
Method Article
In tempo reale analisi del fattore di trascrizione associazione, trascrizione, traduzione e fatturato per visualizzare eventi globali durante l'attivazione cellulare
* Questi autori hanno contribuito in egual misura
In questo articolo
Riepilogo
Questo protocollo descrive l'uso combinatoria di ChIP-seq, 4sU-seq, totale RNA-seq e profilatura per linee cellulari e cellule primarie del ribosoma. Esso consente di tenere traccia delle modifiche in fattore di trascrizione binding, trascrizione de novo , elaborazione di RNA, fatturato e traduzione nel corso del tempo e la visualizzazione il corso generale degli eventi in cellule attivate e/o cambiare rapidamente.
Abstract
Al momento dell'attivazione, le cellule rapidamente cambiano loro programmi funzionali e, quindi, loro profilo di espressione genica. Enormi cambiamenti nell'espressione genica si verificano, ad esempio, durante la differenziazione cellulare, la morfogenesi e stimolazione funzionale (quali l'attivazione di cellule immunitarie), o dopo l'esposizione a farmaci e altri fattori dell'ambiente locale. A seconda del tipo di stimolo e cellula, questi cambiamenti si verificano rapidamente e a qualsiasi livello possibile di regolazione genica. Visualizzazione di tutti i processi molecolari di una cella risponda ad un certo tipo di stimolo/droga è uno dei compiti più difficili nella biologia molecolare. Qui, descriviamo un protocollo che consente l'analisi simultanea di più livelli di regolazione genica. Mettiamo a confronto, in particolare, obbligatoria (cromatina-immunoprecipitazione-sequenziamento (ChIP-seq)) di fattore di trascrizione, trascrizione de novo (4-thiouridine-sequenziamento (4sU-seq)), elaborazione di mRNA e fatturato, nonché traduzione (ribosoma profilatura). Combinando questi metodi, è possibile visualizzare un dettagliata e genoma corso d'azione.
Sequenziamento di RNA recentemente trascritto è particolarmente indicato quando si analizza rapidamente adattare o cambiando sistemi, poiché questo rappresenta l'attività trascrizionale di geni tutti durante il tempo di esposizione 4sU (indipendentemente dal fatto che siano - up o downregulated). L'uso di combinatoria di totale RNA-seq e profilatura ribosoma permette inoltre il calcolo dei tassi di turnover e la traduzione di RNA. Analisi bioinformatica di risultati di sequenziamento ad alte prestazioni permette molti mezzi per l'analisi e interpretazione dei dati. I dati generati consentono inoltre il monitoraggio co-transcriptional e alternative splicing, solo per citare alcuni possibili esiti.
L'approccio combinato qui descritto può essere applicato per organismi di modello diverso o di tipi di cellule, compreso le cellule primarie. Inoltre, forniamo protocolli dettagliati per ogni metodo utilizzato, inclusi i controlli di qualità e discutere i potenziali problemi e insidie.
Introduzione
Negli ultimi anni, la sequenza di RNA (RNA-seq) è diventato lo strumento standard per analizzare tutto espresso RNA all'interno di una cella o a un organismo1. Tuttavia, per comprendere l'intero processo di adattamento in risposta ad una stimolo/droga specifica di cellule, è necessario determinare completamente tutti i processi sottostanti, che vanno dalla trascrizione del mRNA per l'elaborazione, fatturato e traduzione. Cambiamenti a breve termine nella trascrizione del RNA difficilmente possono essere misurate con RNAseq totale, poiché le modifiche di RNA totale dipendono da fattori ad esempio RNA mezza-vita e attività trascrizionale, che sono un povero modello per riflettere l'adattamento delle cellule a effetti ambientali2,3. Infatti, una varietà di nuove tecniche di sequenziamento sono stati sviluppati che permettono un'analisi delle diverse fasi nel processo di gene regolamento4 quando combinati nel modo giusto. Questo protocollo viene descritto come combinare alcune tecniche di sequenziamento piuttosto facilmente applicabile che permettono la regolazione dei livelli essenziali del mRNA di rilevamento in modo comparativo. Per analizzare l'attività trascrizionale, sono state descritte una varietà di metodi, quali Cap-analisi di gene expression (gabbia)5, nativo di allungamento trascrizione sequenziamento (NET-seq)6e genoma nucleare eseguire-sulle (GRO-seq)7 , 8, così come bromouridine-sequenziamento (Bru-seq) e 4-thiouridine-sequenziamento (4sU-seq), che utilizzano metaboliti che sono incorporati in appena trascritto RNA9,10, solo per citarne alcuni. Mentre CAGE identifica il sito di inizio della trascrizione esatta, NET-seq e GRO-seq fornire informazioni più accurate indicazioni di lettura e 4sU-seq (che è il metodo qui descritto) rileva solo recentemente trascritto RNA. Tuttavia, 4sU-seq è altamente sensibile e può essere applicato in tempi diversi per misurare l'attività trascrizionale quantitativa in attivamente cambiando le cellule, così come cambiamenti quantitativi nella trasformazione di mRNA (che avviene all'interno di minuti)9, 11,12. È inoltre ideale per combinare con RNA-seq per calcolare i tassi di fatturato di RNA per geni94sU-seq. La trascrizione di mRNA viene effettuata dalla RNA polimerasi II (RNAPII), che a sua volta è influenzata da una moltitudine di fattori, come fattori di trascrizione, modificazioni istoniche e attivatori/repressori generale che può anche essere parte del trascrizionale complesso. Per verificare quanti gene/promoter/enhancer regioni sono vincolate da un fattore, ChIP-seq è stato sviluppato, che ora è il metodo standard per questo scopo, a causa di molti anticorpi commerciali13. Tuttavia, anche se ChIPseq fornisce informazioni chiare su dove che regolano fattori bind, non riflette se infatti porta a cambiamenti nella trascrizione14. Di conseguenza, l'esecuzione di ChIP-seq con 4sU-seq è la combinazione ideale per tali domande biologiche. Regolazione dell'espressione genica può verificarsi anche in una fase successiva, dal momento che i livelli di mRNA e proteina necessariamente non correlano15,16, che indica il regolamento potenzialmente significativo sul traslazionale o post-traduzionale livello, a seconda del contesto. Durante l'anno 2011, ribosoma profiling erano Stati Uniti in primo luogo con RNA-seq e ora è il metodo di scelta di quantificare i cambiamenti che si verificano rapidamente in proteina, dato che ci sono ancora alcuni limiti di sensibilità con spettrometria di massa17. Infatti, i tassi di conversione ottenuti da tali metodi sono stati indicati per consegnare una relativamente buona stima dei cambiamenti nei livelli della proteina (misurati almeno per cambiamenti a lungo termine) e consente una visuale ancora più dettagliata sul processo di traduzione, ad esempio il determinazione della traduzione alternativa e inizio lato cornici17. L'uso di combinatoria di tutti e quattro i metodi possono essere utilizzati in regime stazionario, tra vari tipi di cellule, o in un tempo-seriale esperimento di un rapido cambiamento di cella11. Questo uso fornisce una panoramica di tutto il genoma dei cambiamenti nell'associazione di fattore di trascrizione che influenzano la trascrizione RNA, elaborazione e traduzione.
Protocollo
Tutti i metodi sono in accordo e in conformità con le linee guida istituzionali, statali e federali di Helmholtz Zentrum München.
1. preparazione
- Fare un piano dettagliato del setup sperimentale tra cui un calendario, quando aggiungere 4sU al terreno di coltura delle cellule e quando raccogliere le cellule per ogni metodo. A seconda della domanda biologica, considerare attentamente i punti di tempo per esempio disegno, tempo 4sU-etichettatura e concentrazione (tabella 1).
Nota: Verificare l'impatto di 4sU sulla vitalità cellulare e risposta allo stress in anticipo (Vedi "Verifica d'etichettatura 4sU condizioni ottimali" nei risultati di rappresentante e Figura 1). È consigliabile eseguire un test preliminare di ogni metodo con almeno un campione. Verificare se la qualità e la quantità di RNA/DNA è sufficiente per sequenziamento profondo (Vedi dedicato le parti del protocollo) e pratica veloce ma manipolazione delicata delle cellule durante l'esperimento. - Calcolare il numero di cellule necessarie per ogni punto di tempo di ciascun metodo (Vedi tabella 2 per una stima approssimativa quando si utilizzano cellule primarie di T). Si consiglia inoltre di sequenziamento meno campioni di RNA totale di solo quella di 4sU RNA (per esempio, solo per tariffe di traduzione di tempo punto o RNA fatturato tariffe dovrebbero essere calcolate). Per confermare e verificare la portata dei risultati (scelta consigliata), è necessario creare almeno una replica biologico.
Nota: Importante, per tutti i metodi e punti di volta consecutiva, i campioni devono essere dallo stesso pool iniziale delle cellule. Almeno un ricercatore dedicato per ogni metodo è raccomandato. - Preparare tutto il necessario in anticipo (per esempio, aliquotati 4sU, cicloesimmide, Buffer di lisi di mammiferi e 1% di formaldeide). Dopo l'aggiunta di 4sU, evitare di esporre le cellule a luce intensa, come questo può portare a reticolazione del 4sU-identificato RNA a proteine cellulari18.
- Piscina tutte le celle di interesse in una boccetta appena prima del trattamento (Figura 2). Contare le celle (ad esempio, con un emocitometro) e utilizzare la quantità necessaria per il controllo non trattato di ciascun metodo (non dimenticate di etichettare anche al controllo non trattato per 4sU-seq con 4sU). Curare le cellule rimanenti e immediatamente diviso il numero di celle per ogni punto nel tempo e metodo. Gestire le cellule più rapidamente possibile per ridurre al minimo lo stress a causa di cambiamenti di temperatura o CO2 livelli.
Nota: ad esempio, vengono prelevati campioni h 1, h 2 e 3 h dopo il trattamento, poi un quarto delle cellule è usato per controllo non trattato, e tre quarti sono utilizzati per controllo trattato.
2. 4sU-etichettatura
Nota: Il presente protocollo viene modificato da Rädle, et al. 19 si riferiscono al loro protocollo per ulteriori informazioni riguardanti l'etichettatura metabolica con 4sU. Per tutti i metodi e punti di volta consecutiva, i campioni devono provenire dallo stesso pool iniziale delle cellule.
-
Inizio di etichettatura
- Disgelo aliquotati 4sU appena prima dell'uso. Aggiungere 4sU in ogni momento direttamente al supporto che contiene le cellule di interesse (vedere tabella 2 per numeri raccomandata delle cellule T, almeno 60 µ g RNA al punto di tempo), mescolare delicatamente e metterli nuovamente dentro l'incubatrice. Metodo Dispose restanti 4sU (non ricongelare).
- Alla fine di etichettatura, raccogliere cellule (ad es., raschietto di cella) e centrifugare a 330 x g per 5 min a 4 ° C in provette di polipropilene (che resistono alle forze g alta). Aspirare il medio e aggiungere il reagente per isolamento del RNA (≥ 1 mL per 3 x 106 cellule, vedi materiali per raccomandazione quale usare) in ogni provetta. Risospendere il pellet (≥ 1 mL per 3 x 106 cellule) completamente, incubare per 5 min a temperatura ambiente (TA) e congelarli a-20 ° C. I campioni possono essere conservati a-20 ° C per almeno 1 mese.
Attenzione: I reagenti utilizzati per l'isolamento di RNA sono estremamente pericolosi quando sempre a contatto con la pelle o gli occhi. Di gestirli con cura e considerare le istruzioni di sicurezza.
-
RNA preparazione utilizzando modificato il protocollo di isolamento del RNA
- Aggiungere 0,2 mL di cloroformio per reagente 1 mL per isolamento del RNA e miscelare accuratamente agitando per 15 s. procedere come indicato nel protocollo d'etichettatura metabolico (passaggio 1-12, 2. Utilizzo di preparazione di RNA modificato protocollo trizol) da Rädle et al. 19
- Misurare la concentrazione di RNA (Vedi Tabella materiali), secondo le istruzioni del produttore. Utilizzare questo RNA per totale RNA-seq pure (vedere il passaggio 3. Totale RNA-seq) o conservare a-80 ° C per almeno 1 mese.
-
Biotinylation tiolo-specifiche di RNA recentemente trascritto
- Iniziare con 30-80 µ g di RNA cellulare totale. 60 µ g di RNA dovrebbe produrre una quantità sufficiente di RNA appena trascritto.
- Preparare la reazione d'etichettatura. Pipetta nel seguente ordine (per µ g di RNA): 1 µ l 10 x Biotinylation Buffer, 7 µ l RNA (contenente 1 µ g di RNA diluito in privo di nucleasi H2O) e 2 µ l biotina-HPDP (1 mg/mL). Aggiungere biotina-HPDP Ultima e miscelare immediatamente pipettando. Avvolgere i tubi con carta stagnola per evitare l'esposizione a luce intensa. Vedere la discussione per un'alternativa alla biotina-HPDP. Incubare a RT per 1,5 h con rotazione.
- Appropriato 2 mL provette (Vedi Tabella materiali) a 15.000 x g per 2 min. Pipettare tutti biotinilati RNA nella provetta 2 mL pre-spun, aggiungere un volume equivalente di cloroformio e mescolano energicamente. Incubare per 2-3 min finchè non separare le fasi e le bolle cominciano a scomparire.
- Centrifuga a 15.000 x g per 15 min a 4 ° C. Trasferire con cautela la fase acquosa superiore in una nuova provetta.
- Ripetere i passaggi da 2.3.3. e 2.3.4. una volta. Aggiungere il 10% del volume di NaCl (5 M) e un uguale volume di isopropanolo alla fase acquosa. Centrifuga a 20.000 x g per 20 min a 4 ° C. Gettare il surnatante.
- Aggiungere che un volume uguale preparata il 75% di etanolo. Centrifugare a 20.000 x g. gettare il surnatante, girare brevemente e rimuovere l'etanolo rimanente. Risospendere completamente in 30-100 µ l di H2O (uso 1 µ l H2O a 1 µ g di RNA input dal punto 2.3.1) pipettando.
- Verificare l'integrità del RNA mediante analisi elettroforetica, o prendere un'aliquota e verificare più tardi.
-
Separazione di appena trascritto (con etichetta) e preesistenza RNA (senza etichetta)
- Rimuovere perline paramagnetici (Vedi Tabella materiali) dal deposito di 4 ° C e lasciar riposare per almeno 30 min per portarli a temperatura ambiente. Tampone di lavaggio di calore 4sU (3 mL per campione) a 65 ° C.
- Preparare la soluzione di ditiotreitolo (DTT) di 100 mM. Pesare DTT su scala ultra-fine e aggiungere la quantità necessaria di acqua priva di nucleasi. Sempre preparare fresco. Utilizzare 200 µ l per campione.
- Campioni di RNA biotinilato (1 µ g / µ l) a 65 ° C per 10 min a denaturare e collocare immediatamente sul ghiaccio di calore. Aggiungere 100 µ l streptavidina branelli di biotinylated RNA e incubare a temperatura ambiente per 15 min con rotazione.
- Inserire una colonna appropriata (Vedi Tabella materiali per consigli) per ogni campione nel supporto magnetico e pre-equilibrare ogni colonna con tampone di lavaggio di 1 mL temperatura 4sU.
Nota: Questo richiederà circa 5-10 min. Se le colonne non avviano il drenaggio dopo 5 min, premere delicatamente alla sommità della colonna con i guanti. - Applicare un mix di RNA/perline al centro di ogni colonna. Scartare il flusso continuo, a meno che non senza etichetta RNA deve essere recuperato. In questo caso, raccogliere il flusso continuo e almeno il primo lavaggio. Eseguire il ripristino di RNA come descritto da Rädle et al. (fase 1-7, 7. Recupero di RNA senza etichetta, non associato)19.
- Lavare tre volte con tampone di lavaggio 4sU 0,9 mL (preriscaldato a 65 ° C dal punto 2.4.2.) e tampone di lavaggio di 0,9 mL RT 4sU, rispettivamente.
- Usare le perline paramagnetiche per recuperare appena trascritti di RNA. Dispensare 400 µ l di ben dispersi perline paramagnetici RT in una provetta per campione e posto sotto ogni colonna. Eluire recentemente trascritto RNA con 100 µ l 100 mM DTT. Attendere 3 minuti ed eseguire un secondo eluizione con 100 µ l 100 mM DTT. (Opzionale: eseguire eluizione e recupero come descritto da Rädle et al.) 19
- Mix di recente trascritti di RNA/perline accuratamente mediante pipetta 10 tempi di miscelazione e procedere secondo le linee guida del produttore. Eluire il RNA in 11 µ l privo di nucleasi H2O.Quantify RNA usando un fluorometro adatto (Vedi Tabella materiali). RNA possa essere conservato a-80 ° C per almeno 1 mese.
Nota: Recentemente trascritto RNA può essere utilizzato per preparare le librerie di cDNA per il sequenziamento di nuova generazione (Vedi Tabella materiali per un suggerimento su quale kit di utilizzare) o ulteriori analisi a valle. 100 - 500 ng RNA sono sufficienti per la maggior parte dei kit di preparazione di biblioteca (vedi discussione).
3. totale RNA-Seq
- Prendere direttamente il RNA da 4sU etichettato RNA dopo la preparazione di RNA utilizzando il reagente modificato per protocollo di isolamento del RNA per totale di RNA-seq (Vedi punto 2.2.2).
- Per la preparazione di biblioteca, diluire una parte aliquota di RNA ad una concentrazione finale di 50-100 ng / µ l. utilizzare lo stesso kit per la preparazione di biblioteca di RNA appena trascritto. 100 - 500 ng RNA sono sufficienti per la maggior parte dei kit di preparazione di biblioteca.
4. ribosoma profilatura
Nota: Per tutti i metodi e punti di volta consecutiva, i campioni devono essere dallo stesso pool iniziale delle cellule. Per consigli su quale kit da utilizzare, fare riferimento alla Tabella materiali.
- Preparazione e l'isolamento del ribosoma protetto frammenti (RPFs):
- Utilizzare adeguate quantità di celle per ogni punto di tempo (Vedi tabella 2 per numeri raccomandata delle cellule di T). Trattare le cellule aderenti con cicloesimmide come descritto nel protocollo del produttore.
Attenzione: Cicloesimmide è altamente tossico e può causare mutazioni. Evitare il contatto con la pelle e l'inalazione. - Raccogliere e piscina non - o semi - adherent celle da ogni volta scegliere in una provetta di polipropilene e regolare la concentrazione finale di 1 x 106 cellule per mL di mezzo di cellula-specifico (ad es., RPMI supplementato per cellule di T). Aggiungere cicloesimmide con una concentrazione finale di 0,1 mg/mL, mescolare capovolgendo la provetta in polipropilene e incubare per 1 min. Centrifugare le cellule per 5 min a 330 x g a 4 ° C. Medio di aspirare e lavare le cellule con almeno 10 mL di PBS completati con cicloesimmide (concentrazione finale di 0,1 mg/mL).
- Centrifugare le cellule per 5 min a 330 x g a 4 ° C. Aspirare il medio e aggiungere 100 µ l tampone di lisi di cellule di mammifero per 10 x 106 cellule. Mescolare pipettando ed espellere attraverso un ago di calibro 25 22 sterile per lisare le cellule completamente.
- Trasferire il lysate delle cellule in una provetta da 1,5 mL preraffreddato. Incubare per 10 minuti sul ghiaccio con inversioni periodiche. Centrifugare per 10 min 20.000 x g a 4 ° C per chiarire il lisato. Trasferire il surnatante in una provetta preraffreddato 1,5 mL.
- Preparare un 01:10 diluizione del lisato con acqua priva di nucleasi e record una A260lettura utilizzando uno spettrofotometro. Utilizzare acqua priva di nucleasi come uno spazio vuoto e un 01:10 diluizione del tampone di lisi della cellula di mammiferi come standard. Calcolare la concentrazione di A260/ml del lisato secondo la seguente equazione:
(Un260 lysate delle cellule - un260 mammiferi tampone di lisi) x fattore di diluizione 10 =un/260ml - Creare 200 aliquote µ l del lisato su ghiaccio e procedere con il trattamento di nucleasi.
Nota: Facoltativamente, preparare un 100 µ l aliquota per RNA totale, aggiungere 10 µ l di 10% SDS e mescolare. Conservare a 4 ° C e procedere con 4.3.2. È consigliabile utilizzare RNA totale da RNA 4sU-etichettati (vedere il passaggio 3. Totale RNA-seq).
- Utilizzare adeguate quantità di celle per ogni punto di tempo (Vedi tabella 2 per numeri raccomandata delle cellule di T). Trattare le cellule aderenti con cicloesimmide come descritto nel protocollo del produttore.
- Footprinting ribosoma
- Eseguire trattamento nucleasi immediatamente senza congelamento lisato. Aggiungere 7,5 unità di nucleasi (incluso nel kit consigliati) per ogni A260 di lisato. Ad esempio: 80 A260/ml lisato x 0,2 mL lisato x 7,5 U/A260nucleasi = 120 nucleasi di U.
Nota: Facoltativamente, titolare della nucleasi per digestione come descritto dal produttore. - Incubare la reazione di nucleasi per 45 min a RT con miscelazione delicata. Congelare 200 aliquote µ l del lisato con azoto liquido e conservare a-80 ° C, o arrestare la reazione di nucleasi con l'aggiunta di inibitore di RNAsi 15 µ l per ogni 200 µ l aliquota e passare al punto 4.3.2.
- Eseguire trattamento nucleasi immediatamente senza congelamento lisato. Aggiungere 7,5 unità di nucleasi (incluso nel kit consigliati) per ogni A260 di lisato. Ad esempio: 80 A260/ml lisato x 0,2 mL lisato x 7,5 U/A260nucleasi = 120 nucleasi di U.
- Purificazione di RPFs
- Sbloccare un campione di RPFs nucleasi digerita e aggiungere inibitore di RNAsi 15 µ l. Mantenere i campioni su ghiaccio.
- Purificare il RPFs secondo il protocollo del produttore (purificazione colonna è consigliato) e misurare la concentrazione di RNA su uno spettrofotometro.
- rRNA svuotamento
- Utilizzare 5 µ g di RPFs purificato per lo svuotamento del rRNA.
- Seguire il protocollo del produttore (fase 1-2, lo svuotamento del rRNA primario) per lo svuotamento del rRNA. Concentrazione di RNA di misura del rRNA impoverito RPFs su uno spettrofotometro.
- Purificazione di pagina di RPFs
- Utilizzo 500 ng di rRNA impoverito RPFs per purificazione di pagina.
- Preparare il controllo della RNA, campioni e scaletta per purificazione di pagina. Mix 5 µ l controllo di RNA e 5 µ l denaturante gel colorante in una microcentrifuga 0,5 mL di caricamento. Miscelare 10 µ l di ogni RPF con 10 µ l di denaturazione, rispettivamente di caricamento del gel. Preparare una scaletta aliquota (4 µ l 20/100 scaletta, acqua priva di nucleasi di 1 µ l e 5 µ l di denaturazione della tintura di caricamento del gel). Caricarla tra ogni campione e controllo per evitare la contaminazione incrociata.
- Denaturare i campioni e scaletta incubando a 95 ° C per 5 min e collocare immediatamente sul ghiaccio. Carico 20 µ l di ciascun campione (facoltativamente, caricare 10 µ l e freeze restanti campioni a 20 ° C) separate da 10 µ l di scaletta preparata su un gel di poliacrilammide-urea 12% o 15%. Caricare 10 µ l di controllo di RNA. Esegua il gel fino a quando la band di blu di bromofenolo raggiunge il fondo del gel (180 V, ~ 70 min) (Figura 3).
- Macchia il gel secondo il protocollo del produttore a 4 ° C. Utilizzare un transilluminatore campo scuro che emette luce blu per visualizzare il RNA. Asportare le fette del gel per ogni campione corrispondente ~ 28 e 30 nt in lunghezza. Prendere il controllo di RNA come riferimento e accise di esso.
Nota: RPFs sono difficilmente visibili. Accise fette con le dimensioni indicate dal controllo RNA - contenente due oligos del nt 28 e 30 di lunghezza - anche se i campioni non sono visibili. - Perforare un foro sul fondo delle provette per microcentrifuga da 0,5 mL con un ago sterile di calibro 20. Trasferire ogni fetta di gel in un tubo separato e posto ricoperto tubi in una provetta da 1,5 mL. Centrifugare per 2 min 12.000 x centrifugazione di ripetizione g. Se fette del gel non sono completamente distruggere nella provetta da 1,5 mL.
- Eluire il RNA da fette del gel perturbato con acqua priva di nucleasi di 400 µ l, 40 µ l di ammonio acetato (5 M) e 2 µ l SDS (10%) ogni notte a 4 ° C.
- Trasferire i residui 1,5 mL provette di filtro (fornite con il kit consigliato) con una punta di Pipetta 1ml (punta di wide-bore o self-made 1 mL con estremità tagliata). Centrifugare per 3 min 2.000 x g per separare eluito RNA da fette del gel. Pipettare delicatamente soluzione acquosa in una provetta da 1,5 mL. Aggiungere 2 glicogeno µ l (fornito con il kit consigliato) e 700 µ l 100% isopropanolo e store a-20 ° C per almeno 1 h.
- Centrifugare a 4 ° C per 20 min a 13.000 x g. Discard surnatante. Lavare la pallina con etanolo di 80% preparata al momento pre-refrigerati a 4 ° C per 10 min a 13.000 x surnatante Discard g. e asciugare all'aria. Risospendere ciascun campione in 20 µ l e il controllo di RNA in acqua priva di nucleasi di 8 µ l. Conservare a-20 ° C se necessario.
- Frammentazione, fine riparazione, 3' adattatore legatura, trascrizione d'inversione
- Eseguire la procedura come descritto dal protocollo del produttore (frammentazione e riparazione fine, 3' adattatore legatura e trascrizione d'inversione).
- PAGINA purificazione del cDNA
- Preparare i campioni, il controllo della RNA e scaletta per purificazione pagina: Mix 10 µ l di ogni campione e controllo di RNA con 10 µ l di denaturare i gel di caricamento della tintura, rispettivamente. Preparare una scaletta aliquota (4 µ l 20/100 scaletta, acqua priva di nucleasi 1 µ l, 5 µ l di denaturazione della tintura di caricamento del gel). Caricarla tra ogni campione e controllo per evitare la contaminazione incrociata.
- Denaturare i campioni e scaletta incubando a 95 ° C per 5 min e collocare immediatamente sul ghiaccio. Carico 20 µ l di ciascun campione (facoltativamente, caricare 10 µ l e freeze restanti campioni a 20 ° C) separate da 10 µ l di scaletta preparata su un 10% poliacrilammide/7 - 8 M gel di urea/TBE. Caricare 10 µ l di controllo di RNA. Esegua il gel fino a quando il blu di bromofenolo migra completamente fuori il gel (180 V, ~ 60 min).
- Macchia il gel secondo il protocollo del produttore a 4 ° C. Utilizzare un transilluminatore campo scuro che emette luce blu per visualizzare il RNA e asportare le fette del gel per ogni campione corrispondente nt ~ 70-80.
- Procedere come descritto al punto 4.5.5 - 4.5.8 e Risospendere ciascun campione in acqua priva di nucleasi di 10 µ l.
- cDNA Circularization
- Preparare abbastanza mix master circularization per tutte le reazioni combinando i seguenti reagenti per ciascun campione sul ghiaccio: 4,0 µ l Circularization Mix di reazione, 2.0 µ l ATP, 2.0 µ l MnCl2e 2.0 µ l ligasi.
- Aggiungere 10 µ l di master mix per ogni campione. Mescolare delicatamente e centrifugare. Incubare i campioni a 60 ° C per 2 h. immediatamente posto campioni su ghiaccio.
- Amplificazione di PCR
- Seguire il protocollo del produttore (passaggio 1-3, l'amplificazione di PCR) per l'amplificazione di PCR. Utilizzare 4 µ l di cDNA circolare per l'amplificazione con 9 cicli PCR per cellule di T primarie, per ottenere migliori risultati.
- Librerie di purificare e controllare la loro granulometria, secondo il protocollo del produttore (passaggio 4-8, l'amplificazione di PCR). La dimensione prevista della biblioteca amplificata è 140-160 bp (Vedi Figura 4).
- Per le librerie di sequenziamento, fare riferimento al protocollo del produttore e l'impianto di sequenziamento per ulteriori indicazioni.
5. chIP-Seq
Nota: Il presente protocollo viene modificato da Blecher-Gonen, et al. 14 si riferiscono al loro protocollo per ulteriori informazioni sul ChIP-seq. Per tutti i metodi e punti di volta consecutiva, i campioni devono essere dallo stesso pool iniziale delle cellule.
- Reticolazione e la raccolta delle cellule
- Crosslink adeguato numero di cellule (fare riferimento alla tabella 2 per numeri raccomandata delle cellule di T) per ciascun punto di tempo con una concentrazione finale di 1% di formaldeide in un mezzo di cellspecific (ad es., RPMI supplementato per cellule di T) per 10 min a temperatura ambiente con dolce dondolio. Fermare la reazione di reticolazione con l'aggiunta di glicina ad una concentrazione finale di 0,125 M.
- Centrifugare le cellule a 330 x g per 5 min a 4 ° C. Scartare il surnatante e lavare le cellule in PBS ghiacciata. Ripetere due volte il punto 5.1.2 e congelare palline delle cellule a-80 ° C. Pellet congelati possono essere memorizzati per almeno 6 mesi.
- Lisi cellulare e sonicazione
Nota: Durante tutti i passaggi delle cellule Lisi e sonicazione, i campioni devono essere tenuti sul ghiaccio o a 4 ° C per ridurre la degradazione della proteina e inversione crosslink.- Risospendere il pellet cellulare in tampone di lisi cellulare ghiacciata 1 mL con inibitori della proteasi appena aggiunto per isolare i nuclei (aggiungere facoltativamente inibitori delle fosfatasi). Incubare per 10 minuti in ghiaccio e centrifugare a 2.600 x g per 5 min a 4 ° C.
- Aspirare il supernatante e risospendere il pellet di nuclei in tampone di lisi ghiacciata nuclei 1 mL con inibitori della proteasi appena aggiunto (facoltativamente: aggiungere inibitori delle fosfatasi). Incubare per 10 minuti in ghiaccio. Sottoporre ad ultrasuoni le cellule per generare una frazione di dimensione media del DNA di 0,2 - 1,0 kb (Vedi Figura 5).
Nota: Sonicazione condizioni necessità di essere ottimizzato in base al tipo di cellula e ulteriori condizioni (ad es., numero di cellulare, volume e buffer). Per cellule di T primarie, sonicazione per 20-25 cicli è raccomandato (Vedi materiali per descrizione dettagliata). - Prendere un 20-50 µ l aliquota della cromatina tranciata e calore per 10 min a 95 ° C e 1000 giri/min agitazione per eseguire un crosslink indietro veloce e verificare la dimensione della cromatina. Aggiungere 2-5 µ l proteinasi K e incubare per 20 minuti a 56 ° C e 1000 giri/min agitazione. Eseguire inattivazione termica per 10 min a 95 ° C e 1000 giri/min agitazione. Purificare la cromatina con un apposito kit (Vedi Tabella materiali). Verifica le dimensioni della cromatina in un gel di agarosio 1% e utilizzare 100 bp Plus marcatore.
- Centrifugare tranciata cromatina con una media di frazione di dimensione del DNA di 0,2 - 1,0 kb per 10 min a 20.000 x g e a 4 ° C per pellet detriti e insolubile della cromatina. Trasferire il surnatante in una provetta nuova e tenere sul ghiaccio.
- Tenere il 5-10% della cromatina lisati mediante come input. Congelare a-20 ° C (utilizzato al punto 5.5.2).
- Anticorpo di coppia per perline
- Anticorpo di coppia 10 µ g (ad es., anti-RNA Pol II; anti-Histon H3K36me3) in 220 µ l PBS (con BSA 0,5% e lo 0,5% Tween 20) a 80 granuli di superparamagnetiche µ l accoppiati alla proteina G (Vedi Tabella materiali) per almeno 1 h a temperatura ambiente con rotazione.
- Posizionare i tubi su un magnete. Attendere che tutte le perle sono vincolati al magnete e rimuovere il surnatante. Ulteriore blocco con 6 µ l sonicato DNA in PBS dello sperma di salmone (con BSA 0.5% e 0.5% Tween 20) per 30 min a temperatura ambiente con rotazione.
- Posizionare i tubi su un magnete. Attendere che tutte le perle sono vincolati al magnete e rimuovere il sovranatante chiaro. Lavare le perle con ChIP IP buffer tre volte.
- Immunoprecipitazione della cromatina
- Diluire la cromatina e il volume totale di 1 mL in tampone di lisi di nuclei con inibitori della proteasi appena aggiunto (facoltativamente, aggiungere inibitori delle fosfatasi). Aggiungere ChIP IP buffer con inibitori della proteasi appena aggiunto (facoltativamente, aggiungere inibitori delle fosfatasi) ad un volume finale di 3 mL. Tenere in ghiaccio o a 4 ° C mentre l'anticorpo è accoppiato ai talloni.
- Aggiungere della cromatina diluita all'anticorpo accoppiato perline dal punto 5.3.3 e incubare tutta la notte a 4 ° C con una leggera rotazione.
- Lavare con i buffer seguenti (1 mL ciascuno, Vedi Tabella materiali) a temperatura ambiente per 5 min con rotazione, inserire le provette indietro sul magnete e rimuovere il surnatante: Wash Buffer I, II di Buffer lavare, lavare Buffer III e 2 x pH TE 8.0 rispettivamente.
- Eliminare il surnatante e asciugare all'aria per ~ 5 min.
- Invertire la reticolazione
- Rimuovere i campioni dal magnete. Aggiungere 50 µ l di tampone di eluizione e Miscelare pipettando per eluire complessi proteina-DNA dalle perle.
- Include ingresso uno o più esemplari da questo passo in avanti. Aggiungere tampone di eluizione per campione (s) per un volume finale di 50 µ l (per mantenere la composizione del buffer simile agli esempi di ChIP) di ingresso e di processo insieme ai campioni di ChIP.
- Mix 3 µ l di tampone di eluizione e 2 µ l di RNAsi (DNasi gratuita). Aggiungere 5 µ l della miscela per ogni campione e incubare per 30 min a 37 ° C.
- Proteinasi di mix 2,5 µ l K, 1 µ l glicogeno e 1,5 µ l di tampone di eluizione per campione. Aggiungere 5 µ l della miscela per ogni campione (1 proteinasi U K e 20 µ g glicogeno per campione) e incubare per 2 ore a 37 ° C.
- Incubare i campioni a 65 ° C durante la notte (almeno 4 ore) con agitazione per eseguire reticolazione inversa.
- Posizionare i tubi sul magnete per almeno 30 s e trasferire il surnatante in una nuova provetta. I campioni possono essere congelati a-20 ° C fino a 12 mesi.
- Purificazione del DNA
- Aggiungere 140 µ l di perline paramagnetici ben dispersi a 60 µ l di campione (2.3: 1 rapporto). Pipettare attentamente su e giù per 25 volte per mescolare accuratamente. Assicurarsi che il liquido in ogni tubo è omogeneo. Incubare a temperatura ambiente per 2 min posto i tubi sul magnete per 4 minuti, o fino a quando tutte le perle sono vincolati al magnete e scartare il surnatante.
- Lasciare le provette sul magnete e aggiungere 200 µ l di etanolo al 70% preparati al momento. Incubare le provette per 30 s senza disturbare le perline. Scartare il surnatante e ripetere questo passaggio una volta di più. Aspirare etanolo completamente e lasciare che il paramagnetico perline asciugare all'aria per 4 min.
Nota: Rimozione incompleta dell'etanolo può seriamente ridurre DNA recupero e resa. Asciugare il pellet solo fino a quando è asciutto. Sovra il pellet può ridurre il recupero di DNA e resa. - Rimuovere i tubi dal magnete e aggiungere 20 µ l 10 mM Tris-HCl (pH 8.0). Pipettare delicatamente l'intero volume su e giù per 25 volte per mescolare accuratamente. Incubare per 2 min a temperatura ambiente. Posizionare i tubi indietro sul magnete per 4 min e trasferire il surnatante in un'altra provetta.
- Misurare la quantità di DNA con un fluorometro adatto (Vedi Tabella materiali).
- Verificare che il ChIP è stato completato da qPCR (diluito 1 µ l a 100 µ l di H2O e uso 2-5 µ l per qPCR). Utilizzare primers specifici per una positiva (sito di legame noto della proteina di interesse) ed il controllo negativo (ad esempio, un gene che è silenzioso e/o non un bersaglio della proteina di interesse).
Nota: Preparazione di libreria può essere effettuato con 2 ng di DNA ChIP a seconda del kit (Vedi Tabella materiali per un suggerimento su quale kit da utilizzare).
Risultati
4 sU etichettatura: verificare ottimale 4 condizioni di etichettatura sU (apoptosi, nucleare stress, lo stress citoplasmatico), tempo e concentrazione: Alti livelli di 4sU possono inibire la produzione e lavorazione di rRNA e indurre stress citoplasmico, nonché nucleare30. Pertanto, le cellule di interesse devono essere testate per lo stress indotto da 4sU così come apoptosi. L'analisi Western blot è consigliato per la visualizzazione di accumulazione di p53, che indica lo sforzo nucleare, aumentando i livelli di phospho-EIF2a che visualizzano stress citoplasmici e fluorescenza-attivato delle cellule ordinamento analisi (FACS) per apoptosi. Alti livelli e l'esposizione prolungata a 4sU o droghe come thapsigargin o arsenito può essere utilizzati per indurre lo stress cellulare. Per indurre apoptosi o delle cellule morte, le cellule sono state trattate con BH3I-1 (500 ng / µ l) o incubate per 5 min a 95 ° C (shock termico). Annessina V/7-AAD macchiatura è stato utilizzato per determinare apoptotica (Annessina V) e cellule morte (7-AAD). Etichettatura di in vitro generato cellule Th1 primarie per 0,5 h con 500 µM 4sU (concentrazione finale) o 1 h con 200 µM 4sU induce né segni di stress cellulare né apoptosi (Figura 1) ma portano a sufficiente 4sU incorporazione.
RNA etichettatura tempo può anche essere ridotto (≤ 5 min) che conduce ad un aumento di breve durata intronic sequenze d'etichettatura rispetto al più volte. Per visualizzare co-transcriptional tariffe d'impionbatura, etichettatura 4sU volte non devono superare 30 min. Per maggiori dettagli riguardo 4sUlabeling, fare riferimento a Rädle et al. 19
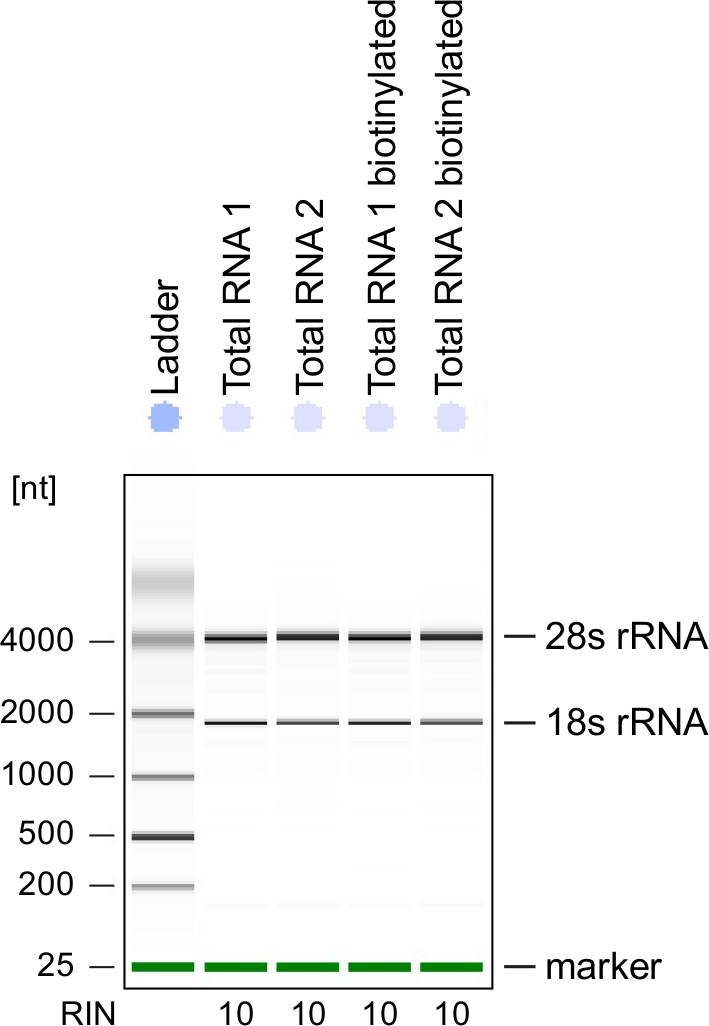
Controllo di qualità: Integrità di RNA è di grande importanza durante l'elaborazione del RNA. È più conveniente controllare la qualità di RNA di RNA con etichetta 4sU dopo biotinylation dall'analisi elettroforetici (Vedi Tabella materiali). Considera verifica RNA isolato dal punto 2.2.2, soprattutto quando lo si utilizza per la sequenza di RNA totale. Numero di RNA integrità (RIN) deve essere ≥ 8 per garantire l'integrità di RNA per un'ulteriore elaborazione (Figura 3).
Analisi elettroforetici possono anche essere utilizzato per verificare appena trascritti di RNA. Essere consapevoli del fatto che recentemente trascritto RNA contiene i rRNAs significativamente meno maturi rispetto a RNA totale con le tipiche bande di rRNA essendo molto meno prominente.
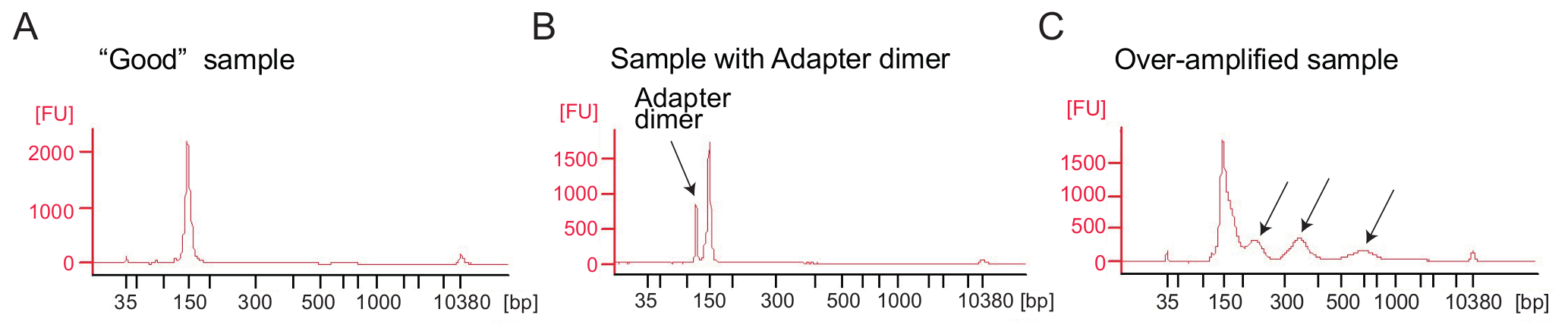
Ribosoma profilatura: amplificazione di PCR di cDNA library: amplificazione del cDNA (passo 4.9.1) è un passo fondamentale per garantire risultati di sequenziamento buona. Analizzare le librerie amplificate dall'analisi elettroforetici. Un buon campione di librerie amplificate Mostra un picco intorno a 140-160 bp (Figura 4A). Una quantità eccessiva di adattatore dimeri dovrebbero essere evitato (Figura 4B) e quei campioni dovrebbero essere ulteriormente purificati utilizzando la procedura di purificazione pagina secondo il protocollo del produttore (purificazione pagina dei prodotti di PCR). Troppo modello o troppi cicli di PCR possono causare eccessiva amplificazione caratterizzata dalla comparsa di bande di peso molecolare più elevato del previsto, i prodotti di PCR spalmati e adattatore dimero prodotti (Figura 4). Per la maggior parte dei campioni 1-5 µ l di cDNA circolare e 9 cicli PCR per l'amplificazione in genere produrrà una quantità sufficiente di prodotto della PCR corretta.
ChIP: cromatina tosatura: Condizioni ottimali di taglio devono essere regolato per ogni tipo di cellula. Determinare in anticipo le condizioni di taglio (ad es., numero di cicli, alta o bassa potenza). Utilizzare lo stesso numero di cellule e lo stesso volume per scopi di test, dal momento che una minore densità di cella aumenta l'efficienza di taglio. Cercate di evitare di sopra - o sotto-tosatura della cromatina. Frammenti di cromatina grande drammaticamente possono influire sui risultati di ChIP di intasamento, e sovra-tosatura può distruggere gli epitopi sulla proteina di interesse, che conduce ad una minore efficienza di associazione dall'anticorpo. In questo esperimento, i migliori risultati sono stati raggiunti quando la frazione principale della cromatina tosata era circa 1.000 bp o leggermente inferiore (Figura 5A).
Verifica ChIP di qPCR: Prima di iniziare il ChIP, si consiglia di verificare se l'anticorpo utilizzato è adatto per ChIP (se possibile, utilizzare gli anticorpi di grado di ChIP) di ChIP-qPCR. Verificare il ChIP per il sequenziamento di qPCR prima di iniziare la preparazione di biblioteca (vedi passo 5.6.5). Disegnare primers che si legano a un sito di destinazione noto della proteina di interesse. Se il luogo di destinazione esatta all'interno di un gene è sconosciuto, diverse coppie di primer consente di eseguire la scansione del gene e associati elementi regolatori. Per RNAPII ChIP di Th1 cellule Ifng, che è transcriptionally upregulated stimolazione e actina primer può essere utilizzato come controllo positivo. SOX9 e insulina servono come controllo negativo, dato che questi geni non sono espressi in cellule Th1 (figura 5B). Ricordate di non usare iniettori essone-spanning, che sono normalmente utilizzati per qPCR di mRNA. Un controllo di IgG è utilizzabile anche per dimostrare la specificità dell'anticorpo utilizzato. DNA immunoprecipitato può essere misurata con un fluorometro adatto (Vedi Tabella materiali). Quantità di DNA non specifico associato dal controllo IgG dovrebbe essere significativamente più basso rispetto alla quantità di DNA associato dall'anticorpo di interesse.
Replica: prova del significato biologico: Si consiglia vivamente di eseguire l'esperimento di cinetica per tutti i metodi a partire dallo stesso pool di cellule affinché le cellule hanno la stessa identità per tutti i campioni non trattati e trattati (Figura 2). Tuttavia, si consiglia di prendere piccole aliquote di intervalli di tempo principale per ogni metodo confrontare i campioni da un replicare biologico (ad es., di qPCR, analisi al FACS). Questo permette una stima approssimativa se il trattamento per entrambi repliche era riproducibile e si potrebbe procedere con sequenziamento. Convalida dei replicati deve essere eseguita mediante analisi bioinformatical. Riproducibilità dei risultati possono essere valutati in termini di correlazione tra valori FPKM tra repliche e visualizzati utilizzando grafici a dispersione (Figura 6).

Figura 1: Verifica delle condizioni ottimali d'etichettatura 4sU senza perturbare la fisiologia cellulare (figura da Dini et al. 11)
(A) rilevazione di apoptosi mediante analisi Citofluorimetrica: In vitro generato Th0 cellule sono state trattate con diverse concentrazioni di 4sU (indicati tra parentesi) per 0,5 h, h 1 e h 2, rispettivamente. Trattamento BH3I-1 è stato usato per indurre apoptosi determinata da Annexin V, mentre shock termico (5 min a 95 ° C) è stata usata per indurre la morte cellulare determinata dal 7-AAD. (B) analisi di Western blot per p53 di 4sU trattati e cellule T attivate: i campioni sono stati etichettati con 200 µM 4sU per il tempo indicato di attivazione, ad eccezione del punto di tempo di 0,5 h che è stato etichettato con 500 µM 4sU. (C) l'analisi Western blot di fosfo-EIF2a ed EIF2a totale nelle cellule Th1 attivate con le stesse condizioni di etichettatura come in (B). Thapsigargin è stato utilizzato come controllo positivo. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Panoramica schematica di un'installazione cinetica per tenere traccia delle modifiche del genoma
Questo schema illustra il programma di installazione di combinando 4sU-seq, totale RNA-seq, ribosoma Profiling e ChIP-seq per studiare i cambiamenti di genoma sopra il trattamento delle cellule. Pool di cellule e mettere da parte il numero di cellule per controllo non trattato. Curare le cellule rimanenti e dividere per ciascun punto di metodo e di tempo. Marcare le cellule non trattate/trattati per 4sU-seq con 4sU come descritto. Punti temporali e campioni per ogni metodo dipendono la questione biologica specifica all'esame. Prelevare campioni per ogni punto nel tempo e metodo e seguire la parte dedicata del protocollo. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: controllo di qualità di RNA 4sU-etichetta
RNA totale e biotinilati RNA ottenuto dalle cellule Th1 attivate sono stati analizzati su un Bioanalyzer. RRNA 18S e 28S rRNA sono mostrati e RNA integrità numero (RIN) è dato dallo strumento per determinare l'integrità del RNA. RIN dovrebbe essere ≥ 8 per garantire l'integrità del RNA. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Bioanalyzer profili del ribosoma profilatura librerie
(A), A buona biblioteca: l'esempio mostra un picco nell'intervallo di dimensioni previste (140-160 bp) e nessun ulteriore purificazione è necessaria. (B), in questo esempio viene illustrato eccessivo adattatore dimero prodotto amplificato (120 bp) rispetto il prodotto desiderato (140-160 bp). Questa libreria richiede ulteriore purificazione. (C), un campione di over-amplificato: picchi di peso molecolare più elevato del previsto e spalmato ampliconi PCR sono visibili (indicati dalle frecce). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Dimensioni di cromatina ottimale dopo la verifica del ChIP di qPCR e tosatura
(A) dell'agarosi gel foto mostra la dimensione del frammento ottimale della cromatina tranciata da tre campioni che erano tranciati per 25 cicli su un sonicatore e purificata come descritto prima nel protocollo. (B) risultati di Q-PCR di un ChIP RNAPII totale (anti-RNA Pol II, 8WG16, ab817) è rappresentato come una percentuale di input. Primer IFNg e actina sono stati utilizzati come un positivo mentre Sox9 e l'insulina sono controlli negativi (entrambi i geni non sono espressi in cellule Th1 attivate). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Confronto del biologico replica (figura da Dini et al. 11)
Rappresentante a dispersione confrontando i valori dell'espressione (FPKM) tra replica di appena trascritto (4sU) RNA 4 h dopo la stimolazione delle cellule Th1 attivate. La linea verde indica valori uguali FPKM e correlazione rigogliosa è indicato in ogni trama.
| Durata di etichettatura (min) | Concentrazione consigliata 4sU (µM) |
| 120 | 100 - 200 |
| 60 | 200 - 500 |
| 15 - 30 | 500 - 1000 |
| < 10 | 500 - 2000 |
Tabella 1: Consigliato 4sU concentrazioni (da Rädle, et al. 19)
L'intervallo di concentrazioni consigliate 4sU è indicato per diverse volte di etichettatura.
| Metodo. | Numero di cellulare | Quantità di RNA |
| 4sU-etichettatura | ≥ 2 x 107 | ≥60µg |
| Profilatura del ribosoma | ≥ 2 x107 | |
| ChIP-seq | ≥ 2 x 107 - 3 x 107 |
Tabella 2: Quantità richiesta primarie delle cellule di T
Quantità minima di cellule T primarie richieste per ciascun metodo. Gli importi possono essere meno quando si utilizzano altri tipi di cella.
Discussione
Analizzando l'intero processo di regolazione genica è necessario comprendere appieno gli adattamenti cellulari in risposta a uno stimolo specifico o trattamento. Combinare totale RNA-seq, 4sU-seq, ribosoma profilatura e ChIP-seq in diversi momenti porta un'analisi completa dei principali processi di regolazione genica nel corso del tempo. Per definire la messa a punto sperimentale, nonché i punti di tempo ottimale è necessaria una profonda comprensione dei processi biologici.
Poiché i metodi per studiare la regolazione genica migliorano rapidamente, questi protocolli possono essere adattati ai rapidi cambiamenti. Tuttavia, essi forniscono i metodi più importanti per lo studio di meccanismi di regolazione genica base in qualsiasi tipo di cellula. Qui, discutiamo alcune delle insidie e fatti si deve considerare quando si utilizzano questi metodi.
Cellule: Cellule devono essere altamente valide e, se utilizza primarie cellule isolate, purezza delle popolazioni delle cellule deve essere garantita (ad es., analisi di FACS per cellule di T primarie). Anche un po ' stressate cellule possono influenzare i risultati di questi metodi di sequenziamento molto sensibile e abbassare la quantità di RNA appena trascritta o tradotta e portare a letture indesiderate della risposta dello stress nei risultati del sequenziamento. La velocità di centrifugazione citata in questo protocollo per agglomerare le cellule è ottimizzata per le cellule T primarie. Quindi, regolare la velocità in funzione del tipo di cella.
4 sU effetti sulla fisiologia cellulare: Oltre alle opzioni di cui sopra per la verifica minima perturbazione di fisiologia cellulare con l'aggiunta di 4sU, ulteriore e/o ulteriori analisi possono essere eseguita, soprattutto quando i numeri di cellulare sono limitati. Effetti sulla proliferazione delle cellule possono essere verificati controllando il tempo di raddoppiamento delle cellule semplicemente contando le cellule marcate e non marcate. Induzione di stress nucleolar poteva essere testati anche analizzando la morfologia delle cellule tramite colorazione di immunofluorescenza di nucleolina e nuclei. Per verificare ulteriormente l'impatto di 4sU, espressione genica globale alterata potrebbe essere misurata correlando leggere conteggi da RNA totale con etichetta a RNA totale senza etichetta.
Numeri di cell: Per in vitro generato le cellule di T, si consiglia di iniziare con almeno la quantità di cellule indicate nella tabella 2. Scegliere un numero adeguato con il metodo in base al tipo di cellule. Poiché le cellule di T hanno meno citoplasma e RNA rispetto ad altre celle, è possibile che gli importi più bassi più probabili di altre cellule sarà adeguati. Per ChIP-seq, numeri di cellulare dipendono altamente l'anticorpo utilizzato e il livello di espressione della proteina di interesse all'interno delle cellule. Per istone o RNAPII ChIP, ambra cellule possono essere utilizzate, mentre i numeri di cellulare devono essere aumentate quando vengono utilizzati fattori di trascrizione, soprattutto se essi sono espressi a livelli bassi.
4 sU-etichettatura e RNA biotinylation: Quando si utilizza cellule aderenti, 4sU etichettatura può essere diretto come descritto da Rädle et al. 19 dal cellule 4sU incorporano molto rapidamente, può essere aggiunto direttamente al mezzo di sospensione, aderente o celle semi-aderente.
Si raccomanda di iniziare la biotinilazione con 60-80 µ g di RNA. Tuttavia, gli importi più bassi di RNA possono essere utilizzati, anche se non abbiamo provato per meno di 30 µ g. aggiungere un coprecipitant (per esempio, GlycoBlue) quando precipitazione RNA se il pellet è difficile da vedere. Duffy et al hanno anche dimostrato che la biotina methylthiosulfonate-attivato (MTS-biotina) reagisce in modo più efficiente con 4sU-identificato RNA oltre HPDP-biotina31. Quindi, potrebbe essere utile in considerazione il passaggio a MTS-biotina, in particolare, per il recupero di piccoli RNA, che tendono ad avere meno residui di uridina (riferisca al protocollo biotinylation menzionato dalla sede di Duffy et al; purificazione di RNA 4sU-etichettati, Procedure sperimentali).
Per il recupero di RNA appena trascritto, è possibile utilizzare perline paramagnetici o RNA pulitura della vostra scelta. Sempre tener conto che questi kit possono o non possono purificare per RNA specifici. Se siete interessati a Mirna, consideri, ad esempio, utilizzando i kit specifici per l'acquisizione di miRNA e sequenziamento.
Quantificazione del RNA trascritto recentemente: Per quantificare con precisione appena trascritto RNA, misurazione deve essere eseguita da un fluorometro adatto (Vedi Tabella materiali). Entro 1 h di esposizione 4sU, recentemente trascritto RNA rappresenta circa 1-4% di RNA totale. Recentemente trascritto RNA di 1 h con l'etichetta, le cellule di T attivate è costituito di ~ 90-94% di rRNA11.
Ribosoma profilatura: Quando si stabilisce il metodo, abbiamo determinato che utilizzando 1,5 x la quantità di nucleasi rispetto a quanto suggerito nel protocollo originale garantisce una corretta digestione. Inoltre, gli effetti contrari non sono stati segnalati per importi elevati di nucleasi. Poiché è molto difficile overdigest la RPFs mentre fanno parte del RNA vincolato dalle proteine ribosomiali, è possibile aumentare ancora leggermente la quantità per titolo la digestione ottimale nucleasi.
Se meno di 500 ng di RNA RPF sono stati recuperati nel passaggio 4.4.2, ripetere lo svuotamento del rRNA e piscina depurata RPFs con RNA Clean & concentratore-5 colonne. In alternativa, è possibile caricare campioni identici due vicino a vicenda sul gel (punto 4.5.3) e fette del gel piscina durante l'eluizione di RNA dal gel (passo 4.5.6).
Si consiglia di tagliare il RPFs su un gel più strettamente possibile alle bande nt 28 e 30. Questo aiuta a eliminare frammenti indesiderati da rRNA e tRNA, che più tardi diventerà parte della tua libreria e ridurre letture di sequenziamento per il tuo RPFs.
Si consiglia inoltre di evitare luce durante la purificazione del gel UV. Questo può creare Nick in frammenti di RNA, come pure i dimeri di pirimidina, che alla fine possono compromettere seriamente la libreria preparazione e risultati di sequenziamento.
Preparazione di biblioteca e l'ordinamento dei dati: Ribosoma protocollo di profilatura consente di generare una libreria di cDNA adatta per il sequenziamento. Campioni generati da 4sU-etichettatura possono essere direttamente utilizzati per la preparazione di libreria con qualsiasi kit di sequenziamento di RNA appropriato. Dal RNA appena trascritto, soprattutto quando si usa bruscamente etichettatura volte, potrebbe non essere ancora poliadenilazione, nessuna selezione di poly-A deve essere eseguita. Invece, è consigliabile lo svuotamento di rRNA per prevenire la riduzione della profondità sequenziamento per il campione reale. Utilizzando cellule di T, abbiamo iniziato con 400 ng di RNA totale e recentemente trascritto (a seconda del kit, vedi materiali), eseguita rRNA svuotamento e ridotti cicli di amplificazione di PCR di minimizzare bias della PCR. Preparazione di libreria può essere effettuato con meno materiale di partenza. Per tenere conto di numeri di complessità di libreria di cicli PCR deve essere ottimizzato.
Per ChIP-seq ci sono anche molti kit disponibili per la preparazione di biblioteca. Nella nostra biblioteca di mani preparazione lavorato bene a partire con 2 ng di DNA ChIP (Vedi materiali per un suggerimento su quale kit da utilizzare). Assicuratevi di controllare gli indici per il bilanciamento del colore durante la sequenziazione. Si consiglia una profondità di sequenziamento di ≥ 40 x 106 letture ogni per 4sU-seq, totale RNA-seq e campioni di ChIP-seq e ≥ 80 x 106 letture per ribosoma campione di profilo. La profondità di sequenziamento dipende il campione e l'analisi bioinformatica a valle e dovrebbe essere considerata attentamente. Per analizzare introniche letture per l'impionbatura cotranscriptional, 100 sequenziamento accoppiato-fine bp deve essere scelto.
Sequencing bias: Sequenziamento è diventata il gold standard nel determinare cambiamenti globali nella trascrizione, traduzione o associazione di fattore di trascrizione. In anni recenti, i metodi esistenti sono stati spinti al limite o nuove tecniche sono state sviluppate per la sequenziazione di partenza sempre più piccole quantità di RNA. Ciò richiede amplificazione del cDNA, che introduce rumore o pregiudizi. Recentemente, identificatori univoci molecolari (UNMIS) sono stati sviluppati per sperimentalmente identificare duplicati introdotti dalla PCR. Recentemente, è stato indicato che l'UNMIS solo leggermente migliorare potenza sequenziamento e tasso di falsi scoperta per di espressione genica differenziale32. Tuttavia, considerare l'utilizzo di identificatori univoci molecolari (UNMIS) per tutte le librerie di sequenziamento al controllo per complessità di biblioteca, soprattutto quando si inizia con basse quantità di RNA e quando sono necessari molti cicli di PCR.
Buffer e soluzioni di riserva: Tutti i buffer per 4sU-seq e profilatura ribosoma devono essere preparati a condizioni rigorose di RNAsi-libera utilizzando acqua priva di nucleasi. È consigliabile acquistare pre-fatte senza nucleasi NaCl, Tris-HCl, EDTA, citrato di sodio e acqua. Per garantire condizioni di nucleasi-free, una soluzione decontaminante RNasi utilizzabile per pulire pipette o superfici. Tutti i buffer per ChIP-seq devono essere almeno dnasi-libero e può essere conservato a temperatura ambiente. Sempre aggiungere gli inibitori della proteasi e, facoltativamente, inibitori delle fosfatasi bene prima dell'uso e conservare il ghiaccio.
Bioinformatica: Analisi di tutti i dati di sequenziamento (cioè, ChIP-seq, RNA-seq e profilatura ribsosome) comporta il controllo di qualità (ad esempio, utilizzando FastQC, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), rimozione di adattatore (ad es., con cutadapt20) seguita da mappatura del genoma di riferimento per le cellule in fase di studio. Per dati di RNA-seq (sia totale e 4sU-seq), come pure ribosoma profiling dei dati, un impiombato mapper di RNA-seq è richiesto, come ContextMap 221. Per gli allineamenti di dati unspliced ChIP-seq, è sufficiente utilizzare BWA-MEM22 . Espressione genica può essere calcolato utilizzando il modello (letture per Kilobase dell'essone per milione frammenti mappato) RPKM1, dopo la determinazione leggere conteggi per gene usando un programma, ad esempio featureCounts23. Per la chiamata di picco da ChIP-seq dati, una serie di programmi è disponibile, ad esempio, Mac24 o GEM25. Ulteriori analisi a valle possono essere eseguite in R26, in particolare utilizzando gli strumenti forniti dal progetto Bioconductor27.
Qui, una sfida importante nell'integrazione dei livelli di RNA 4sU - e totale e attività traduzionale dal ribosoma profilatura è normalizzazione. Un classico approccio per affrontare questo problema è quello di normalizzare a livelli di geni. Per ridurre il rumore dovuto le fluttuazioni casuali per singoli geni, si raccomanda di utilizzare non solo poche geni ma livelli mediani per un insieme più ampio, ad esempio il > 3.000 geni compilati da Eisenberg e Levanon28 . Per il calcolo dei tassi di fatturato di RNA da rapporti di 4sU-al che RNA totale, normalizzazione è basata sul fatturato mediano di prezzo (ad es., supponendo un'emivita RNA 5 h)29. Tuttavia, poiché questo non si assume nessun cambiamenti complessivi per geni, si consiglia di utilizzare approcci di analisi indipendente di normalizzazione, ad esempio, di una serie temporale dei tipi di dati diversi per identificare gruppi di geni di clustering basato su correlazione con comportamento distinto nella trascrizione e nella traduzione durante l'attivazione. Per una descrizione dettagliata sull'integrazione di bioinformatical dei tipi di dati diversi, ci riferiamo al originale pubblicazione11.
Analisi di integrazione di dati e tariffe di fatturato: Un libro recentemente pubblicato33 confrontando emivite determinate da un controllo di gene multiplex (MGC) ai metodi globali potrebbe mostrare che emivite correlato meglio con quelli ottenuti mediante metodi di etichettatura metaboliche rispetto ad altri metodi (ad esempio, generale inibizione della trascrizione di farmaci). Tuttavia, va ricordato che le differenze tra calcoli di Half-Life possono sorgere e sono stati descritti 15,34. Ci conto per la maggior parte dei problemi e le differenze introdotte dalla risposta allo stress dovuto all'esposizione prolungata 4sU. Pertanto, è indispensabile escludere la risposta allo stress introdotta da 4sU-etichettatura. Per convalidare ulteriormente i tassi di fatturato, si consiglia l'uso di MGCs.
Inoltre, un set di dati come generato qui potrebbe essere utilizzato anche per un più integrante dati analisi (ad es., regolamento di RNA lunghi non codificanti)35,36.
Divulgazioni
Gli autori dichiarano di non avere nessun concorrenti interessi finanziari.
Riconoscimenti
Si ringrazia Lars Dölken per consigli per stabilire 4sU etichettatura per cellule di T primarie; Elisabeth Graf e Thomas Schwarzmayr per aiuto critico nelle generazioni di biblioteca e sequenziamento; Dirk Eick e Andrew Flatley per fornire gli anticorpi delle cellule T e RNAPII; N. Henriette Uhlenhaut e Franziska Greulich per aiuto nella preparazione di libreria per ChIP-seq; Caroline C. Friedel è stata sostenuta da sovvenzioni FR2938/7-1 e CRC 1123 (Z2) dalla Deutsche Forschungsgemeinschaft (DFG); Elke Glasmacher è stata sostenuta dalla concessione GL 870/1-1 dalla Deutsche Forschungsgemeinschaft (DFG) e il centro tedesco per ricerca di diabete (DZD), Helmholtz Zentrum München.
Materiali
| Name | Company | Catalog Number | Comments |
| 4sU-labeling | |||
| 4-thiouridine (100 mg) | Carbosynth | 13957-31-8 | Prepare 50 mM stock in sterile H2O/PBS; store at –20°C in aliquots of 50-500 µl; do not refreeze. |
| 1.5 ml safe-lock tubes | Eppendorf | 30121589 | Optional |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72692005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72694005 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| Agencourt RNAClean XP | Beckman Coulter | A63987 | We recommend to use these paramagnetic beads. Aliquot and store at 4°C |
| Chloroform | Sigma Aldirch | 372978 | WARNING – HAZARDOUS TO HEALTH |
| Dimethylformamide | Sigma Aldrich | D4551 | |
| Dithiothreitol (DTT) | Roth | 6908.1 | Prepare as 100 mM DTT in nuclease-free H2O; always prepare fresh |
| Ethanol | Merck | 1.00983.1000 | |
| EZ-Link Biotin-HPDP (50 mg) | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg Biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4°C in aliquots of 1 ml. DMF dissolves some plastic materials. We recommend to use glass pipettes to transfer DMF from ist stock glass bottle to 50 ml Falcon tubes. |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 | |
| Isopropanol | Merck | 1.09634.1011 | |
| NaCl (5M) | Sigma Aldrich | 71386 | Stock solution |
| nuclease-free EDTA (500 mM ), pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| Nuclease-free H2O | Sigma Aldrich | W4502 | Stock solution |
| nuclease-free Tris Cl (1M), pH 7.4 | Lonza | 51237 | Stock solution |
| Phase Lock Gel Heavy tubes (2.0 ml) | 5Prime | 2302830 | Use in step 1.3.4. |
| Polypropylene 15 ml centrifuge tubes | Greiner Bio-One | 188271 | Or equivalent; they have to tolerate up to 15,000 × g |
| QIAzol Lysis Reagent (200 ml) | Qiagen | 79306 | Use this or equivalent TRI reagent for RNA isolation, WARNING – CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol) |
| Qubit RNA HS assay kit | Life Technologies | Q32852 | Use this kit for quantifying RNA quantity in step 1.4.11 |
| RNeasy MinElute Kit | Qiagen | 74204 | Optional; includes Buffer RLT |
| Sodium citrat | Sigma Aldrich | C8532 | Prepare 1.6 M stock solution using nuclease-free water |
| Tween 20 | Sigma Aldrich | P1379 | |
| µMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store at 4°C, includes µMacs columns used in step 1.4.6. (store at RT) |
| Cell viability and stress assay | |||
| PE Annexin V Apoptosis Detection Kit I | BD Biosciences | 559763 | Optional |
| Thapsigargin | Sigma-Aldrich | T9033 | Optional |
| p53 | abcam | ab26 | Optional |
| p-EIF2a (Ser51) | Cell Signaling | 9721 | Optional |
| BH3I-1 | Sigma-Aldrich | B 8809 | Optional |
| Buffers | |||
| 4sU Washing Buffer | store at RT | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O | |
| Biotinylation Buffer (10x) | store at 4 °C | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water; make aliquots of 1 ml; store at 4°C | |
| RNA precipitation buffer | store at RT | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | |
| Equipment | |||
| 2100 Bioanalyzer instrument | Agilent | G2939BA | |
| RNA 6000 Nano Kit | Agilent | 5067-1511 | Use this kit to verify RNA integrity in step 1.3.10 |
| RNA 6000 Pico Kit | Agilent | 5067-1513 | Optional |
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use in step 1.2.2/3.1.8/3.3.3/3.4.3 |
| High-speed centrifuge | Thermo Scientific | Heraeus Multifuge X3R | Or equivalent equipment capable of reaching 13,000 × g |
| High-speed rotor | Thermo Scientific | Fiberlite F15-6 x 100y | |
| Adaptors for 15 ml tubes | |||
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer C | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 µMacs columns. |
| Ultra-fine scale | Mettler Toledo | ML204T | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
| DynaMag-2 Magnet-1 each | Life Technologies | 12321D | |
| RNaseZap | Sigma | R2020 | Optional |
| TruSeq stranded total RNA library prep kit | Illumina | RS-122-2201 | Or equivalent. For T cells we used 400 ng 4sU and Total RNA with 11 cycles for PCR amplification. rRNA depletion is included in this kit |
| Nanodrop | Thermo Scientific | use a Nanodrop or equivalent instrument to measure RNA concentration | |
| Ribosome Profiling | |||
| TruSeq Ribo Profile kit (Mammalian or Yeast) | Illumina | RPYSC12116 (Yeast) | |
| TruSeq Ribo Profile kit (Mammalian or Yeast) | Illumina | RPHMR12126 (Mammalian) | |
| Illustra MicroSpin S-400 HR Columns | GE Healthcare | 27-5140-01 | |
| RNA Clean & Concentrator-25 kit | Zymo Research | R1017 | |
| RNA Clean & Concentrator-5 kit | Zymo Research | R1015 | |
| Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat) | Illumina | MRZG126 or MRZG12324 | |
| (High Sensitivity DNA Kit) | Agilent Technologies | 5067-4626 | Already needed for 4sU-seq |
| All other consumables and equipment are listed in the User guide | !!! | Carefully read the user guide and order required consumables in advance (consider a long delivery time for some consumables e.g. gels) | |
| ChIP | |||
| 10 mM Tris-HCl (pH 8.0) | gereral lab supplier | ||
| 100 bp Plus Marker | Thermo Fisher | SM0323 | |
| 16 % Formaldehyde | Thermo Fisher | 28908 | Add to a final concentration of 1 % |
| 70% EtOH | gereral lab supplier | Always prepare fresh | |
| Agarose | gereral lab supplier | ||
| Agencourt RNAClean XP beads | Beckman Coulter | A63987 | We recommend to use these paramagnetic beads. Aliquot and store at 4°C |
| ChIP library preparation kit | KapaBiosystems | KK8504 | Or use the kit of your choice |
| DNA low bind microcentrifuge tubes | Eppendorf | Z666548-250EA | or equivalent |
| Dynabeads Protein G | Invitrogen | 10004D | Use these superparamagnetic beads coupled to protein G in step 4.3.1.; Bring to RT before use |
| Glycine | gereral lab supplier | Prepare a 2M stock solution | |
| Glycogen | Roche | 10-901-393-001 | |
| MinElute PCR Purification Kit | Qiagen | 28004 | Use this kit (or equivalent) to purify chromatin in step 4.2.4. |
| Phosphatase Inhibitor (PhosStop) | Roche | 4906837001 | Add freshly to the buffer and keep on ice |
| Power SYBRgreen Master mix | Thermo Fisher | 4367659 | |
| Protease Inhibitor (cOmplete, EDTA-free) | Roche | 11873580001 | Add freshly to the buffer and keep on ice |
| Proteinase K | Invitrogen | 25530049 | |
| Qubit dsDNA HS Assay kit | Invitrogen | Q32851 | Use this kit for quantifying DNA quantity in step 4.6.4. on a Qubit Fluorometer |
| Rnase, DNase free | Roche | 11-119915001 | |
| Salmon sperm (sonicated to around 100bp) | Sigma | D1626 | |
| TE pH 8.0 | gereral lab supplier | ||
| Antibodies (ChIP grade if possible) | |||
| anti-RNA Pol II [8WG16] | abcam | ab817 | |
| anti-Histon H3K36me3 | abcam | ab9050 | |
| or antibody of interest | |||
| Buffers | |||
| Binding/Blocking buffer | Store at RT | PBS with 0.5 % BSA and 0.5 % Tween 20 | |
| Cell-Lysis buffer | Store at RT | 5 mM Pipes [pH 8.0], 85 mM KCl, and 0.5 % NP40 | |
| ChIP IP buffer | Store at RT | 0.01 % SDS; 1. 1% Triton X-100;1.2 mM EDTA; 16.7 mM Tris-HCl, pH 8.1; 16.7 mM NaCl | |
| Elution buffer | Store at RT up to 6 months | 10 mM Tris-HCl (pH 8.0), 5 mM EDTA (pH 8.0), 300 mM NaCl and 0.5 % SDS | |
| Nuclei-Lysis buffer | Store at RT | 50 mM Tris [pH 8.0], 10 mM EDTA, and 1 % SDS | |
| Wash buffer I | Store at RT | 0.1 % SDS; 1 % Triton X-100; 2 mM EDTA; 20mM Tris-HCL pH 8.1; 150 mM NaCl | |
| Wash buffer II | Store at RT | 0.1 % SDS; 1 % Triton X-100; 2 mM EDTA; 20 mM Tris-HCL pH 8.1; 500 mM NaCl | |
| Wash buffer III | Store at RT | 0.25 M LiCl; 1% NP-40; 1 mM EDTA; 10 mM Tris-HCl, pH 8.1 | |
| Equipment | |||
| 2100 Bioanalyzer instrument | Agilent | G2939BA | use this instrument for electrophoretical analysis |
| Nanodrop | Thermo Scientific | ||
| Bioruptor TBX microtubes 1.5 ml | Diagenode | C30010010 | |
| or tubes special for your sonication device | |||
| Bioruptor sonication device or sonication device of your choice | Sonication of T cells with Bioruptor: 20 - 25 cycles (30 s on, 30 s off at high in two 1.5 ml bioruptor microtubes with 500 µl each tube) | ||
| Magnetic stand for tubes | |||
| Thermomixer | |||
| Agarose gel electrophoresis | |||
| Qubit Fluorometer | Thermo Scientific | Use this Fluorometer for quantifying low amounts of RNA/DNA |
Riferimenti
- Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 5 (7), 621-628 (2008).
- Chavez, S., Garcia-Martinez, J., Delgado-Ramos, L., Perez-Ortin, J. E. The importance of controlling mRNA turnover during cell proliferation. Curr Genet. 62 (4), 701-710 (2016).
- Rutkowski, A. J., et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat Commun. 6, 7126 (2015).
- Ozsolak, F., Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 12 (2), 87-98 (2011).
- Carninci, P., et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 38 (6), 626-635 (2006).
- Churchman, L. S., Weissman, J. S. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 469 (7330), 368-373 (2011).
- Core, L. J., Waterfall, J. J., Lis, J. T. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 322 (5909), 1845-1848 (2008).
- Hah, N., et al. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 145 (4), 622-634 (2011).
- Dolken, L., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14 (9), 1959-1972 (2008).
- Paulsen, M. T., et al. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 67 (1), 45-54 (2014).
- Davari, K., et al. Rapid Genome-wide Recruitment of RNA Polymerase II Drives Transcription, Splicing, and Translation Events during T Cell Responses. Cell Rep. 19 (3), 643-654 (2017).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. 22 (10), 2031-2042 (2012).
- Park, P. J. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 10 (10), 669-680 (2009).
- Blecher-Gonen, R., Barnett-Itzhaki, Z., Jaitin, D., Amann-Zalcenstein, D., Lara-Astiaso, D., Amit, I. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat Protoc. 8 (3), 539-554 (2013).
- Schwanhausser, B., et al. Global quantification of mammalian gene expression control. Nature. 473 (7347), 337-342 (2011).
- Larsson, O., Tian, B., Sonenberg, N. Toward a genome-wide landscape of translational control. Cold Spring Harb Perspect Biol. 5 (1), a012302 (2013).
- Brar, G. A., Weissman, J. S. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol. 16 (11), 651-664 (2015).
- Hafner, M., et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 141 (1), 129-141 (2010).
- Radle, B., Rutkowski, A. J., Ruzsics, Z., Friedel, C. C., Koszinowski, U. H., Dolken, L. Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J Vis Exp. (78), (2013).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 17 (1), 10-12 (2011).
- Bonfert, T., Kirner, E., Csaba, G., Zimmer, R., Friedel, C. C. ContextMap 2: fast and accurate context-based RNA-seq mapping. BMC Bioinformatics. 16, 122 (2015).
- Li, H., Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 26 (5), 589-595 (2010).
- Liao, Y., Smyth, G. K., Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 30 (7), 923-930 (2014).
- Zhang, Y., et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9 (9), R137 (2008).
- Guo, Y., Mahony, S., Gifford, D. K. High resolution genome wide binding event finding and motif discovery reveals transcription factor spatial binding constraints. PLoS Comput Biol. 8 (8), e1002638 (2012).
- Team, R. D. C. . R: A Language and Environment for Statistical Computing. , (2016).
- Huber, W., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nature Methods. 12 (2), 115-121 (2015).
- Eisenberg, E., Levanon, E. Y. Human housekeeping genes, revisited. Trends Genet. 29 (10), 569-574 (2013).
- Friedel, C. C., Dolken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol Biosyst. 5 (11), 1271-1278 (2009).
- Burger, K., et al. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol. 10 (10), 1623-1630 (2013).
- Duffy, E. E., Rutenberg-Schoenberg, M., Stark, C. D., Kitchen, R. R., Gerstein, M. B., Simon, M. D. Tracking Distinct RNA Populations Using Efficient and Reversible Covalent Chemistry. Mol Cell. 59 (5), 858-866 (2015).
- Parekh, S., Ziegenhain, C., Vieth, B., Enard, W., Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci Rep. 6, 25533 (2016).
- Baudrimont, A., et al. Multiplexed gene control reveals rapid mRNA turnover. Sci Adv. 3 (7), e1700006 (2017).
- Rabani, M., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 29 (5), 436-442 (2011).
- Schlackow, M., Nojima, T., Gomes, T., Dhir, A., Carmo-Fonseca, M., Proudfoot, N. J. Distinctive Patterns of Transcription and RNA Processing for Human lincRNAs. Mol Cell. 65 (1), 25-38 (2017).
- Mukherjee, N., Calviello, L., Hirsekorn, A., de Pretis, S., Pelizzola, M., Ohler, U. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat Struct Mol Biol. 24 (1), 86-96 (2017).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati