
Method Article
In Vivo Direct Reprogramming of Resident Glial Cells into Interneurons by Intracerebral Injection of Viral Vectors
In This Article
Summary
This protocol aims at generating directly reprogrammed interneurons in vivo, using an AAV-based viral system in the brain and a FLEX synapsin-driven GFP reporter, which allows for cell identification and further analysis in vivo.
Abstract
Converting resident glia in the brain into functional and subtype-specific neurons in vivo provides a step forward towards the development of alternative cell replacement therapies while also creating tools to study cell fate in situ. To date, it has been possible to obtain neurons via in vivo reprogramming, but the precise phenotype of these neurons or how they mature has not been analyzed in detail. In this protocol, we describe a more efficient conversion and cell-specific identification of the in vivo reprogrammed neurons, using an AAV-based viral vector system. We also provide a protocol for functional assessment of the reprogrammed cells’ neuronal maturation. By injecting flip-excision (FLEX) vectors, containing the reprogramming and synapsin-driven reporter genes to specific cell types in the brain that serve as the target for cell reprogramming. This technique allows for the easy identification of newly reprogrammed neurons. Results show that the obtained reprogrammed neurons functionally mature over time, receive synaptic contacts and show electrophysiological properties of different types of interneurons. Using the transcription factors Ascl1, Lmx1a and Nurr1, the majority of the reprogrammed cells have properties of fast-spiking, parvalbumin-containing interneurons.
Introduction
The overall goal of this method is to efficiently convert brain resident glia in vivo into functional and subtype-specific neurons such as parvalbumin-expressing interneurons. This provides a step forward towards the development of an alternative cell replacement therapy for brain diseases without the need for an exogenous cell source. It also creates a tool to study cell fate switches in situ.
The brain has only a limited capacity for generating new neurons. Therefore, in neurological diseases, there is a need of exogenous cell sources for brain repair. For this, different sources of cells have been subjected to intense research over the years, including cells from primary tissue, stem cell-derived cells and reprogrammed cells1,2,3. Direct reprogramming of resident brain cells into neurons is a recent approach that could provide an attractive method for brain repair as it uses the patient’s own cells for generating novel neurons inside the brain. To date, several reports have shown in vivo reprogramming through viral vector delivery in the brain4,5,6,78,9 in different brain regions such as the cortex, spinal cord, striatum and the midbrain5,10,11, as well as in intact and lesioned brain5,8,11,12. Both inhibitory and excitatory neurons have been obtained4,8, but the precise phenotype or functionality of these cells has not yet been analyzed in detail.
In this protocol, we describe a more efficient reprogramming and cell-specific identification of in vivo reprogrammed neurons. We provide a protocol for functional assessment of the neuronal maturation and phenotype characterization based on functionality and immunohistochemical traits.
We used a Cre-inducible AAV vector and a GFP reporter to identify the in vivo reprogrammed neurons. This choice of viral vectors has the advantage of infecting both dividing and non-dividing cells of the brain, increasing the number of targeted cells, as an alternative to the use of retrovirus7,8. A neuron-specific synapsin-driven FLEX reporter (GFP), enabled us to specifically detect the newly generated neurons. Previous studies have used subtype-specific promoters for in vivo reprogramming7,9, that also allow the expression of reprogramming genes and reporters in specific cell types. However, that method requires further identification of reprogrammed neurons by postmortem analysis of the co-expression of reporter and neuronal markers. The use of a neuron-specific reporter, such as the one described herein, allows for a direct identification. This provides a direct proof of a successful conversion and allows a live cell identification that is required for patch-clamp electrophysiology.
Protocol
All experimental procedures were carried out under the European Union Directive (2010/63/EU) and approved by the ethical committee for the use of laboratory animals at Lund University and the Swedish Department of Agriculture (Jordbruksverket). Mice are housed in a 12 h light/dark cycle with ad libitum access to food and water.
1. Viral Vectors
- Cloning of AAV vectors
- To create Cre-inducible AAV5 vectors, insert cDNA for GFP, Ascl1, Lmx1a and NR4A2 (Nurr1) in a reverse orientation flanked by two pairs of heterotypical, antiparallel LoxP flip-excision (FLEX) sequences (Table of Materials). For the cDNA insertion, use a backbone like pAAV-Cba-FLEX or one with a similar structure, containing FLEX sequences and a chicken beta actin (CBA) promoter.
- Insert each single cDNA (e.g., Ascl1) in the backbone by polymerase chain reaction (PCR) and restriction enzymes. Express GFP under the control of a synapsin promoter and Ascl1, Lmx1a, Nurr1 under the control of a ubiquitously expressed CBA promoter.
NOTE: Ensure to have endotoxin free DNA using specific Endotoxin-Free Plasmid DNA Isolation Kits. - Perform sequencing and restriction analysis on the constructs before use, in order to check the success of the cloning step.
- AAV5 viral vector production
CAUTION: Refer to local biosafety guidelines when handling adeno-associated virus (AAV). In Sweden, the AAV used in this protocol requires Biosafety Level 2 (BSL-2).- Seed HEK293T cells with standard culture media (DMEM+Glutamax + 10% FBS + penicillin (100 U/ml) streptomycin (100 μg/mL), see Table of Materials) in T175 flasks at a density of 3 x 106 cells per flask. Account for 5 flasks per batch of AAV and plan for 6 batches at a time.
- When the cells reach 50-70% confluency, prepare the following mix for transfection (per 175 cm2 flask).
- In a 50 mL centrifuge tube, add equimolar amounts of vector plasmid and pDG series helper plasmid (pDP5, pDP6), with a total amount of 72 µg per 175 cm2 flask.
- Add Tris-EDTA buffer (TE buffer, 10 mM Tris-HCl, 1 mM EDTA) to a final volume of 144 μL.
- Add ultrapure water so that the total volume becomes 1296 μL and mix.
- Add 144 μL of 2.5 M CaCl2 and mix. Add 1.92 μL of HEPES Buffered Saline (HBS) (1.5 mM Na2HPO4 ,140 mM NaCl, 50 mM HEPES) to the DNA solution and mix immediately by vortexing.
- Incubate at room temperature (RT) for exactly 60 s. Transfer the solution to 28 mL fresh cell culture media and mix.
- Replace the medium in the flasks with cell culture medium containing transfection mix.
- Three days after transfection, harvest the cells by pouring off thr medium from the flasks into a disposable container for waste and add 5 mL of harvest buffer (EDTA added to Phosphate-Buffered Saline, see Table of Materials, DPBS to a final concentration of 5 mM) to a concentration of 5 mM) in each flask to allow cell detachment.
- Pour the cell solution in a 50 mL centrifuge tube. Add another 4 mL of DPBS to each flask to rinse the remaining cells and pool with first cell solution. Centrifuge the harvested cells at 1,000 x g for 5 min at 4 °C.
- After centrifugation, remove the supernatant and dissolve the pellets in 15 mL of lysis buffer (50 mM Tris-HCl pH 8.5, 150 mM NaCl, 1 mM MgCl2) by vortexing.
- Freeze them in CO2-ice/ethanol bath for 15 min and store in a -20 °C freezer. Thaw the harvested cell lysate in a 37 °C water bath before use.
- AAV5 viral vector purification
- Perform AAV Purification by Iodixanol Gradient Ultracentrifugation13 and use ultracentrifuge sealing tubes with centrifugation at 350,000 x g for 1 h and 45 min at RT.
- Use a 10 mL syringe with an 18G needle and insert about 2 mm below the 40/60% phase border with the bevel facing upwards in order to extract the AAV-containing phase. Make sure to stop before reaching the protein band after 5-6 mL has been extracted.
- Store the gradient extracts in autoclaved glass bottles at 4 °C. Avoid storing times longer than overnight.
- Dilute the Iodixanol gradient extract 3-fold by slowly pipetting in 12 mL of Iodixanol Elution (IE) buffer (20 mM Tris-HCl pH 8.0, 15 mM NaCl) while swirling.
- Purify and concentrate the diluted Iodixanol gradient through an anion exchange filter. Push it through slowly with a speed no faster than 1 drop/s. Push 3 mL of IE buffer slowly through the filter to wash it.
- Elute into a centrifugal filter unit with 1-2 mL of elution buffer (20 mM Tris-HCl pH 8.0;250 mM NaCl). Add DPBS to the device to a final volume of 4 mL. Centrifuge at 2,000 x g at RT until less than 0.5 mL is left in the filter. Remove the liquid from the bottom of the tube, refill with 4 mL of DPBS and centrifuge again. Repeat this step two more times. Make sure that the volume of concentrated vector on the filter is about 200 µL after the last centrifugation step.
- Remove the 200 µLconcentrated vector using a pipette and push the concentrate through a 0.22 μm filter to sterilize it. Aliquot 200 µLinto a 9 mm glass vial with interlocked insert (Table of Materials).
NOTE: The AAV5 vectors stocks can be stored in -80 °C freezers for long storage, or at 4 °C if used within 2 weeks.
- AAV5 viral vector titer determination
- Determine AAV5 titer using standard quantitative polymerase chain Reaction (qPCR) with primers for Inverted Terminal Repeat (ITR) sequence and a 5´FAM / 3´BHQ1 probe for ITR sequence (Table of Materials). Use a standard curve achieved with known amounts of ITR containing plasmid. Each AAV vector should have a range of 1E+14 – 1E+15 genome copies per milliliter if ITR sequence is used for titer determination.
NOTE: A successful AAV5 virus production gives stocks with titers in the minimum range of 3E+13 – 7E+13 Units/mL.
- Determine AAV5 titer using standard quantitative polymerase chain Reaction (qPCR) with primers for Inverted Terminal Repeat (ITR) sequence and a 5´FAM / 3´BHQ1 probe for ITR sequence (Table of Materials). Use a standard curve achieved with known amounts of ITR containing plasmid. Each AAV vector should have a range of 1E+14 – 1E+15 genome copies per milliliter if ITR sequence is used for titer determination.
2. Injection of Reprogramming Factors into the Brain
- Animal setup, stereotaxic placement and drilling
NOTE: This protocol focuses on the use of Ascl1, Lmx1a and Nurr1 (ALN) for the reprogramming of NG2 glia into interneurons. In our experience, interneurons of a similar phenotype can be obtained using other factor combinations11.- Prior to surgery, prepare the viral mix containing the vectors Cba-FLEX-Ascl1, Cba-FLEX-Lmx1a, Cba-FLEX-Nurr1 and the reporter vector Syn-FLEX-GFP. Add each one of the stocks prepared in section 1 to the final mix, so that the final viral solution has 5% of each one of the reprogramming factors (Ascl1, Lmx1a and Nurr1) and 10% of the reporter construct (5% A, 5% L, 5% N and 10% GFP).
NOTE: The AAV5 vector mixes can be stored at 4 °C and kept for future use in vivo. - Anaesthetize the mouse using 2% isoflurane in a mix of air and nitrous oxide (N2O) at a 4:1 ratio. Monitor the animal’s breathing by observing the movements of the diaphragm. During the surgery, maintain anesthesia using 1–1.5 % isoflurane.
NOTE: The mouse model hereby described consists of a mouse strain that specifically expresses Cre in NG2 glia. In vivo reprogramming can be achieved using different mouse strains that express Cre in other populations of glial cells (e.g., astrocytes14). - Once the animal is completely anesthetized (e.g., complete muscle relaxation and no response to a pinch in the foot pad), shave the area around the incision site and bring the animal to the stereotaxic frame.
- To maintain animal’s body temperature during surgery, attach a heating pad to the base of the stereotaxic frame. Place the mouse on a clean, dry paper towel.
- Carefully place the mouse’s head into the ear bars. If correctly placed, no lateral movement of the head should be observed. Set the left ear bar to 4 mm, prior to starting placing the mouse in the stereotaxic frame.
- Secure the tooth bar in place, and then tighten the nose bar. Make sure that the head does not move in any dimension and point straight ahead (the midline being perpendicular to the plane of the ear bars). Apply ophthalmic ointment for eye protection.
- Before the beginning of surgery, administer the appropriate analgesia (e.g., 0.05 mg/kg of Buprenorphine, sub-cutaneous).
- Clean the incision area with a cotton gauze or a wipe soaked with 70% EtOH, without approaching the eye area.
NOTE: For intracerebral viral injection, a 5 or 10 μL syringe adapted with a pulled glass capillary is used. Glass capillaries are pulled using a micropipette puller, resulting in a capillary with a very fine tip, which will minimize the invasiveness of the procedure. To adapt the capillary into the syringe, use a piece of rubber tubing over the connection between the needle of the syringe and the glass capillary and melt it using a heat source (e.g., a lighter). A tight connection between the two pieces will make sure there are no fluid leaks during the injection. Before starting the surgery, test this by filling the syringe with saline and pushing the liquid out of the syringe. - Make an incision of approximately 0.5-0.8 cm along the midline of the head. Cut through both cutaneous and sub-cutaneous layers, with a scalpel.
- Widen the skin flaps on each side of the incision. Clean the incision of any blood and scrape back the subcutaneous layers with a cotton bud.
- Move the M/L-D/V arm of the stereotaxic frame into place (above the animal) and secure it.
NOTE: The stereotaxic frame allows adjustment of the syringe along the Anterior/Posterior (A/P, Y axis), Medial/Lateral (M/L, X axis) and Dorsal/Ventral (D/V, Z axis) axis. - Move the syringe along the different axis of the stereotaxic frame, to bring the tip of the glass capillary just above bregma (the junction point where the different skull plates meet).
- Make sure the capillary tip is perfectly straight in both A/P and M/L planes. In case of an ambiguous bregma, take the average of the lateral and midline sutures.
- When the tip of the capillary is correctly placed above bregma, reset both the M/L and A/P values to 0.0 on the digital coordinate counter.
- To make sure the head of the animal is in a perfectly flat position, use the digital coordinate counter to measure the D/V coordinate value, when the A/P arm is at +2.0 and -2.0 (M/L = 0.0), as well as when M/L arm is at +2.0 and -2.0 (A/P = 0.0). Adjust the height of tooth bar and ear bars accordingly.
- Move the syringe to the desired coordinates for injection of viral vectors in the striatum (A/P = +1.0; M/L = -2.0, relative to bregma).
- Raise the syringe slightly and, by looking at the injection site through the microscope, drill a hole using a dental drill at the injection coordinates. Start to drill at the site, working in circular and gentle manner.
NOTE: Do not put too much downward pressure, as the drill-bit should be sharp enough to go through the bone without additional force. Avoid long, sustained drilling as this creates heat. - At the end of the drilling, check that the dura mater remain intact and exposed for injection.
- Prior to surgery, prepare the viral mix containing the vectors Cba-FLEX-Ascl1, Cba-FLEX-Lmx1a, Cba-FLEX-Nurr1 and the reporter vector Syn-FLEX-GFP. Add each one of the stocks prepared in section 1 to the final mix, so that the final viral solution has 5% of each one of the reprogramming factors (Ascl1, Lmx1a and Nurr1) and 10% of the reporter construct (5% A, 5% L, 5% N and 10% GFP).
- Syringe setup preparation
- Place a piece of cotton gauze over the open incision and flush the syringe with saline solution.
- After flushing, take up an air bubble of 1–2 μL, followed by 1 μL of solution containing the viral vectors, avoiding any unintentional bubbles. Make sure that the viral solution can easily be visualized below the air bubble, whilst being injected.
- Viral injection
- Remove the cotton gauze from over the incision site, and lower the syringe using the D/V arm of the stereotaxic frame. As the surface of the skull is approached, carefully look through the microscope and measure the D/V level of dura mater — it should bulge slightly under gentle pressure.
- While touching the dura mater with the tip of the capillary, set the D/V coordinate to 0.0.
- Lower the syringe, progressing slowly to the desired depth (D/V = -2.7, relative to dura mater). Make sure that the trajectory is clear of bone fragments, so that no bending of the needle/capillary is observed.
NOTE: The presented coordinates refer to an injection of the reprogramming factors into the striatum of NG2-Cre mice. Coordinates corresponding to other brain regions may be used. Always test the coordinates for injection first using a dye (e.g. Trypan Blue), colored bead injection or a reporter viral vector prior to injection of reprogramming factors into the brain of a new mouse strain. If injecting Trypan Blue, the animals need to be sacrificed immediately after surgery and the brain dissected out. Fresh brains can be cut using a microtome, while frozen, and the site of injection determined by visualization of the dye position in the brain. If testing coordinates using colored beads, it is possible to determine the site of injection on perfused and cut brains a week after injection. Alternatively, an injection with a viral vector carrying a reporter gene can be used, and the site of injection determined in perfused cut brains. - Inject 1 μL of the viral solution at a rate of 0.4 μL/min. When the whole volume is injected, allow for a diffusion period of 2 min before syringe withdrawal.
- After diffusion, slowly retract the syringe until the tip of the capillary is completely out of the brain.
- Place a piece of cotton gauze over the wound and flush the syringe with saline solution.
- Move the M/L-D/V arm of the stereotaxic frame out of the working area.
- Wound closing and post-operative procedures
- Carefully suture the incision using suture thread.
NOTE: All suture threads used in injected animals should be disposed in a cup/vial containing anti-viral detergent solution. The same solution is used to clean all surfaces surrounding the surgery area that have been in contact with surgical materials. All surgical tools are thoroughly washed and autoclaved at the end of each surgery day. - Remove the animal from the stereotaxic frame and place it in a post-operative station, which includes a clean, heated cage, access to food and water, and where the animal stays until fully awake. During this period, monitor the animal closely until consciousness is regained.
- Do not house operated animals with non-operated animals until the former have fully recovered from surgery.
- Supervise operated animals daily. Depending on the sutures used, make sure they are removed if necessary. All animals were given Bupenorphinum (Temgesic at 1ml/kg) subcutaneous as post operative care.
NOTE: If operating using the same syringe in two consecutive days, flush it with water, followed by ethanol 70% and water again. Leave the syringe filled with water over-night, to allow dissolution of any possible residues.
- Carefully suture the incision using suture thread.
3. Electrophysiological Recordings
- Tissue slice preparation for electrophysiology
CAUTION: A well-executed tissue preparation is necessary in order to achieve good electrophysiological recordings. Prepare the room carefully and place tools for perfusion and dissection on ice.- Prepare ice-cold and oxygenized (95% O2 and 5% CO2) Krebs solution for perfusion, dissection and cutting (prepare this on the same day from the 10x stock by diluting the stock solution in ultrapure water and adding NaHCO3 and glucose). The components in the Krebs solution in mM (after dilution to 1x) are: 126 NaCl, 2.5 KCl, 1.2 NaH2PO4·H2O, 1.3 MgCl2·6H2O, and 2.4 CaCl2·6H2O, 22 NaHCO3, 10 glucose. Adjust the pH of the solution to 7.4.
- Perform a calibration (Vibracheck) for the vibratome with a new razor blade.
- Start the cooling block of the vibratome (or fill the surrounding chamber with ice), fill the cutting chamber with Krebs solution for oxygenation with 95 % O2 and 5 % CO2 at least 30 min before use.
- Put a Petri dish on ice and fill it with oxygenized Krebs solution. Place the blade, scissors and mounting plate on ice.
CAUTION: Bring the cage containing the mouse to the room at least 1 h before starting the procedure for acclimatization. Stress has a negative effect on the condition of the brain tissue sections. - Terminally anesthetize the mouse by injecting an overdose of Pentobarbital and let the animal fall asleep. When the blink reflex is out and animal do not respond to pain stimuli, transcardially perfuse the animal with ice-cold Krebs solution for 2-3 min (at a rate of 10-20 mL/min).
- Rapidly, but carefully, dissect out the brain and put it upside down in the Petri-dish that is placed on ice (containing Krebs solution).
NOTE: In order to compare the functional maturation of the reprogrammed neurons, sacrifice the animals for recordings at different time-points post-viral injection. Assess the firing properties of distinct subtypes of neurons based on existing literature15 . - Make a coronal cut along the mid cerebellum and glue this side onto the mounting plate (also ice-cold) for vibratome cutting.
- Dip the glued brain carefully into the buffer chamber in the vibratome.
CAUTION: Be careful not to touch the razor blade for your own safety, and so that the blade will remain calibrated. - Cut from the most rostral part of the brain down to the striatal level at a high speed. Then cut the striatum coronally at 275 μm at slow speed (0.10 mm/s).
- After each cut, carefully remove the non-injected striatal side (e.g., with a bended needle) and transfer the injected side into another vial in a water bath, containing a bottom net (oxygenized Krebs at RT) placed in a water bath. Keep this at RT until all sections are cut.
- Slowly increase the temperature of the water bath to 37 °C and leave it for 30 min. Then turn off the heater and let cool down until RT.
NOTE: At this point, you can pause until you start recording. The sections last for 4-6 h.
- Whole-cell patch-clamp recordings
- Transfer the first tissue section to the recording chamber submerged in a continuous flow of Krebs solution. Mount the section using light weights and submerge the objective.
- Identify the striatal region in the microscope and search for GFP-positive (reprogrammed) neurons. Select a neuron that is extensive in morphology and not covered by fiber bundles or blood vessels.
- Prepare borosilicate glass pipettes (3–7 MΩ) for patching and fill with the following intracellular solution (in mM): 122.5 potassium gluconate, 12.5 KCl, 0.2 EGTA, 10 HEPES, 2 MgATP, 0.3 Na3GTP and 8 NaCl, adjusted to pH 7.3 with KOH.
- For biocytin filling, add 1 mg of biocytin salt to 1 mL of internal solution and vortex.
CAUTION: Make sure to filter the internal solution with biocytin as this can otherwise clog the electrode. - Attach the glass pipette to the recording electrode and dip into the solution. Double-check the resistance of the electrode. Then slowly approach the cell with the pipette, keeping a slight positive pressure in the electrode to avoid plugging the tip.
CAUTION: Be careful to keep track of your cell while descending the electrode and not to bleach the fluorescence in the cell (i.e., turn off the fluorescence lamp when you do not need it). - Rinse the surrounding tissue carefully using positive pressure of the electrode and approach the cell with the electrode. Locate the electrode right on top of the cell and descend until the electrode touchd the membrane. Make a Giga-Ω seal between the electrode and cell membrane, and with negative pressure pulses, rupture the membrane to create a whole-cell patch.
CAUTION: Reprogrammed neurons are sensitive. Be careful when patching and do not put too much negative pressure when reaching a Giga-Ω seal or opening the cell membrane. Also, patching onto animals that are older requires practice and patience as their connective tissue is thicker and it is harder to visualize the neurons. - Check the resting membrane potentials immediately after breaking-in in current-clamp mode and write down for analysis.
- In current clamp, maintain the cell between -60 mV to -80 mV and inject 500 ms currents from -20 pA to +90 pA, with 10 pA increments to induce action potentials.
- Transfer into voltage-clamp and measure the inward sodium and delayed rectifying potassium currents at depolarizing steps of 10 mV.
NOTE: Voltage-clamp recordings are better assessed using a different internal solution, composed of chemicals that more efficiently clamp the membrane. However, for reprogrammed neurons, the number of cells that you can record from is few and the number of tissue sections with these neurons is limited. Hence, it is an advantage to record both in current-clamp and voltage-clamp from the same cell. - In voltage-clamp mode, record spontaneous postsynaptic activity at -70 mV. Distinguish inhibitory postsynaptic events at a membrane potential of 0 mV from excitatory events at -70 mV.
- After reaching a stable baseline, add Picrotoxin, the GABAA receptor antagonist, to the Krebs solution that flows into the recording chamber, to extract excitatory events (add a stock solution of Picrotoxin to a final concentration of 50 mM in separate buffer syringe that is connected to the chamber perfusion. Connect the outflow of buffer from the chamber to a vacuum bag for safe disposal).
- After 20 min, add CNQX (20 mM), an AMPA antagonist, to the Krebs solution that flows into the recording chamber, to block the inhibitory events (using the same procedure as for Picrotoxin). Leave in for another 20 min and then wash out with Krebs solution. Remove the tissue section from the chamber.
- After finishing the recordings, do an offline analysis of spontaneous excitatory post-synaptic currents (EPSC) and inhibitory post-synaptic currents (IPSC) using a threshold-event detection (>5 pA) in the analysis program.
- During the whole recording, the cell will be slowly filled with the biocytin-containing internal solution. Keep the patch for at least 20 min before slowly removing the electrode in order to achieve complete filling of the cell.
CAUTION: Be careful not to have any positive pressure in the electrode that could destroy the consecutive morphological analysis of the cells afterwards. - For biocytin-visualization, put the tissue section in 4 % paraformaldehyde (PFA) overnight at 4 °C. Rinse the section in 0.02 M potassium phosphate buffer (KPBS) with 0.1 % Triton. Then, stain for streptavidin- Cy3 (1: 600 in KPBS-T, 2 h), for identification of the biocytin-filled cell and morphological visualization of the reprogrammed neuron.
NOTE: Biocytin filling can be used for morphological analysis and illustrations of the patched neurons.
4. Immunohistochemistry, Stereology, and Quantification
NOTE: Dedicate a specific group of mice for immunohistochemistry, as tissue sections used for electrophysiology are not optimal for immunohistochemistry.
- Anaesthetize mice with an i.p. injection of an overdose of pentobarbital and mount the animal for perfusion.
- Transcardially perfuse the mice, first with a saline solution to remove blood, and then with ice-cold 4% PFA.
- Dissect the brains and post-fix for at least 12 h in 4% PFA.
- Put the brains in 25% sucrose solution (for cryoprotection) for approximately 12 h.
NOTE: The brains are ready to be cut when they sink in the 25% sucrose solution, indicating that the sucrose has penetrated the entire mouse brain. - Make a coronal cut across the cerebellum, use the flat, most caudal part of the brain to place the brain on a microtome and fix it in place using optimal cutting temperature compound (OCT).
- Cut the brain into coronal slices with a thickness of 35 μm and divide the brain into consecutive series placed in vials or wells containing 0.02 M KPBS.
- Process the sections for immunohistochemistry using antibodies against GFP and interneuron markers such as GAD65/67, PV, Chat (choline acetyltransferase) and NPY (Neuropeptide Y), according to standard protocols16, 17.
NOTE: Brain slices can be kept at 4 °C or -20 °C in anti-freeze solution for long periods. - After staining is complete, mount the sections on a glass slide and cover with a glass coverslip, using a mounting solution to fix it in place. Leave the coverslipped slides to dry over-night.
NOTE: For our studies, whole brains are cut in 1:8 series (i.e., each vial containing sections will hold one eighth of the mouse brain11). - Analyze the sections using an inverted fluorescence and/or confocal microscope.
CAUTION: Fluorescent microscopy is useful for a general overview of results and capturing images for calculating reprogramming efficiency. For a more detailed analysis of phenotypic identity by observation of double-positive cells, confocal imaging should be used. - For quantification of different markers expressed in GFP+ neurons in the striatum, count the number of double-positive cells in relation to the total number of GFP+ cells (i.e., the reprogrammed neurons), in at least two fields of each striatal section.
- For determining the approximate extrapolated total number of reprogrammed neurons per brain, count the GFP+ neurons present in the striatum of one of the brain series cut earlier, and then multiply by the total number of series.
NOTE: Different methods of quantification can be used to express reprogramming efficiency, number of reprogrammed neurons per animal and percentage of neurons that express certain phenotypic markers. For more information, see previous publication11. - Analyze the differences between conditions using Graph Pad Prism or similar.
NOTE: For efficiency calculation, we injected NG2-Cre mice with the CRE-dependent ALN conversion vectors and the GFP reporter. To estimate the conversion efficiency, we also injected animals with a Cre-dependent GFP under the ubiquitous cba promoter, rendering all targeted cells GFP+. Using this comparison, we estimated that 66.81% ± 38.38% of targeted cells converted into neurons. For validation of expression of each one of the genes, complementary double immunofluorescent-staining can be performed for GFP/Ascl1, GFP/Lmx1a and GFP/Nurr1. Additionally, genome sequencing such as single cell RNA sequencing can reveal the presence of each one of the genes in the reprogrammed cells.
Results
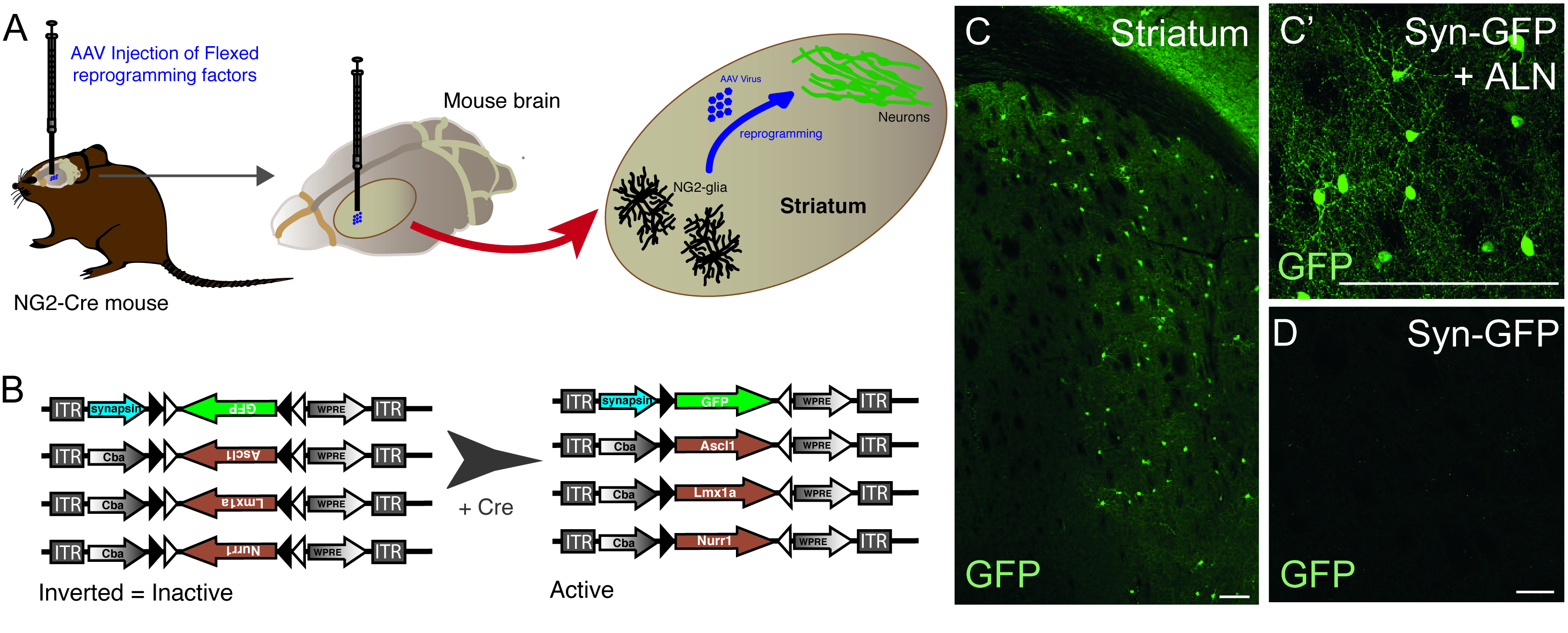
The injection of AAV vectors is used to successfully reprogram resident NG2 glia cells into neurons in the NG2-Cre mouse striatum (Figure 1A). To specifically target NG2 glia, FLEX vectors with reprogramming/reporter genes, are inserted in an antisense direction and flanked by two pairs of antiparallel, heterotypic loxP sites (Figure 1B). Each of the three reprogramming genes (Ascl1, Lmx1a and Nurr1) is placed under the control of the ubiquitous cba promoter on individual vectors. In order to make sure GFP expression is restricted to reprogrammed neurons originating from a Cre-expressing cell, GFP is placed under the control of the neuron-specific synapsin promoter, (also in a FLEX vector).
The use of the combination of reprogramming and reporter constructs allows for the generation of GFP-positive neurons in the mouse striatum (Figure 1C,C’). The use of the reporter construct without the presence of reprogramming genes does not yield GFP-positive neurons (Figure 1D).
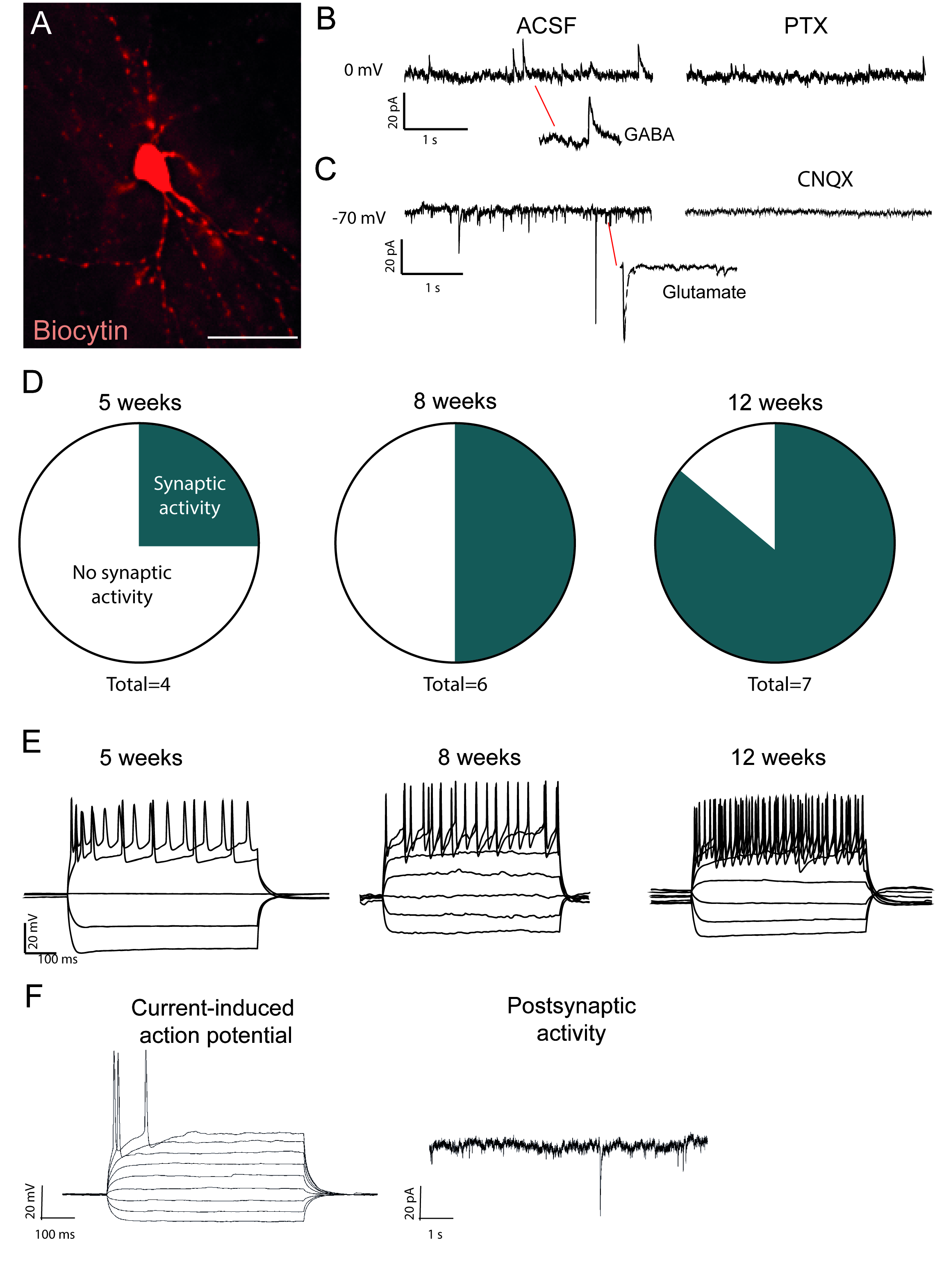
The reprogrammed neurons that are biocytin-filled are visible after post-mortem immuno-staining (Figure 2A). If conversion is successful, there should be extensive neuronal morphology. The electrophysiological recordings of reprogrammed neurons show presence of postsynaptic functional connections with spontaneous activity measures (Figure 2B,C). This can be blocked with either ionotropic GABAergic or glutamatergic blocker (Picrotoxin or CNQX), suggesting both excitatory and inhibitory synaptic input to the reprogrammed neurons. The occurrence of spontaneous activity increases with time post viral injection (Figure 2D), indicating a gradual maturation.
Current-induced action potentials are present in functional neurons. The action potentials increase in number over time after conversion (Figure 2E). This further indicates maturation in neuronal function. In an immature neuron, current will induce either none or very few action potentials (Figure 2F).
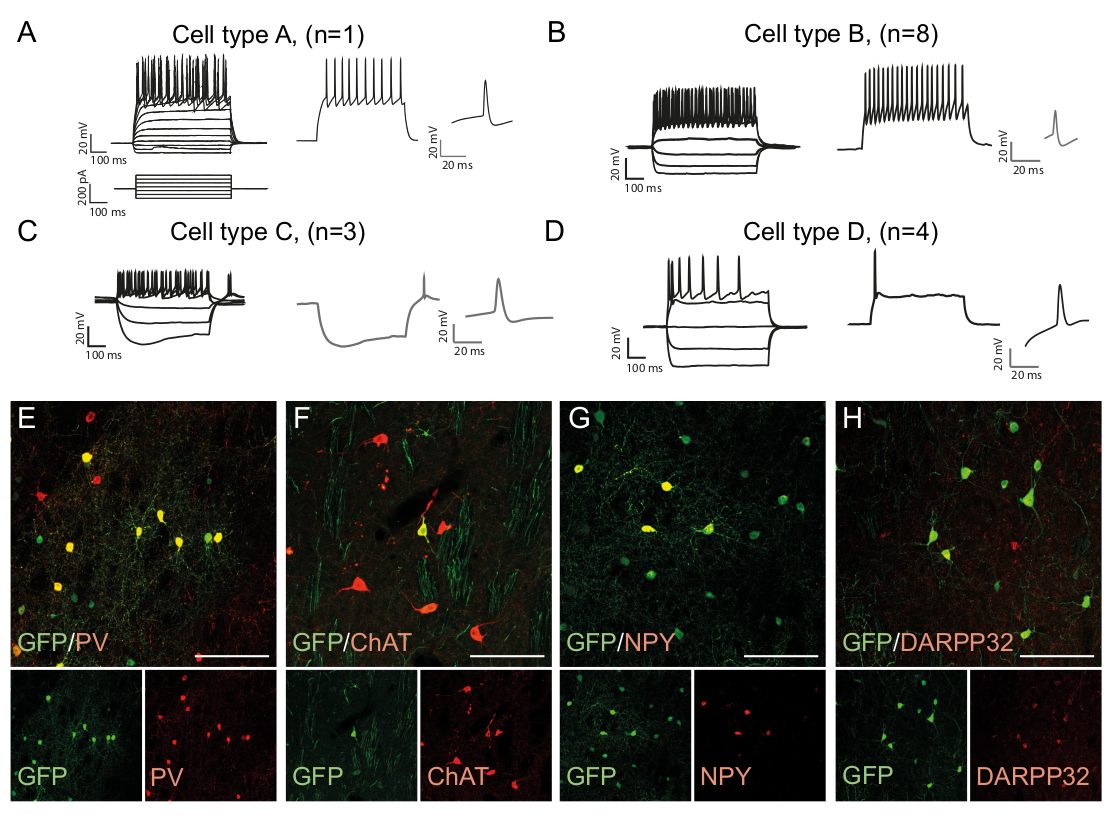
The firing patterns of a neuron is cell type specific as it depends on factors such as the morphology of the cells and channel expression15. The recorded patterns in the in vivo reprogrammed neurons can be distinguished into groups and compared to that of endogenous neuronal subtypes, e.g. fast-spiking interneurons (Cell Type B, Figure 3B) or other cell types (Figure 3A,C,D). The observed electrophysiological differences can be confirmed by the presence of specific subtype markers and co-expression with GFP (Figure 3E-H). Altogether, this data indicates that the reprogrammed neurons present in the striatum have properties of different types of interneurons, such as Parvalbumin-, ChAt- and NPY- expressing interneurons, as well as striatal medium spiny neuron identity (DARPP32+) (Figure 3E-H).

Figure 1: In vivo reprogramming of resident NG2 glia into neurons. (A) Schematic representation of AAV virus-mediated in vivo reprogramming of striatal NG2 glia. (B) Schematic representation of AAV5 FLEX constructs used for in vivo reprogramming, in which gene expression is regulated by Cre expression in the targeted cells. (C and C’) In vivo reprogrammed neurons, resulting from Syn-GFP + ALN injection into the Striatum. (D) Absence of reprogrammed neurons when no reprogramming factors are added into the viral cocktail, and only reporter construct is injected in vivo. Scale bars = 100 μm (C),25 μm (C'), 25 μm (D). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: In vivo reprogrammed neurons are functional and show maturation over time. (A) Biocytin-filled reprogrammed neuron, shows mature neuronal morphology, including dendritic spines. Traces shows (B) inhibitory (GABAergic) activity that is blocked with picrotoxin, a GABAA receptor antagonist and (C) excitatory activity that is blocked with CNQX, an AMPA receptor antagonist. (D) The number of neurons with postsynaptic activity increases over time. (E) Patched neurons show repetitive firing already at 5 weeks post-injection (w.p.i.) and continue to show that at 8 and 12 w.p.i. (F) Current-induced action potential and postsynaptic activity of an immature neuron, showing few synaptic events and few action potentials compared to B and D. Scale bar = 25 μm Please click here to view a larger version of this figure.
{kind=link}

Figure 3: In vivo reprogrammed neurons show immunohistochemical and electrophysiological properties of striatal interneurons. (A-D) The firing patterns of in vivo reprogrammed neurons can be of distinct types: (A) Type A is similar to endogenous medium spiny neuron (DARPP32+); (B) similar to fast-spiking (PV+) interneurons; (C) similar to low-threshold spiking neurons with prominent sag (NPY+); (D) neurons firing with large after-hyperpolarization (Chat+). (E-H) Confocal images showing co-localization of GFP and the interneuron markers PV (E), ChAT (F), NPY (G), and projection neuron marker DARPP32 (H). All scale bars = 50 μm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
In vivo direct reprogramming can be achieved using AAV FLEX vectors in Cre-expressing mouse strains. It is important to note that differences between mouse strains with regards to reprogramming efficacy have been observed. For in vivo reprogramming in the striatum, the NG2-Cre mouse line has proved to be the more efficient when compared with other strains. Prior to starting to use a new animal strain, it is important to check the mouse provider guidelines regarding Cre expression over time, as the age of animals often impacts the specificity of this protein expression. In our studies, animals older than 12 weeks were not used for in vivo reprogramming as there was a risk for Cre expression in cells other than NG2 glia. The constant presence and monitoring of control animals injected only with Synapsin-FLEX-GFP construct is advised. This allows monitoring of the animals for GFP-positive cells that should not be present if no reprogramming genes (ALN) are used.
To identify the newly reprogrammed neurons, a neuron-specific identification method such as the one described in this protocol is of utmost importance. This allows for a proper identification and distinction of the reprogrammed neurons from the endogenous surrounding cells that is of particular relevance when the reprogramming in a homotopic region.
It is also important to target the correct structure for viral injection. Therefore, the stereotaxic surgery for viral injection is important and needs to be approached with accuracy, especially when targeting smaller structures of the brain.
We have previously shown11 that it is not reliable to predict the outcome of in vivo reprogramming based on in vitro reprogramming experiments using the same reprogramming factors. All factors of interest therefore need to be tested in vivo. In our hands, many different factor combinations give the same subtype of neurons (i.e., interneurons11 in vivo) despite the fact that these genes have been implicated in the development of other neurons.
Whole-cell patch clamp for reprogrammed neurons is a delicate technique and tissue processing is important for a good outcome. Perfusion with ice-cold Krebs solution improves tissue quality. Also, patched neurons need to be treated carefully. Even if maturation and phenotypic identity of reprogrammed neurons can be somewhat assessed using whole-cell patch clamp, these cells are not fully comparable to their endogenous counterparts. Additional types of analysis, such as genome sequencing (e.g., RNA sequencing) should be used to further confirm reprogrammed cell identity.
The technique described herein is could be considered for the development of future therapies where neuronal replacement is needed in the brain. Although in vivo reprogramming is still at its early stages and the translation into humans is not yet foreseen, this technique could provide a method to assess exogenous gene function in the brain and study cell maturation in vivo.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Marcella Birtele has been funded by European Union Horizon 2020 Program (H2020-MSCA-ITN-2015) under the Marie Skłodowska-Curie Innovative Training Networks and Grant Agreement No. 676408. Daniella Rylander Ottosson has been funded by Swedish Research Council (2017-01234).
Materials
| Name | Company | Catalog Number | Comments |
| REAGENTS FOR AAV5 CLONING AND VIRAL VECTOR PREPARATION | |||
| pAAV-CA-FLEX | AddGene | 38042 | |
| Ascl1 | AddGene | 67291 | NM_008553.4 |
| Lmx1a | AddGene | 33013 | NM_0033652.5 |
| Nurr1 | AddGene | 35000 | NM_013613.2 |
| GFP-syn | AddGene | 30456 | |
| LoxP (FLEX) sequence 1 | GATCTccataacttcgtataaagtatcctatac gaagttatatcaaaataggaagaccaatgcttc accatcgacccgaattgccaagcatcaccatcg acccataacttcgtataatgtatgctatacgaa gttatactagtcccgggaaggcgaagacgcgga agaggctctaga | ||
| LoxP (FLEX) sequences 2 | tactagtataacttcgtataggatactttatac gaagttatcattgggattcttcctattttgatc caagcatcaccatcgaccctctagtccacatct caccatcgacccataacttcgtatagcatacat tatacgaagttatgtccctcgaagaggttcgaa ttcgtttaaacGGTACCCTCGAC | ||
| pDP5 | Plasmid Factory | PF435 | |
| pDP6 | Plasmid Factory | PF436 | |
| Phosphate-Buffered Saline (PBS) | Thermo Fisher Scientific | 10010023 | |
| FBS (Fetal bovine serum) | Thermo Fisher Scientific | 10500064 | |
| Penicillin streptomycin | Thermo Fisher Scientific | 15140122 | |
| DMEM (Dulbecco's Modified Eagle Medium)+ Glutamax | Thermo Fisher Scientific | 61965026 | |
| DPBS (Dulbecco's Phosphate Buffer Saline) | Thermo Fisher Scientific | 14190094 | |
| HEK293 cells | Thermo Fisher Scientific | 85120602-1VL | |
| Flasks | BD Falcon | 10078780 | 175 cm2 |
| Tris H-CL | Sigma Aldrich | 10812846001 | For TE buffer use 10 mM, pH 8.0; for lysis buffer use 50mM, pH 8.5; for IE buffer use 20 mM, pH 8.0; for elution buffer use 20 mM, pH 8.0 |
| EDTA | EDTA: Invitrogen | EDTA: AM9260G | For TE buffer use 1 mM EDTA |
| Ultrapure water | see Ultrapure water system | ||
| CaCl2 | SigmaAldrich | C5080 | 2.5 M |
| Dulbecco´s phosphate-buffered saline (DPBS) | Thermo Fisher Scientific | 14190136 | |
| NaCl | Sigma-Aldrich | S3014 | FOR HBS use 140 mM; for Lysis Buffer use 150 mM; for IE buffer use 15 mM; for elution buffer use 250 mM |
| MgCl2 | Sigma-Aldrich | M8266-100G | For lysis Buffer: 1 mM |
| Ultracentrifuge sealing tubes | Beckman Coulter | Quick-Seal® Polypropylene Tube | |
| OptiPrep™ Density Gradient Medium | Sigma Aldrich | D1556-250ML | |
| 10-mL syringe-18G needle | BD | 305064 | |
| Laboratory glass bottles | VWR | ? | |
| Anion exchange filter | PALL laboratory | MSTG25Q6 | Acrodisc unit with Mustang Q membrane |
| Centrifugal filter unit | Merck | Z740210-24EA | Amicon Ultra-4 device |
| Endotoxin-Free Plasmid DNA Isolation Kits | Thermo Fisher Scientific | A33073 | |
| Na2PO4 | Sigma-Aldrich | S7907 | |
| HEPES | Sigma-Aldrich | H7523 | 15 mL |
| Falcon tube | Thermo Fisher Scientific | Corning 352196 | |
| Falcon tube | Thermo Fisher Scientific | Corning 352070 | |
| Glass Vials | Novatech | 30209-1232 | CGGCCTCAGFGAGCGA |
| Forward Primer for Inverted Terminal Repeat (ITR) sequence | GGAACCCCTAGTGATGGAGTT | ||
| Reverse Primer for Inverted Terminal Repeat (ITR) sequence | CACTCCCTCTCTGCGCGCTCG | ||
| 5´FAM / 3´BHQ1 probe | Jena Bioscience | ||
| 0.22 mm filter | Merck | SLGV004SL | Millex-GV filter |
| EQUIPMENT AAV5 VIRAL VECTOR PREPARATION | |||
| Freezer -20 °C | |||
| Freezer -80 °C | |||
| Fridge +4 °C | |||
| AAV viral room | |||
| Ultrapure Water system | Merck | Milli-Q® IQ 7000 | |
| Vortex mixer | VWR | 444-0004 | |
| Ultracentrifuge | Beckman Coulter | Beckman Optima LE-80K Ultracentrifuge | |
| Autoclave | Tuttnauer | 2540 EL | |
| Polymerase Chain Reaction (PCR) | BioRad | C1000 Touch Thermal Cycler | |
| Quantitative PCR (qPCR) | Roche | LightCycler® 480 System | |
| Centrifuge | Thermo Fisher Scientific | Sorvall ST16 | |
| Water Bath | Thermo Fisher Scientific | TSGP02 | |
| ANIMAL MODEL | |||
| NG2-Cre mice | Jackson | NG2-CrexB6129, Stock #008533 | |
| REAGENTS FOR INJECTION OF REPROGRAMMING FACTORS INTO THE BRAIN | |||
| Water | |||
| Saline | Apoteket AB | 70% | |
| Ethanol | Solveco | ||
| Isolfurane | Apoteket AB | Dilute to 1% solution with warm water. | |
| Virkon | Viroderm | 7511 | |
| Pentobarbital | Apoteket AB | P0500000 | i.p. for terminal procedure at the dose of 60 mg/ml. |
| Sucrose | Merck | S0389-500G | |
| Trypan Blu | Thermo Fisher Scientific | 15250061 | |
| Retrobeads | Lumafluor | R170 | |
| Buprenorphine | Apoteket AB | ||
| EQUIPMENT FOR INJECTION OF REPROGRAMMING FACTORS INTO THE BRAIN | From RSG Solingen. | ||
| Scissors | VWR | 233-1552 | From Biochem. |
| Tweezers | VWR | 232-0007 | |
| Forcep | VWR | 232-0120 | |
| Scalpel holder | VWR | RSGA106.621 | Number 20. |
| Scalpel | VWR | RSGA106.200 | |
| Stereotaxic frame | Stoelting Europe | 51500D | |
| Mouse ear bars | Stoelting Europe | 51648 | 5 ul |
| Syringe with Removable needle | Hamilton Company | 65 | 0,75 inner diameter and 1.5 outer diameter. |
| Glass capilaries | Stoelting | 50811 | |
| Glass capillary puller | Sutter company | P-1000 | |
| Dental drill | Agnthos AB | 1464 | |
| Shaver | Agnthos AB | GT420 | |
| Isoflurane Chamber and pump | Agnthos AB | 8323101 | 2 mL |
| Syringes | Merck | Z118400-30EA | 25G |
| Needles | Merck | Z192414 Aldrich | |
| Mouse and neonatal rat adaptor for stereotaxic fram | Stoelting | 51625 | |
| Heating pad | Braintree scientific, inc | 53800M | From Covidien 2187. |
| Cotton gauze | Fisher Scientific | 22-037-902 | |
| Rubber tube | Elfa Distrelec | HFT-A-9.5/4.8 PO-X BK 150 | |
| Cotton swabs | Fisher Scientific | 18-366-473 | |
| REAGENTS FOR WHOLE-CELL PATCH CLAMP RECORDINGS | |||
| NaCl | Sigma-Aldrich | S3014 | |
| KCl | Sigma-Aldrich | P9333 | |
| NaH2PO4-H2O | Sigma-Aldrich | S9638 | |
| MgCl2-6 H2O | Sigma-Aldrich | M2670 | |
| CaCl2-6 H2O | Sigma-Aldrich | C8106 | |
| MgSO4-7 H2O | Sigma-Aldrich | 230391 | |
| NaHCO3 | Sigma-Aldrich | S5761 | |
| Glucose | Sigma-Aldrich | G7021 | see Ultrapure Water system |
| Ultrapure Water | Prepare 1 or 2M. | ||
| KOH | Sigma-Aldrich | P5958-500G | |
| K-D-gluconate | Sigma-Aldrich | G4500 Sigma | |
| KCl | Sigma-Aldrich | P9541 | |
| KOH-EGTA (Etilene glycol-bis-N-tetracetic acid) | Sigma-Aldrich | E3889 Sigma | |
| KOH- Hepes acid (N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid) | Sigma-Aldrich | H7523 | |
| NaCl | Sigma-Aldrich | S3014 | |
| Mg2ATP | Sigma-Aldrich | A9187 | |
| Na3GTP | Sigma-Aldrich | G8877 | |
| Biocytin | Sigma-Aldrich | B4261 | |
| Picrotoxin | Merck | P1675 Sigma | |
| CNQX | Merck | C239 Sigma | |
| Ice | |||
| EQUIPMENT FOR WHOLE-CELL PATCH CLAMP RECORDINGS | |||
| Borosilicate glass pipette | Sutter Company | B150-86-10 | |
| Glass capillary puller | Sutter company | P-1000 | |
| Vibratome | Leica | Leica VT1000 S | |
| WaterBath | Thermo Fisher Scientific | TSGP02 | |
| Clampfit software | Molecular Devices | ||
| Multiclamp software | Molecular Devices | ||
| REAGENTS FOR IMMUNOHISTOCHEMISTRY | Use at a concentration of 4%. CAUTION: PFA is a potent fixative. Avoid ingestion and contact with skin. | ||
| Paraformaldehyde (PFA) | Merck Millipore | 1040051000 | Use at a concentration of 0.1%. |
| Triton X-100 | Fisher Scientific | 10254640 | Donkey.Use at a concentration of 1 : 400. |
| Serum | Merck Millipore | S30-100ML | Reconstitute the powder in Milli-Q water to 1 mg/mL. Aliquot and store at -20°C, light sensitive. Use at a concentration of 1 : 500. |
| 4′,6-Diamidino-2′-phenylindole dihydrochloride (DAPI) | Sigma Aldrich | D9542 | 1: 600 in KPBS-T. |
| streptavidin- Cy3 | Thermo Fisher Scientific | 434315 | 1:5000, rabbit. |
| Anti-GAD65/67 | Abcam | 1:2000, mouse. | |
| Anti-PV | Sigma | P3088 | 1:200, goat. |
| Anti-Chat | Merk | AB143 | 1:5000, rabbit. |
| Anti-NPY | Immunostar | 22940 | |
| K2HPO4 | Sigma-Aldrich | P3786 | |
| NaCl | Sigma-Aldrich | S3014 | |
| KH2PO4 | Sigma-Aldrich | NIST200B | 1:1000, chicken. |
| Anti-GFP | Abcam | ab13970 | |
| Mounting solution | Merck | 10981 | Polyvinyl alcohol mounting medium with DABCO®, antifading |
| OCT | Agar Scientific | ||
| Glycerol | Sigma-Aldrich | G9012-100ML | |
| Distilled water | |||
| Anti-freeze solution | Tissue Pro Technology | AFS05-1000N, 1000 mL/ea. | |
| EQUIPMENT FOR IMMUNOHISTOCHEMISTRY | |||
| Inverted fluorescence microscope | Leica | DMI6000 B | |
| Confocal microscope | Leica | TCS SP8 laser scanning confocal microscope. | |
| Prism | GraphPad | ||
| Microtome | Leica | SM2010R |
References
- Barker, R. A., Parmar, M., Studer, L., Takahashi, J. Human Trials of Stem Cell-Derived Dopamine Neurons for Parkinson's Disease: Dawn of a New Era. Cell Stem Cell. 21 (5), 569-573 (2017).
- Southwell, D. G., et al. Interneurons from embryonic development to cell-based therapy. Science. 344 (6180), 1240622 (2014).
- Parmar, M., Torper, O., Drouin-Ouellet, J. Cell-based therapy for Parkinson's disease: A journey through decades toward the light side of the Force. European Journal of Neuroscience. , (2018).
- Torper, O., et al. In Vivo Reprogramming of Striatal NG2 Glia into Functional Neurons that Integrate into Local Host Circuitry. Cell Reports. 12 (3), 474-481 (2015).
- Su, Z., Niu, W., Liu, M. L., Zou, Y., Zhang, C. L. In vivo conversion of astrocytes to neurons in the injured adult spinal cord. Nature Communication. 5, 3338 (2014).
- Liu, Y., et al. Ascl1 Converts Dorsal Midbrain Astrocytes into Functional Neurons In Vivo. Journal of Neuroscience. 35 (25), 9336-9355 (2015).
- Guo, Z., et al. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer's disease model. Cell Stem Cell. 14 (2), 188-202 (2014).
- Grande, A., et al. Environmental impact on direct neuronal reprogramming in vivo in the adult brain. Nature Communication. 4, 2373 (2013).
- Niu, W., et al. In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nature Cell Biology. 15 (10), 1164-1175 (2013).
- Weinberg, M. S., Criswell, H. E., Powell, S. K., Bhatt, A. P., McCown, T. J. Viral Vector Reprogramming of Adult Resident Striatal Oligodendrocytes into Functional Neurons. Molecular Therapy. 25 (4), 928-934 (2017).
- Pereira, M., et al. Direct Reprogramming of Resident NG2 Glia into Neurons with Properties of Fast-Spiking Parvalbumin-Containing Interneurons. Stem Cell Reports. 9 (3), 742-751 (2017).
- Rivetti di Val Cervo, P., et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson's disease model. Nature Biotechnology. 35 (5), 444-452 (2017).
- Crosson, S. M., Dib, P., Smith, J. K., Zolotukhin, S. Helper-free Production of Laboratory Grade AAV and Purification by Iodixanol Density Gradient Centrifugation. Molecular Therapy Methods & Clinial Development. 10, 1-7 (2018).
- Torper, O., et al. Generation of induced neurons via direct conversion in vivo. Proceedings of the National Academy of Sciences of the United States of America. 110 (17), 7038-7043 (2013).
- Kawaguchi, Y., Kubota, Y. Correlation of physiological subgroupings of nonpyramidal cells with parvalbumin- and calbindinD28k-immunoreactive neurons in layer V of rat frontal cortex. Journal of Neurophysiology. 70 (1), 387-396 (1993).
- Grealish, S., et al. The A9 dopamine neuron component in grafts of ventral mesencephalon is an important determinant for recovery of motor function in a rat model of Parkinson's disease. Brain. 133 (Pt 2), 482-495 (2010).
- Thompson, L., Barraud, P., Andersson, E., Kirik, D., Bjorklund, A. Identification of dopaminergic neurons of nigral and ventral tegmental area subtypes in grafts of fetal ventral mesencephalon based on cell morphology, protein expression, and efferent projections. Journal of Neuroscience. 25 (27), 6467-6477 (2005).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved