
Method Article
In Vivo Imaging of Transduction Efficiencies of Cardiac Targeting Peptide
In This Article
Summary
We describe protocols for assessing the degree of transduction by cell-penetrating peptides utilizing ex vivo imaging systems followed by paraffin embedding, sectioning, and confocal fluorescent microscopy using cardiac targeting peptide as an example. In our protocol, a single animal can be used to acquire both types of imaging assessment of the same organs, thereby cutting the number of animals needed for studies by half.
Abstract
Since the initial description of protein transduction domains, also known as cell penetrating peptides, over 25 years ago, there has been intense interest in developing these peptides, especially cell-specific ones, as novel vectors for delivering diagnostic and therapeutic materials. Our past work involving phage display identified a novel, nonnaturally occurring, 12 amino acid-long peptide that we named cardiac targeting peptide (CTP) due to its ability to transduce normal heart tissue in vivo with peak uptake seen in as little as 15 min after an intravenous injection. We have undertaken detailed biodistribution studies by injecting CTP labeled with fluorophore cyanine5.5, allowing it to circulate for various periods of time, and euthanizing, fixing, and sectioning multiple organs followed by fluorescent microscopy imaging. In this publication, we describe these processes as well as ex vivo imaging of harvested organs using an in vivo imaging system in detail. We provide detailed methodologies and practices for undertaking transduction as well as biodistribution studies using CTP as an example.
Introduction
The cell plasma membrane is a semipermeable barrier that is essential for cell integrity and survival and serves to regulate the interior of the cell by controlling movement of substances into the cell. Although vital for survival, it also presents a barrier to delivery of cargo to the cell. In 1988, the trans-activator of transcription (Tat) protein of the human immunodeficiency virus was shown to enter cultured cells and promote viral gene expression1,2, with the domains responsible for this transduction limited to an arginine and lysine rich 13-amino acid region of the third helix3 These were thus named cell penetrating peptides (CPPs). This was followed by research showing the ability of the Tat peptide to carry functional β-galactosidase into multiple cell types4. Since the initial description, the number of cell-penetrating peptides has increased dramatically. These transduction domains are naturally occurring or synthetic short peptides, typically 6−20 amino acids long, that are able to carry functional cargoes across cell membranes. These cargoes can range from other small peptides, full-length proteins, nucleic acids, nanoparticles, viral particles, fluorescent molecules, and radioisotopes5. The initial CPPs described were not cell specific, with Tat being taken up by multiple cell types and even crossing the blood-brain barrier4, hence limiting its therapeutic potential. In order to identify cell-specific CPPs, investigators have undertaken phage display utilizing large, commercially available phage libraries6. Our own work using a combinatorial in vitro and in vivo phage display methodology led to the identification of a CPP named cardiac targeting peptide (CTP)7, a 12-amino acid, nonnaturally occurring peptide (NH2-APWHLSSQYSRT-COOH) that targets the heart with peak uptake at 15 min after a peripheral intravenous injection8. Using immunofluorescent colocalization with actin, an intracellular marker, and exclusion of laminin, a cell membrane marker, we showed that CTP is internalized into mouse cardiomyocytes after an intravenous injection7. Additionally, we incubated human induced pluripotent stem cell-derived beating cardiomyocytes with dual-labeled CTP, labeled with 6-carboxyfluorescein at its C-terminus and rhodamine at its N-terminus through an ester linkage that could only be cleaved off by intracellular esterases. Rapid accumulation of rhodamine into beating cardiomyocytes was observed within 15 min on confocal microscopy8.
In this article, we present two complimentary methodologies that can be used to track biodistribution and confirm tissue-specific internalization of CPPs using CTP as an example. For these methodologies, CTP was synthesized, fluorescently labeled at the N-terminus with cyanine5.5 (CY5.5), and amide-capped at the C-terminus for greater peptide stability. The two methodologies used are in vivo fluorescent optical imaging systems and fluorescent microscopy of tissue sections. Both methods are very useful in studying biodistribution, uptake, and elimination of fluorescently labeled CPPs. The advantage of assessing biodistribution using these methods over others, such as single-photon emission tomography (SPECT) or positron emission tomography (PET), is that there is no need for the time-intensive radiolabeling of CPPs compared with fluorescent labeling, which is relatively easy and in routine use at all peptide synthesis facilities. The use of in vivo imaging quickly produces biodistribution data in the context of a living system, while sectioning provides greater in-depth information about peptide uptake and localization within cells via the preservation of morphological detail. This protocol can be applied to a wide variety of organs and tissues such as the heart, lung, liver, kidney, brain, spleen, stomach, large intestine, small intestine, skeletal muscle, bone, testes/ovaries, and eyes.
Protocol
The University of Pittsburgh's Institutional Animal Care and Use Committee approved all animal protocols specified in this publication prior to undertaking any of these animal experiments.
NOTE: This protocol details how to perform ex vivo biodistribution studies utilizing ex vivo optical fluorescent imaging followed by in vivo biodistribution studies by embedding the organs in paraffin, sectioning, and performing fluorescent microscopy. Although CTP is used as an example, this method can be applied to any fluorescently labeled CPP.
1. In vivo imaging
- Synthesize CTP using solid phase synthesis techniques9,10 from a peptide synthesis facility using L-amino acids with an N-terminus labeled with Cy5.5 and C-terminus amide-capped for increased stability.
NOTE: All synthesized CPPs should be characterized using high-performance liquid chromatography (HPLC) and/or matrix assisted laser desorption/ionization (MALDI) prior to use11. - Make a 1 mM or 10 mM stock solution of CTP in dimethyl sulfoxide (DMSO), aliquot, and store at -80 °C, light-protected.
NOTE: Peptides are delivered as lyophilized powder. Lyophilized powder can be stored long-term (6 months-2 years) in -20 °C, light-protected, and under hygroscopic conditions. When aliquoted in DMSO and stored, freeze-thaw cycles should be avoided. - Weigh and anesthetize 6-week-old CD1 female mice using 2 µL/g of tissue weight of a ketamine/xylazine solution (100 mg/kg ketamine and 20 mg/kg xylazine) administered intramuscularly (hind leg) or intraperitoneally (lower left quadrant).
NOTE: Ketamine/xylazine solutions should be made fresh on the day of use. Unused ketamine/xylazine should be disposed of appropriately and volumes used versus volumes discarded recorded in a log, as ketamine is a controlled substance. Adequate level of anesthesia will be achieved in 5−7 min, as assessed by lack of response to a toe pinch. - Calculate a 10 mg/kg dose of Cy5.5-CTP, dilute with phosphate buffered saline (PBS) to no more than 200 µL, and inject either retro-orbitally or through tail vein injection using an insulin syringe (Figure 1A).
- Allow peptide to circulate for the prespecified time, ranging from 15 min-6 h.
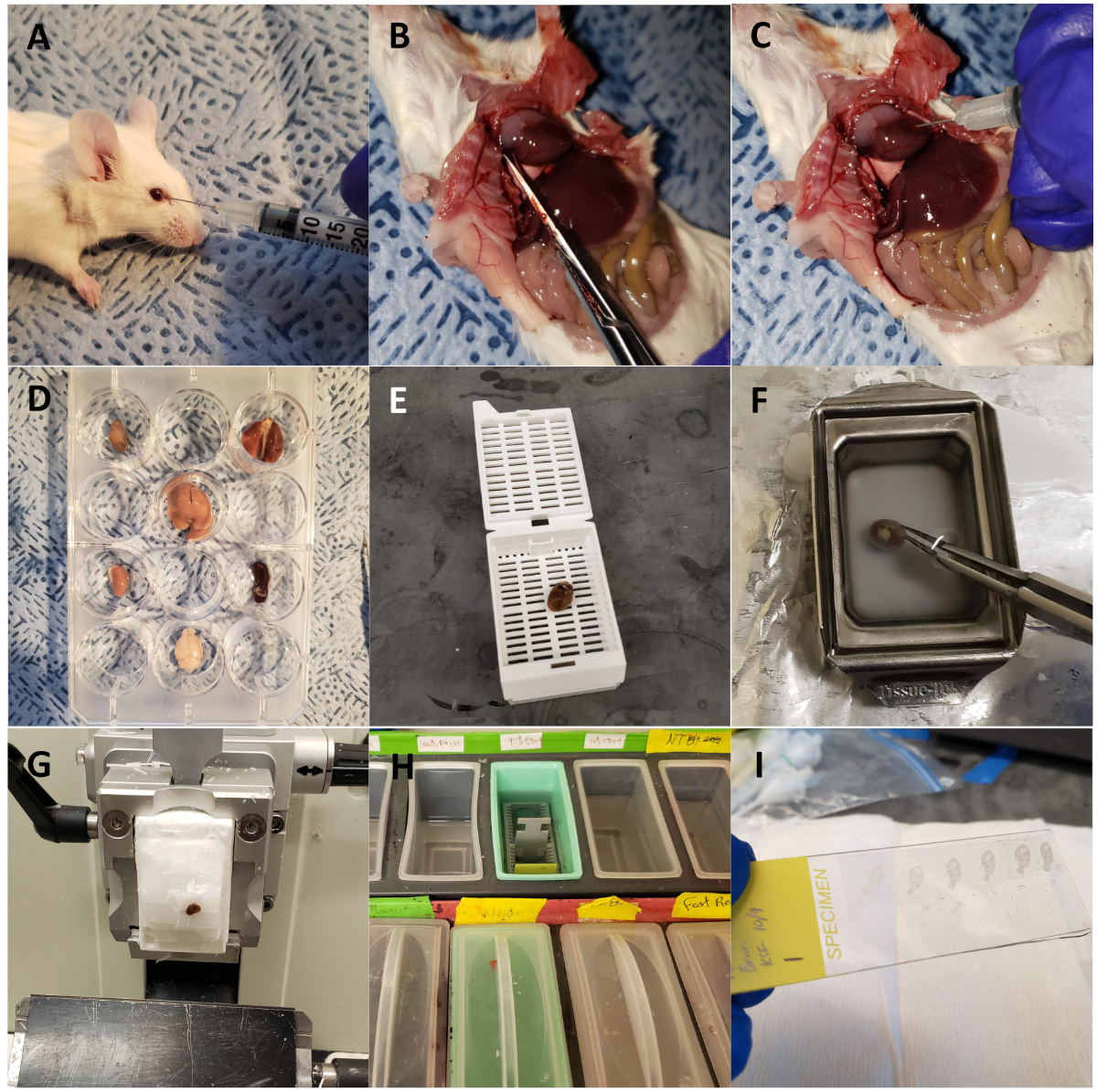
- Euthanize the mouse using the high CO2 inhalational method as specified by the Institutional Animal Care and Use Committee and open the chest cavity using scissors (Figure 1B).
- Use scissors to place a small nick in the lateral, free wall of the right atrium and inject 3 mL of 10% buffered formalin phosphate using a 26 G needle for the dual purpose of perfusion fixing the organs of the mouse and flushing out red blood cells (Figure 1C).
- Dissect out heart, lung, liver, kidneys, spleen, large intestines, small intestines, bladder, ovaries/testes, and brain and place into individual wells of a 12 well plate for ex vivo optical imaging (Figure 1D).
- Start the image acquisition software and click the Initialize button to prepare the system for imaging samples. Open the imaging wizard, select fluorescence, then filter pair, then select the Cy5.5 dye from the list of dyes to apply 580 nm excitation and 620 nm emission filters. Select manual settings for exposure parameters then set the stage position to B, set exposure time to 1 second, set the F/stop to 8, and set the binning to small.
NOTE: The excitation emission filters will vary depending on the label used. The excitation and emission of the label used can be manually entered for the software to assign excitation and emission filters. Ensure that the counts are between 600-60,000 in order to avoid saturation. Depending on the system being used, automatic acquisition optimization might be available. If the system has compensation to compare images acquired with different settings, the auto settings can be used. If no compensation is available, settings will need to be determined, saved, and used consistently across samples. - When the system is completely initialized, arrange the organs of interest in a 12 well plate and place it inside of the optical imaging chamber, ensuring that the samples are arranged as desired for the images. Select acquire, then select a save location for the images. Information about the mouse can be written into the edit image labels pop-up window.
- After imaging, remove the samples from the chamber and store in 10% buffered formalin phosphate in a volume at least 20 times the volume of the tissue in scintillation vials with light protection at room temperature (RT).
- Quantify the images by setting units to Radiant Efficiency then selecting the 4x3 ROI and adjusting to fit each well into one square of the ROI evenly, followed by clicking measure. Open the grid ROI measurements tab, select export, and save the measurements as a .cvs file.
2. Histology
NOTE: Follow steps 1.1−1.8 to obtain different organs. Alternatively, the same organs that were imaged can be placed in formalin immediately and used for sectioning as detailed below.
- Store each organ individually in 10% buffered formalin phosphate for a minimum of 48 h before processing for histology.
- When the organs are sufficiently fixed after 48 h, transfer the organs into tissue processing and embedding cassettes and place into a tissue processing machine (Figure 1E).
- Set the processing machine to dehydrate the tissue in 70% ethanol for 30 min, followed by 80% ethanol for 30 min, 95% ethanol for 30 min, 95% ethanol for 30 min, 100% ethanol for 15 min, 100% ethanol for 20 min, and finally 100% ethanol for 20 min.
- Clear the tissue in xylene 2x, with 30 min for each xylene treatment.
- Infiltrate the cleared tissue with paraffin wax 4x at 60 °C for 30 min with each treatment.
- Embed in paraffin using metal molds. Fill the mold with molten paraffin (kept at 65 °C), and transfer to a cold plate. As the paraffin at the bottom of the mold begins to solidify, place the organ into the paraffin (Figure 1F).
- Place a labeled cassette on top of the mold as a backing and overfill with molten paraffin. Allow to cool until solid. Then store the block in a -20 °C freezer overnight, allowing the wax to shrink further for easy removal from the molds.
NOTE: The blocks can now be stored at RT with light protection. - Prepare a 38 °C water bath with distilled water. Set up the microtome with a blade angle of 6° and a section thickness of 10 µm.
- Mount the tissue blocks in the microtome and begin cutting until sections containing tissue are obtained. Then place the blocks face down in the water for 5 min or until the tissue has absorbed some moisture (e.g., when a thin white outline of the tissue appears in the block [Figure 1G]).
NOTE: The blade should be replaced frequently to ensure the quality of the sections. - Place the tissue onto a flat ice block for 10 min, then return it to the microtome in the same orientation as in step 2.9. Begin taking sections and allow truncated sections to form long ribbons of 6−10 sections each. Discard suboptimal paraffin ribbons until a high-quality ribbon of sufficient length to cover a slide is produced. Optimize sectioning by adjusting cutting speeds, tissue moisturization, and water bath temperature in order to avoid shearing or wrinkling of the tissue sections.
NOTE: Sections that have holes where tissue fell out, rips in the tissue, or tight wrinkles and folds in the tissue should be discarded. - Carefully pick the quality ribbon of choice with blunt edge forceps and float on the surface of the 38 °C water bath. Let the sections sit on the surface until they smooth out, taking care to not leave them too long to prevent the paraffin from disintegrating and tearing the section apart.
- Float the flattened sections onto the surface of clean glass slides. Place the slides into a 65 °C oven for 30 min to melt the wax.
NOTE: These slides can be stored at RT with light protection. - Deparaffinize the slides in xylene 3x, 10 min for each treatment.
- Rehydrate the tissue in 100% ethanol for 5 min, followed by 95% ethanol for 5 min, 70% ethanol for 5 min, 50% ethanol for 5 min, and finally 1x tris-buffered saline (TBS) for 5 min (Figure 1H). Allow the slides to dry overnight.
NOTE: These slides can be stored long-term at RT as long as they are light-protected. - Mount the slides with coverslips using 125 µL of mounting media containing 4′,6-diamidino-2-phenylindole (DAPI), a nuclear fluorescent probe (Figure 1I). Dry slides overnight at RT, light-protected.
NOTE: Dried slides can be stored long-term at 4 °C, light-protected. - Image slides using fluorescent microscopy.
NOTE: Saturated images should be avoided, as they are not quantitatively useful. Adjusting the exposure and gain settings can be lowered to avoid saturation. Take care to use the same settings across the samples being compared. Avoid bleaching of the tissue by limiting exposure times.
Results
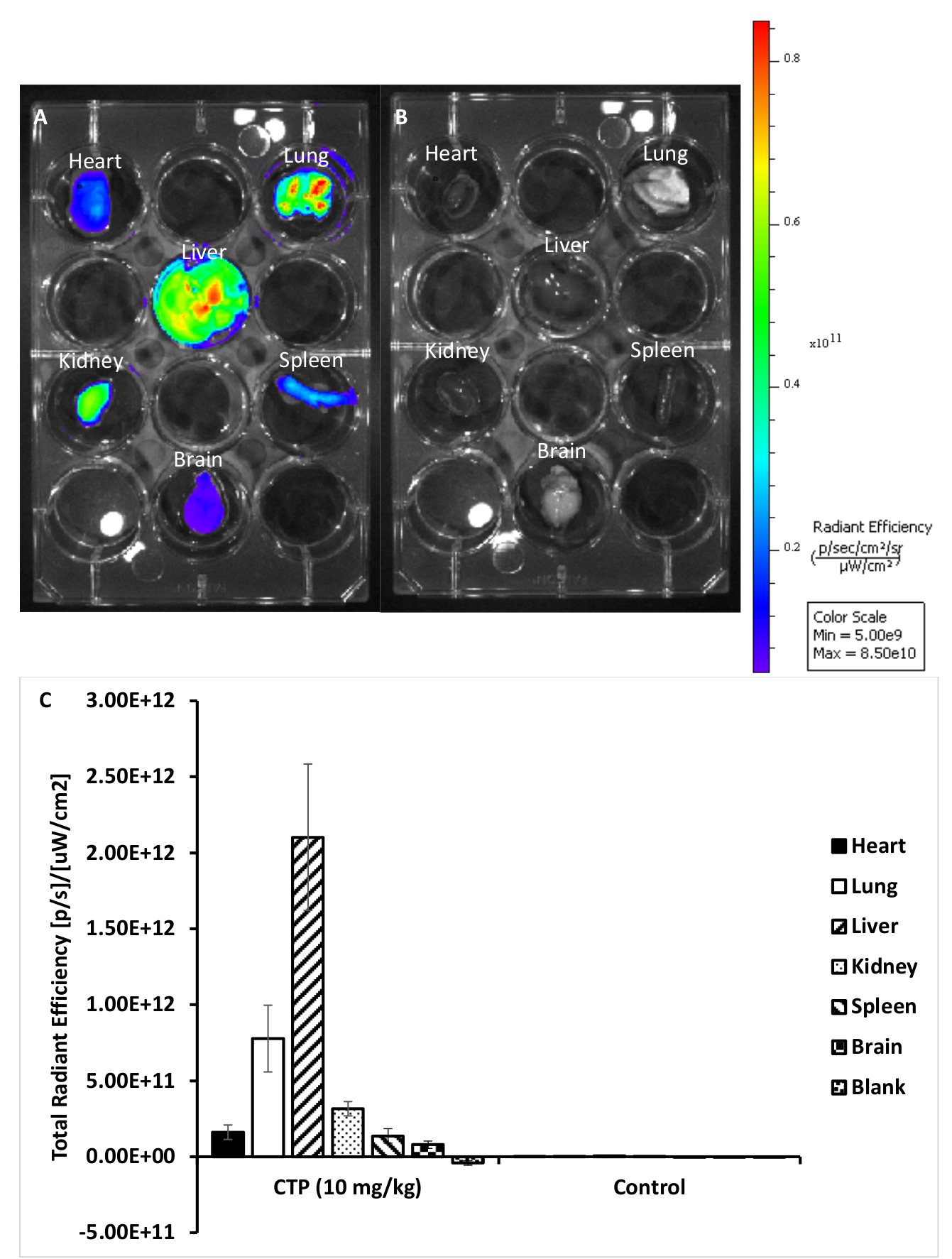
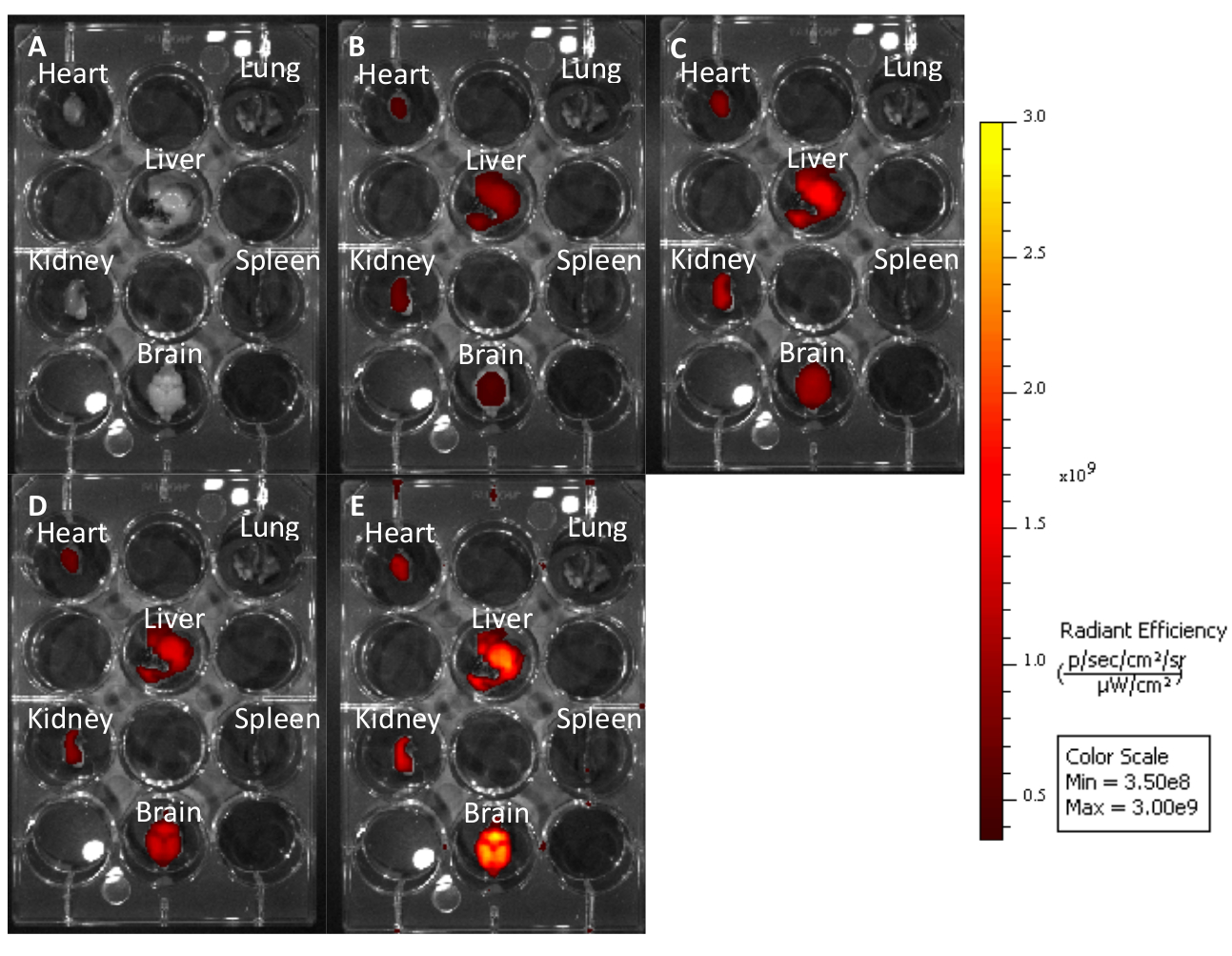
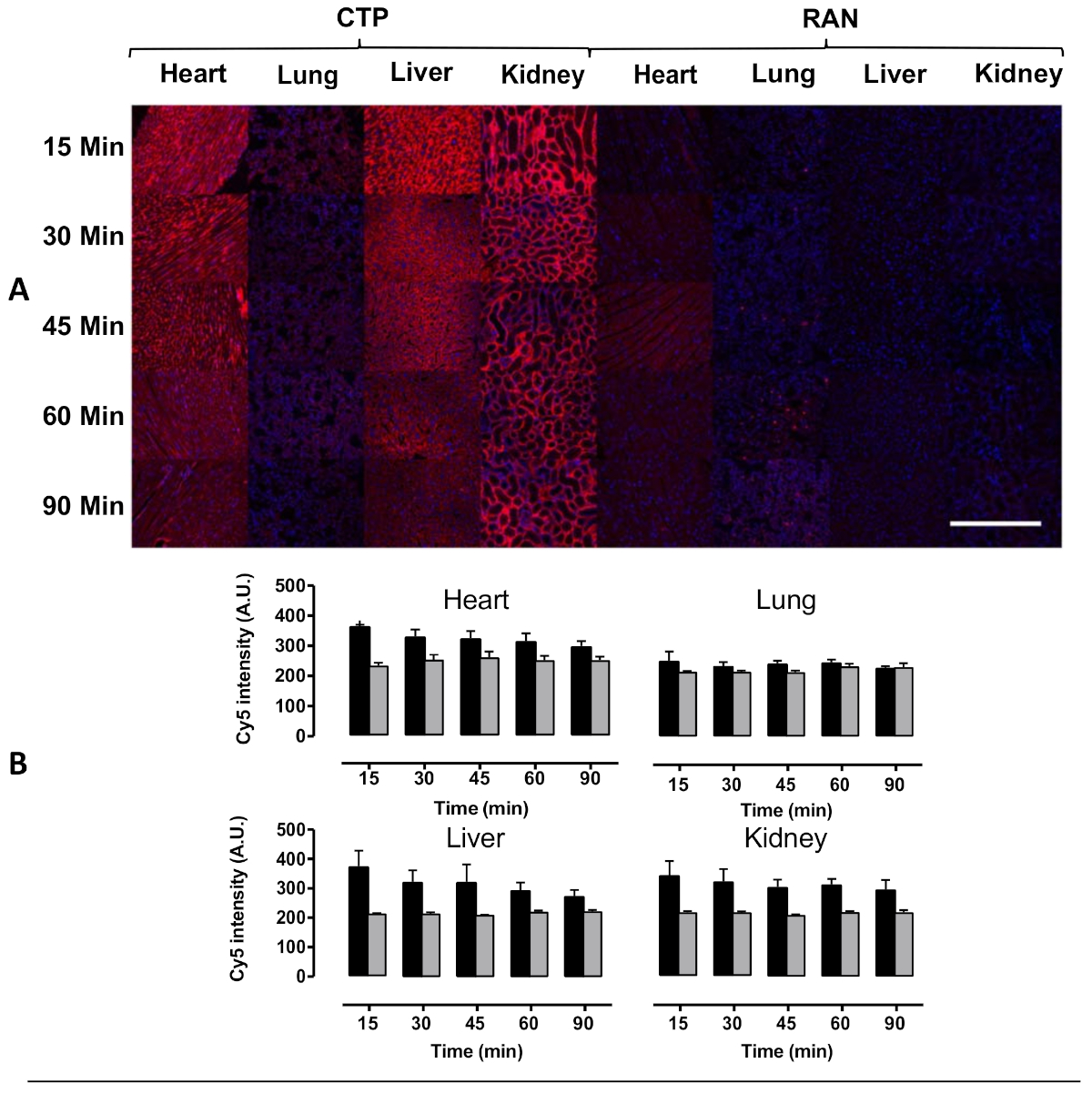
Using this protocol, we treated three mice with a 10 mg/kg dose of Cy5.5-CTP through a retro-orbital injection (Figure 1A). After allowing the peptide to circulate for 15 min, the mice were euthanized using a CO2 chamber, the chest opened through a median sternotomy incision, the right atrium was nicked, and the mice perfusion fixed using 3 mL of 10% phosphate-buffered formalin (Figure 1B,C). After fixation, the heart, lungs, liver, kidney, spleen, and brain were dissected out and arranged in a 12 well plate for ex vivo optical fluorescent imaging (Figure 1D and Figure 2A). Three control mice with no injections were also perfused, dissected, and imaged (Figure 1B−D and Figure 2B). The fluorescence data acquired for each set of organs can be compared due to the counts being converted to radiant efficiency, the relative fluorescence unit of the imaging software. This data can be quantified to yield both qualitative images and quantitative data for each organ in question (Figure 2C) across different time points for biodistribution studies. The autofluorescence of different organs in response to different excitation wavelengths is demonstrated in Figure 3A−E. Shorter excitation wavelengths, such as enhanced green fluorescent protein (EGFP), are associated with higher autofluorescence, especially in liver and brain, than far-red (Cy5.5) or near infrared (Cy7) excitation wavelengths (Figure 3). The imaged organs were fixed in 10% phosphate buffered formalin at RT for a minimum of 48 h, followed by a transfer to tissue processing and embedding cassettes (Figure 1E). The organs were processed and paraffinized in a tissue processing machine, positioned in molds filled with paraffin, frozen on a -20 °C stage, and stored overnight at -20 °C (Figure 1F). The samples were sectioned (Figure 1G) and treated with solution exchanges to rehydrate the tissue (Figure 1H). The prepared slides were then mounted with DAPI fluorescent mounting medium (Figure 1I). Once dry, usually overnight, the slides were imaged using fluorescent microscopy. Representative images of each organ from a mouse are shown in Figure 4A. The images from different organs were acquired using identical settings to allow for comparison across mice, and to allow for quantification (Figure 4B).

Figure 1: Harvesting mouse organs for ex vivo optical fluorescent imaging and sectioning. (A) Wild type mouse injected retro-orbitally with CTP-Cy5.5. (B) Mouse dissected with chest opened, right atrium snipped, for perfusion fixation. (C) Heart injected with 3 mL of 10% buffered formalin phosphate for perfusion fixation of the animal. (D) Organs of interest harvested and arranged in a 12 well plate for fluorescent optical imaging. (E) Heart loaded into a cassette and processed using a tissue processor. (F) Heart embedded in paraffin. (G) Heart sectioned on a microtome. (H) Slides deparaffinized through a series of solution exchanges. (I) Sections mounted with coverslips using DAPI. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative images from treated and control organs. (A) Organs from a mouse treated with 10 mg/kg CTP-Cy5.5. (B) Organs from an untreated control mouse. (C) Quantification of fluorescence for each set of organs. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Untreated mouse organs imaged at different excitation emission wavelengths to demonstrate how longer wavelengths produce less autofluorescence. (A) Cy7: 740−790. (B) Cy5.5: 660−710. (C) Cy5: 620−670. (D) Cy3: 520−570. (E) EGFP: 480−520. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Transduction of heart, lung, liver, and kidney after intravenous injection in mice. Wild type mice were injected with 10 mg/kg of Cy5.5-CTP, or a random peptide (RAN-Cy5.5), and euthanized at the indicated time points. (A) Peak transduction of heart tissue was seen at 15 min with steady decrease in fluorescence over time. Some capillary uptake was noted in the lungs with robust transduction in the liver as well at the kidney glomerular capillaries, the latter implying a renal mechanism of excretion. (B) Quantification of fluorescent intensity shows significantly increased heart uptake of CTP-Cy5.5 over RAN-Cy5.5. Scale bar = 500 µm. This figure has been modified from Zahid et al.8. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Animal models are essential for preclinical drug development at every stage of the process from identification of novel CPPs, biodistribution studies, mechanism of transduction, to ultimately testing for efficacy of the delivered cargo using these novel CPPs as vectors. There are many commonly used methods available to assess biodistribution such as histology, nuclear medicine imaging (SPECT and PET), and in vivo optical imaging12. Nuclear imaging methods can be cumbersome due to the limited availability of small animal SPECT and PET systems, as well as the ability to produce radiolabel-drug conjugates, which require the expertise of a radiochemist. In contrast, fluorescent labels are much simpler and can be cost effective to work with. The protocol described in this paper allows for rapid analysis of biodistribution using multiple methods. Ex vivo whole organ fluorescent optical imaging allows for the immediate comparison of fluorescence across different tissues and treatment groups and can identify organ uptake and time to peak uptake in organ of interest in a semiquantitative manner. Quantitative tissue histology requires more extensive processing to treat, section, image, and analyze tissues, but it provides more data on the microscopic level and is a current standard technique. It is worthwhile to note that direct, quantitative comparison across the two techniques is not possible, because the autofluorescence seen with each technique for different organs and excitation wavelengths varies significantly.
An important part of this protocol is selecting the right fluorophore to label candidate CPPs for experiments. One potential issue is spectra overlap, which can be problematic when multiple fluorophores are needed. The DAPI fluorescent mounting media and Cy5.5 do not have overlapping spectra. However, for certain applications where multiple fluorophores are needed, the risk of spectral overlap needs to be carefully considered. Depending on the system being used, fluorophore selection may be limited. Therefore, knowledge of the system's capabilities is key. Fluorescent optical systems are best utilized with far-red or near-infrared fluorophores due to the high tissue absorption of shorter wavelengths13. Fluorophores in the range of enhanced green fluorescent protein have a major limitation, because there is significant organ autofluorescence seen at its excitation wavelength, specifically in brain and liver tissue. Depending on the conditions of an experiment, some fluorophores are best avoided. Some water-soluble organic fluorophores have a strong interaction with lipid bilayers, which can cause false positives. Hence, taking steps to determine if a fluorophore has strong affinity to the tissue of interest is advisable14. Another factor to consider is selecting the appropriate method of fluorophore conjugation, which can be an important parameter affecting the results. CPPs can be labeled fluorescently at the N- or C-terminus through a covalent bond between the N-terminus of the peptide and the carboxyl group of the dye such as Cy5.5-NHS. Care should be taken, because the mechanism of transduction of most CPPs is not understood in detail and conjugation at one end may affect the uptake mechanism more so than at the other end. Another possibility for labeling CPPs is through biotinylating the N-terminus for conjugation to fluorescently labeled streptavidin. Using this strategy has the convenience of allowing different fluorescent streptavidin conjugates to be utilized. However, a possible limitation of this strategy is that a biotin-streptavidin complex is a large construct, which could potentially interfere with transduction.
Fluorescent optical imaging systems are an effective strategy for generating comparison of fluorescence across different organs and treatments efficiently but are incapable of producing a quantitative measure of absolute concentrations in tissue. This is due to light scattering effects within the tissue, which is further compounded by the naturally occurring variety in tissue sizes and densities, and differences in vascularity, with variable fluorescence scattering. Tissue autofluorescence can be a factor as well, due to naturally occurring biochemical sources such as collagen, or dietary sources like chlorophyll in food13.
Histology is the most commonly used method of measuring biodistribution and can potentially be used to accurately measure and compare uptake across different tissues over time. Light scattering issues are avoided using this method because all tissues are sectioned to the same thickness15. A major advantage of this method is the ability to include additional fluorescent labels postsectioning for immunohistochemistry. Although the addition of another fluorophore could make imaging more challenging, the use of fluorescent labels can be useful for localization of a CPP to particular intracellular compartments, like lysosomes or mitochondria. An antibody could be used in a confocal microscopy experiment to determine if a transduced candidate CPP colocalizes with a structure of interest, which can show the potential of a CPP as a delivery agent. One limitation of this method is that preparation of slides from organ samples can be time consuming, labor intensive, and prone to human error12,15. When imaging slides, care should be taken to not image the same location for too long to avoid photobleaching. Some photobleaching will be inevitable, depending on the sensitivity of the fluorophore. Care should be taken at every step of this protocol to protect the samples from ambient light and store them properly16. We recommend that slides be stored at 4 °C, light-protected, for future imaging.
There are a variety of methods available for measuring the biodistribution of a candidate CPP that require specialized equipment and can produce comparable results, though they may require more complex CPP labeling. The protocol described in this paper uses two compatible methods to efficiently produce biodistribution data in the context of a living system while allowing for the acquisition of greater in-depth information about peptide internalization within cells from the same sample, thus cutting the number of animals needed for a study by half. These methods were used to generate the above data, which demonstrate that both methods can be utilized sequentially in the same animal, and the quality of the data generated by each. Our results also highlight the inability to directly correlate results between the two techniques in a quantitative manner.
Disclosures
M.Z. and Paul D. Robbins (University of Minnesota, Minnesota, MN, USA) hold a patent on the use of cardiac targeting peptide as a cardiac vector (Cardiac-specific protein targeting domain, U.S. Patent Serial No. 9,249,184). M.Z. also serves as Chief Scientific Officer and is on the Board of Directors of the startup Vivasc Therapeutics Inc. and holds substantial future equity in it.
Acknowledgements
M.Z. and K.S.F. are supported by American Heart Association Scientist Development Award 17SDG33411180, and by a grant awarded under the Pitt Innovation Challenge (PInCh®), through the Clinical and Translational Science Institute at the University of Pittsburgh.
Materials
| Name | Company | Catalog Number | Comments |
| 10% Buffered Formalin Phosphate | ThermoFisher | SF100-4 | |
| 10x Tris Buffered Saline (TBS) | ThermoFisher | BP2471-1 | |
| 12-well Cell Culture Plate | ThermoFisher | 353043 | |
| 1x TBS Solution | 10x TBS is diluted 1:10 with deionized water. This solution can be stored at room temperature. | ||
| 20 mL Scintillation Vials | Wheaton | 334173 | |
| 26G Needles | Becton Dickinson | 305110 | |
| 28G 0.5ml Insulin syringes | Becton Dickinson | 329461 | |
| 3mL Syringe | Becton Dickinson | 309657 | |
| CD1 Mice | Charles River | 022 | 6 to 12 week old albino, female mice |
| Cover Glass | ThermoFisher | 12-544-14 | |
| Cy5.5-CTP-amide | Prepared by peptide synthesis, conjugated with Cy5.5 fluorophore, and purified using HPLC. Lyophilized powder is reconstituted in DMSO at 10mM concentration. After reconstituting, store at -80 °C. | ||
| Dapi Fluoromount G | SouthernBiotech | 0100-20 | |
| Dumont #5 Forceps | Fine Science Tools | 99150-20 | |
| Ethanol | |||
| Extra Fine Bonn Scissors | Fine Science Tools | 14084-08 | |
| Ketamine HCl 100mg/mL | KetaVed | NDC 50989-161-06 | |
| Ketamine/Xylazine Solution | Ketamine HCl is mixed with Xylazine 1:1 to produce a stock solution containing 50mg/mL ketamine and 10mg/mL Xylazine. This solution is made fresh before each use. | ||
| Leica RM2235 Rotary Microtome | Leica | RM2235 | |
| Microscope Slides | ThermoFisher | B9992000TN | |
| Paraplast X-TRA | Sigma | P3808-1KG | |
| Perkin Elmer Lumina S5 IVIS | Perkin Elmer | CLS148588 | |
| Sparkle Optical Lens Cleaner | ThermoFisher | NC0090079 | |
| Tissue-Tek Processing/Embedding Cassettes | ThermoFisher | NC9499605 | |
| Tissue-Tek VIP Processing machine | Tissue-Tek | VIP 5A-F1 | |
| Xylazine 20 mg/mL | AnaSed | NDC 59399-110-20 | |
| Xylenes | ThermoFisher | X5-4 |
References
- Green, M., Loewenstein, P. M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 55 (6), 1179-1188 (1988).
- Frankel, A. D., Pabo, C. O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 55 (6), 1189-1193 (1988).
- Derossi, D., Joliot, A. H., Chassaing, G., Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. Journal of Biological Chemistry. 269 (14), 10444-10450 (1994).
- Schwarze, S. R., Ho, A., Vocero-Akbani, A., Dowdy, S. F. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 285 (5433), 1569-1572 (1999).
- Zahid, M., Robbins, P. D. Cell-type specific penetrating peptides: therapeutic promises and challenges. Molecules. 20 (7), 13055-13070 (2015).
- Zahid, M., Robbins, P. D. Identification and characterization of tissue-specific protein transduction domains using peptide phage display. Methods in Molecular Biology. 683, 277-289 (2011).
- Zahid, M., et al. Identification of a cardiac specific protein transduction domain by in vivo biopanning using a M13 phage peptide display library in mice. PLoS One. 5 (8), e12252 (2010).
- Zahid, M., et al. Cardiac Targeting Peptide, a Novel Cardiac Vector: Studies in Bio-Distribution, Imaging Application, and Mechanism of Transduction. Biomolecules. 8 (4), E147 (2018).
- Amblard, M., Fehrentz, J. A., Martinez, J., Subra, G. Methods and protocols of modern solid phase Peptide synthesis. Molecular Biotechnology. 33 (3), 239-254 (2006).
- Katritzky, A. R., Yoshioka, M., Narindoshvili, T., Chung, A., Johnson, J. V. Fluorescent labeling of peptides on solid phase. Organic and Biomolecular Chemistry. 6 (24), 4582-4586 (2008).
- Prabhala, B. K., Mirza, O., Hojrup, P., Hansen, P. R. Characterization of Synthetic Peptides by Mass Spectrometry. Methods in Molecular Biology. 1348, 77-82 (2015).
- Arms, L., et al. Advantages and Limitations of Current Techniques for Analyzing the Biodistribution of Nanoparticles. Frontiers in Pharmacology. 9, 802 (2018).
- Leblond, F., Davis, S. C., Valdes, P. A., Pogue, B. W. Pre-clinical whole-body fluorescence imaging: Review of instruments, methods and applications. Journal of Photochemistry and Photobiology. 98 (1), 77-94 (2010).
- Hughes, L. D., Rawle, R. J., Boxer, S. G. Choose your label wisely: water-soluble fluorophores often interact with lipid bilayers. PLoS One. 9 (2), e87649 (2014).
- McGowan, J. W., Bidwell, G. L. The Use of Ex Vivo Whole-organ Imaging and Quantitative Tissue Histology to Determine the Bio-distribution of Fluorescently Labeled Molecules. Journal of Visualized Experiments. (118), e54987 (2016).
- Waters, J. C. Accuracy and precision in quantitative fluorescence microscopy. Journal of Cell Biology. 185 (7), 1135-1148 (2009).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved