный дизайн на рисунке 1) легкие были собраны, и приток иммунных клеток был определен с помощью проточной цитометрии в соответствии со стратегией стробирования, показанной на рисунке 3. (A) Среднее процентное содержание каждого указанного типа клеток в затворе CD45 отображается для всех групп обработки на тепловой карте. (B) Репрезентативные точечные графики типов клеток, которые показали значительные различия между обработками, показаны для каждой группы мышей. (C) Показано процентное содержание указанных типов иммунных клеток. Каждый круг соответствует одной мышке. (А,С) Показаны объединенные данные от n = 5 мышей в группе. *, указывает на достоверную разницу (p < 0,05) между Co-inf и неинфицированными; $, указывает на значимость между IAV и неинфицированным; #, указывает на существенную разницу между Co-inf и IAV в одиночку. Значимые различия между проблемными группами для каждого типа клеток были определены с помощью ANOVA с последующим тестом Тьюки. Сокращения: NK = природный киллер; ПМН = полиморфноядерный лейкоцит; DC = дендритная клетка; TCR = Т-клеточный рецептор; IAV = вирус гриппа А. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
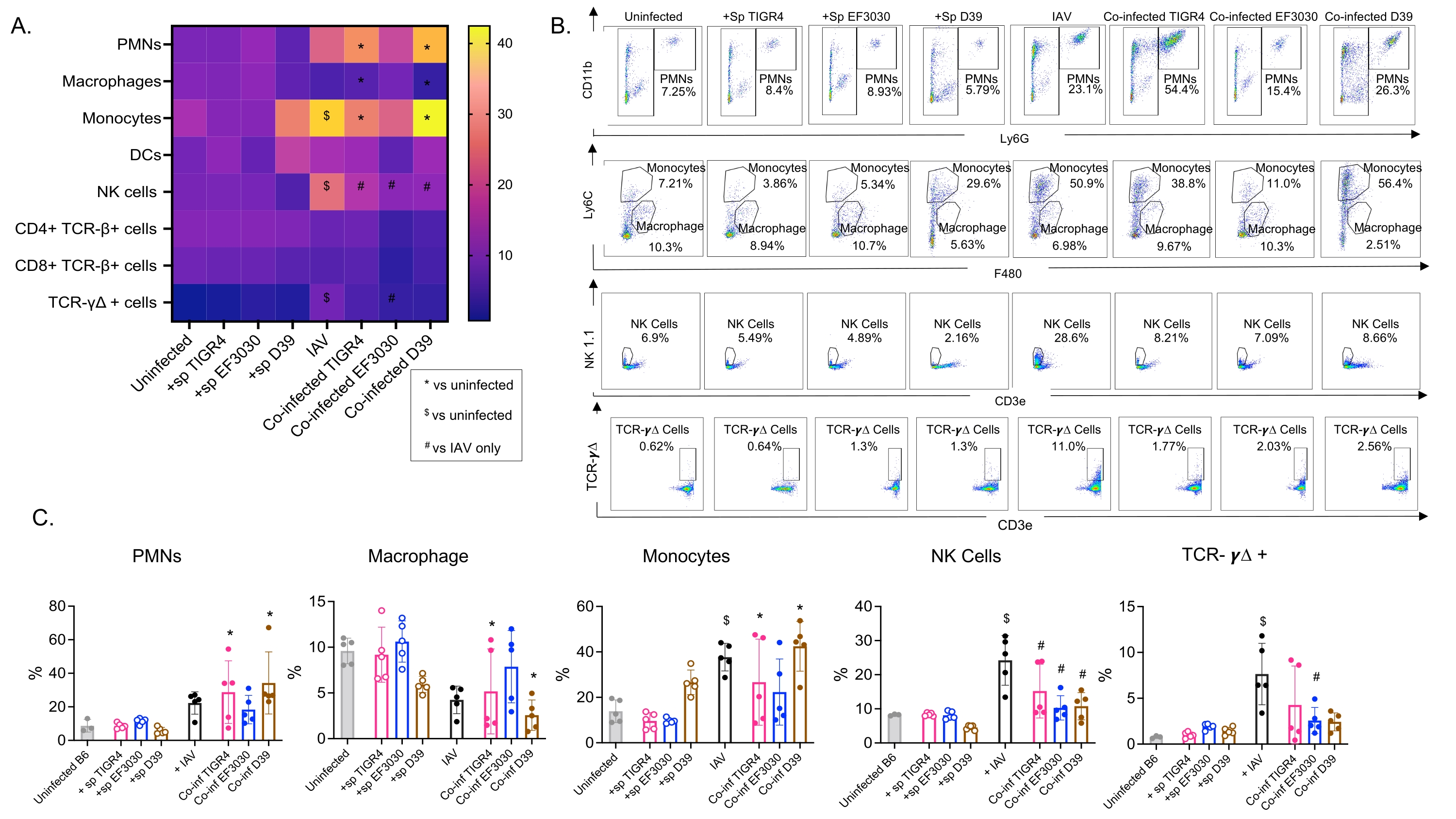{kind=link}

Рисунок 5: Старение и повышенная восприимчивость хозяина к коинфекции IAV/Streptococcus pneumoniae . Молодые (10-12 недель) и в возрасте (21-22 месяца) самцы мышей C57BL/6 были коинфицированы S. pneumoniae TIGR4 i.n. и IAV i.n. и i.t. (как показано на рисунке 1) или по отдельности подвергались воздействию только IAV. (A) Вирусные титры определяли через 48 часов. Звездочки указывают на статистическую значимость (p < 0,05), определяемую t-критерием Стьюдента. Данные объединены из n = 4 мышей в группе. (B) Клиническая оценка и (C) выживаемость контролировались с течением времени. (B) Показаны средние ± SEM, объединенные из n = 6 мышей в группе. Звездочки указывают на статистическую значимость (p < 0,05) между молодыми и старыми мышами в указанный момент времени, как определено тестом Манна-Уитни. (C) Данные объединяются из n = 6 мышей в группе. Звездочки указывают на статистическую значимость (p < 0,05) между молодыми и старыми мышами, как определено по критерию логарифмического ранга (Мантел-Кокс). Сокращения: IAV = вирус гриппа А; и.н. = интраназально; и.т. = интратрахеально; SEM = стандартная погрешность среднего. Рисунок 5А перепечатан с разрешения Joma et al.23. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
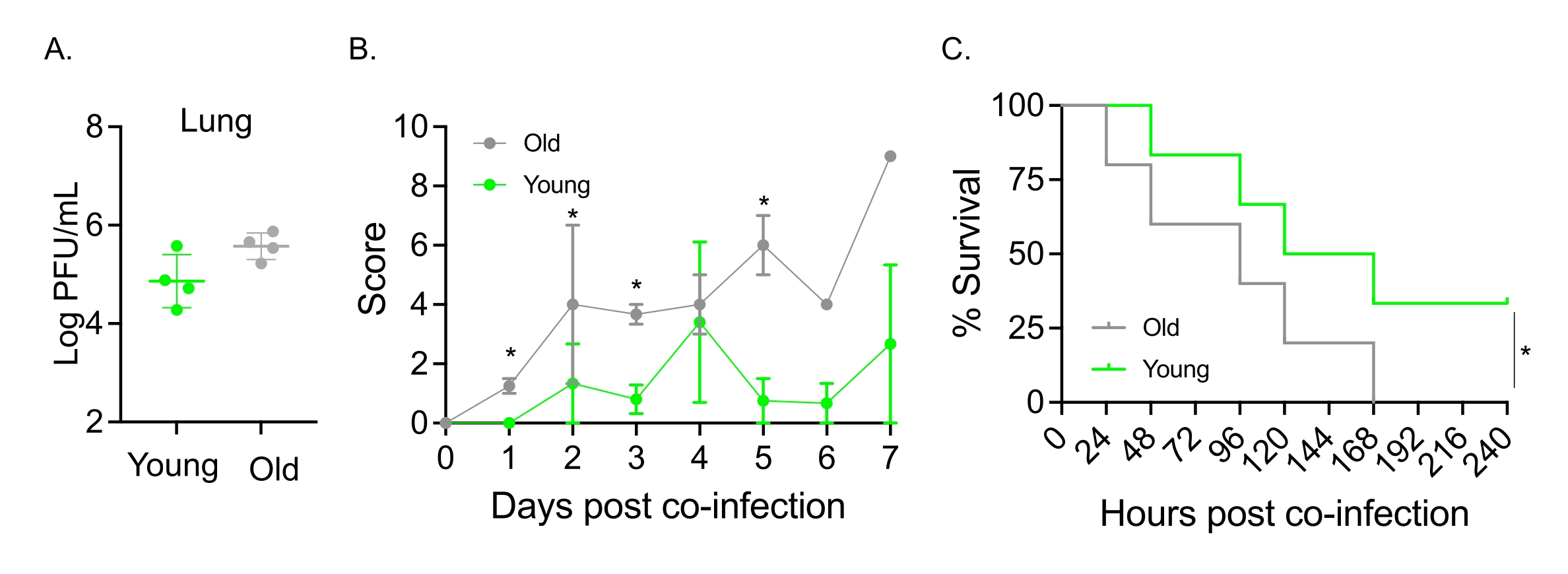{kind=link}
| Запас Mix I для CDM | |
| Аденин | 0,1 г |
| D-аланин | 0,25 г |
| CaCl2 безводный | 0,025 г |
| Сульфат марганца | 0,03 г |
| Цианокобаламин | 100 мкл запаса 10 мг/мл |
| Парааминобензойная кислота | 400 мкл 5 мг/мл |
| Пиридоксамин 2HCl | 100 мкл запаса 10 мг/мл |
| Запас Mix II для МЧР | |
| Гуанин | 0,05 г |
| Урацил | 0,05 г |
| Запас Mix III для МЧР | |
| Нитрат железа 9H2O | 50 мг/мл |
| Сульфат железа 7H2O | 10 мг/мл |
| Запас смеси IV для МЧР | |
| Бета-никотинамидадениндинуклеотид | 25 мг/мл |
Таблица 1: Смешанные запасы I, II, III и IV для МЧР. Аббревиатура: CDM = химически определенные среды.
| Витаминная смесь для CDM | |
| Пиридоксаль гидрохлорид | 0,8 г |
| Тиамин Cl2 | 0,4 г |
| Рибофлавин | 0,4 г |
| Ка-пантотенат | 0,4 г |
| Биотин | 0,04 г |
| Фолиевая кислота | 0,4 г |
| Ниацинамид | 0,4 г |
Таблица 2: Запас витаминной смеси для CDM. Аббревиатура: CDM = химически определенные среды.
| Аминокислотный запас для CDM | |
| L-аланин | 0.480 г |
| L-аргинин | 0,250 г |
| L-аспарагин | 0.700 г |
| L-аспарагиновая кислота | 0.600 г |
| L-цистеин | 1.000 г |
| L-цистин | 0,100 г |
| L-глутаминовая кислота | 0.200 г |
| L-глютамин | 0.780 г |
| L-глицин | 0,350 г |
| L-гистидин | 0.300 г |
| L-изолейцин | 0.430 г |
| L-лейцин | 0.950 г |
| L-лизин | 0.880 г |
| L-метионин | 0,250 г |
| L-фенилаланин | 0,550 г |
| L-пролин | 1.350 г |
| L-серин | 0.680 г |
| L-треонин | 0,450 г |
| L-триптофан | 0,100 г |
| L-валин | 0,650 г |
Таблица 3: Аминокислотный запас для CDM. Аббревиатура: CDM = химически определенные среды.
| Стартовый запас для CDM | |
| Глюкоза | 1,0 г |
| Сульфат магния-7-гидрат | 0,070 г |
| Двузамещенный фосфат калия | 0,02 г |
| Фосфат калия одноосновный | 0,1 г |
| Ацетат натрия безводный | 0,45 г |
| Бикарбонат натрия | 0,25 г |
| Двузамещенный фосфат натрия | 0.735 г |
| Фосфат натрия одноосновный | 0,32 г |
| Заключительные дополнения для МЧР | |
| Холина хлорид | 0,1 г |
| L-цистеин гидрохлорид | 0,075 г |
| Бикарбонат натрия | 0,25 г |
Таблица 4: Стартовый запас и конечные добавки для МЧР. Аббревиатура: CDM = химически определенные среды.
| Антитело/флуорофор | Клон | Коэффициент разбавления |
| L/D для возбуждения ультрафиолета | Н/Д | 0.38888889 |
| Ly6G AF 488 | 1А8 | 0.25 |
| CD11b APC | М1/70 | 0.25 |
| CD11c PE | Н418 | 0.18055556 |
| Мышь Fc Block | 2.4Г2 | 0.11111111 |
| F4/80 PE Cy7 | БМ8 | 0.18055556 |
| Ли6С БВ605 | АЛ-21 | 0.25 |
| CD103 BV 421 | М290 | 0.18055556 |
| CD45 APC-eF-780 | 30-Ф11 | 0.18055556 |
Таблица 5: Панель антител 1.
| Антитело/флуорофор | Клон | Коэффициент разбавления |
| L/D для возбуждения ультрафиолета | Н/Д | 0.388888889 |
| TCR-β БТР Cy7 | Х57-597 | 0.180555556 |
| CD4 V450 (Тихоокеанский синий) | РМ4-5 | 0.25 |
| CD8 BV650 | 53-6.7 | 0.180555556 |
| Мышь Fc Block | 2.4Г2 | 0.111111111 |
| CD45 PE | 30-Ф11 | 0.180555556 |
| CD3 AF488 | 145-2С11 | 0.180555556 |
| TCR- γΔ APC | ГЛ-3 | 0.180555556 |
| NK1.1 AF 700 | ПК136 | 0.180555556 |
Таблица 6: Панель антител 2.
Обсуждение
Большинство существующих экспериментальных исследований коинфекции S. pneumoniae / IAV основаны на доставке бактерий в легкие мышей, предварительно инфицированных IAV. Эти модели помогли выявить изменения в легочной среде и системном иммунном ответе, которые делают хозяина восприимчивым к вторичной бактериальной инфекции 15,16,17,32,33,34,35,36,37. Однако эти модели не смогли имитировать переход S. pneumoniae от бессимптомного колонизатора к патогену, способному вызывать серьезные легочные и системные инфекции. Кроме того, эти модели не подходят для изучения факторов хозяина и взаимодействия хозяина и патогена в верхних дыхательных путях, которые способствуют восприимчивости к инфекции. Предыдущая модель перемещения пневмококков из носоглотки в легкие после инфекции IAV основывалась на бактериальной инфекции носоглотки с последующей вирусной инфекцией. Однако он не смог воспроизвести тяжелые признаки заболевания, наблюдаемые у пациентов21. Модифицированная модель мышиной инфекции, описанная здесь, повторяет переход S. pneumoniae от бессимптомного носительства к патогену, вызывающему тяжелое клиническое заболевание.
Важнейшим этапом этой модели является установление инфекции S. pneumoniae в носоглотке. Streptococcus pneumoniae образуют биопленки и колонизируют носоглотку с разной эффективностью21,38. Для установления последовательной инфекции требуется не менее 5 × 106 КОЕ бактериальных штаммов, выращенных в биопленке, протестированных до сих пор23. Рекомендуется, чтобы любой новый бактериальный штамм был протестирован на стабильную инфекцию носоглотки до вирусной инфекции. Что касается вирусной коинфекции, предыдущие исследования показали, что интраназальная инфекция IAV необходима для дисперсии бактерий из носоглотки21,22,23. В этих предыдущих исследованиях использовалось 500 БОЕ IAV для интраназальной доставки, в то время как в этом исследовании 200 БОЕ было достаточно для увеличения количества бактерий в носоглотке. Инфекция IAV не ограничивается верхними дыхательными путями и может распространяться на легкие39,40, что является ключом к тому, чтобы сделать легочную среду более благоприятной для бактериальной инфекции15,16,41. Доставка IAV в легкие может быть достигнута либо интраназальной доставкой, либо интратрахеальной установкой анестезированных мышей. Предыдущая работа с мышами BALB/cByJ показала, что интраназальная доставка приводит к вирусной пневмонии21; однако доступ инокулята в легкие после интраназальной инокуляции более ограничен у мышей C57BL / 6. У мышей C57BL/6 для последовательной доставки вируса23 требуется интратрахеальная установка. В этой модели предшествующая бактериальная колонизация ускоряет проявление симптомов заболевания после вирусной инфекции23. Поскольку вирусная инфекция сама по себе может вызывать симптомы заболевания с потенциальными изменениями кинетики, рекомендуется сначала протестировать диапазон доз для любого нового тестируемого вирусного штамма и выбрать дозу, которая выявляет ускоренную кинетику у коинфицированных хозяев.
Легкие обеспечивают еще один критический показатель для оценки заболевания в этой модели. Для оценки патогенной нагрузки и притока иммунных клеток можно использовать легкое той же мыши. Однако, поскольку тяжесть инфекции и воспаления может различаться между долями, рекомендуется не брать разные доли одного и того же легкого для различных оценок. Скорее, все доли можно измельчить на мелкие кусочки, хорошо перемешать вместе, а затем одинаково разобрать для разных оценок. Аналогичным образом, носоглотка может быть использована для подсчета бактериальных КОЕ или вирусного КОЕ и иммунного ответа. Однако количество клеток, полученных из промывок и тканей, слишком мало, чтобы выполнить проточную цитометрию без объединения образцов мышей в одной группе. Кроме того, воспаление в носоглотке можно оценить гистологически23.
Важной особенностью этой модели является то, что она повторяет клиническое заболевание, наблюдаемое у пациентов. У людей вторичная пневмококковая пневмония после инфекции IAV часто приводит к очевидным признакам заболевания, включая кашель, одышку, лихорадку и мышечные боли, которые могут привести к госпитализации, дыхательной недостаточности и даже смерти 8,15,42,43. Эта модель повторяет тяжелые признаки клинического заболевания, наблюдаемые у людей, с точки зрения затруднения дыхания (отраженного в оценке дыхания) и общего недомогания (отраженного в оценках осанки и движения), демонстрируемого мышами, а также смерти у некоторых здоровых молодых людей из контрольной группы. Обострение симптомов заболевания у коинфицированных мышей, вероятно, является результатом как бактериальной диссеминации в легкие, так и нарушения вирусного клиренса у мышей с пневмококковым носительством23. Ограничением модели является то, что частота клинического заболевания и распространения бактерий из носоглотки варьируется между мышами и зависит от бактериального штамма, возраста хозяина и генотипа21,22,23. Отражая это, для инвазивных штаммов прогрессирование от локализованной инфекции (без обнаруживаемой бактериемии) до смерти может произойти в течение 24 часов. Поэтому для истинной оценки системного распространения бактериемию следует наблюдать через более короткие промежутки времени (каждые 6-12 ч). Аналогичным образом, оценка заболевания может быстро меняться, особенно в первые 72 часа после коинфекции. Поэтому, чтобы внимательно отслеживать симптомы заболевания, рекомендуется контролировать мышей три раза в день в течение 1-3 дней после заражения IAV.
Таким образом, эта модель воспроизводит перемещение S. pneumoniae от бессимптомного колонизатора носоглотки к патогену, способному вызывать легочные и системные заболевания при инфекции IAV. В этой модели IAV запускает переход S. pneumoniae путем изменения поведения бактерий в носоглотке, увеличения распространения бактерий в легкие и изменения антибактериального иммунитета23. Аналогичным образом, бактериальный носитель притупляет противовирусный иммунный ответ и ухудшает клиренс IAV из легких23. Это делает эту модель идеальной для анализа изменений иммунных реакций при единичных и полимикробных инфекциях. Кроме того, течение заболевания после коинфекции частично зависит от штамма пневмококков, присутствующих в носоглотке. Таким образом, модель подходит для анализа бактериальных факторов, необходимых для бессимптомной колонизации, по сравнению с патогенным переходом S. pneumoniae. Наконец, эта модель воспроизводит восприимчивость старения к коинфекциям, и, хотя это не было проверено здесь, ее можно легко использовать для оценки влияния фона хозяина на течение заболевания. В заключение, разделение носительства и заболевания на отдельные этапы дает возможность проанализировать генетические варианты как патогенов, так и хозяина, что позволяет детально изучить взаимодействия важного патобионта с хозяином на разных фазах прогрессирования заболевания. В дальнейшем эта модель может быть использована для адаптации вариантов лечения уязвимых хозяев.
Раскрытие информации
У авторов нет конфликтов интересов, которые необходимо раскрывать.
Благодарности
Мы хотели бы поблагодарить Ника Ленхарда за критическое прочтение и редактирование этой рукописи. Мы также хотели бы поблагодарить Эндрю Камилли и Энтони Кампаньяри за бактериальные штаммы и Брюса Дэвидсона за вирусные штаммы. Эта работа была поддержана грантом Национального института здравоохранения (R21AG071268-01) для J.L. и грантами Национального института здравоохранения (R21AI145370-01A1), (R01AG068568-01A1), (R21AG071268-01) для E.N.B.G.
Материалы
| Name | Company | Catalog Number | Comments |
| 4-Aminobenzoic acid | Fisher | AAA1267318 | Mix I stock |
| 96-well round bottom plates | Greiner Bio-One | 650101 | |
| 100 µm Filters | Fisher | 07-201-432 | |
| Adenine | Fisher | AC147440250 | Mix I stock |
| Avicel | Fisher | 501785325 | Microcyrstalline cellulose |
| BD Cytofix Fixation Buffer | Fisher | BDB554655 | Fixation Buffer |
| BD Fortessa | Flow cytometer | ||
| BD Intramedic Polyethylene Tubing | Fisher | 427410 | Tubing for nasal lavage |
| BD Disposable Syringes with Luer-Lok Tips (1 mL) | Fisher | 14-823-30 | |
| BD Microtainer Capillary Blood Collector and BD Microgard Closure | Fisher | 02-675-185 | Blood collection tubes |
| Beta-Nicotinamide adenine dinucleotide | Fisher | AAJ6233703 | Mix IV stock |
| Biotin | Fisher | AC230090010 | Vitamin stock |
| C57BL/6J mice | The Jackson Laboratory | #000644 | Mice used in this study |
| Calcium Chloride Anhydrous | Fisher Chemical | C77-500 | Mix I stock |
| CD103 BV 421 | BD Bioscience | BDB562771 | Clone: M290 DF 1:200 |
| CD11b APC | Invitrogen | 50-112-9622 | Clone: M1/70, DF 1:300 |
| CD11c PE | BD Bioscience | BDB565592 | Clone: N418 DF 1:200 |
| CD3 AF 488 | BD Bioscience | OB153030 | Clone: 145-2C11 DF 1:200 |
| CD4 V450 | BD Horizon | BDB560470 | Clone: RM4.5 DF 1:300 |
| CD45 APC eF-780 | BD Bioscience | 50-112-9642 | Clone: 30-F11 DF 1:200 |
| CD45 PE | Invitrogen | 50-103-70 | Clone: 30-F11 DF 1:200 |
| CD8α BV 650 | BD Horizon | BDB563234 | Clone: 53-6.7 DF 1:200 |
| Choline chloride | Fisher | AC110290500 | Final supplement to CDM |
| Corning Disposable Vacuum Filter/Storage Systems | Fisher | 09-761-107 | Filter sterilzation apparatus |
| Corning Tissue Culture Treated T-25 Flasks | Fisher | 10-126-9 | |
| Corning Costar Clear Multiple Well Plates | Fisher | 07-201-590 | |
| Corning DMEM With L-Glutamine and 4.5 g/L Glucose; Without Sodium Pyruvate | Fisher | MT10017CM | |
| Cyanocobalamin | Fisher | AC405925000 | Mix I stock |
| D39 | National Collection of Type Culture (NCTC) | NCTC 7466 | Streptococcus pneumoniae strain |
| D-Alanine | Fisher | AAA1023114 | Mix I stock |
| D-Calcium pantothenate | Fisher | AC243301000 | Vitamin stock |
| Dextrose | Fisher Chemical | D16-500 | Starter stock |
| Dnase | Worthington Biochemical | LS002147 | |
| Eagles Minimum Essential Medium | ATCC | 30-2003 | |
| EDTA | VWR | BDH4616-500G | |
| EF3030 | Center for Disease Control and Prevention | Available via the isolate bank request | Streptococcus pneumoniae strain, request using strain name |
| F480 PE Cy7 | BD Bioscience | 50-112-9713 | Clone: BMB DF 1:200 |
| Falcon 50 mL Conical Centrifuge Tubes | Fisher | 14-432-22 | 50 mL round bottom tube |
| Falcon Round-Bottom Polypropylene Test Tubes With Cap | Fisher | 14-959-11B | 15 mL round bottom tube |
| Falcon Round-Bottom Polystyrene Test Tubes (5 mL) | Fisher | 14-959-5 | FACS tubes |
| FBS | Thermofisher | 10437-028 | |
| Ferric Nitrate Nonahydrate | Fisher | I110-100 | Mix III stock |
| Fisherbrand Delicate Dissecting Scissors | Fisher | 08-951-5 | Instruments used for harvest |
| Fisherbrand Disposable Inoculating Loops | Fisher | 22-363-602 | Inoculating loops |
| Fisherbrand Dissecting Tissue Forceps | Fisher | 13-812-38 | Forceps for harvest |
| Fisherbrand Premium Microcentrifuge Tubes: 1.5 mL | Fisher | 05-408-137 | Micocentrifuge tubes |
| Fisherbrand Sterile Syringes for Single Use (10 mL) | Fisher | 14-955-459 | |
| Folic Acid | Fisher | AC216630500 | Vitamin stock |
| Gibco RPMI 1640 (ATCC) | Fisher | A1049101 | |
| Gibco DPBS, no calcium, no magnesium | Fisher | 14190250 | |
| Gibco HBSS, calcium, magnesium, no phenol red | Fisher | 14025134 | |
| Gibco MEM (Temin's modification) (2x), no phenol red | Fisher | 11-935-046 | |
| Gibco Penicillin-Streptomycin (10,000 U/mL) | Fisher | 15-140-122 | |
| Gibco Trypan Blue Solution, 0.4% | Fisher | 15-250-061 | |
| Gibco Trypsin-EDTA (0.25%), phenol red | Fisher | 25-200-056 | |
| Glycerol (Certified ACS) | Fisher | G33-4 | |
| Glycine | Fisher | AA3643530 | Amino acid stock |
| Guanine | Fisher | AAA1202414 | Mix II stock |
| Invitrogen UltraComp eBeads Compensation Beads | Fisher | 50-112-9040 | |
| Iron (II) sulfate heptahydrate | Fisher | AAA1517836 | Mix III stock |
| L-Alanine | Fisher | AAJ6027918 | Amino acid stock |
| L-Arginine | Fisher | AAA1573814 | Amino acid stock |
| L-Asparagine | Fisher | AAB2147322 | Amino acid stock |
| L-Aspartic acid | Fisher | AAA1352022 | Amino acid stock |
| L-Cysteine | Fisher | AAA1043518 | Amino acid stock |
| L-Cysteine hydrochloride monohydrate | Fisher | AAA1038914 | Final supplement to CDM |
| L-Cystine | Fisher | AAA1376218 | Amino acid stock |
| L-Glutamic acid | Fisher | AC156211000 | Amino acid stock |
| L-Glutamine | Fisher | O2956-100 | Amino acid stock |
| L-Histidine | Fisher | AC166150250 | Amino acid stock |
| LIFE TECHNOLOGIES LIVE/DEAD Fixable Blue Dead Cell Stain Kit, for UV excitation | Invitrogen | 50-112-1524 | Clone: N/A DF 1:500 |
| L-Isoleucine | Fisher | AC166170250 | Amino acid stock |
| L-Leucine | Fisher | BP385-100 | Amino acid stock |
| L-Lysine | Fisher | AAJ6222514 | Amino acid stock |
| L-Methionine | Fisher | AAA1031822 | Amino acid stock |
| Low endotoxin BSA | Sigma Aldrich | A1470-10G | |
| L-Phenylalanine | Fisher | AAA1323814 | Amino acid stock |
| L-Proline | Fisher | AAA1019922 | Amino acid stock |
| L-Serine | Fisher | AC132660250 | Amino acid stock |
| L-Threonine | Fisher | AC138930250 | Amino acid stock |
| L-Tryptophan | Fisher | AAA1023014 | Amino acid stock |
| L-Valine | Fisher | AAA1272014 | Amino acid stock |
| Ly6C BV 605 | BD Bioscience | BDB563011 | Clone: AL-21 DF 1:300 |
| Ly6G AF 488 | Biolegend | NC1102120 | Clone: IA8, DF 1:300 |
| Madin-Darby Canine Kidney (MDCK) cells | American Type Culture Collection (ATCC) | CCL-34 | MDCK cell line for PFU analuysis |
| Magnesium Sulfate 7-Hydrate | Fisher | 60-019-68 | CDM starter stock |
| Manganese Sulfate | Fisher | M113-500 | Mix I stock |
| MilQ water | Ultra-pure water | ||
| Mouse Fc Block | BD Bioscience | BDB553142 | Clone: 2.4G2 DF 1:100 |
| MWI VETERINARY PURALUBE VET OINTMENT | Fisher | NC1886507 | Eye lubricant for infection |
| NCI-H292 mucoepidermoid carcinoma cell line | ATCC | CRL-1848 | H292 lung epithelial cell line for biofilm growth |
| Niacinamide | Fisher | 18-604-792 | Vitamin stock |
| NK 1.1 AF 700 | BD Bioscience | 50-112-4692 | Clone: PK136 DF 1:200 |
| Oxyrase For Broth 50Ml Bottle 1/Pk | Fisher | 50-200-5299 | To remove oxygen from liquid cultures |
| Paraformaldehyde 4% in PBS | Thermoscientific | J19932-K2 | |
| Pivetal Isoflurane | Patterson Veterinary | 07-893-8440 | Isoflurane for anesthesia during infection |
| Potassium Phosphate Dibasic | Fisher Chemical | P288-500 | Starter stock |
| Potassium Phosphate Monobasic | Fisher Chemical | P285-500 | Starter stock |
| Pyridoxal hydrochloride | Fisher | AC352710250 | Vitamin stock |
| Pyridoxamine dihydrochloride | Fisher | AAJ6267906 | Mix I stock |
| Riboflavin | Fisher | AC132350250 | Vitamin stock |
| Sodium Acetate | VWR | 0530-500G | Starter stock |
| Sodium Azide | Fisher Bioreagents | BP922I-500 | For FACS buffer |
| Sodium Bicarbonate | Fisher Chemical | S233-500 | Starter stock and final supplement to CDM |
| Sodium Phosphate Dibasic | Fisher Chemical | S374-500 | Starter stock |
| Sodium Phosphate Monobasic | Fisher Chemical | S369-500 | Starter stock |
| TCR APC | BD Bioscience | 50-112-8889 | Clone: GL-3 DF 1:200 |
| TCRβ APC-Cy7 | BD Pharmigen | BDB560656 | Clone: H57-597 DF 1:200 |
| Thermo Scientific Blood Agar with Gentamicin | Fisher | R01227 | Blood agar plates with the antibiotic gentamicin |
| Thermo Scientific Trypsin, TPCK Treated | Fisher | PI20233 | |
| Thiamine hydrochloride | Fisher | AC148991000 | Vitamin stock |
| TIGR4 | ATCC | BAA-334 | Streptococcus pneumoniae strain |
| Uracil | Fisher | AC157300250 | Mix II stock |
| Worthington Biochemical Corporation Collagenase, Type 2, 1 g | Fisher | NC9693955 |
Ссылки
- Kadioglu, A., Weiser, J. N., Paton, J. C., Andrew, P. W. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nature Reviews Microbiology. 6 (4), 288-301 (2008).
- Obaro, S., Adegbola, R. The pneumococcus: Carriage, disease and conjugate vaccines. Journal of Medical Microbiology. 51 (2), 98-104 (2002).
- Chong, C. P., Street, P. R. Pneumonia in the elderly: A review of the epidemiology, pathogenesis, microbiology, and clinical features. Southern Medical Journal. 101 (11), 1141-1145 (2008).
- Kadioglu, A., Andrew, P. W. Susceptibility and resistance to pneumococcal disease in mice. Briefings in Functional Genomics and Proteomics. 4 (3), 241-247 (2005).
- Ganie, F., et al. Structural, genetic, and serological elucidation of Streptococcus pneumoniae serogroup 24 serotypes: Discovery of a new serotype, 24C, with a variable capsule structure. Journal of Clinical Microbiology. 59 (7), 0054021 (2021).
- Centers for Disease Control and Prevention. Estimates of deaths associated with seasonal influenza --- United States. MMWR. Morbidity and Mortality Weekly Report. 59 (33), 1057-1062 (2010).
- Shrestha, S., et al. Identifying the interaction between influenza and pneumococcal pneumonia using incidence data. Science Translational Medicine. 5 (191), (2013).
- McCullers, J. A. Insights into the interaction between influenza virus and pneumococcus. Clinical Microbiology Reviews. 19 (3), 571-582 (2006).
- Pneumococcal Disease Global Pneumococcal Disease and Vaccine. Centers for Disease Control and Prevention Available from: https://www.cdc.gov/pneumococcal/global.html (2018)
- Grudzinska, F. S., et al. Neutrophils in community-acquired pneumonia: Parallels in dysfunction at the extremes of age. Thorax. 75 (2), 164-171 (2020).
- Boe, D. M., Boule, L. A., Kovacs, E. J. Innate immune responses in the ageing lung. Clinical and Experimental Immunology. 187 (1), 16-25 (2017).
- Krone, C. L., van de Groep, K., Trzcinski, K., Sanders, E. A., Bogaert, D. Immunosenescence and pneumococcal disease: An imbalance in host-pathogen interactions. The Lancet Respiratory Medicine. 2 (2), 141-153 (2014).
- Cho, S. J., et al. Decreased NLRP3 inflammasome expression in aged lung may contribute to increased susceptibility to secondary Streptococcus pneumoniae infection. Experimental Gerontology. 105, 40-46 (2018).
- Disease Burden of Influenza. Centers for Disease Control and Prevention Available from: https://www.cdc.gov/flu/about/burden/index.html (2018)
- McCullers, J. A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nature Reviews Microbiology. 12 (4), 252-262 (2014).
- McCullers, J. A., Rehg, J. E. Lethal synergism between influenza virus and Streptococcus pneumoniae: Characterization of a mouse model and the role of platelet-activating factor receptor. The Journal of Infectious Diseases. 186 (3), 341-350 (2002).
- Metzger, D. W., Sun, K. Immune dysfunction and bacterial coinfections following influenza. Journal of Immunology. 191 (5), 2047-2052 (2013).
- Chao, Y., Marks, L. R., Pettigrew, M. M., Hakansson, A. P. Streptococcus pneumoniae biofilm formation and dispersion during colonization and disease. Frontiers in Cellular and Infection Microbiology. 4, 194 (2014).
- Bogaert, D., De Groot, R., Hermans, P. W. Streptococcus pneumoniae colonisation: The key to pneumococcal disease. The Lancet Infectious Diseases. 4 (3), 144-154 (2004).
- Simell, B., et al. The fundamental link between pneumococcal carriage and disease. Expert Review of Vaccines. 11 (7), 841-855 (2012).
- Marks, L. R., Davidson, B. A., Knight, P. R., Hakansson, A. P. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio. 4 (4), 00438 (2013).
- Reddinger, R. M., Luke-Marshall, N. R., Sauberan, S. L., Hakansson, A. P., Campagnari, A. A. Streptococcus pneumoniae modulates Staphylococcus aureus biofilm dispersion and the transition from colonization to invasive disease. mBio. 9 (1), 02089 (2018).
- Joma, B. H., et al. A murine model for enhancement of Streptococcus pneumoniae pathogenicity upon viral infection and advanced age. Infection and Immunity. 89 (8), 0047120 (2021).
- Andersson, B., et al. Identification of an active disaccharide unit of a glycoconjugate receptor for pneumococci attaching to human pharyngeal epithelial cells. Journal of Experimental Medicine. 158 (2), 559-570 (1983).
- Avery, O. T., Macleod, C. M., McCarty, M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: Induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. The Journal of Experimental Medicine. 79 (2), 137-158 (1944).
- Tettelin, H., et al. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. 293 (5529), 498-506 (2001).
- Tothpal, A., Desobry, K., Joshi, S. S., Wyllie, A. L., Weinberger, D. M. Variation of growth characteristics of pneumococcus with environmental conditions. BMC Microbiology. 19 (1), 304 (2019).
- Bou Ghanem, E. N., et al. Extracellular adenosine protects against Streptococcus pneumoniae lung infection by regulating pulmonary neutrophil recruitment. PLoS Pathogens. 11 (8), 1005126 (2015).
- Bou Ghanem, E. N., et al. The alpha-tocopherol form of vitamin E boosts elastase activity of human PMNs and their ability to kill Streptococcus pneumoniae. Frontiers in Cellular and Infection Microbiology. 7, 161 (2017).
- Tait, A. R., Davidson, B. A., Johnson, K. J., Remick, D. G., Knight, P. R. Halothane inhibits the intraalveolar recruitment of neutrophils, lymphocytes, and macrophages in response to influenza virus infection in mice. Anesthesia & Analgesia. 76 (5), 1106-1113 (1993).
- Aaberge, I. S., Eng, J., Lermark, G., Lovik, M. Virulence of Streptococcus pneumoniae in mice: A standardized method for preparation and frozen storage of the experimental bacterial inoculum. Microbial Pathogenesis. 18 (2), 141-152 (1995).
- McCullers, J. A., Bartmess, K. C. Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. The Journal of Infectious Diseases. 187 (6), 1000-1009 (2003).
- Smith, A. M., McCullers, J. A. Secondary bacterial infections in influenza virus infection pathogenesis. Current Topics in Microbiology and Immunology. 385, 327-356 (2014).
- Cundell, D. R., Gerard, N. P., Gerard, C., Idanpaan-Heikkila, I., Tuomanen, E. I. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. 377 (6548), 435-438 (1995).
- Ballinger, M. N., Standiford, T. J. Postinfluenza bacterial pneumonia: Host defenses gone awry. Journal of Interferon & Cytokine Research. 30 (9), 643-652 (2010).
- Sun, K., Metzger, D. W. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nature Medicine. 14 (5), 558-564 (2008).
- Nakamura, S., Davis, K. M., Weiser, J. N. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. Journal of Clinical Investigation. 121 (9), 3657-3665 (2011).
- Blanchette-Cain, K., et al. Streptococcus pneumoniae biofilm formation is strain dependent, multifactorial, and associated with reduced invasiveness and immunoreactivity during colonization. mBio. 4 (5), 00745 (2013).
- Rello, J., Pop-Vicas, A. Clinical review: Primary influenza viral pneumonia. Critical Care. 13 (6), 235 (2009).
- Torres, A., Loeches, I. M., Sligl, W., Lee, N. Severe flu management: A point of view. Intensive Care Medicine. 46 (2), 153-162 (2020).
- Bakaletz, L. O. Viral-bacterial co-infections in the respiratory tract. Current Opinion in Microbiology. 35, 30-35 (2017).
- Palacios, G., et al. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS One. 4 (12), 8540 (2009).
- Dhanoa, A., Fang, N. C., Hassan, S. S., Kaniappan, P., Rajasekaram, G. Epidemiology and clinical characteristics of hospitalized patients with pandemic influenza A (H1N1) 2009 infections: The effects of bacterial coinfection. Virology Journal. 8, 501 (2011).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены