
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Collection and Processing of Lymph Nodes from Large Animals for RNA Analysis: Preparing for Lymph Node Transcriptomic Studies of Large Animal Species
In This Article
Summary
This protocol provides an overview of procedures for the isolation of RNA for the transcriptomic profiling of lymph node tissues from large animals, including steps in the identification and excision of lymph nodes from livestock and wildlife, sampling approaches to provide consistency across multiple animals, and considerations plus representative results for the post-collection preservation and processing for RNA analysis.
Abstract
Large animals (both livestock and wildlife) serve as important reservoirs of zoonotic pathogens, including Brucella, Mycobacterium bovis, Salmonella, and E. coli, and are useful for the study of pathogenesis and/or spread of the bacteria in natural hosts. With the key function of lymph nodes in the host immune response, lymph node tissues serve as a potential source of RNA for downstream transcriptomic analyses, in order to assess the temporal changes in gene expression in cells over the course of an infection. This article presents an overview of the process of lymph node collection, tissue sampling, and downstream RNA processing in livestock, using cattle (Bos taurus) as a model, with additional examples provided from the American bison (Bison bison). The protocol includes information about the location, identification, and removal of lymph nodes from multiple key sites in the body. Additionally, a biopsy sampling methodology is presented that allows for a consistency of sampling across multiple animals. Several considerations for sample preservation are discussed, including the generation of RNA suitable for downstream methodologies like RNA-sequencing and RT-PCR. Due to the long delays inherent in large animal vs. mouse time course studies, representative results from bison and bovine lymph node tissues are presented to describe the time course of the degradation in this tissue type, in the context of a review of previous methodological work on RNA degradation in other tissues. Overall, this protocol will be useful to both veterinary researchers beginning transcriptome projects on large animal samples and to molecular biologists interested in learning techniques for in vivo tissue sampling and in vitro processing.
Introduction
RNA-sequencing analysis of the transcriptome of lymph nodes provides the opportunity to characterize the immune response of animals to a variety of pathogens. While this methodology has been utilized extensively in mice, analyses have recently been expanding into larger mammals1,2. Livestock/large animal lymph nodes can be used to characterize host responses to an infection, not only for their use in vaccine or genetic susceptibility studies and for the identification of targets for drug development, but also as model systems for human studies on zoonotic diseases. For example, in the case of brucellosis (a zoonotic bacterial disease that impacts half a million people around the world each year), despite significantly increased costs, studies in sheep or goats are more relevant to the human infection and human vaccine development than laboratory animal models. Mouse infection models recapitulate the reticuloendothelial system infection but not the characteristic clinical signs3.
In large animal experiments as compared to laboratory animal studies, the process of tissue harvesting necessarily involves a longer delay between the euthanasia and the tissue collection, which presents a potential challenge for the preservation of high-quality RNA. Intact RNA is essential for the generation of biologically relevant transcriptomic data. The generation of high-quality RNA from tissue samples is particularly critical for large animal pathogen studies conducted in containment facilities. Such studies are inherently more difficult to perform as they not only require approved facilities and highly trained personnel but also carry significant financial costs, which, depending on the work, can range from tens to hundreds of thousands of dollars. These types of studies also involve a cross-disciplinary collaboration and cross-disciplinary knowledge for their completion, adding to their complexity. Therefore, training on, development of, and adherence to a streamlined system for the sample collection and preservation provides significant benefits for downstream molecular studies of tissues from infected animals.
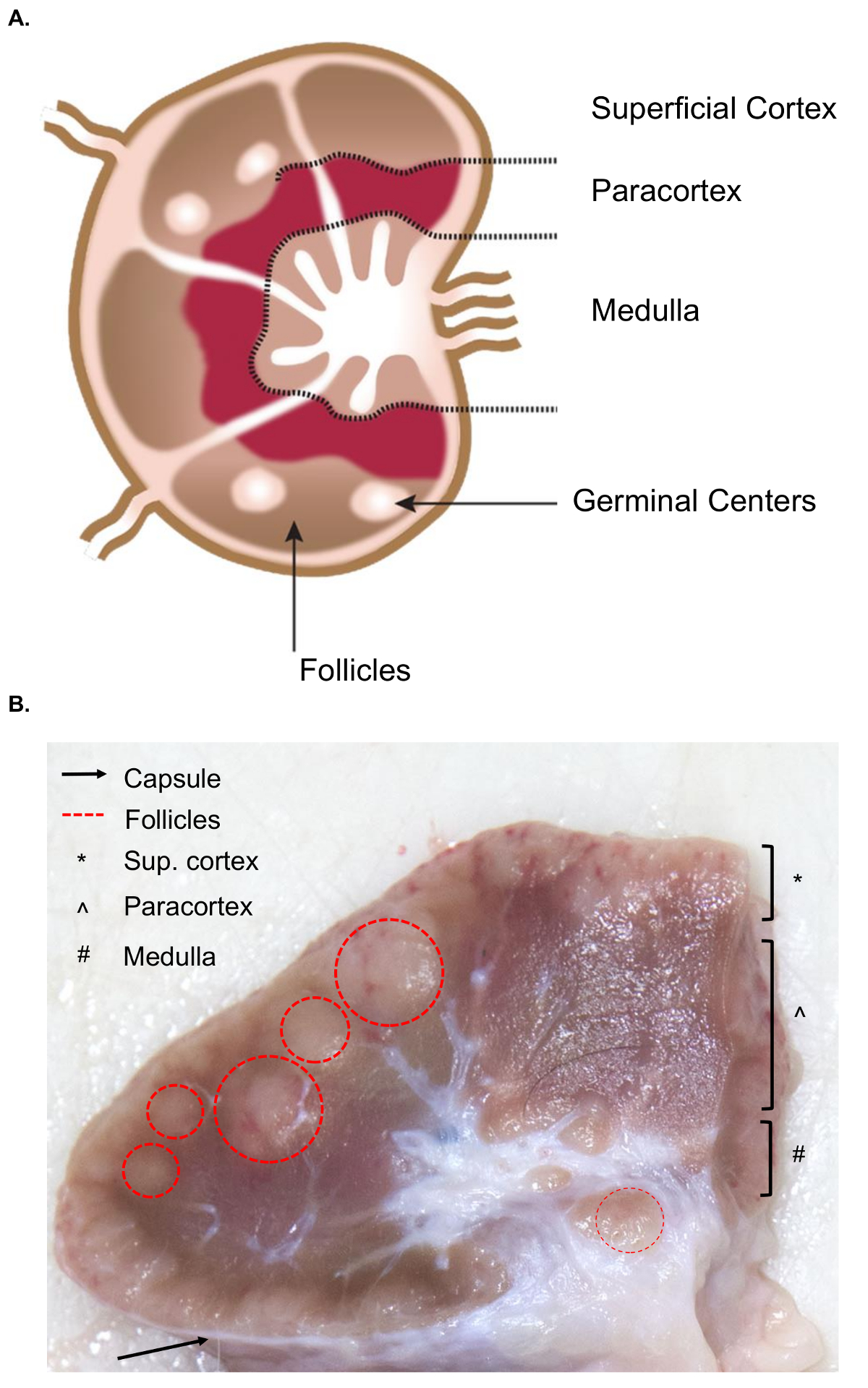
The collection of larger lymph nodes presents additional challenges for the tissue collection compared to the similar sampling of murine lymph nodes. The preparation for the sample excision necessitates a basic understanding of the anatomy of the lymph node, including the relevant internal structures. The structure of a lymph node is comprised of lymphoid lobules surrounded by sinuses filled with lymph. These structures are enclosed within a tough, fibrous capsule.4 A lymphoid lobule is the "basic anatomical and functional unit of the lymph node" and is composed of follicles, a deep cortical unit, and medullary cords and sinuses4 (Figure 1A). B and T lymphocytes are home to the follicles and deep cortical units, respectively. These structures provide a 3D scaffold and facilitate the interaction between the lymphocytes and antigen or antigen presenting cells.
Grossly, follicles and deep cortical units can be identified on cut surface as they contain a denser reticular meshwork and appear darker than the sinuses, which are comprised of a more delicate reticular meshwork and appear lighter (Figure 1B). By convention, pathologists refer to the regions of the lymph nodes as the superficial cortex (follicles), the paracortex (deep cortical units) and the medulla (medullary cords and sinuses). A proper examination of all three regions has been deemed as best practice in routine pathological examination guidelines for lymph nodes5. Note that there is a considerable variation in the consistency, size, and color of lymph nodes, even within a single animal. As animals age, their lymph nodes will tend to decrease in size and become firmer than those of younger animals, typically due to an increase in their connective tissue and a reduction of the normal lymphoid structure6,7.

Figure 1. Anatomy of the lymph node. (A) This cartoon image shows the anatomy of the lymph node, depicting key structures. (B) This still image shows a bovine lymph node cut in cross-section. The relevant structures/layers that are visible to the naked eye are highlighted. Please click here to view a larger version of this figure.
{kind=link}
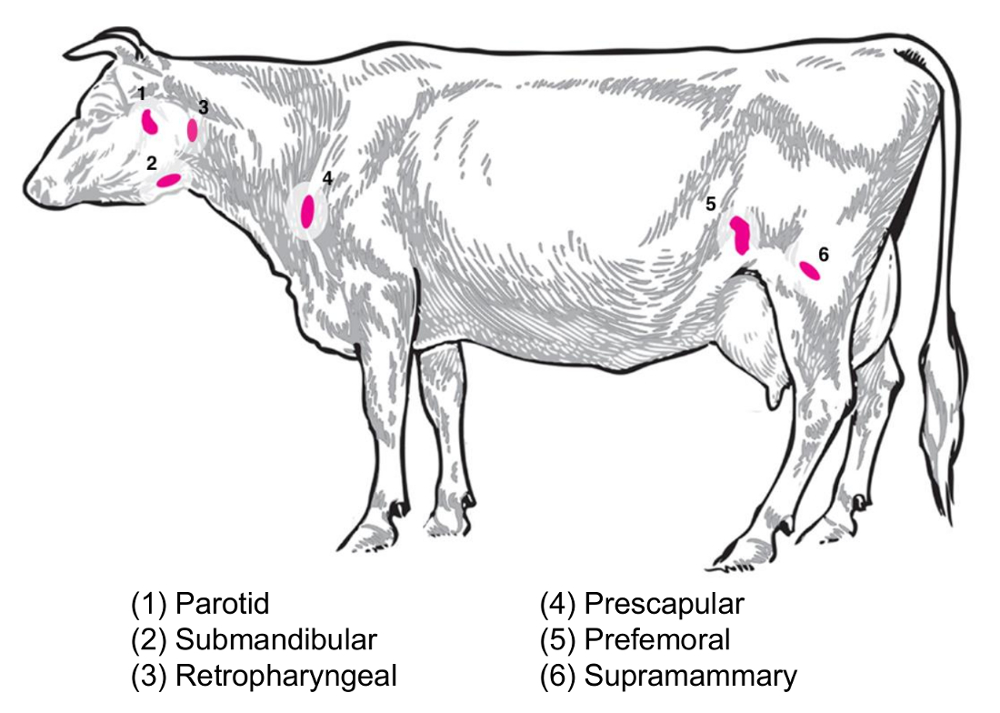
Depending on the experimental question, different lymph nodes will be of interest for the collection and analysis. Peripheral lymph nodes are those located deep in the subcutaneous tissue. In cattle, peripheral or superficial lymph nodes often used in clinical and experimental practice include parotid, submandibular, retropharyngeal, prescapular, prefemoral (precrural) and superficial inguinal (supramammary in females, scrotal in males) (Figure 2). In Table 1, the properties of key superficial lymph nodes, as described in the cattle system8, are outlined. Below, some potential lymph node collection plans for infectious bacterial diseases of cattle are presented as a starting point for the investigation.
Brucella abortus/Brucella melitensis: Standard necropsies for B. abortus-infected cattle and B. melitensis-infected goats at the National Animal Disease Center recover supramammary, prescapular, and parotid lymph node tissue, both for the grinding for the bacterial enumeration and for the RNA preparation for the host RNA expression profiling. B. abortus can be regularly recovered in each of these lymph nodes in experimentally infected cattle9. The presence of bacteria in each of these lymph node types can be detected in B. melitensis-infected goats up to at least nine months post-infection using the RNA-based methodologies from our studies (Boggiatto et al., unpublished). Salmonella sp.: The prescapular, subiliac (prefemoral), and mesenteric lymph nodes have been useful during the profiling of cattle carcasses for a Salmonella prevalence10,11,12 and would be of potential interest for transcriptomic studies. E. coli O157:H7: Mesenteric lymph nodes (at the middle small intestine and distal small intestine locations) can be the sites of an occasional recovery of the bacteria in infected calves (but not in infected adult cattle)13. Leptospirosis (Leptospira sp.): A chronic persistence of the bacteria has been observed in the lymph nodes draining the mammary gland14. Mycobacterium bovis: In cattle, the bacteria have been recovered post-experimental infection from the mediastinal and tracheobronchial lymph nodes of calves15. Additionally, lymph node RNA has been utilized to examine large animal host responses to viruses, such as the porcine reproductive and respiratory syndrome virus2. Figure 2 depicts the location of a subset of these major lymph nodes in the cattle body.

Figure 2: Cartoon depicting selected lymph node locations in Bos taurus. The numbered lymph nodes are annotated. Please click here to view a larger version of this figure.
{kind=link}
In this paper and the associated video, we present a protocol for the isolation of large animal lymph nodes for RNA studies, designed to be informative for molecular biologists involved in transcriptomic studies of large animal infections. First, we provide an overview of the isolation procedure for the lymph nodes, using sampling from bovine and bison tissues as examples. Paired with this demonstration, as displayed in the video, is a workflow for a reproducible tissue sampling for RNA isolation. Next, we describe important considerations for the processing of an infected lymph node, with a focus on safety, consistency, and RNA quality.
The preparation of RNA from the tissue with an acidified phenol-guanidine isothiocyanate reagent is based on the original method of Chomczynski and Sacchi16,17, with a purification over silica-based spin columns in the presence of chaotropic agents based on the original work of Vogelstein and Gillespie18. We also examine the potential for the recovery of RNA for transcriptomics from cattle lymph nodes preserved by alternative methods. Finally, we explore the impact of the time variable on the RNA quality in large animal necropsies, including a representative experiment depicting the effect of an increase in time between the euthanasia and the sampling on the recovered RNA profile from bison and bovine lymph nodes. This article will be useful not only to molecular biologists but also to veterinary researchers commencing transcriptomic studies.
Protocol
The animal necropsy procedures depicted here are covered under approved IACUC protocols at the National Animal Disease Center, Ames, IA. All experiments were conducted in accordance with the approved guidelines for animal care and welfare.
1. Pre-planning Before Necropsy
- Before the necropsy, prepare the following materials for transport to the necropsy suite:
- 1.5 or 2 mL microcentrifuge tubes filled with ~ 1.0 mL of RNA preservation solution, in a rack or transport container. Be sure to follow the manufacturer's guidelines for the ratio of the volume of the preservation solution to the volume of the tissue.
- Disposable scalpels and clean, autoclaved forceps: one set for dissecting the skin and a different set for collecting the lymph nodes (one for each lymph node, per animal), thus preventing a cross-contamination of the skin and environmental flora with the lymph node tissues.
- 3 mm punch biopsy tools, necropsy knives, a sharps container for the used scalpels and biopsy tools, and cutting boards or disposable trays for use in the lymph node dissection.
- Use personal protective equipment, including PPE required for work at the appropriate BSL level for the system.
- Autoclave reusable metal tools, including the forceps, in metal pans prior to the necropsy, using the utensils/dry goods setting.
2. Identification and Sampling of Lymph Nodes in Cattle and Bison

Figure 3. Lymph nodes of the bovine head and neck. (A) This cartoon image shows selected lymph nodes of the head and neck of Bos taurus. (B) This image shows the parotid lymph node in cross-section (left) as compared to the parotid salivary gland in cross-section (right). Note the difference in textures between the two tissue types. Please click here to view a larger version of this figure.
{kind=link}
- Isolation of prescapular lymph nodes in cattle and bison
- Euthanize the animal (cattle or bison of any age, as appropriate for the experiment) based on the local IACUC protocol. Confirm the death by methods specifically described in the institutional IACUC protocol, including the lack of a heartbeat, the lack of respiration, and the absence of a corneal reflex. Move the animal to the necropsy floor.
Note: There are various methods of humane euthanasia used in the laboratory practice of large animals. However, intravenous administration of pentobarbital sodium and phenytoin sodium solution is the most commonly approved and utilized form. Consult the AVMA Guidelines for the euthanasia of animals.19
CAUTION: Follow all relevant biosafety protocols for a necropsy at the host institution, including the guidelines for the safe disposal of sharps in a puncture-proof container, and the decontamination of reusable metal knives and forceps. This protocol generates sharps waste from the use of disposable scalpels. Wear personal protective equipment as dictated by the institutional guidelines, including boots, coveralls, gloves, and eye protection. - Identify 3 participants to work on the animal tissues.
Note: Here, dissector 1 will be in charge of the large-scale sectioning of the desired animal tissues, dissector 2 will recover the lymph nodes from the different animal sections, and dissector 3 will work at a separate table to isolate sections from each lymph node and transfer them to tubes with a preservative solution. - Begin by placing the animal in lateral recumbency.
- Extend the shoulder by moving the front limb back at the level of the knee (carpus); this motion will bring the shoulder back and allow for an easier palpation of the lymph node. Feel for the presence of the lymph node "lump".
Note: During disease conditions, lymphadenopathy may be present, which makes the identification of lymph nodes easier, but this process can also affect the consistency, composition, and architecture of the lymph node. - Once located, and with the shoulder still extended, use a scalpel or necropsy knife to make a skin incision along the outline of the shoulder.
Note: Do not restrict the size of the incision, even if the lymph node has been located via palpation. A larger incision provides easier access to deeper tissues and makes the excision of the lymph node easier for the dissector. - Using a pair of forceps, retract the skin to reveal the point of the shoulder and neck musculature.
Note: The prescapular lymph node is often found in subcutaneous fat, especially in overconditioned animals. - Once again, palpate the location of the lymph node (Figure 3A).
Note: Unlike the subcutaneous fat, the lymph node will hold its shape when palpated. Additionally, lymph nodes are not contiguous with any surrounding tissue, as they are enclosed by a thin fibrous capsule. - After locating the prescapular lymph node, carefully dissect out the subcutaneous fat, using a scalpel and forceps, and isolate the lymph node.
- Look for the presence of a thin fibrous capsule, as depicted in Figure 1B, to distinguish between the lymph node and adipose tissue; lymph nodes are not contiguous with any surrounding tissue.
- Transfer the lymph node to the lymph node dissection table, using a plastic tray for the tissue transfer, for further sectioning.
Note: The appearance of these lymph nodes in bison is similar. The associated video depicts the appearance of the prescapular lymph node as dissected out of the right shoulder of an American bison.
- Euthanize the animal (cattle or bison of any age, as appropriate for the experiment) based on the local IACUC protocol. Confirm the death by methods specifically described in the institutional IACUC protocol, including the lack of a heartbeat, the lack of respiration, and the absence of a corneal reflex. Move the animal to the necropsy floor.
- Isolation of parotid lymph nodes in cattle and bison
- Begin by placing the animal in lateral recumbency on the necropsy floor. Locate the temporomandibular joint (TMJ).
- If this is difficult to locate, move the mandible slowly and determine the location at which the mandible attaches to the skull; this is the TMJ.
- Using a scalpel or necropsy knife, carefully make an incision in the skin. Start the incision dorsal to the joint and extend it ventrally/vertically along the jawline.
- Dissect the skin away to reveal the subcutaneous tissue. The parotid salivary gland will be visible first, due to its large size, lobulated surface appearance, and red coloration.
- Using forceps and a scalpel, move the parotid salivary gland aside to find the parotid lymph node sitting just cranial and medial to the salivary gland (Figure 3A). Examine the parotid lymph node tissue for texture before further processing.
Note: Lymph nodes of the face (parotid and submandibular) are associated with salivary glands, which can confound the lymph node isolation. In comparison to lymph nodes, salivary glands are much larger and have a lobulated surface appearance. Moreover, on cut surface, salivary glands are homogenous in nature and do not exhibit the classic cortical and medullary architectural pattern of lymph nodes (Figure 3B). - Excise the gland with a scalpel. Transfer it to the lymph node dissection table, using a plastic tray for the tissue transfer, for further sectioning.
- Begin by placing the animal in lateral recumbency on the necropsy floor. Locate the temporomandibular joint (TMJ).
- Isolation of supramammary lymph nodes in female cattle and bison
- Place the animal in lateral recumbency.
- Elevate the hind leg to expose the inguinal region and locate the udder.
Note: In a healthy, non-lactating animal, palpation of these lymph nodes may not be possible. In a lactating animal, these lymph nodes may be palpable at the caudal border of the base of the udder. - Using a scalpel, make an incision just dorsal to the udder, running along the upper leg, to expose the supramammary glands.
- Dissect the subcutaneous tissue and find the supramammary lymph node embedded in a thin layer of subcutaneous fat. If not visually distinguishable, palpate the exposed tissue for the presence of a firm structure within the subcutaneous fat.
- Using forceps and a scalpel, carefully dissect out the lymph node from the fat. Transfer it to the lymph node dissection table, using a plastic tray for the tissue transfer, for further sectioning.
3. Sectioning and Storage of Lymph Nodes
- Pass the isolated lymph nodes from dissector 2 to dissector 3 at a dissection table adjacent to the main necropsy space. Place each lymph node on a fresh section of a cutting board. Pre-label the cutting boards in sections by lymph node, to facilitate the receipt of multiple lymph nodes in succession. For infectious samples, use disposable trays.
- Using forceps and a disposable scalpel or necropsy knife, remove a section of the lymph node. Using a sharp knife, make a sagittal cut to open up the lymph node.
- Examine the interior of the lymph node, especially in specimens from infected animals, looking for asymmetry, lesions, color differences, etc. Record the observations.
- (Option 1) Small tissue sections
- Use disposable scalpels to excise pieces of each lymph node that are less than 5 mm in thickness and transfer each piece to a tube with an RNA preservation solution with clean forceps.
- (Option 2) Lymph node biopsy or sectioning methodologies
Note: Biopsies or sectioning methodologies, when applied to parallel lymph node sections across different nodes and animals, provide consistency of sampled tissue for a bulk RNA analysis.- Wedge sectioning method
- After examining the sagittal section to capture cells across the profile of the lymph node, excise a pie-shaped wedge from the lymph node, from the center to the outer capsule. For a node 2 cm in width (standard size; see Table 1), a wedge cut to the center that is 4 mm in outer arc length and 5 mm in thickness will have a wet weight of ~ 100 mg.
- Using the scalpel, cut the wedge into small pieces, no thicker than 5 mm, and approximately 50 - 100 mg each in wet tissue weight.
Note: For consistency, process all wedge pieces from a single lymph node either in one batch or in separate tubes followed by RNA pooling to provide a representative RNA profile from the lymph node of interest. - Place the tissue pieces with forceps into tubes containing at least 10 volumes of RNA preservation solution, as compared to the volume of the tissue.
- Punch biopsy method
- Select a punch biopsy tool if the goal is to collect specific sections, such as specific follicles from the node. Select a tool consistent with the size of the sample desired for collection. Punch biopsy tools are available in a range of sizes, from 1 to 8 mm in diameter.
- Visually locate the regions of interest to sample in the lymph node.
- Use the punch biopsy tool to excise the sections of interest, pressing and turning the tool into the tissue to create the core. Transfer the tissue piece with forceps to a tube containing at least 10 volumes of RNA preservation solution. If thicker than 5 mm, use forceps and a scalpel to further divide the tissue into smaller pieces.
- Wedge sectioning method
- Once the samples have been transferred to preservation solution (e.g., RNALater), transport them back to the laboratory at room temperature.
- Store the samples at 4 °C overnight to allow for tissue penetration.
- The next day, pour off the excess RNA preservative solution and store the tissue samples at -80 °C. Remove as much preservative as possible before freezing the samples. Use a P1000 and/or P200 micropipette tip for an efficient RNA preservative solution removal.
CAUTION: Discard the decanted preservation solution appropriately, based on the infective properties of the pathogen utilized as well as the chemical and environmental hazards of the preservative solution as specified on the Safety Data Sheet.
Note: The guidelines provided here are provided for the RNA preservation solution described in the Table of Materials. Consult the manufacturer's guidelines if using alternative preservation solutions.
4. Processing from RNA Lymph Nodes
CAUTION: Wear a lab coat, gloves, and proper eye protection for the processing steps.
Note: The phenol-based reagent used here is described in the Table of Materials (and the protocol is based on the manufacturer's guidelines)20. The use of alternative phenol-based reagents may necessitate a modification of the procedure, based on the manufacturer's recommendations for the specific product purchased.
- Prior to accessing the samples, pre-aliquot the chilled (4 °C) phenol-based reagent to homogenization tubes (1.5 mL/tube), using a pipette.
CAUTION: Phenol is toxic, corrosive, and a health hazard. Chloroform is toxic and a health hazard. Complete processing steps 4.1 - 4.9 in a chemical fume hood, and wear gloves to prevent any contact with the chemicals. In the United States, chloroform is a hazardous waste; dispose of tubes containing hazardous waste according to local and institutional guidelines. Process tissue samples from animals infected with pathogens under the corresponding BSL-2, BSL-3, or BSL-4 guidelines. - Once a sample can be loosened from the microcentrifuge tubes with clean forceps, immediately transfer approximately 50 - 100 mg of the preserved tissue to the homogenization tube containing the phenol-based reagent.
- Limit the thawing prior to the addition to the phenol-based reagent. Use storage on dry ice when multiple samples are removed from the freezer for processing.
- Tightly cap the tube, place it on the homogenizer, and homogenize the sample with a rotor-stator tissue homogenizer in the phenol-based reagent. Complete the processing at room temperature. When complete, check for a cloudy appearance to ensure the sample was successfully homogenized; the tissue should be dispersed into a cloudy suspension.
Note: A 2 min RNA extraction setting is used (the pre-set RNA_02_1 setting for the homogenizer described in the Table of Materials; designed for frozen tissues). The rotor-stator specified in this protocol (see the Table of Materials) is large-toothed and adapted for the homogenization of a range of tissue types, including fibrous material. The RNA_02_01 setting is designed specifically for frozen (as opposed to fresh) tissues. As lymph nodes have fibrous connective tissue components, a similarly optimized rotor-stator homogenizer is recommended to achieve an effective dissociation. See the National Institute of Environmental Health Sciences21 for additional information about homogenization recommendations.- If large chunks of tissue remain after the homogenization, repeat the homogenization procedure. In order to efficiently recover the RNA, the tissue must be well dissociated.
- For more fibrous lymph node pieces, use forceps and scissors to pre-chop the lymph node piece into several smaller pieces prior to the transfer to the homogenization tube. As described in the introduction, lymph node tissues from older animals may be more fibrous.
Note: If a dual analysis of the host and pathogen RNA is desired, additional treatment (such as bead homogenization) may be necessary for the recovery of bacterial RNA, especially if Gram-positive pathogens are present in the tissues.
- Immediately remove the homogenized sample from the tubes and transfer it to RNase-free microcentrifuge tubes (2.0 mL in size) with a P1000 micropipette.
- Centrifuge the sample for 5 min at 12,000 x g at 4 °C to remove any sample debris. Using a P1000 micropipette tip, transfer the supernatant to a fresh microcentrifuge tube, leaving the pellet behind.
- Incubate the supernatant for 5 min at room temperature. Split each sample between two 1.5 microcentrifuge tubes.
- Add 0.2 mL of chloroform per 1 mL of the phenol-based reagent (i.e., 0.3 mL for a 1.5 mL sample); invert it by hand for 1 - 2 min to mix the phases, but do not vortex the sample. Incubate it for 2 min at room temperature.
- Centrifuge it for 15 min at 12,000 x g at 4 °C.
- Carefully remove the upper, clear aqueous phase with a micropipette tip (P1000 or P200) which will form the upper layer, without disrupting the interphase or the red phenol-chloroform layer. Transfer it to a new microcentrifuge tube.
- Mix it with an equal volume of 100% ethanol; mix it thoroughly by inverting the tube 4 - 5 times. The tube will often appear cloudy.
- With a micropipette tip, transfer the liquid to a silica-based commercial spin column, to further purify and elute the RNA, following the manufacturer's instructions22. Elute the RNA generated from ~ 50 mg of tissue into 50 - 100 µl of RNase-free water.
Note: While the phenol-based reagent extraction removes DNA in the organic phase, for the downstream usage of RNA in qRT-PCR and/or RNA-sequencing applications, an additional treatment of RNA samples with DNase is recommended. - Aliquot the eluted RNA to separate tubes for use in a quantification and quality assessment, to reduce any freeze-thawing of the main RNA sample. Transfer the tubes to -80 °C for storage.
- In the case of lymph node samples derived from animals infected with zoonotic pathogens, especially at the BSL-3 containment level, validate the resulting RNA sample for the absence of pathogens if the sample will be moved to a lower biosafety level containment area for quality analysis, RT-PCR, and/or RNA-sequencing library preparation.
Note: It is critical to consider local regulations, and in the case of the CDC (Centers for Disease Control and Prevention) inactivation guidelines for work with select agents, it is necessary to validate all inactivation procedures at the researcher's local site.- Remove 1/10th volume of each eluted RNA sample from step 4.12 (i.e., 5 µl from 50 µl of the total RNA). Using a sterile technique, mix the sample with 100 µl of bacterial growth broth in a sterile 1.5 mL microcentrifuge tube. Using a sterile plastic spreader, spread the RNA-broth mixture across the surface of an agar plate.
Note: Select a broth type and agar plate consistent with the growth of the pathogen with which the animals were challenged. - Incubate the agar plate under conditions specific to the growth of the pathogen with which the animals were challenged. Check for the absence of recovered bacteria before removing the samples from the BSL-3 containment.
Note: The resulting eluted RNA, after quantification and tests of integrity, is suitable for downstream applications, including an RNA-sequencing and RT-PCR analysis.
- Remove 1/10th volume of each eluted RNA sample from step 4.12 (i.e., 5 µl from 50 µl of the total RNA). Using a sterile technique, mix the sample with 100 µl of bacterial growth broth in a sterile 1.5 mL microcentrifuge tube. Using a sterile plastic spreader, spread the RNA-broth mixture across the surface of an agar plate.
- Assess the RNA recovery and purity using a spectrophotometer. To assess the RNA quality, load 1 - 2 µl of the eluted RNA sample to the spectrophotometer for an analysis. Record a spectrum of the sample in the ultraviolet (UV) range.
- From the UV spectrum, record the A260/A280 ratio, the A260/A230 ratio, and the A260 value for the sample (A = Absorbance).
- As single-stranded RNA at a concentration of 40 µg/mL has an absorbance of 1.0 at 260 nm, calculate the RNA concentration (µg/mL) by multiplying the A260 x 40 for the purified samples.
- As a quick method of assessment of RNA integrity, to exclude any degraded samples from further analysis, mix 2 µl of each RNA sample with 4 µl of 1.5x formamide loading dye [95% deionized formamide, 0.025% (w/v) bromophenol blue, 0.025% (w/v) xylene cyanol, 5 mM EDTA (pH 8.0), 0.025% (w/v) SDS] in 1.5 mL microcentrifuge tubes23,24.
- Heat the tubes at 70 °C for 1 min, and then transfer the tubes to ice for 1 min.
- Load the samples to a 1% agarose gel prepared in 1x TBE (for a full description of a native agarose gel electrophoresis for RNA, see Rio, Ares Jr., Hannon, and Nilsen)24. Separate the samples by standard gel electrophoresis methods and confirm the presence of 28S and 18S ribosomal RNA bands.
- Follow up the gel electrophoresis with quantitative analysis methods for the RNA integrity determination [e.g., a Bioanalyzer and RIN (RNA Integrity Number) analysis] as described in the Representative Results and in Mueller, O. et al.25 and Schroeder, A. et al.26.
5. Alternative Extraction Method from Formalin-fixed, Paraffin-embedded (FFPE) Tissues
Note: Although FFPE tissue preservation does not represent the most robust method of nucleic acid preservation, the protocol presented below can be a way to study some transcriptional changes when other preserved tissues are unavailable.
- Prepare formalin reagents for the tissue preservation by dispensing formalin into plastic containers. Tissues should be preserved in formalin with a ratio of 20:1 formalin:tissue volume/weight, respectively.
CAUTION: Formalin is a known human carcinogen and although not acutely toxic, chronic exposure (typically through inhaled fumes) can represent a significant health hazard. Proper ventilation and PPE (i.e., the use of nitrile gloves, to be changed within 15 min after the initial exposure) are required to minimize the health risks. Refer to the OSHA Formaldehyde Standard (29 CFR 1910.1048) for additional information on exposure monitoring and risk assessment.
Note: Using too little formalin can impact the ability of the tissue to become fully preserved when submerged in preservative. - After the isolation of the tissue of interest, carefully trim the tissue to less than 5 mm in thickness.
- Using forceps, carefully submerge the trimmed tissue into formalin to minimize any splashing.
Note: The tissues should be completely submerged in formalin to allow for a complete fixation. It is critical that all surfaces of the tissue are completely covered with formalin.
- Using forceps, carefully submerge the trimmed tissue into formalin to minimize any splashing.
- Once the tissues have been submerged in formalin, allow for the tissue fixation to occur for at least 24 h at room temperature.
Note: Formalin penetrates tissues slowly, at a rate of 1 mm per hour from all surfaces27,28. Therefore, keeping the thickness of the tissue sections to less than 5 mm results in a rapid fixation of the entire tissue. - After the fixation, transfer the tissues to an appropriate reagent for the paraffin embedding.
Note: For example, the tissues examined in this manuscript were transferred to 70% ethanol prior to the embedding. - Embed the tissues into paraffin.29
- Section the tissue to 10 - 80 µm in thickness (use 10-20 µm if miRNA is desired to be extracted)29.
- Using scalpel and forceps, remove a 40 mg tissue piece from each sample to be processed and place it into a sterile, nuclease-free microfuge tube.
- Utilize a commercial kit designed for deparaffinization and nucleic acid recovery of FFPE tissue samples to extract RNA from the preserved samples.
Note: The kit referenced in the Table of Materials utilizes xylene and ethanol washes to remove paraffin, a protease treatment step, and a glass-fiber filter to purify RNA.
Results
The use of the considerations presented in this article (steps 1 - 4 of the protocol) will aid in the recovery of RNA from large animal samples that is suitable for a downstream analysis in host gene expression studies. The RNA quality for downstream applications is assessed by multiple standard measures. For spectrophotometry, the A260/A280 ratio provides a measure of the protein contamination, and the A260/A230 ratio provides another means of purity assessment ...
Discussion
The majority of transcriptomic studies and the associated protocols focus on mouse, rat, or post-mortem human samples. However, investigations in livestock and wildlife provide a wide range of opportunities for the characterization of the immune response to disease, both as applicable to veterinary medicine and, in regard to zoonotic diseases, to human public health. This protocol provided an outline of key considerations for high-integrity RNA extraction from tissues from large animals, such as cattle, bison, goats, and...
Disclosures
The authors have no conflicts of interest to disclose. All research is funded with intramural funds from the U.S. Department of Agriculture, Agricultural Research Service. All references to specific products are provided for the purpose of experimental reproducibility and do not represent any endorsement of these products by the federal government.
Acknowledgements
The authors would like to thank James Fosse for his excellent work on all videography and video processing; Michael Marti for his excellent work in the generation of digitized cattle images; Lilia Walther for her help with RNA extraction and Bioanalyzer runs; Mitch Palmer and Carly Kanipe for their helpful review and feedback on lymph node images; and the animal care and veterinary staff at the National Animal Disease Center for all of their hard work and assistance with animal husbandry and the preparation for necropsies.
Materials
| Name | Company | Catalog Number | Comments |
| RNA preservation solution (we used RNALater for all experiments) | ThermoFisher | AM7020 | |
| 1.5 ml or 2 ml polypropylene microcentrifuge tubes | Fisher Scientific | 05-408-129 | |
| Disposable scalpels | Daigger Scientific | EF7281 | |
| Tissue forceps, rat tooth | Fisher Scientific | 12-460-117 | Other tissue forceps available including curved tip, tapered edge, etc. , depends on user preference |
| 3 mm punch biopsy needles | Fisher Scientific | NC9949469 | |
| Sharps container (small and transportable for necropsy) | Stericycle | 8900SA | 1 qt. size shown here |
| Cutting boards or disposable trays | Fisher Scientific | 09-002-24A | Available in a variety of sizes, depends on user preference |
| Personal protective equipment | Varies with pathogen (gloves, respirator masks, goggles, etc.) | ||
| Phenol-based RNA extraction reagent (we used TRIzol Reagent for all experiments) | ThermoFisher | 15596026 | |
| Silica column-based RNA extraction kit (we used the PureLink RNA Mini kit for all experiments) | ThermoFisher | 12183018A | Designed for up to 100 mg tissue |
| 100% Ethanol (200 proof for molecular biology) | Sigma-Aldrich | E7023 | |
| Tissue homogenizer with enclosed homogenization tubes (we used the gentleMACS dissociator for all experiments) | Miltenyi Biotec | 130-093-235 | |
| Agarose (General, for gel electrophoresis) | Sigma-Aldrich | A9539 | |
| 1X TBE | Fisher Scientific | BP24301 | Can also make from scratch in the laboratory |
| Deionized formamide | EMD Millipore | S4117 | |
| Sodium dodecyl sulfate | Sigma-Aldrich | L3771 | |
| Bromophenol blue | Sigma-Aldrich | 114391 | |
| Xylene cyanol | Sigma-Aldrich | X4126 | |
| EDTA (Ethylenediaminetetraacetic acid) | Sigma-Aldrich | EDS | |
| UV-Vis Spectrophotometer (we used the NanoDrop Spectrophotometer) | ThermoFisher | ND-2000 | |
| Device for quantitative RNA assessment (we used the Bioanalyzer, with associated components and protocols) | Agilent | G2939BA | |
| FFPE RNA extraction kit (we used the RecoverAll Total Nucleic Acid Isolation Kit for Formalin Fixed, Paraffin Embedded Tissue) | ThermoFisher | AM1975 | |
| Plastic spreader (L-shaped spreader) | Fisher Scientific | 14-665-231 | Only needed for sterility testing for samples from infected animals |
| Necropsy knives | Livestock Concepts | WI-0009209 |
References
- Tizioto, P. C., et al. Immunological response to single pathogen challenge with agents of the bovine respiratory disease complex: an RNA-sequence analysis of the bronchial lymph node transcriptome. PLoS One. 10 (6), e0131459 (2015).
- Miller, L. C., et al. Analysis of the swine tracheobronchial lymph node transcriptomic response to infection with a Chinese highly pathogenic strain of porcine reproductive and respiratory syndrome virus. BMC Veterinary Research. 8, 208 (2012).
- Silva, T. M. A., Costa, E. A., Paixao, T. A., Tsolis, R. M., Santos, R. L. Laboratory animal models for brucellosis research. Journal of Biomedicine and Biotechnology. 2011, 518323 (2011).
- Willard-Mack, C. L. Normal structure, function, and histology of the lymph nodes. Toxicologic Pathology. 34, 409-424 (2006).
- Elmore, S. A. Histopathology of the lymph nodes. Toxicologic Pathology. 34 (5), 425-454 (2006).
- Luscieti, P., Hubschmid, T. h., Cottier, H., Hess, M. W., Sobin, L. H. Human lymph node morphology as a function of age and site. Journal of Clinical Pathology. 33 (5), 454-461 (1980).
- Hadamitsky, C., et al. Age-dependent histoarchitectural changes in human lymph nodes: an underestimated process with clinical relevance?. Journal of Anatomy. 216 (5), 556-562 (2010).
- Sisson, S., Grossman, J. D., Getty, R. . Sisson and Grossman’s The Anatomy of Domestic Animals, Volume 1. , (1975).
- Olsen, S. C., Johnson, C. Comparison of abortion and infection after experimental challenge of pregnant bison and cattle with Brucella abortus strain 2308. Clinical and Vaccine Immunology. 18 (12), 2075-2078 (2011).
- Brichta-Harhay, D. M., et al. Microbiological analysis of bovine lymph nodes for the detection of Salmonella enterica. Journal of Food Protection. 75 (5), 854-858 (2012).
- Samuel, J. L., O’Boyle, D. A., Mathers, W. J., Frost, A. J. Isolation of Salmonella from mesenteric lymph nodes of healthy cattle at slaughter. Research in Veterinary Science. 28 (2), 238-241 (1980).
- Arthur, T. M., et al. Prevalence and characterization of Salmonella in bovine lymph nodes potentially destined for use in ground beef. Journal of Food Protection. 71 (8), 1685-1688 (2008).
- Cray, W. C., Moon, H. W. Experimental infection of calves and adult cattle with Escherichia coli O157:H7. Applied and Environmental Microbiology. 61 (4), 1586-1590 (1995).
- Thiermann, A. B. Experimental leptospiral infections in pregnant cattle with organisms of the Hebdomadis serogroup. American Journal of Veterinary Research. 43 (5), 780-784 (1982).
- Palmer, M. V., Waters, W. R., Whipple, D. L. Investigation of the transmission of Mycobacterium bovis from deer to cattle through indirect contact. American Journal of Veterinary Research. 65 (11), 1483-1489 (2004).
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA, and proteins from cell and tissue samples. BioTechniques. 15 (3), 532-537 (1993).
- Chomczynski, P., Sacchi, N. Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 162 (1), 156-159 (1987).
- Vogelstein, B., Gillespie, D. Preparative and analytical purification of DNA from agarose. Proceedings of the National Academy of Sciences of the United States of America. 76 (2), 615-619 (1979).
- . . AVMA Guidelines for the Euthanasia of Animals. , (2013).
- . . TRIzol Reagent Manual. , (2016).
- . . Disruption and Homogenization of Tissue for the Extraction of RNA. , (2014).
- . . PureLink RNA Mini Kit Protocol . , (2012).
- . . RNA Gel-loading Buffer. , (2006).
- Rio, D. C., Ares, M., Hannon, G. J., Nilsen, T. W. Nondenaturing Agarose Gel Electrophoresis of RNA. Cold Spring Harbor Protocols. , (2010).
- Mueller, O., et al. A microfluidic system for high-speed reproducible DNA sizing and quantitation. Electrophoresis. 21 (1), 128-134 (2000).
- Schroeder, A., et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Molecular Biology. 7 (3), (2006).
- Suvarna, K. S., Layton, C., Bancroft, J. D. . Bancroft’s Theory and Practice of Histological Techniques. , (2012).
- Medawar, P. B. The rate of penetration of fixatives. Journal of Microscopy. 61 (1-2), 46-57 (1941).
- Canene-Adams, K. Preparation of formalin-fixed paraffin-embedded tissue for immunohistochemistry. Methods in Enzymology. 533, 225-233 (2013).
- Fleige, S., Pfaffl, M. W. RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine. 27, 126-139 (2006).
- Fajardy, I., et al. Time course analysis of RNA stability in human placenta. BMC Molecular Biology. 10 (21), (2009).
- Bahar, B., et al. Long-term stability of RNA in post-mortem bovine skeletal muscle, liver and subcutaneous adipose tissues. BMC Molecular Biology. 8, 108 (2007).
- Yamagishi, A., et al. Gene profiling and bioinformatics analyses reveal time course differential gene expression in surgically resected colorectal tissues. Oncology Reports. 31 (4), 1532-1538 (2014).
- Choi, S., Ray, H. E., Lai, S. H., Alwood, J. S., Globus, R. K. Preservation of multiple mammalian tissues to maximize science return from ground based and spaceflight experiments. PLoS One. 11 (12), e0167391 (2016).
- Almeida, A., Thiery, J. P., Magdelenat, H., Radvanyi, F. Gene expression analysis by real-time reverse transcription polymerase chain reaction: influence of tissue handling. Analytical Biochemistry. 328 (2), 101-108 (2004).
- Lee, S. M. L., Schelcher, C., Thasler, R., Schiergens, T. S., Thasler, W. E. Pre-analytical determination of the effect of extended warm or cold ischaemia on RNA stability in the human ileum mucosa. PLoS One. 10 (9), e0138214 (2015).
- Kap, M., et al. The influence of tissue procurement procedures on RNA integrity, gene expression, and morphology in porcine and human liver tissue. Biopreservation and Biobanking. 13 (3), 200-206 (2015).
- Hong, S. H., et al. Effects of delay in the snap freezing of colorectal cancer tissues on the quality of DNA and RNA. Journal of the Korean Society of Coloproctology. 26 (5), 316-325 (2010).
- Lee, S. M., et al. RNA stability in human liver: comparison of different processing times, temperatures and methods. Molecular Biotechnology. 53 (1), 1-8 (2013).
- Morrison, P. K., et al. Post-mortem stability of RNA in skeletal muscle and adipose tissue and the tissue-specific expression of myostatin, perilipin and associated factors in the horse. PLoS One. 9 (6), e100810 (2014).
- Seear, P. J., Sweeney, G. E. Stability of RNA isolated from post-mortem tissues of Atlantic salmon (Salmo salar L.). Fish Physiology and Biochemistry. 34 (1), 19-24 (2008).
- Marchuk, L., Sciore, P., Reno, C., Frank, C. B., Hart, D. A. Postmortem stability of total RNA isolated from rabbit ligament, tendon, and cartilage. Biochimica et Biophysica Acta. 1379 (2), 171-177 (1998).
- Inoue, H., Kimura, A., Tuji, T. Degradation profile of mRNA in a dead rat body: basic semi-quantification study. Forensic Science International. 130 (2-3), 127-132 (2002).
- Micke, P., et al. Biobanking of fresh frozen tissue: RNA is stable in nonfixed surgical specimens. Laboratory Investigation. 86 (2), 202-211 (2006).
- Srinivasan, M., Sedmak, D., Jewell, S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. The American Journal of Pathology. 161 (6), 1961-1971 (2002).
- Ahmed, A. M., et al. Variation in the ovine abomasal lymph node transcriptome between breeds known to differ in resistance to the gastrointestinal nematode. PLoS One. 10 (5), e0124823 (2015).
- Russell, G. C., et al. Host gene expression changes in cattle infected with Alcelaphine herpesvirus 1. Virus Research. 169 (1), 246-254 (2012).
- Avraham, R., et al. A highly multiplexed and sensitive RNA-seq protocol for simultaneous analysis of host and pathogen transcriptomes. Nature Protocols. 11, 1477-1491 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved