
Internal Standards
Обзор
Source: Laboratory of Dr. B. Jill Venton - University of Virginia
The goal of many chemical analyses is a quantitative analysis, where the amount of a substance in a sample is determined. In order to accurately calculate the concentration of an unknown from a sample, careful sample preparation is key. Every time a sample is handled or transferred, some of the sample can be lost. There are strategies however, for minimizing sample loss. There are also strategies for coping with sample loss and still making accurate measurements of concentration.
To minimize sample loss, the ideal is to minimize the number of sample handling and transfer steps. For example, massing a solid sample directly into a flask that a solution will be made in reduces a transfer step. If it's necessary to transfer from one flask to another and a dilution is being made, then triple rinsing the glassware helps ensure all the sample is transferred. Other strategies are more specific to the sample. For example, samples that adsorb to glass, such as proteins, might better be handled in polypropylene disposable tubes. The tubes are not hydrophilic, so if a small amount of sample is to be pipetted in water, it is best to have already added the water to the tube, so the sample can be pipetted directly into the solvent. It may be better to concentrate, rather than completely dry a sample, due to losses from insolubilities after rehydration.
Another source of sample loss is through incomplete sample manipulations. For example, if a derivatization procedure is used and the derivatization is incomplete, then the full amount of sample is not observed. Errors such as this are systematic errors, and can be solved by correcting the problem, such as changing the derivatization procedure. Another cause of systematic error in measurements is matrix effects. These can interfere with measurement of certain substances and performing calibrations in the same matrix as the sample can reduce this effect.
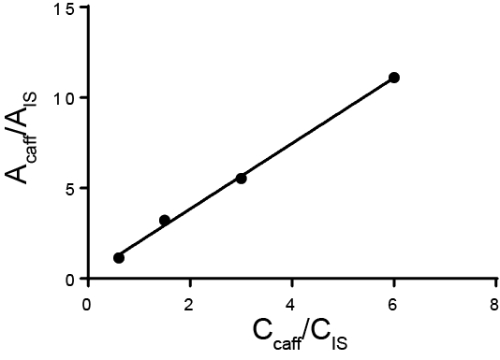
Quantitative analysis is typically carried out using either external or internal standards. For external standards, a calibration curve is made by measuring different known concentrations of the analyte of interest. Then, the sample is run separately from the standard. For internal standards, the standard is in the same sample as the analyte of interest, allowing the measurement to be taken simultaneously. Typically, a different species is added called the internal standard and the ratio of the response for that internal standard and the analyte is calculated. The idea is that the ratio of the response, called the response factor, is proportional to their concentration. While the method must be able to distinguish between the analyte of interest and the internal standard, any sample losses that occur after the internal standard is added should be similar for both substances and thus the ratio of the response stays the same. A special case of using internal standards is the method of standard additions, where increasing amounts of the analyte is added to the solution and the original amount of analyte is back-calculated. Internal standards can be used in chromatography, electrochemistry, and spectroscopy.
Процедура
1. Proper Sample Handling: Making a Solution
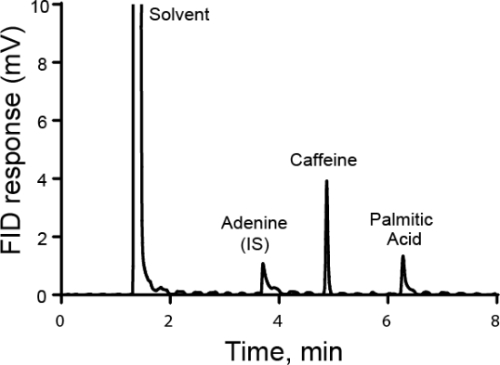
- Take a clean beaker and mass the correct amount of sample into it. Record the actual mass used. In this example, a solution of adenine is made in a volumetric flask for use as an internal standard for the next analysis. The mass of adenine is 100 mg. Do not directly mass into a volumetric flask because it has a long neck and the adenine cannot be easily added or removed.
- Add roughly 25 mL of solvent (in this case dimethyl sulfoxide (DMSO)) to the beaker a
Заявка и Краткое содержание
Internal standards are used in many fields, including spectroscopy and chromatography. In spectroscopy, internal standards can help correct for random errors due to changes in light source intensity. If a lamp or other light source has variable power, it will affect the absorption and consequently, emission of a sample. However, the ratio of an internal standard to analyte will stay constant, even if the light source does not. One example of this is using lithium (Li) as an internal standard for the analysis of sodium in
Теги
Перейти к...
Видео из этой коллекции:

Now Playing
Internal Standards
Analytical Chemistry
205.4K Просмотры

Sample Preparation for Analytical Characterization
Analytical Chemistry
85.3K Просмотры

Method of Standard Addition
Analytical Chemistry
320.8K Просмотры

Calibration Curves
Analytical Chemistry
798.7K Просмотры

Ultraviolet-Visible (UV-Vis) Spectroscopy
Analytical Chemistry
625.3K Просмотры

Raman Spectroscopy for Chemical Analysis
Analytical Chemistry
51.4K Просмотры

X-ray Fluorescence (XRF)
Analytical Chemistry
25.9K Просмотры

Gas Chromatography (GC) with Flame-Ionization Detection
Analytical Chemistry
283.1K Просмотры

High-Performance Liquid Chromatography (HPLC)
Analytical Chemistry
386.0K Просмотры

Ion-Exchange Chromatography
Analytical Chemistry
265.1K Просмотры

Capillary Electrophoresis (CE)
Analytical Chemistry
94.5K Просмотры

Introduction to Mass Spectrometry
Analytical Chemistry
112.9K Просмотры

Scanning Electron Microscopy (SEM)
Analytical Chemistry
87.6K Просмотры

Electrochemical Measurements of Supported Catalysts Using a Potentiostat/Galvanostat
Analytical Chemistry
51.8K Просмотры

Cyclic Voltammetry (CV)
Analytical Chemistry
126.0K Просмотры
Авторские права © 2025 MyJoVE Corporation. Все права защищены