
Method Article
Quantitative Measurement of Relative Retinoic Acid Levels in E8.5 Embryos and Neurosphere Cultures Using the F9 RARE-Lacz Cell-based Reporter Assay
In This Article
Summary
Methods to accurately measure retinoic acid (RA) levels in small amounts of tissue do not exist. This protocol describes the easy, quantitative measurement of relative RA levels in E8.5 embryos and neurospheres using an RA reporter cell line.
Abstract
Retinoic acid (RA) is an important developmental morphogen that coordinates anteroposterior and dorsoventral axis patterning, somitic differentiation, neurogenesis, patterning of the hindbrain and spinal cord, and the development of multiple organ systems. Due to its chemical nature as a small amphipathic lipid, direct detection and visualization of RA histologically remains technically impossible. Currently, methods used to infer the presence and localization of RA make use of reporter systems that detect the biological activity of RA. Most established reporter systems, both transgenic mice and cell lines, make use of the highly potent RA response element (RARE) upstream of the RAR-beta gene to drive RA-inducible expression of reporter genes, such as beta-galactosidase or luciferase. The transgenic RARE-LacZ mouse is useful in visualizing spatiotemporal changes in RA signaling especially during embryonic development. However, it does not directly measure overall RA levels. As a reporter system, the F9 RARE-LacZ cell line can be used in a variety of ways, from simple detection of RA to quantitative measurements of RA levels in tissue explants. Here we describe the quantitative determination of relative RA levels generated in embryos and neurosphere cultures using the F9 RARE-LacZ reporter cell line.
Introduction
Retinoic acid (RA) is a metabolite derivative of retinol or vitamin A and is produced through the sequential activity of several cellular dehydrogenases1. Because of its amphiphatic chemical nature, it readily crosses lipid membranes and can act as a soluble morphogenetic factor1. It is an essential molecule that regulates numerous developmental and adult cellular processes by acting as a ligand for nuclear retinoid receptors and activating gene expression2-10. Without RA, these RA receptors (RARs) bind RAREs in DNA and act as repressors; RA binding to these receptors turns them into transcriptional activators resulting in target gene expression1,5,6.
Although the RA signaling pathway is well characterized, the small size (~ 300 Da) and amphipathic nature of RA makes direct histological visualization and measurement of RA currently technically unfeasible11. The only method for direct measurement of RA levels is through isolation of RA from tissue samples using high performance liquid chromatography (HPLC)11. This method typically requires large amounts of tissue, facilitated by pooling different samples12. Thus, this technique is poorly suited for measurement of RA levels in individual embryos or small amounts of sample/tissue.
In order to investigate the cellular function of RA, several methods have been developed to indirectly infer the presence of RA. These methods make use of reporter systems containing the highly responsive RAR-beta RARE driving the expression of beta-galactosidase or luciferase11,13,14. The transgenic RARE-LacZ reporter mouse generated by Rossant et al.13 is an ideal system for visualizing spatiotemporal regions of RA signaling in situ11,13,15,16 but is unsuitable for quantitative measurements. The F9 RARE-LacZ cell line14, on the other hand, is useful for detection of RA and quantitative measurements of RA levels in tissue explants11,12,14,17,18.
F9 teratocarcinoma cells express endogenous alpha-, beta-, and gamma-retinoic acid receptors 19, and initiate differentiation into parietal endoderm after exposure to RA19-21. F9 cells have long been established as a model for RA-induced cellular differentiation, which is why it was chosen as an ideal cell line for an RA reporter assay. The F9-derived Sil-15 RA reporter cell line generated by Wagner et al.14 contains an E. coli'LacZ gene downstream of one copy of the 64-bp retinoic acid response element (RARE) of the human beta-retinoic acid receptor (RAR-beta) gene. In order to select and maintain stable clones, the construct also contains the aminoglycoside phosphotransferase (NEOr) gene as a selectable marker in the presence of G418. This construct confers inducible expression of b-galactosidase in the presence of RA, which can be visualized through LacZ staining and this response can be subsequently quantified using colorimetric methods11,12,14,18.
This extremely versatile reporter cell line has been widely used in the detection of endogenous RA production through co-culture of tissue samples with the reporter cells, such as cochlear17 and embryonic floor plate explants14. In addition, this reporter line has been used for quantification of RA levels in the developing spinal cord by culturing pooled sections of embryonic spinal cords separately and adding conditioned media from these cultures to F9 RARE-LacZ cells18. Quantification was performed after LacZ staining through colorimetric reading using a standard ELISA plate reader11,12,18. Lastly, this reporter cell line has been used in the detection of the presence of RA metabolic enzymes by monitoring changes in RA levels12,18.
Here we report that the sensitivity of this reporter cell line also allows for the measurement of RA levels generated from individual co-cultured E8.5 embryos. This enables the comparison between individual embryos of different genotypes. As a specific example, Gpr161 is an orphan GPCR that regulates neurulation in part through the RA signaling pathway22, and we report using this reporter cell line to investigate the effect of a recessive mutation in Gpr161 (Gpr161vl) on overall RA levels in the embryo. In addition to this, the versatility and ease-of-use of this reporter system allows the freedom to measure and compare RA levels in different tissue samples, even in neurosphere cultures obtained from adult spinal cord stem cells. Here we describe the quantitative determination of relative RA levels in embryos and neurosphere cultures through direct co-culture with the F9 RARE-LacZ RA reporter cell line.
Protocol
All animal work was done according to approved Rutgers-RWJMS IACUC methods.
1. Solution and Media Preparation
- Prepare stock and buffer solutions as per Table 1. Prepare working solutions as per Table 2.
2. Culture and Maintenance of F9 RARE-LacZ Cells
- Thawing Frozen Cells
- Prepare F9 RARE-LacZ cell culture medium as detailed in Table 2.
- Coat 10 cm culture dishes (cell culture treated) with 10 ml of 0.2% gelatin (refer to Table 2) for 30 min at RT. Wash off excess gelatin with two rinses of 10 ml distilled water. Immediately add 9 ml of F9 RARE-LacZ cell culture medium into the flask to prevent the gelatin from drying.
- Thaw a vial of F9 RARE-LacZ cells14 in a 37 °C water bath for 2 - 3 min. Plate cells (around 1 x 106 cells/ml) into the coated dish containing 9 ml of culture medium. Culture at 37 °C, 5% CO2. Change the medium the next day.
- Maintenance of F9 RARE-LacZ Cells
- Maintain a regular passage every 2 - 3 days at a ratio of 1:10.
Note: Do not let cells grow into confluency as this will result in their differentiation. - Split the cultures at a confluency of 80-90%. Remove the media and wash the cells with 10 ml of 1x phosphate buffered saline (PBS). Remove the PBS and add 1 ml of trypsin, incubate for 5 min. Add 9 ml of culture medium, pipette up and down and split cells 1:10 for passage.
- Maintain a regular passage every 2 - 3 days at a ratio of 1:10.
- Freezing F9 RARE-LacZ Cells
- Trypsinize a 10 cm dish containing F9 RARE-LacZ cells at 80 - 90% confluence as detailed in step 2.2.2. Transfer the dissociated cells into a 15 ml centrifuge tube and spin at 200 x g for 5 min.
- Remove supernatant and resuspend cell pellet in 10 ml of F9 RARE-LacZ freezing medium (see Table 2). Aliquot 1 ml into labeled cryovials and transfer vials into freezing containers. Place freezing container in -80 °C freezer O/N and transfer vials to liquid nitrogen tanks the next day.
3. Dissection of E8.5 Embryos
- Mating Cage Set-up and Plug Check
- Set-up a mating cage containing one mating pair. The morning of the next day, check for the presence of a vaginal plug and designate the day a plug is found as day 0.523. At day E8.5, harvest embryos.
- Dissection of Embryos
- Sacrifice the dam through cervical dislocation and spray the abdominal area with 70% ethanol (see Table 2). Make a transverse cut on the skin above the abdomen using surgical scissors and pull the two flaps apart (anterior and posterior) to expose the abdominal area. Make a similar cut on the peritoneum to expose the abdomen.
- Locate the uteri and release uteri from the oviducts and mesometrium using a pair of scissors23. Place the uteri in a 6 cm dish (non-cell culture treated) with 10 ml 1x PBS to wash off excess fat and blood. Transfer to another 6 cm dish containing fresh 10 ml 1x PBS.
- Separate one uterus containing one embryo by cutting it off using surgical scissors. Remove the uterus and decidua and release the yolk sac containing the embryo using two pairs of no. 5 forceps23.
- Separate the yolk sac from the embryo, and transfer the yolk sac to a 1.5 ml microcentrifuge tube for genomic DNA extraction to be used for genotyping. Use a P20 pipette with a wide-bore tip, transfer the whole embryo into a 1.5 ml microcentrifuge tube containing 10 µl of trypsin. Place the tube on ice.
- Repeat steps 3.2.3 - 3.2.4 until all embryos have been dissected.
- Embryo Dissociation
- Incubate the 1.5 ml tubes containing embryos in trypsin at 37 °C for 5 min to facilitate enzymatic dissociation.
- Triturate gently using a P20 pipettor for mechanical dissociation of the embryo, and plate the whole 10 µl volume containing one dissociated embryo over one well of F9 RARE-LacZ cells in a 96-well plate (see step 5.1 for plating F9 RARE-LacZ cells in a 96-well plate). Culture O/N at 37 °C and 5% CO2.
- Incubate the 1.5 ml tubes containing embryos in trypsin at 37 °C for 5 min to facilitate enzymatic dissociation.
4. Laminectomy, Dissection of Adult Lumbar Spinal Cord and Neurosphere Culture
- Laminectomy
- Sacrifice an adult (P60) mouse through cervical dislocation.
- Spray the dorsal side with 70% ethanol and make a transverse cut using surgical scissors. Pull both flaps (anterior and posterior) apart to expose the back and vertebral column.
- Using surgical scissors, trim off the muscles covering the spine. Locate the lumbar region of the spinal cord, below the ribs. Using the scissors, make a deep cut to sever the spine at this point to expose the spinal cord.
- Pull up the lumbar region of the spine with no. 5 forceps so that the exposed transverse section of the spinal cord is facing the experimenter. Using the tips of fine scissors, snip the vertebra and muscle at the 3 o'clock and 9 o'clock positions of the exposed end. Grasp the dorsal flap of the cut vertebra with forceps. Continue these cuts caudally until the length of the lumbar spinal cord is exposed.
- Release the spinal cord from the vertebrae using no. 5 as well as blunt-ended forceps. Transfer the spinal cord to a 15 ml tube containing 3 ml of DMEM/F12 culture medium. Keep on ice until all lumbar spinal cords have been isolated.
- Spinal Cord Dissection
- Pour the culture medium with the spinal cord into a 6 cm dish (non-cell culture treated).
- Under a stereomicroscope, remove the dorsal root ganglia and blood vessels around the spinal cord segment by grasping the ganglia and blood vessels using two pairs of no. 5 forceps and gently pulling them away from the spinal cord. Transfer the spinal cord segment to a 6 cm dish with no medium and mince the tissue finely using a scalpel blade.
- Transfer minced tissue to 15 ml tube containing 1 ml of neurosphere dissociation media (see Table 2). Put tube on ice and repeat steps 4.2.1 - 4.2.2 until all spinal cords have been processed.
- Incubate tubes containing minced tissue in dissociation medium at 37 °C for 10 min, flick the tube on the side to mix and then incubate at 37 °C for another 10 min. After enzymatic digestion, triturate with fire-polished pipettes of decreasing diameters for mechanical dissociation.
Note: Prepare fire-polished pipettes of decreasing diameters prior to performing experiments. Take a glass Pasteur pipette and plug the wide end with clean cotton. Using an alcohol lamp, fire polish the tips of the pipettes in the flame to create different diameters: "regular", "fine", and "extra fine". For "regular" polished pipettes, twirl the end of the pipette in the flame to round of the edges without decreasing the diameter (~ 1 mm). Rotate the end of the pipette in the flame for a slightly longer periods to create "fine" (~ >1 mm and > 5 mm diameter) and "extra fine" (~ 0.5 mm) polished pipettes. Do not use pipettes with diameters less than 0.5 mm.
- Neurosphere Culture
- Spin down tubes containing dissociated spinal cord tissue at 200 x g for 10 min. Remove supernatant and resuspend cell pellet in 1 ml of neurosphere culture media (see Table 2). Triturate with a P1,000 pipette followed by a P200 pipette to facilitate dissociation.
Note: Avoid introducing bubbles, which would decrease cell viability. - Transfer 10 µl of dissociated cells into a hemocytometer and obtain a cell count. Plate cells at a density of 1x105/10 ml in a T25 flask with 10 ml of neurosphere culture media. Incubate at 37 °C and 5% CO2 for 7 days until neurospheres appear 24.
- Spin down tubes containing dissociated spinal cord tissue at 200 x g for 10 min. Remove supernatant and resuspend cell pellet in 1 ml of neurosphere culture media (see Table 2). Triturate with a P1,000 pipette followed by a P200 pipette to facilitate dissociation.
- Neurosphere Dissociation
- Collect neurosphere culture and transfer to 15 mL tubes. Spin at 200 x g for 10 min. Remove most of the supernatant leaving 1 ml behind. Triturate with a P1,000 pipette to resuspend the cell pellet and dissociate neurospheres into single cells. Transfer 10 µl of dissociated cells into a hemocytometer and get a cell count.
- Plate 1 x 105 dissociated neurosphere cells over one well of F9 RARE-LacZ in a 96-well plate (see section 5.1 for plating F9 RARE-LacZ in 96-well plate). Plate 3 wells as replicates. Culture O/N at 37 °C and 5% CO2.
5. RA Assay of E8.5 Embryos and Spinal Cord Neurospheres
- Plating F9 RARE-LacZ in 96-well plate
- One day prior to harvesting E8.5 embryos or dissociation of neurospheres, coat a 96-well plate with 100 µl of 0.2% gelatin per well for 30 min at RT. Aspirate out the gelatin and rinse twice with 200 µl of distilled water.
- Trypsinize F9 RARE-LacZ cells maintained in 10 cm dishes as detailed in step 2.2.2. Plate 1 x 105 cells/well of F9-RARE LacZ cells, but this time using culture medium without G418.
- Prepare enough wells for co-culture of embryos (~ 12 - 20, depending on litter size), neurospheres (3 wells/genotype) as well as for a standard curve in triplicate (27 wells).
- Co-culture of F9 RARE-LacZ and E8.5 Embryos or Spinal Cord Neurospheres
- Harvest embryos as detailed in section 3.2 and plate on top of F9 RARE-LacZ in the 96-well plate from section 5.1. In a similar manner, dissociate neurospheres and plate on top of F9 RARE-LacZ in the 96-well plate as detailed in step 4.4. Culture O/N at 37 °C and 5% CO2.
- Generating a Standard Curve
- To generate a standard curve, prepare all-trans RA (at-RA) solutions with the following concentrations: 100 nM, 10 nM, 1 nM, 100 pM, 10 pM, 1 pM, 100 fM, by serial dilution of the at-RA stock (see Table 1) with F9 RARE-LacZ culture medium.
- Add 100 µl of these different at-RA concentrations to F9 RARE-LacZ cells from section 5.1. Assign wells containing untreated F9 RARE-LacZ cells as negative control. Prepare triplicates of each RA concentration and negative control wells. Culture O/N at 37 °C and 5% CO2.
Note: F9 RARE-LacZ cells respond in a dose-dependent linear manner to varying concentrations of all-trans RA.
- RA Assay
- After O/N culture of the 96-well plate containing the standard curves and co-cultures, remove the media and fix the cultures by adding 100 µl of 2.5% glutaraldehyde (see Table 2) for 15 min, RT. Remove the fixative and wash twice with 200 µl 1x PBS, 10 min per wash.
- Remove PBS and wash three times with 200 µl of the LacZ wash solution (see Table 2), 10 min per wash. During the third wash, prepare the X-gal staining solution in a 15 ml tube wrapped in foil (see Table 2). Calculate how much staining solution is needed and prepare the appropriate amount (200 µl/well).
Note: X-gal staining solution is light sensitive. Use the staining solution within 15 min after preparation. - Prepare a humidified chamber by placing a paper towel dampened with distilled water into a plastic container big enough to hold a 96-well plate. Add 200 µl of the X-gal staining solution per well of the 96-well plate and place the plate in a humidified chamber.
- Incubate humidified chamber containing the 96-well plate at 37 °C to allow for development of blue-colored precipitate. For E8.5 embryos, incubate the plate O/N (12 - 16 hr). For neurosphere cultures, incubate the plate for 4 - 8 hr.
- Reading Absorbance at OD610 and Imaging
- After color reaction, remove the LacZ staining solution and replace with 200 µl 1x PBS. Measure the absorbance at 610 nm using a standard ELISA plate reader to quantify the reporter response to RA. Image individual wells using a standard stereomicroscope equipped with a camera.
6. Subcloning of F9 RARE-LacZ Cells
- Checking F9 RARE-LacZ rResponse to RA
- Before starting any experiments, test the response of F9 RARE-LacZ cells to RA. Plate F9 RARE-LacZ cells in 96-well plate (see section 5.1), and add 1 nM all-trans retinoic acid (see step 5.3). Incubate in a humidified chamber at 37 °C O/N.
- Visualize development of blue colored precipitate using a microscope. If color development is not uniform throughout the cells in the well (see Figure 1B, left), perform subcloning detailed below.
- Subcloning of F9 RARE-LacZ Cells
- Split the F9 RARE-LacZ cells cultured in 10 cm dishes as detailed in step 2.2.2. Instead of a 1:10 ratio, plate a low seeding density of 1 x 105cells/10 ml in a 10 cm dish to allow for growth of isolated colonies such that each colony arises from a single F9 RARE-LacZ cell.
- After two days, coat a 24-well plate with 500 µl of 0.2% gelatin per well for 30 min. Remove gelatin solution and rinse wells twice with 500 µl distilled water. Replace water with 500 µl of F9 RARE-LacZ cell culture medium.
- Pick a colony using a sterile P10 pipette tip and transfer into one well; do this 24 times to obtain 24 individual colonies. Culture at 37 °C, 5% CO2 for 3 - 4 days.
- Split the individual colonies into two 24-well plates. After 2 days in culture, add 1nM of RA to each well of one 24-well plate and culture O/N. Perform LacZ staining as detailed in section 5.4 to test for high responders. Visualize color reaction of each well under a microscope to determine strong and uniform responders.
- Once a strong subclone responder is determined, expand the corresponding culture from the other 24-well plate. Further expand that colony in a 10 cm dish, freeze down several vials as detailed in section 2.3, and discard all other colonies.
Note: Only about one-fourth of the original colonies result in high responders. Typically, the first round of subcloning does not result in uniform LacZ staining and a subsequent round of subcloning needs to be done to achieve uniform strong responders.
Results
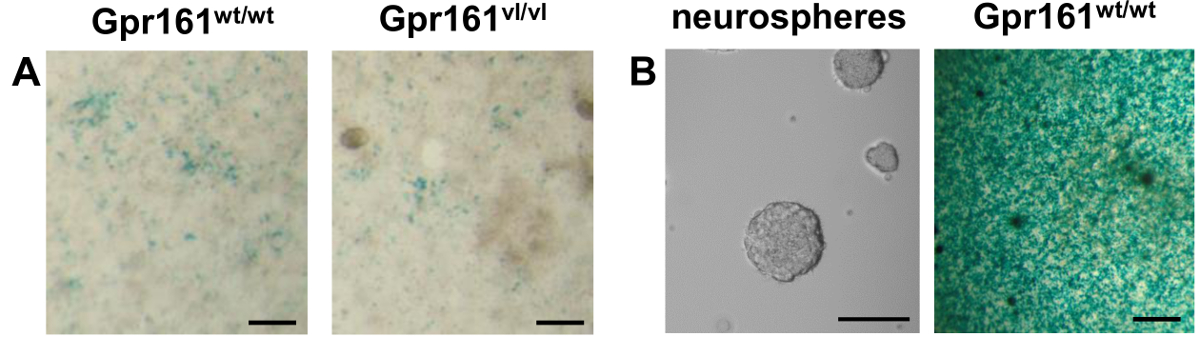
The F9 RARE-LacZ reporter is an adherent embryonic carcinoma cell line grown on gelatin-coated surface. The reporter construct allows the cells to respond quantitatively to RA with a proportional induction of beta-galactosidase. 11,12,14,18. These cells display an epithelial morphology (Figure 1A). Because of their high doubling rate, it is recommended that these cells be split every 2 - 3 days at a ratio of 1:10. During our work with this reporter cell line, we have observed that even with the addition of G418 into the culture medium, the response of these cells to RA weakens after several passages (Figure 1B, left), and this can affect the reporter capacity of this cell line. This loss of responsiveness may be due to partial loss of the transgene at later passages. As such, periodic subcloning is necessary to ensure strong and uniform responders to RA (Figure 1B, right).
While various uses of this cell line has been previously published12,14-18, reported here is the use of this cell line to measure endogenous RA levels from individual E8.5 embryos with different genotypes. The Gpr161vl/vl mutation results in decreased embryonic RA signaling22. As shown by co-culture of dissociated embryos with F9 RARE-LacZ cells, these Gpr161vl/vl embryos have reduced endogenous RA compared to wild-type littermates (Figure 2A). Because of the versatility of this reporter cell line, also reported here is a novel use of these cells to detect RA levels produced by neurosphere cultures obtained from adult Gpr161wt/wt mice (Figure 2B). This reporter cell line was able to demonstrate that neurosphere cultures from adult spinal cord stem cells produce endogenous RA. The great difference in staining intensity indicates a greater reporter cell response to spinal cord neurospheres compared to E8.5 embryos. This may be due to much greater RA levels generated by the neurospheres over time compared to E8.5 embryos, which could be due to the fact that neurosphere cultures are more homogenous than E8.5 embryos and thus contain more RA-producing cells.
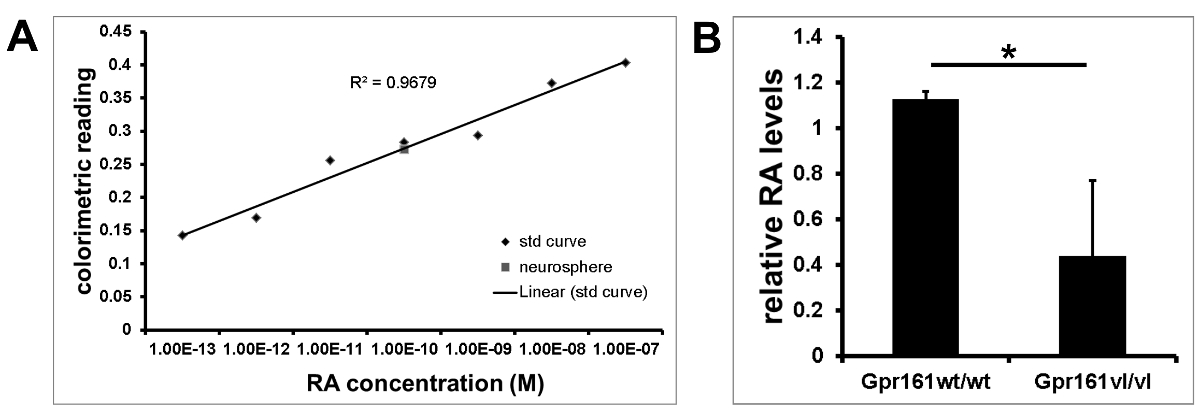
Addition of different concentrations of at-RA results in a dose-dependent linear response of the reporter cells. This response can be measured colorimetrically by reading the absorbance at 610 nm. The generated standard curve can then be used to quantify RA levels in samples, such as in wild-type neurosphere cultures (Figure 3A) or E8.5 embryos (Figure 3B).

Figure 1. Maintenance and Subcloning of F9 RARE-LacZ Cells. (A) A phase-contrast image of F9 RARE-LacZ cells is shown. These cells have a doubling time of ~10 hr and must be passed every 3 days (roughly 70 - 80% confluence) at a 1:10 ratio. (B) Periodic testing of cultures with addition of 1 nM at-RA and subsequent LacZ staining must be done to ensure cultures remain strong and uniform responders to RA (right). In case of poor and non-uniform responders (left), subcloning must be performed. Scale bars 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. LacZ Staining of Co-cultured Samples (Dissociated E8.5 Embryos/Neurospheres) and F9 RARE-LacZ cells. (A) E8.5 mouse embryos of various genotypes were dissociated and plated on top of F9 RARE-LacZ cells. LacZ staining 24 hr later allows visualization of response of the F9 RARE-LacZ cells to endogenous RA produced by co-cultured samples. The Gpr161vl mutation results in decreased endogenous RA as visualized by the reduced response of the reporter cells. (B) Left. A phase-contrast image of neurospheres cultured from adult spinal cord neural stem cells is shown. Right. Neurospheres derived from adult Gpr161wt/wt spinal cord were dissociated and plated on top of F9 RARE-LacZ cells. The presence of blue precipitate following LacZ staining indicate the presence of endogenous RA in these neurospheres. The difference in staining intensity between neurosphere and E8.5 embryo co-cultures indicate greater levels of RA present in neurospheres than E8.5 embryos. Scale bars 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Standard Curve and Colorimetric Quantification of RA Levels. The response of the F9 RARE-LacZ reporter cell line can be measured colorimetrically at 610 nm using a standard ELISA plate reader. (A) Representative standard curve used to quantify relative RA levels of wild-type neurosphere cultures. (B) Quantification of relative RA levels from co-cultured E8.5 embryos. N= 3; *p< 0.05, student's t-test. Error bars SEM. Please click here to view a larger version of this figure.
{kind=link}
| Stock Buffers/Solutions | Components | Storage conditions |
| 10% Sodium deoxycholate monohydrate (C24H39NaO4 · H2O) | 10 ml in 100 ml dH2O | 4 ºC |
| 1 M magnesium chloride (MgCl2) | RT | |
| 100x potassium ferrocyanide (K4[Fe(CN)6]·3H6O) | 2.12 g in 10 ml dH2O | RT in the dark |
| 100x potassium ferricyanide (K3[Fe(CN)6]) | 1.64 g in 10 ml dH2O | RT in the dark |
| 20 mg/ml X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) | add 5 ml dimethyl formamide to make 20 mg/ml stock solution | -20 ºC |
| 10 mM (3 mg/ml) all-trans retinoic acid | Dissolve 50 mg in 16.67 ml absolute ethanol. Prepare 1 ml aliquots. | -80 ºC in the dark, in 1.5 ml amber microcentrifuge tubes |
| Papain | dissolve one vial in 7 ml HBSS | 4 ºC |
Table 1. Buffers and Solution Compositions.
| Working Buffers/Solutions | Components | Storage conditions |
| F9 RARE-LacZ cell culture medium | 50 ml fetal bovine serum (FBS) (final concentration 10%) | Filter-sterilize. |
| 5 ml 100x Penicillin Streptomycin (final conc 1X) | Store at 4 ºC. | |
| 4 ml 50 mg/ml G418 (final conc 0.4 mg/ml) | ||
| to 500 ml DMEM/F-12 (with L-glutamine and HEPES) | ||
| 0.2% gelatin | 25 ml 2.0% gelatin | Store at 4 ºC. |
| 80 ml distilled water | ||
| Mix well | ||
| 70% ethanol | 30 ml 95% ethanol | Place in spray bottle. |
| 70 ml distilled water | Store at RT | |
| Neurosphere dissociation medium | 1 ml 20U papain | prepare fresh every time |
| 100 μl 13 mg/ml trypsin | ||
| 100 μl 7 mg/ml hyaluronic acid | ||
| 28 μl 10 mg/ml DNaseI | ||
| 50 μl 4mg/ml kynurenic acid | ||
| Neurosphere culture medium | DMEM/F-12 (with L-glutamine and HEPES) | prepare fresh every time |
| 2% B-27 supplement | ||
| 20 ng/ml fibroblast growth factor (FGF) | ||
| 20ng epidermal growth factor (EGF) | ||
| 2.5% glutaraldehyde | 250 μl 50% glutaraldehyde | prepare fresh every time |
| 4750 μl 1x PBS | ||
| LacZ wash solution | 0.5 ml 10% deoxycholate (final conc 0.1%) | Store at 4 ºC. |
| 1.0 ml 100% NP-40 (final conc 0.2%) | ||
| 50 ml 10X PBS | ||
| distilled water to 500 ml | ||
| X-gal staining solution | 1x potassium ferrocyanide | prepare fresh every time |
| 1x potassium ferricyanide | ||
| 1 mg/ml X-gal | ||
| to appropriate volume using LacZ wash solution | ||
| F9 RARE-LacZ freezing medium | F9 RARE-LacZ culture medium + 5% DMSO | prepare fresh every time |
Table 2. Working Buffers and Solution Compositions.
Discussion
The F9 RARE-LacZ cell line14 is a powerful system that can be used for detection of RA produced by tissue explants12,14,17,18. It is sensitive to RA concentrations as low as 100 fM, with no non-specific response detected in cells untreated with RA12,14,18. Furthermore, the reporter cells respond to all-trans RA in a linear manner within a specific range of concentration, thus allowing the LacZ reaction to be quantified and used to determine the magnitude of RA levels12,14,18. The quick and easy RA reporter assay presented here utilizes the F9 RARE-LacZ reporter cell line for visualization (Figure 2) and relative quantitative measurement (Figure 3) of embryos and cultured adult neural stem cells. This reporter system allows comparison of endogenous RA levels between individual samples, as shown in Figure 2. It is sensitive enough to be able to detect RA generated by individual E8.5 embryos (Figure 2A) as well as RA produced by cultured neurospheres (Figure 2B). The dose-dependent response of this cell line to varying concentrations of RA allows for the quantification of RA levels (Figure 3A) in the samples as well as comparison of relative RA levels between samples (Figure 3B). It is important to note that the F9 RARE reporter cell line measures the overall amount of RA generated by the tissue samples over the time of incubation. This is equivalent to the net balance of RA synthesis and catabolism.
While the F9 RARE-LacZ cell reporter system is highly sensitive and versatile, there are several critical troubleshooting steps that we have incorporated in order to ensure the reliability of this system. First, as shown in Figure 1, it is important to maintain the health of these cells by regular passages without letting them grow to confluency. Second, we have observed later passages tend to result in inconsistent RA responses, even if the cultures are constantly maintained under selection with G418. To troubleshoot this, it is recommended to discard cultures after 20 passages and thaw a new vial. In addition, regular checks must be performed to ensure that the reporter cells are still strong responders to the presence of RA in the media. Third, if the early passages display poor or non-uniform response to RA (Figure 1B, left), one or several rounds of subcloning may be necessary until strong responders are obtained (Figure 1B, right). Lastly, prior to co-culture, it is recommended that tissue samples such as embryos and neurospheres are dissociated to single cells. We have observed that dissociation of the samples results in more consistent colorimetric measurements. This may be due to a more even distribution of RA in the culture wells, as the dissociated cells are in uniform contact with the reporter cells.
Several modifications were also made to the original protocol14. First, RA response of the F9 RARE-LacZ cells were quantified by directly fixing the cells and measuring absorbance at 610 nm instead of collecting cells and analyzing lysate for b-galactosidase activity14. Another modification is the direct co-culture of dissociated samples with F9 RARE-LacZ prior to quantification of RA response. Previous methods reported culturing samples separately and adding only the media from these cultures to F9 RARE-LacZ16,18.
Despite its being a powerful system, the F9 RARE-LacZ reporter assay has several limitations. One limitation of this reporter cell line is that although it responds strongly to the presence of all-trans RA, it also responds to other retinoic acid isomers including 9-cis-RA and 13-cis-RA; however, they are 100-fold more sensitive to all-trans RA compared to the other isomers18. Another limitation to this system is that the linear dose-dependent response only falls between 10-13 M to 10-7 M at-RA14,18; thus, if comparing RA levels outside these limits, it might be necessary to perform a serial dilution of the sample until the measurements fall within the linear range. Lastly, since this is primarily a colorimetric assay, the endpoint of color development will vary between samples and experiments. Thus, it is important that a standard curve always be plated in parallel for every single experiment in order to use it for quantification of the reporter cell response.
In conclusion, we have reported the use of the F9 RARE-LacZ RA reporter cell line to measure RA levels quantitatively in E8.5 embryos and, previously unreported, in neurospheres. The versatility of this reporter system may allow the quantification of RA levels in virtually any cell or tissue type, and may result in the development of varied applications in the future.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to acknowledge Dr. Michael Wagner (SUNY Downstate Medical Center) and Ben Thiede (Dr. Jeff Corwin lab, University of Virginia) for providing us with the F9 RARE-LacZ cells and technical support. This study is funded by the New Jersey Commission on Spinal Cord Research (NJCSCR) (Grantnumber:10-3092-SCR-E-0) and the New Jersey Commission on Brain Injury Research (NJCBIR) (Grant number: CBIR13FEL006).
Materials
| Name | Company | Catalog Number | Comments |
| C3H/HeSn-Gpr161vl/J mouse strain | Jackson Laboratory | 000316 | |
| F9 RARE-LacZ reporter cell line | from Dr. Michael Wagner (SUNY Downstate Medical Center) and Ben Thiede (Dr. Jeff Corwin lab, University of Virginia) | ||
| DMEM/F-12 (with L-glutamine and HEPES) | ThermoFisher Scientific | 11330-057 | |
| Fetal Bovine Serum (FBS), qualified | ThermoFisher Scientific | 26140-079 | Store at -20 °C in 10-ml aliquots to avoid repeated freeze-thaw; warm in 37 °C for media preparation |
| Penicillin/Streptomycin | ThermoFisher Scientific | 15140-122 | Store at -20 °C in 10-ml aliquots to avoid repeated freeze-thaw; light-sensitive |
| G418 | ThermoFisher Scientific | 10131-027 | Store at 4 °C; light-sensitive |
| 2% Gelatin | Sigma Aldrich | G1393 | Store at 4 °C |
| B-27 supplement (2 % stock solution) | ThermoFisher Scientific | 17504-044 | Store at -20 °C in 1-ml aliquots to avoid repeated freeze-thaw; warm at room temperature for media preparation |
| Murine Epidermal Growth Factor (EGF) | PeproTech | 315-09 | Prepare 100 μg/ml stock solution in 0.1% bovine serum albumin (BSA) in 1xPBS; prepare working concentrations by diluting with 1x PBS; store at -20 °C |
| Murine Fibroblast Growth Factor (FGF)-basic | PeproTech | 450-33 | Prepare 100 μg/ml stock solution in 0.1% bovine serum albumin (BSA) in 1x PBS; prepare working concentrations by diluting with 1x PBS; store at -20 °C |
| 50% glutaraldehyde | Amresco | M155 | Store at 4 °C |
| Sodium deoxycholate monohydrate (C24H39NaO4 · H2O) | Sigma Aldrich | D5670 | Store at RT |
| X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) | Promega | V3941 | Store stock at -20 °C |
| Dimethyl sulfoxide (DMSO) | Sigma Aldrich | 41639 | Store at RT |
| all-trans Retinoic Acid (at-RA) | Sigma Aldrich | R-2625 | Store 1 ml aliquots of stock in -80 °C; extremely sensitive to light |
| TrypLE Express | ThermoFisher Scientific | 12605-010 | Store at RT |
| 0.25% trypsin-EDTA | ThermoFisher Scientific | 25200-056 | alternative to TrypLE Express; aliquot and store at -20 °C |
| Hank's Balanced Salt Solution (HBSS) | ThermoFisher Scientific | 14170-112 | Store at 4 °C |
| 10x phosphate buffered solution (PBS) | ThermoFisher Scientific | 10010-023 | Store at 4 °C |
| Papain | Worthington | LK003176 | Store at 4 °C. |
| Trypsin | Worthington | LS003703 | Store in 1 ml aliquots at -20 °C. |
| Hyaluronic acid | Calbiochem | 385908 | Store 200 μl aliquots at -20 °C. |
| DNaseI | Worthington | LS002139 | Store in 200 μl aliquots at -20 °C |
| Kynurenic acid | Sigma Aldrich | K3375 | Store in 200 μl aliquots at -20 °C |
| 95% ethanol | |||
| Mr. Frosty Freezing container | ThermoFisher Scientific | 5100-0001 | |
| 10 cm culture dish (non-cell culture treated) | |||
| 10 cm culture dish (cell culture treated) | |||
| 6 cm culture dish (non-cell culture treated) | |||
| 96-well cell culture plates | |||
| 1.5 ml microcentrifuge tubes | |||
| 1.5 ml amber microcentrifuge tubes | |||
| 1.5 ml cryovials | |||
| 37 °C water bath | |||
| surgical scissors | |||
| fine surgical scissors | |||
| no. 5 forceps | |||
| blunt-ended forceps | |||
| scalpel blade | |||
| glass pasteur pipettes | |||
| cotton | |||
| alcohol lamp | |||
| ELISA plate reader | |||
| stereomicroscope |
References
- Vilhais-Neto, G. C., Pourquie, O. Retinoic acid. Curr Biol. 18 (5), R191-R192 (2008).
- Drager, U. C. Retinoic acid signaling in the functioning brain. Science Signalling. 324, 1-3 (2006).
- Lane, M. A., Bailey, S. J. Role of retinoid signalling in the adult brain. Prog Neurobiol. 75 (4), 275-293 (2005).
- Maden, M. Retinoid signalling in the development of the central nervous system. Nat Rev Neurosci. 3 (11), 843-853 (2002).
- Maden, M. Retinoids and spinal cord development. J Neurobiol. 66 (7), 726-738 (2006).
- Rhinn, M., Dolle, P. Retinoic acid signalling during development. Development. 139 (5), 843-858 (2012).
- Ang, H. L., Deltour, L., Hayamizu, T. F., Zgombic-Knight, M., Duester, G. Retinoic acid synthesis in mouse embryos during gastrulation and craniofacial development linked to class IV alcohol dehydrogenase gene expression. J Biol Chem. 271 (16), 9526-9534 (1996).
- Ang, H. L., Duester, G. Stimulation of premature retinoic acid synthesis in Xenopus embryos following premature expression of aldehyde dehydrogenase ALDH1. Eur J Biochem. 260, 227-234 (1999).
- Bastien, J., Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene. 328, 1-16 (2004).
- Chatzi, C., Cunningham, T. J., Duester, G. Investigation of retinoic acid function during embryonic brain development using retinaldehyde-rescued Rdh10 knockout mice. Dev Dyn. 242 (9), 1056-1065 (2013).
- Sakai, Y., Drager, U. C. Detection of retinoic acid catabolism with reporter systems and by in situ hybridization for CYP26 enzymes. Methods Mol Biol. 652, 277-294 (2010).
- Yamamoto, M., Drager, U. C., McCaffery, P. A novel assay for retinoic acid catabolic enzymes shows high expression in the developing hindbrain. Dev Biol Res. 107, 103-111 (1998).
- Rossant, J., Zirngibl, R., Cado, D., Shago, M., Giguere, V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes & Dev. 5, 1333-1344 (1991).
- Wagner, M., Han, B., Jessel, T. M. Regional differences in retinoid release from embryonic neural tissue detected by an in vitro reporter assay. Development. 116, 55-66 (1992).
- Luo, T., Wagner, E., Crandall, J. E., Drager, U. C. A retinoic-acid critical period in the early postnatal mouse brain. Biol Psychiatry. 56 (12), 971-980 (2004).
- Luo, T., Wagner, E., Grun, F., Drager, U. C. Retinoic acid signaling in the brain marks formation of optic projections, maturation of the dorsal telencephalon, and function of limbic sites. J Comp Neurol. 470 (3), 297-316 (2004).
- Kelley, M. W., Xu, X. M., Wagner, M. A., Warchol, M. E., Corwin, J. T. The developing organ of Corti contains retinoic acid and forms supernumerary hair cells in response to exogenous retinoic acid in culture. Development. 119, 1041-1053 (1993).
- McCaffery, P., Drager, U. C. Hot spots of retinoic acid synthesis in the developing spinal cord. Proc Natl Acad Sci. 91, 7194-7197 (1994).
- Hu, L., Gudas, L. J. Cyclic AMP analogs and retinoic acid influence the expression of retinoic acid receptor alpha, beta, and gamma mRNAs in F9 teratocarcinoma cells. Mol Cell Biol. 10 (1), 391-396 (1990).
- Martin, G. R. Teratocarcinomas and mammalian embryogenesis. Science. 209 (4458), 768-776 (1980).
- Strickland, S., Mahdavi, V. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell. 15 (2), 393-403 (1978).
- Li, B. I., et al. The orphan GPCR, Gpr161, regulates the retinoic acid and canonical Wnt pathways during neurulation. Dev Biol. 402 (1), 17-31 (2015).
- Hogan, B., Constantini, F., Lacy, E. Manipulating the Mouse Embryo: A Laboratory Manual. , 1st, Cold Spring Harbor Laboratory. (1986).
- Deleyrolle, L. P., Reynolds, B. A. Isolation, expansion, and differentiation of adult Mammalian neural stem and progenitor cells using the neurosphere assay. Methods Mol Biol. 549, 91-101 (2009).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved