
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
宿主細胞と嫌気性菌の相互作用を評価するためのモデル生物としてポルフィロモナス・ジンジバリス
要約
This article presents two protocols: one to measure anaerobic bacteria that can successfully invade and survive within the host, and the other to visualize anaerobic bacteria interacting with host cells. This study can be applied to any cultivable anaerobe and any eukaryotic cell type.
要約
嫌気性細菌は、これまで、このような腸、口、膣などの多くのヒトのニッチで好気性菌数を上回ります。また、嫌気性感染は一般的であり、しばしば先住民起源のものです。ヒトの細胞に侵入するためにいくつかの嫌気性病原体の能力は、彼らに先天性免疫を逃れるためだけでなく、宿主細胞の挙動を調節するために、適応策を提供します。しかし、嫌気性細菌は、イベントの実験的調査中に生きていることを確保することが課題を提起することができる。 ポルフィロモナス・ジンジバリス 、グラム陰性嫌気性菌は、真核生物の非食細胞の多様な侵入が可能です。この記事では、成功した文化とは、Pの能力を評価する方法について説明ジンジバリスは、ヒト臍帯静脈内皮細胞(HUVEC)を浸潤します。宿主細胞と相互作用する細菌を可視化するために成功裏に侵入し、宿主内で生存することができる細菌を測定するためのものを、その他:2つのプロトコルが開発されました。これらの技術はanaeの使用を必要とP.を供給する robic室最適な成長のための嫌気性環境でジンジバリス 。
第一のプロトコルは、主に細菌による宿主細胞の浸潤を研究するために使用される抗生物質保護アッセイに基づいています。しかし、抗生物質保護アッセイは限られています。抗生物質処置および宿主細胞の溶解後に培養可能である唯一の細胞内細菌が測定されます。生と死の両方の宿主細胞と相互作用するすべての細菌を評価するために、我々は、宿主 - 病原体相互作用を調べるために、蛍光顕微鏡を使用するプロトコルを開発しました。細菌は蛍光2 '、7'-ビス - (2-カルボキシエチル)-5-(および-6) - カルボキシフルオレセインアセトキシメチルエステル(BCECF-AM)で標識し、嫌気性条件下で真核細胞を感染させるために使用されます。 0.2%トリトンX-100で透過処理し、パラホルムアルデヒドで固定した後、宿主細胞は、それぞれ、細胞骨格と細胞核を標識するためのTRITCファロイジンおよびDAPIで標識されます。複数のIMA異なる焦点(Zスタック)で撮影されたGESは、細菌の時空間視覚化のために得られます。本研究で使用した方法は、任意の培養可能嫌気性菌及び任意の真核細胞型に適用することができます。
概要
嫌気性細菌は、人間の体のほぼすべての面にコロニーを形成します。酸素濃度が低い腸管および尿生殖路の細菌叢において優勢が、それらはまた、皮膚、口、鼻、喉1上に高レベルで存在します。嫌気性細菌は、内因性感染症の一般的な原因であり、病気のサイトから頻繁に分離されています。しかし、その気難しい性質上、嫌気性菌が分離し、培養することは困難です。嫌気性細菌が関与する研究が制限された条件の下で行われなければなりません。現代嫌気培養技術は、研究者は、多くの嫌気性実験室株、あるいは臨床分離2,3を研究するために必要な嫌気性の設定を模倣することができます。
病原性嫌気性細菌は、それらが存在する宿主細胞とのダイナミックな関係と共進化を開発しました。ほとんどの嫌気性菌はinfectiに到達する前に宿主免疫応答による殺傷に影響されやすいです組織単位(OU)レベル。しかし、いくつかの病原性細菌から逃れるか、宿主の免疫応答を破壊するメカニズムを開発しました。彼らは、 ポルフィロモナス・ジンジバリス 、両方の経口に関与し、グラム陰性嫌気性菌。4に信号を送るような免疫認識の回避、免疫メディエーターの中和、細胞性免疫の変化、宿主細胞の浸潤、および免疫の変更などのメカニズムを通じてこの目標を達成し、口外疾患、 ホスト5-7の病原性の変化を引き起こすことができる非常に適合細菌性病原体の一例です。
歯と歯肉粘膜組織との間に形成された深い裂け目に計上バイオフィルムプラークのポケットには、大気中の酸素8から保護されている嫌気性細菌を抱くことができます。これらの歯周ポケットは、P.など、様々な嫌気性病原体のためのニッチとして機能ジンジバリス 9。P.ジンジバリス改造が可能であるキーストン病原体であります歯周病10の発症と進行を促進する方法で、口腔微生物のコミュニティをる。これは、宿主タンパク質の広いスペクトルに対して活性である毒性因子を大量に生成し、宿主防御の回避11ためのメカニズムを提供します。また、上皮細胞、内皮細胞、線維芽細胞、 および in vitro および in vivo 12-14 15 で歯根膜細胞に侵入することが可能です。効果的に宿主細胞に侵入することにより、P。ジンジバリスは、宿主の免疫を逃れることができます。宿主細胞の効果的な侵入だけでなく、細菌が宿主防御をエスケープすることを可能にするだけでなく、将来の再感染するためのリザーバとして働くだけでなく、宿主細胞を変化させます。宿主細胞が細菌の接着および内在化に関与する分子機構の研究が必要とされています。いくつかの研究室での研究は 、Pの内部に関連する分子事象を理解することに焦点を当てています宿主細胞によってジンジバリス同様に抑制し、免疫応答をハイジャックし、敵対的な宿主防御機構を生き残るために使用されるメカニズムとして。
宿主細胞に侵入することができる病原体を同定し、特徴付けることが可能な多くのアッセイがあります。これは、酸素の不存在下でかさばる器具に依存して研究を行うことが困難であるためしかし、嫌気性病原体によるインビトロ研究は、主に研究者のために多くの実験的な問題を引き起こします。これは、真核細胞が増殖するために酸素を必要とし、従って、組織培養インキュベーター中で別途用意しなければならないという事実によって悪化します。このような障害物を回避する一つの方法は、大気中の酸素の下で研究を行うことであろうが、それは、嫌気性細菌の増殖を不可能にするであろう。別の方法は、感染して宿主細胞の相互作用を研究するために加熱殺菌細菌を使用することであろう。しかし、違いは、ホスト病原体interactiの関連性を低下させる熱殺菌し、生菌の間に存在します16に。これは、宿主細胞との相互作用変更のない表現で生菌を研究するために中央です。 Pを培養するため、方法嫌気性環境におけるジンジバリスが与えられます 。また、2つの簡単なコスト効率の高いプロトコルがPの能力を評価するための実証されヒト臍帯静脈内皮細胞(HUVEC)によって内在するジンジバリス 。最初のプロトコルは、人気のある抗生物質保護アッセイに基づいています。アッセイは簡単であるが、嫌気性微生物を用いた検討事項が挙げられます。第二プロトコルは、相互作用可視化する蛍光顕微鏡の使用を必要とし、Pを内在化ジンジバリス 。各アッセイは、制限および嫌気性細菌の侵襲性を研究するための研究者に概要を提供するために説明される利点を有します。現在の原稿は 、P を研究しているがジンジバリス及びHUVECを、これらのプロトコルは、他の多くの嫌気性細菌のために使用することができます宿主細胞の他のタイプのように。
プロトコル
以下のプロトコルは、Pを嫌気性種による侵入を培養し、研究するための方法を説明しますジンジバリス ;しかしながら、これらのプロトコルは、嫌気性病原体の数のために使用することができます。たHUVECを用いているが、このプロトコルは、免疫および非免疫の両方の他の真核細胞のために使用することができます。
1.嫌気性チャンバーを使用するとメンテナンス
注意:ポルフィロモナス・ジンジバリスは、周囲の空気中に遭遇酸素の正常なレベルに敏感嫌気性菌です。制御嫌気性環境は、Pの栽培のために不可欠ですジンジバリス 。
- ここで、(80%N 2、10%H 2、10%のCO 2)ビニール嫌気性チャンバー( 図1A)内の混合嫌気性ガスとして指定された人工的な雰囲気を維持します。嫌気性チャンバーに実験室環境から項目を転送するためのエアロック( 図1B)を使用します。エアロックは、手動でTWを運営混合嫌気性ガスを導入する前に、N 2ガ スと氷のパージ。
- 望ましくない硫化水素のメンテナンスフリーを除去するための硫化水素除去カラム( 図1C)を使用します。触媒によって作成されたH 2 Oを除去し、汚染の拡大を促進するエアロゾルを回避するために、チャンバ内の除湿器を置きます。
注:硫化水素は、多くの嫌気性細菌の天然の代謝副産物であり、その蓄積は細菌に有毒であり、電子機器に損傷を与えると、触媒の寿命を低下させることができます。 - 水素の存在下( 図1D)中の酸素を除去し、パラジウム触媒、を介してチャンバの雰囲気を循環させるためにファンボックスを使用してください。
注:再循環の大気(HEPA)フィルターは0.22ミクロン以上の大きさで空気中の汚染物質を除去します。 - 嫌気性の内側に位置する37℃のインキュベーターで培養嫌気性細菌室。嫌気性チャンバー内で作業するときは、標準的な無菌技術を使用してください。

図1.嫌気性ビニル室およびその構成要素(A)大気中の酸素から完全に密封されたビニール嫌気性チャンバーは、一度に(32×78でに)二人の個人のためのワークスペースを提供します。これは、(バック中央)37℃に設定したインキュベーターが含まれています。 (B)は、嫌気性チャンバーエアロックのラボ環境からのアイテムの転送に使用されます。 。写真は自動的に嫌気性環境を作成するために必要な真空及びパージ手順を実行するようにプログラムすることができるコントローラを介して操作する自動エアロックです。 (C)硫化水素除去カラムは、望ましくない硫化水素のメンテナンスフリーの高容量の除去を提供します。 (D)2つの触媒ファンボックスであります水素の存在下で、酸素を除去し、パラジウム触媒を介してチャンバの雰囲気を循環を助けるために嫌気性チャンバー全体に置きました。嫌気性チャンバーは、製造元の指示に従って設定されている。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
嫌気性菌の調製
注:P。ジンジバリスは耐気で、好気的条件で保存することができますが、それは6%17,18よりも高いレベルで酸素の存在下で成長しません。嫌気性チャンバーは、Pの適切な培養のために必要ですジンジバリスおよびその他の嫌気性の種( 図1)。嫌気性チャンバーの使用に関する適切な研修や教育がmicroanaerobes 19を使用する前に必要とされます。
- 嫌気コンディットにすべての液体培地、プレートを平衡化残留酸素を除去するために、実験前に少なくとも12時間のイオン。
- Pを転送嫌気性チャンバーに-80℃の冷凍庫からジンジバリスは 、解凍してみましょう。
- ストリークP.トリプチケースソイ血液寒天プレート上ジンジバリス (5%ヒツジ血液とTSA II)。 4-7日間嫌気培養器中で37℃でパラフィルムや店舗でプレートを包みます。
- Pを接種3ミリリットルブレインハートインフュージョンにジンジバリスは (BHI)培養液は、滅菌ループを使用して、ヘミンおよびメナジオン、嫌気性および選好性微生物の単離および培養のために濃縮された非選択液体培地を補充しました。
注意:長期保存のため、-80℃の冷凍庫にグリセロールまたはDMSO(10〜20%の最終濃度)と場所をBHIで製造した細菌培養物を混ぜます。 - Pのスターターカルチャーを準備1:10希釈を作り、細菌が対数中期まで増殖させることによってジンジバリス 。
注:BACの光学密度をterial懸濁液が決定され、検査されるべき各株のための細菌濃度が調整されています。 Pのジンジバリス対数中期および〜7×10 8細胞/ mlに相当する0.7のOD 660でサスペンション。上記プロトコルに記載の成長条件は 、Pに対して特異的ですジンジバリスおよび他の細菌株に適合させることが必要になる場合があります。
3.内皮細胞の培養
注:購入は、製造業者の説明書に従って、5%CO 2中、37℃で、血管内皮増殖因子(VEGF)を含む基本培地中での一次HUVECを培養をプールしました。
- 15ミリリットルのVEGF培地中に2.5×10 5細胞 /フラスコでT-75フラスコ内の種子のHUVEC。
注:4.0%トリパンブルーで1希釈:1を介して実行可能性を確認してください。双眼光顕微鏡下で見たときに、トリパンブルーを保持します妥協膜を有する細胞は、無傷の膜を有する健康な細胞は、白い表示されます。 COUN100 T細胞は、細胞の80%が生存し20であることを確認してください。 - 細胞は〜80%の集密度に達するまで予め温めておいた新鮮なVEGF培地で2日ごとにメディアを交換してください。
- 予め温めておいたPBSで細胞を1回洗浄します。 2 mlのトリプシン中和溶液、続いて5分間、2 mlのトリプシン-EDTA(0.25%)を用いてインキュベートすることにより、T75フラスコから細胞を遊離させます。
- 50mlコニカルチューブ中の浮遊たHUVECを収集します。 PBSでT-75フラスコから余分な細胞を洗浄し、50mlコニカルチューブに移します。
- 10分間200×gで遠心細胞。
- 上清を除去し、予め温めておいた10ミリリットル中のVEGFメディアを細胞ペレットを中断。
- 血球計または類似の細胞計数装置を用いて細胞濃度を決定します。
- カバースリップで6ウェルプレート(40万/ウェル)または12ウェルプレートのいずれかに追加する細胞懸濁液の量を計算し(50,000 /ウェル)。 HUVECを、次の日の実験のための準備が整います。
4.生存アッセイ侵攻/インタラクション(メッキ)
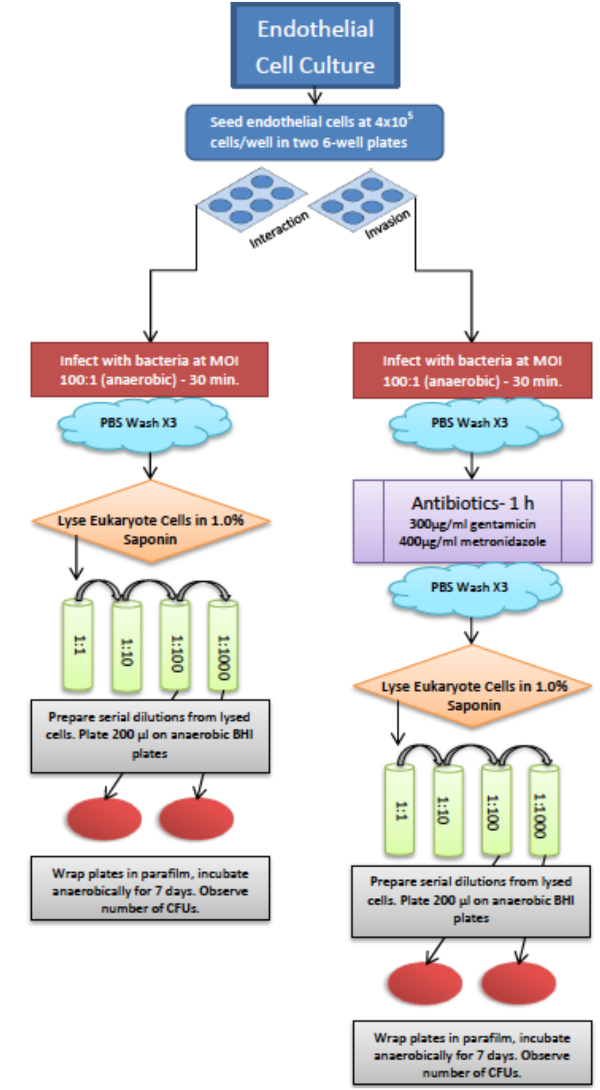
注:このアッセイを行う場合、/ウェル40万細胞で播種した内皮細胞の2つの6ウェルプレートを準備します。一方のプレートは、細菌に取り付けられ、宿主細胞によって内在化を評価するために使用されます。他方のプレートは、細胞内細菌を占めることになります。 6ウェルプレートの1つの実験において実行される2つのサンプルの三連を可能にします。このプロトコルの概要については生存アッセイのフローチャート( 図2)を参照してください。
- 彼らは中期対数増殖(OD 660 0.5から0.7)に達するまで(セクション1を参照)は、上述したように、嫌気性細菌を準備します。
- 10分間5000×gで遠心分離菌。
注:遠心外部嫌気性チャンバーである場合に、密封15mlチューブ中の細菌試料を運ぶ、酸素の漏れを防止するためにパラフィルムで蓋を包みます。 - ペレット化したPを配置しますバックチャンバー内ジンジバリス 、上清を捨てます。 VEGF私の中で再懸濁する前に再度、PBSでペレット細菌を洗浄DIA。すべての菌株は対数中期(〜7×10 8細胞/ ml)に相当する0.7のOD 660でテストされるために懸濁液を準備します。細菌は現在、感染のための準備が整いました。
- 嫌気性チャンバー内に組織培養インキュベーターからのHUVECを含むトランスファー6ウェルプレート。メディアを削除し、嫌気性PBSで3回洗浄します。各ウェルに嫌気VEGFメディアの2ミリリットルを追加し、感染の温度を平衡化するために20分間嫌気培養器で37℃でプレートを配置します。
注意:感染のために使用されるものを確実にするために、血液寒天プレート上にプレート細菌が均一で感染時に汚染されていません。 - 1:100の:感染の多重度(MOIホスト細菌)で細菌を宿主細胞に感染。
注:HUVEC細胞の数は、感染の前に単一のウェルにトリパン排除試験を実施することによって決定されます。細菌細胞の数を光学密度(0.5 = 5×10 8細胞/ mlの、例えば、OD)を介して決定されます。 Bacterial濃度は、HUVECを濃度21に基づいて、適切なMOIに調整されます。 - 嫌気性インキュベーターに感染したHUVECで6ウェルプレートを置き、細菌は30分間の宿主細胞と相互作用することを可能にします。
- 嫌気性チャンバー内BHI(w / vの1.0%)でサポニンを準備し、0.2μmのフィルターでろ過します。
- 両方が結合し、内在化細菌の生存。
- 、インキュベーター、吸引メディアからプレートを取り外し嫌気PBSで3回洗浄し、2ml(ステップ4.8に記載のように調製)1.0%のサポニンを濾過加えます。宿主細胞の溶解を可能にするために15分間インキュベートします。
- セルスクレーパーで各ウェルの底をこすり。各ウェルからの細胞混合物を収集し、1にする:BHIで1希釈。
- サンプルの連続希釈液を作るために進んでください。細菌種および濃度に応じて、連続希釈液を調整します。 100または1:1,000希釈液1で開始します。
- 血液寒天プレート上の所望の希釈液200μlをプレート。ラップトン37℃の嫌気性インキュベーターでパラフィルムと場所で彼のプレート。
- 37℃でのインキュベーションの7日後、プレートを取り外し、手動でコロニーをカウントするライトボックスを使用してコロニー形成単位(CFUの)を数えます。
注:のCFUが列挙されています。 CFUの大量のため、画像を撮影することができ、コンピュータソフトウェアは、のCFUの計数を容易にするために使用することができます。
- 内在化細菌の生存。
- インキュベーターからプレートを外します。嫌気性PBSで3回洗浄し、補足し、抗生物質(ゲンタマイシン300μgの/ mlのメトロニダゾール400μgの/ mlの)で2 mlのVEGFのメディアを追加。
- 1時間インキュベートします。それらが所望の細菌株を殺すで100%効果的であるように、抗生物質をテストし、それらが宿主細胞22,23を貫通していないことを確認するようにしてください。
- 培地を吸引は、フィルター1.0%サポニンの2ミリリットルを追加します。宿主細胞の溶解を可能にするために15分間インキュベートします。
- 各WELの底をこすりセルスクレーパーでリットル。各ウェルから細胞混合物を収集し、1にする:BHIで1希釈。
- (:100、1:1,000 1)試料の連続希釈液を調製します。
- 血液寒天プレート上の所望の希釈液200μlをプレート。嫌気性インキュベーターにパラフィルム、所定の位置にプレートを包みます。
- 37℃でのインキュベーションの7日後、プレートを取り外して、CFUの数をカウントC。

図2.真核細胞での嫌気性細菌の生存のために使用されるプロトコルの概略図。全細菌の生存および内在化細菌の生存のための両方のアッセイを同時に行うことができます。 この図の拡大版をご覧になるにはこちらをクリックしてください。
{kind=link}
ホストのCeへの細菌の5内在LLS(蛍光顕微鏡)
注:P。ジンジバリスは -カルボキシフルオレセイン、アセトキシメチルエステル(BCECF-AM)、2 'で標識7'-ビス- (2-カルボキシエチル)-5-(および-6)されます。 BCECF-AMは非蛍光膜透過性色素です。細胞内エステラーゼの作用を介してフルオレセインBCECFへの変換は、細胞の生存率を示すことができる。P.をジンジバリスは、BCECF-AM色素で標識した後、真核生物細胞を感染させるために使用されます。感染後、細胞を固定し、DAPIおよびTRITCファロイジンで標識しました。真核細胞の核を染色するために使用DAPI染色はまた、代謝的に切断するのBCECF-AMできない非生存細菌を同定するための対策を提供する細菌の細胞核を標識します。宿主細胞はTRITCファロイジン、赤アクチン染料で強調表示されます。
- オートクレーブカバースリップ。無菌/ウェル5×10 4個の細胞で内皮細胞を播種する前に、12ウェルプレートにカバースリップを追加します。 (実験前に準備日)
- は上記のように12ウェルプレートに18ミリメートル(#1.5厚さ)の円形カバーガラス上で作成した内皮細胞を持っています。
- セクション1で説明したように対数期中期(OD 660 = 0.5〜0.7)まで増殖させ、嫌気性細菌を準備します。
- ウォッシュ細菌は5,000×gで遠心分離し、5-7×10 8細胞/ mlでPBSにペレットを懸濁させることによって、嫌気性PBSで2倍。
- 2μmのBCECF-AMの最終濃度になるように細菌懸濁液(5-7×10 8細胞/ ml)の2 mlに0.2 mMのBCECF-AMの20μLを加えます。
- 暗所で30分間37℃でインキュベートします。
- 内皮細胞での転写プレートを嫌気性チャンバー内に18ミリ(#1.5厚さ)の組織培養インキュベーターから円形のカバースリップ上に播種しました。 PBSおよび嫌気性のVEGFメディアとの交換で洗浄します。
注:HUVECを、光顕微鏡下で健康であることを確認します。 HUVECSは形態がメーカーに匹敵する必要があり、〜80%コンフルエントにする必要があります。 - で遠心ラベル細菌残留BCECF-AM色素を除去するために10分間、5,000×gで。 2ミリリットル嫌気VEGF培地に懸濁します。
- 1(細菌:ホスト)100のMOIで標識細菌を宿主細胞に感染。
- 30分間、37℃で嫌気性チャンバー内でインキュベートします。
- そしてPBSで3回感染洗浄後、細胞を10分間、新たに調製した4.0%パラホルムアルデヒド中で固定します。
注:細胞を固定した後、実験を嫌気性チャンバーの外で行うことができます。 - 洗浄は、PBSで3回カバースリップ。
- 10分間、0.2%トリトンX-100の1ミリリットルを追加します。
- 洗浄は、PBSで3回カバースリップ。
- 45分間カバーガラスにTRITCファロイジン(50μg/ mlの)の50μLを追加します。
- 洗浄は3回カバースリップ、DAPIを含むソフトセット封入剤でスライド上で12ウェルプレートと場所から削除します。マニキュアと側面をシール。
注意:スライドは、暗所で数ヶ月のために保存することができます。光退色を防止するために、光の露出を避けてください。 - 表示スライド共焦点顕微鏡を使用して。
- ここで、(反転)AxioObserver周りに構成された34チャンネル分光システム(32チャネルアレイ検出器と両サイドのPMT検出器に加えて、透過光検出器)、電動XYステージとスタンドを使用しています。青色ダイオード(405 nm)で、マルチラインアルゴン(458、488、514 nm)で、緑ダイオード(561 nm)で、赤のヘリウムネオン(633 nm)を440 nmのパルスレーザー:システムは、5つのレーザーを持っています。 (FRET-FLIM用)2ハイブリッドのGaAsP検出器と蛍光寿命イメージングシステムを装備。
- 340から380 nmおよび540〜560 nmでの励起波長、それぞれ435から485 nmおよび570から590ナノメートルの発光フィルターとデュアルバンドフィルタを使用して、1つのチャンネルにDAPIおよびTRITCからの蛍光を検出します。 440から500 nmでの励起波長および510から590ナノメートルの発光フィルターを用いてフィルターを用いBCECF-AMからの蛍光を検出します。
注意:BCECF-AMのコントロールは、生菌に関する表示の適正化を確保するために研究されているすべての細菌株に行われるべきです。まずそのnonviabを検証ル細菌は、DAPI陽性およびBCECF-陰性です。第二に、生きた細菌はフルオレセインBCECFにBCECF-AMを代謝できることを確認します。細菌やBCECF-AM色素の種々の濃度は、最適なラベル付けをテストする必要があるかもしれません。
結果
上記で概説したプロトコルはPとの間の宿主-病原体相互作用を研究に使用しましたジンジバリスおよび内皮細胞。P.ジンジバリス W83 と P. PG0228の欠失を保有するジンジバリス V3150を研究に使用しました。 PG0228は、最終的にPの相互作用に影響を与える可能性があるRNAおよびタンパク質のレベルを変化させることができるタンパク質をコードすることが予?...
ディスカッション
上記のすべての方法は、真核細胞で嫌気性細菌の相互作用を評価するために、特定のアッセイを設計することができます。しかし、成功した実験を行うには、いくつかの考慮事項があります。最初の研究で使用される菌株です。
それは彼らが同じような成長段階にあり、上記の違いと同様の細胞濃度を達成侵入効率13に影響を与えることができること生存アッセ?...
開示事項
Authors have nothing to disclose.
謝辞
We would like to thank Dr. Hiroshi Miyazaki, Dr. Scott Henderson, Dr. Todd Kitten, Dr. Justin Hutcherson, Dr. Catherine Jauregui, and Collin R. Berry. This work was supported by NIH NIDCR grants R01DE016124, R01DE018039, and R01DE023304 to J.P. Lewis.
Microscopy was performed at the VCU Department of Anatomy and Neurobiology Microscopy Facility, supported, in part, with funding from NIH-NINDS Center core grant (5P30NS047463).
資料
| Name | Company | Catalog Number | Comments |
| Vinyl Anaerobic Chamber-Type B | Coy Laboratory Products | Model 2000 incubator | |
| TSA II Trypticase Soy Agar with 5% Sheep Blood | BBL | 221261 | |
| Human Umbilical Vein Endothelial Cells 10-donor Pool | LifeLine Technology | FC-0044 | |
| VascuLife VEGF Medium Complete Kit | LifeLine Technology | LL-0003 | |
| TrypKit | LifeLine | LL-0013 | |
| Saponin | Riedel-de Haen | 16109 | |
| Gentamicin Sulfate Salt | Sigma-Aldrich | G-1264 | |
| Metronidazole | Sigma-Aldrich | M-3761 | |
| BCECF-AM | LifeTechnologies | B1150 | |
| TRITC Phalloidin | Sigma-Aldrich | P1951 | |
| 18 mm Circular Coverslips | Electron Microscopy Sciences | 72222-01 | |
| VectaShield Mounting Medium with DAPI | Vector Laboratories | H-1200 |
参考文献
- Hentges, D. J. The Anaerobic Microflora of the Human Body. Clin. Infect. Dis. 16 (4), S175-S180 (1993).
- Willis, A. T. . Anaerobic bacteriology: clinical and laboratory practice. , (2014).
- Wren, M. W., Baldwin, A. W., Eldon, C. P., Sanderson, P. J. The anaerobic culture of clinical specimens: a 14-month study. J. Med. Microbiol. 10 (1), 49-61 (1977).
- Woolard, M. D., Frelinger, J. A. Outsmarting the host: bacteria modulating the immune response. Immunol. Res. 41 (3), 188-202 (2008).
- Mayrand, D., Holt, S. C. Biology of asaccharolytic black-pigmented Bacteroides species. Microbiol. Rev. 52 (1), 134-152 (1988).
- Lamont, R. J., Jenkinson, H. F. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62 (4), 1244-1263 (1998).
- Haffajee, A. D., Socransky, S. S. Microbial etiological agents of destructive periodontal diseases. Periodontol. 2000. 5 (1), 78-111 (1994).
- Listgarten, M. A. Structure of the microbial flora associated with periodontal health and disease in man. A light and electron microscopic study. J. Periodontol. 47 (1), 1-18 (1976).
- Ximénez-Fyvie, L. A., Haffajee, A. D., Socransky, S. S. Comparison of the microbiota of supra- and subgingival plaque in health and periodontitis. J. Clin. Periodontol. 27 (9), 648-657 (2000).
- Darveau, R. P., Hajishengallis, G., Curtis, M. A. Porphyromonas gingivalis as a potential community activist for disease. J. Dent. Res. 91 (9), 816-820 (2012).
- Holt, S. C., Kesavalu, L., Walker, S., Genco, C. A. Virulence factors of Porphyromonas gingivalis. Periodontol. 2000. 20 (1), 168-238 (1999).
- Lamont, R. J., Yilmaz, &. #. 2. 4. 6. ;. Z. In or out: the invasiveness of oral bacteria. Periodontol. 2000. 30 (1), 61-69 (2002).
- Lamont, R. J., et al. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect. Immun. 63 (10), 3878-3885 (1995).
- Belton, C. M., Izutsu, K. T., Goodwin, P. C., Park, Y., Lamont, R. J. Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell. Microbiol. 1 (3), 215-223 (1999).
- Rautemaa, R., et al. Intracellular localization of Porphyromonas gingivalis thiol proteinase in periodontal tissues of chronic periodontitis patients. Oral Dis. 10 (5), 298-305 (2004).
- Kaufmann, S. H. Immunity to intracellular bacteria. Annu. Rev. Immunol. 11 (1), 129-163 (1993).
- Diaz, P., Rogers, A. The effect of oxygen on the growth and physiology of Porphyromonas gingivalis. Oral Microbiol. Immunol. 19 (2), 88-94 (2004).
- Lewis, J. P., Iyer, D., Anaya-Bergman, C. Adaptation of Porphyromonas gingivalis to microaerophilic conditions involves increased consumption of formate and reduced utilization of lactate. Microbiology. 155, 3758-3774 (2009).
- Edwards, A. N., Suarez, J. M., McBride, S. M. Culturing and maintaining Clostridium difficile in an anaerobic environment. J. Vis. Exp. (79), e50787 (2013).
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. , (2001).
- Koch, A. L., Crandall, M. Photometric measurement of bacterial growth. The American Biology Teacher. 30 (6), 481-485 (1968).
- Wikins, T. D., Holdeman, L. V., Abramson, I. J., Moore, W. E. Standardized single-disc method for antibiotic susceptibility testing of anaerobic bacteria Antimicrob. Agents Chemother. 1 (6), 451-459 (1972).
- Bauer, A. W., Kirby, W. M., Sherris, J. C., Turck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 45 (4), 493-496 (1966).
- Mandell, G. L. Interaction of intraleukocytic bacteria and antibiotics. J. Clin. Invest. 52 (7), 1673-1679 (1973).
- Menzies, B. E., Kourteva, I. Internalization of Staphylococcus aureus by endothelial cells induces apoptosis. Infect. Immun. 66 (12), 5994-5998 (1998).
- Naito, M., et al. Determination of the genome sequence of Porphyromonas gingivalis strain ATCC 33277 and genomic comparison with strain W83 revealed extensive genome rearrangements in P. gingivalis. DNA Res. 15 (4), 215-225 (2008).
- Goebel, W., Kuhn, M. Bacterial replication in the host cell cytosol. Curr. Opin. Microbiol. 3 (1), 49-53 (2000).
- Gospodarowicz, D. C. Extracellular matrix and control of proliferation of vascular endothelial cells. J. Clin. Invest. 65 (6), 1351-1364 (1980).
- DeQuach, J. A., et al. Simple and high yielding method for preparing tissue specific extracellular matrix coatings for cell culture. PloS One. 5 (9), e13039 (2010).
- Sellers, J. R., Cook, S., Goldmacher, V. S. A cytotoxicity assay utilizing a fluorescent dye that determines accurate surviving fractions of cells. J. Immunol. Methods. 172 (2), 255-264 (1994).
- Van Veen, H. W., et al. Generation of a proton motive force by the excretion of metal-phosphate in the polyphosphate-accumulating Acinetobacter johnsonii strain 210A. J. Biol. Chem. 269 (47), 29509-29514 (1994).
- Jackson, V. N., Halestrap, A. P. The kinetics, substrate, and inhibitor specificity of the monocarboxylate (lactate) transporter of rat liver cells determined using the fluorescent intracellular pH indicator, 2',7'-bis(carboxyethyl)-5(6)-carboxyfluorescein. J. Biol. Chem. 271 (2), 861-868 (1996).
- He, J., et al. Role of Porphyromonas gingivalis FeoB2 in metal uptake and oxidative stress protection. Infect. Immun. 74 (7), 4214-4223 (2006).
- Anaya-Bergman, C., et al. Porphyromonas gingivalis ferrous iron transporter FeoB1 influences sensitivity to oxidative stress. Infect. Immun. 78 (2), 688-696 (2010).
- Ueshima, J., et al. Purification, gene cloning, gene expression, and mutants of Dps from the obligate anaerobe Porphyromonas gingivalis. Infect. Immun. 71 (3), 1170-1178 (2003).
- Johnson, M. B., Criss, A. K. Fluorescence microscopy methods for determining the viability of bacteria in association with mammalian cells. JoVE. (79), (2013).
- Cordes, T., Maiser, A., Steinhauer, C., Schermelleh, L., Tinnefeld, P. Mechanisms and advancement of antifading agents for fluorescence microscopy and single-molecule spectroscopy. Physical Chemistry Chemical Physics. 13 (14), 6699-6709 (2011).
- Pawley, J. . Handbook of biological confocal microscopy. , (2010).
- Gursoy, U., Könönen, E., Uitto, V. Prevotella intermedia ATCC 25611 targets host cell lamellipodia in epithelial cell adhesion and invasion. Oral Microbiol. Immunol. 24 (4), 304-309 (2009).
- Sengupta, D., et al. Interaction of Prevotella intermedia strain 17 leucine-rich repeat domain protein AdpF with eukaryotic cells promotes bacterial internalization. Infect. Immun. 82 (6), 2637-2648 (2014).
- Reyes, L., Herrera, D., Kozarov, E., Roldán, S., Progulske-Fox, A. Periodontal bacterial invasion and infection: contribution to atherosclerotic pathology. J. Clin. Periodontol. 40, S30-S50 (2013).
- Grant, M. M., et al. Oxygen tension modulates the cytokine response of oral epithelium to periodontal bacteria. J. Clin. Periodontol. 37 (12), 1039-1048 (2010).
- Biedermann, A., Kriebel, K., Kreikemeyer, B., Lang, H. Interactions of Anaerobic Bacteria with Dental Stem Cells: An In Vitro Study. PloS One. 9 (11), e110616 (2014).
- Kriebel, K., Biedermann, A., Kreikemeyer, B., Lang, H. Anaerobic Co-Culture of Mesenchymal Stem Cells and Anaerobic Pathogens-A New In Vitro Model System. PloS One. 8 (11), e78226 (2013).
- Peyyala, R., Kirakodu, S. S., Novak, K. F., Ebersole, J. L. Oral microbial biofilm stimulation of epithelial cell responses. Cytokine. 58 (1), 65-72 (2012).
- Halldorsson, S., Lucumi, E., Gòmez-Sjöberg, R., Fleming, R. M. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens Bioelectro. 63, 218-231 (2015).
- Iyer, D., et al. AdpC is a Prevotella intermedia 17 leucine-rich repeat internalin-like protein. Infect. Immun. 78 (6), 2385-2396 (2010).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved