Method Article
Анализ расплавов и жидкостей из моделирования молекулярной динамики Ab Initio с помощью пакета UMD
В этой статье
Резюме
Расплавы и жидкости являются вездесущими векторами массового переноса в природных системах. Мы разработали пакет с открытым исходным кодом для анализа ab initio молекулярно-динамического моделирования таких систем. Мы вычисляем структурные (связывание, кластеризация, химическое видообразование), перенос (диффузия, вязкость) и термодинамические свойства (вибрационный спектр).
Аннотация
Мы разработали пакет с открытым исходным кодом на основе Python для анализа результатов, полученных в результате моделирования ab initio молекулярной динамики жидкостей. Пакет лучше всего подходит для применения в природных системах, таких как силикатные и оксидные расплавы, жидкости на водной основе и различные сверхкритические жидкости. Пакет представляет собой коллекцию скриптов Python, которые включают в себя две основные библиотеки, работающие с форматами файлов и кристаллографией. Все скрипты выполняются в командной строке. Мы предлагаем упрощенный формат для хранения атомных траекторий и соответствующей термодинамической информации моделирования, которая сохраняется в файлах UMD, что означает универсальная молекулярная динамика. Пакет UMD позволяет вычислять ряд структурных, транспортных и термодинамических свойств. Начиная с функции распределения пар, он определяет длины связей, строит матрицу межатомной связности и в конечном итоге определяет химическое видообразование. Определение времени жизни химических видов позволяет провести полный статистический анализ. Затем специальные скрипты вычисляют среднеквадратичные смещения для атомов, а также для химических видов. Реализованный самокорреляционный анализ атомных скоростей дает коэффициенты диффузии и вибрационный спектр. Тот же анализ, примененный к напряжениям, дает вязкость. Пакет доступен через веб-сайт GitHub и через собственную специальную страницу проекта ERC IMPACT в виде пакета открытого доступа.
Введение
Жидкости и расплавы являются активными химическими и физическими транспортными векторами в природных средах. Повышенные скорости атомной диффузии благоприятствуют химическим обменам и реакциям, низкая вязкость в сочетании с изменяющейся плавучестью способствует большому массопереносу, а отношения плотности кристаллов и расплава благоприятствуют наслоению внутри планетарных тел. Отсутствие периодической решетки, типичные высокие температуры, необходимые для достижения расплавленного состояния, и трудность закалки делают экспериментальное определение ряда очевидных свойств, таких как плотность, диффузия и вязкость, чрезвычайно сложным. Эти трудности делают альтернативные вычислительные методы сильными и полезными инструментами для исследования этого класса материалов.
С появлением вычислительной мощности и доступностью суперкомпьютеров в настоящее время используются два основных метода численного атомистического моделирования для изучения динамического состояния некристаллической атомистической системы, Monte Carlo1 и молекулярной динамики (MD)1,2. В моделировании Монте-Карло конфигурационное пространство выбирается случайным образом; Методы Монте-Карло показывают линейное масштабирование при распараллеливании, если все наблюдения выборки независимы друг от друга. Качество результатов зависит от качества генератора случайных чисел и репрезентативности выборки. Методы Монте-Карло показывают линейное масштабирование при распараллеливании, если выборка независима друг от друга. В молекулярной динамике (МД) конфигурационное пространство отбирается по зависящим от времени атомным траекториям. Начиная с заданной конфигурации, атомные траектории вычисляются путем интегрирования ньютоновских уравнений движения. Межатомные силы могут быть вычислены с использованием модельных межатомных потенциалов (в классическом MD) или с использованием методов первых принципов (in ab initio, или first-principles, MD). Качество результатов зависит от длины траектории и ее способности не притягиваться к локальным минимумам.
Моделирование молекулярной динамики содержит множество информации, связанной с динамическим поведением системы. Термодинамические средние свойства, такие как внутренняя энергия, температура и давление, довольно стандартны для расчета. Они могут быть извлечены из выходного файла (файлов) моделирования и усреднены, тогда как величины, связанные непосредственно с движением атомов, а также с их взаимосвязью, должны быть вычислены после извлечения атомных положений и скоростей.
Следовательно, много усилий было посвящено визуализации результатов, и сегодня доступны различные пакеты, на разных платформах, с открытым исходным кодом или нет [Ovito3, VMD4, Vesta5, Travis6 и т. Д.]. Все эти инструменты визуализации эффективно справляются с межатомными расстояниями, и поэтому они позволяют эффективно вычислять функции распределения пар и коэффициенты диффузии. Различные группы, выполняющие крупномасштабное моделирование молекулярной динамики, имеют проприетарное программное обеспечение для анализа различных других свойств, возникающих в результате моделирования, иногда в условно-бесплатных или других формах ограниченного доступа к сообществу, а иногда ограниченных по объему и использованию некоторыми конкретными пакетами. Сложные алгоритмы извлечения информации о межатомных связях, геометрических закономерностях и термодинамике разработаны и реализованы в некоторых из этих пакетов3,4,5,6,7 и т.д.
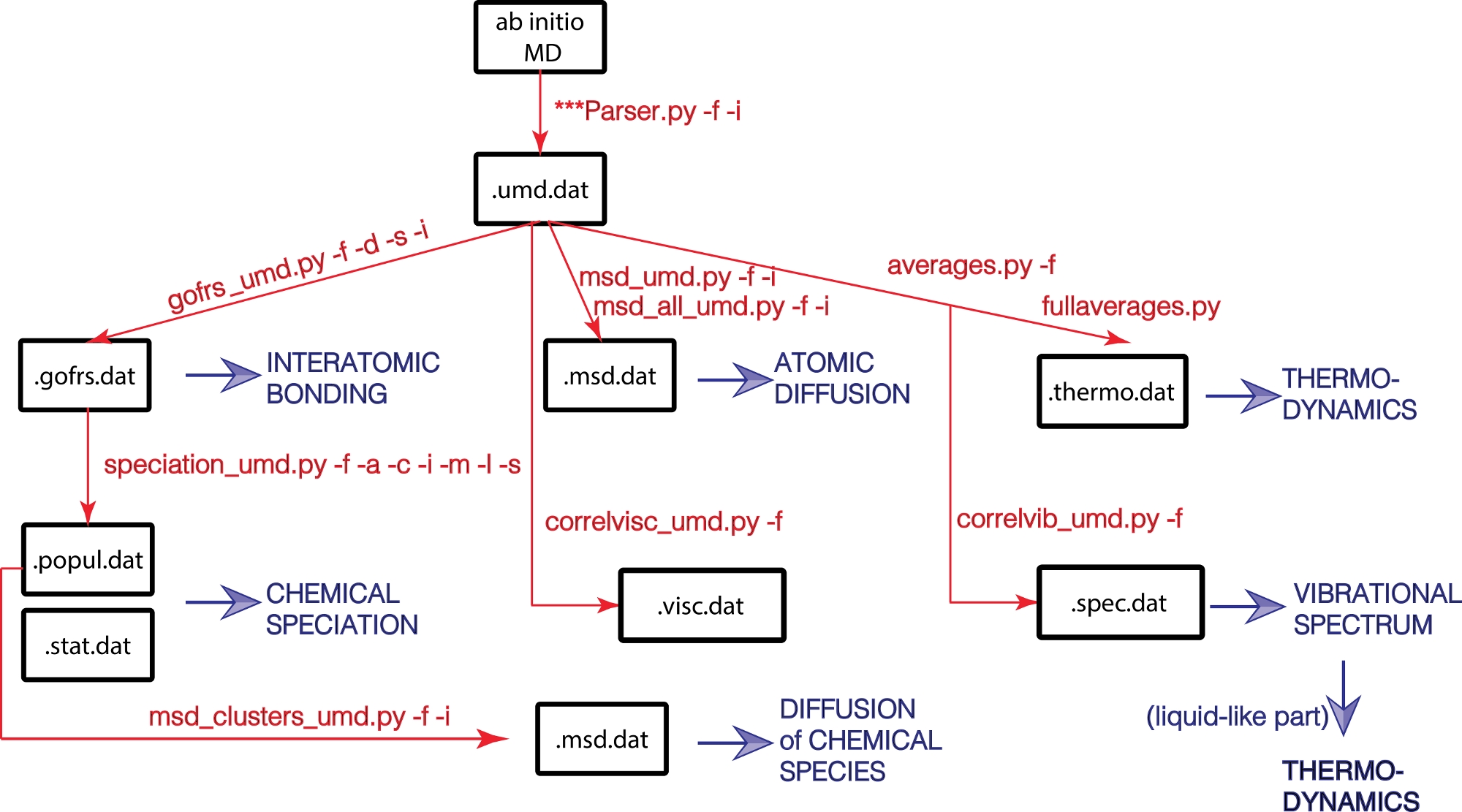
Здесь мы предлагаем пакет UMD - пакет с открытым исходным кодом, написанный на Python для анализа результатов моделирования молекулярной динамики. Пакет UMD позволяет вычислять широкий спектр структурных, динамических и термодинамических свойств (рисунок 1). Пакет доступен через веб-сайт GitHub (https://github.com/rcaracas/UMD_package) и через специальную страницу (http://moonimpact.eu/umd-package/) проекта ERC IMPACT в качестве пакета открытого доступа.
Чтобы сделать его универсальным и простым в обращении, наш подход заключается в том, чтобы сначала извлечь всю информацию, связанную с термодинамическим состоянием и атомными траекториями, из выходного файла фактического молекулярно-динамического запуска. Эта информация хранится в выделенном файле, формат которого не зависит от исходного пакета MD, в котором выполнялось моделирование. Мы называем эти файлы «umd» files, что расшифровывается как Universal Molecular Dynamics. Таким образом, наш пакет UMD может быть легко использован любой группой ab initio с любым программным обеспечением, и все это с минимальными усилиями по адаптации. Единственным требованием для использования настоящего пакета является запись соответствующего парсера из выходных данных конкретного программного обеспечения MD в формат файла umd, если этого еще не существует. В настоящее время мы предоставляем такие парсеры для пакетов VASP8 и QBox9 .

Рисунок 1: Блок-схема библиотеки UMD.
Физические свойства выделены синим цветом, а основные скрипты Python и их параметры — красным. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Файлы umd являются файлами ASCII; Типичным расширением является "umd.dat", но не обязательно. Все компоненты анализа могут считывать ASCII-файлы формата umd, независимо от фактического расширения имени. Тем не менее, некоторые из автоматических скриптов, предназначенных для выполнения быстрой крупномасштабной статистики в нескольких симуляциях, специально ищут файлы с расширением umd.dat. Каждое физическое свойство выражается в одной строке. Каждая строка начинается с ключевого слова. Таким образом, формат легко адаптируется и позволяет добавлять новые свойства в файл umd, сохраняя при этом его читабельность во всех версиях. Первые 30 строк umd-файла моделирования пиролита при 4,6 ГПа и 3000 К, использованные ниже в обсуждении, показаны на рисунке 2.

Рисунок 2: Начало файла umd, описывающего моделирование жидкого пиролита при 4,6 ГПа и 3000 К.
За заголовком следует описание каждого снимка. Каждое свойство записывается в одну строку, содержащую имя физического свойства, значение (значения) и единицы, разделенные пробелами. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Все файлы umd содержат заголовок, описывающий содержимое ячейки моделирования: количество атомов, электронов и атомных типов, а также детали для каждого атома, такие как его тип, химический символ, количество валентных электронов и его масса. Пустая строка отмечает конец заголовка и отделяет его от основной части файла umd.
Затем каждый шаг моделирования детализируется. Во-первых, задаются мгновенные термодинамические параметры, каждый на отдельной строке, указывая (i) название параметра, например, энергию, напряжения, эквивалентное гидростатическое давление, плотность, объем, параметры решетки и т. Д., (ii) его значение (значения) и (iii) его единицы. Далее следует таблица, описывающая атомы. Строка заголовка дает различные меры, такие как декартовы позиции, скорости, заряды и т. Д., И их единицы. Затем каждый атом детализируется на одной строке. Группами по три, соответствующие трем осям x, y, z , записи следующие: уменьшенные позиции, декартовы позиции, сложенные в ячейку моделирования, декартовы положения (которые должным образом учитывают тот факт, что атомы могут пересекать несколько единичных ячеек во время моделирования), атомные скорости и атомные силы. Последние две записи являются скалярами: заряд и магнитный момент.
Две основные библиотеки обеспечивают надлежащее функционирование всего пакета. Библиотека umd_process.py имеет дело с файлами umd, такими как чтение и печать. Библиотека crystallography.py имеет дело со всей информацией, связанной с фактической структурой атома. Основная философия библиотеки crystallography.py заключается в том, чтобы рассматривать решетку как векторное пространство. Параметры ячейки единицы вместе с их ориентацией представляют собой базовые векторы. «Пространство» имеет ряд скалярных атрибутов (удельный объем, плотность, температура и удельное число атомов), термодинамические свойства (внутренняя энергия, давление, теплоемкость и т. д.) и ряд тензорных свойств (напряжение и упругость). Атомы заполняют это пространство. Класс «Решетка» определяет этот ансамбль, наряду с различными короткими вычислениями, такими как конкретный объем, плотность, получение обратной решетки из прямой и т. Д. Класс "Atoms" определяет атомы. Они характеризуются рядом скалярных свойств (имя, символ, масса, количество электронов и др.) и рядом векторных свойств (положение в пространстве либо относительно векторного базиса, описанного в классе Решётки, либо относительно универсальных декартовых координат, скоростей, сил и т.д.). Помимо этих двух классов, библиотека crystallography.py содержит ряд функций для выполнения различных тестов и вычислений, таких как атомные расстояния или умножение ячеек. Периодическая таблица элементов также включена в качестве словаря.
Различные компоненты пакета umd записывают несколько выходных файлов. Как правило, все они представляют собой файлы ASCII, все их записи разделены вкладками, и они сделаны максимально понятными. Например, в них всегда четко указывается физическое свойство и его единицы. Файлы umd.dat полностью соответствуют этому правилу.
протокол
1. Анализ молекулярно-динамических прогонов
ПРИМЕЧАНИЕ: Пакет доступен через веб-сайт GitHub (https://github.com/rcaracas/UMD_package) и через специальную страницу (http://moonimpact.eu/umd-package/) проекта ERC IMPACT в качестве пакета открытого доступа.
- Извлеките каждый определенный набор физических свойств с помощью одного или нескольких выделенных скриптов Python из пакета. Запустите все скрипты в командной строке; все они используют серию флагов, которые максимально последовательны от одного сценария к другому. Флаги, их значение и значения по умолчанию приведены в таблице 1.
| Флаг | Значение | Скрипт с его помощью | Значение по умолчанию |
| -ч | Краткая справка | все | |
| -ф | Имя файла UMD | все | |
| -и | Этапы термоизации, подлежащие отбрасыванию | все | 0 |
| -и | Входной файл, содержащий межатомные связи | видообразование | bonds.input |
| -с | Выборка частоты | msd, видообразование | 1 (учитывается каждый шаг) |
| -а | Список атомов или анионов | видообразование | |
| -с | Список катионов | видообразование | |
| -л | Длина связи | видообразование | 2 |
| -т | Температура | вибрации, реология | |
| -v | Дискретизация ширины окна выборки траектории для анализа среднеквадратичного смещения | msd | 20 |
| -з | Дискретизация начала окна выборки траектории для анализа среднеквадратичного смещения | msd | 20 |
Таблица 1: Наиболее распространенные флаги, используемые в пакете UMD, и их наиболее распространенное значение.
- Начните с преобразования выходных данных МОДЕЛИРОВАНИЯ MD, выполненного в коде первых принципов, таком как VASP8 или QBox9, в файл UMD.
- Если MD моделирование проводилось в VASP, то в командной строке введите:
VaspParser.py -f -i
где флаг –f определяет имя файла VASP OUTCAR, а –i — длину температуризации.
ПРИМЕЧАНИЕ: Начальный шаг, определяемый –i, позволяет отказаться от первых шагов моделирования, которые представляют собой температуру. В типичном молекулярно-динамическом прогоне первая часть расчета представляет собой температуризацию, то есть время, необходимое системе для всех атомов, чтобы описать гауссовоподобное распределение температуры, и для всей системы, чтобы демонстрировать колебания температуры, давления, энергии и т. Д. Вокруг равновесных значений. Эта тепловизирующая часть моделирования не должна учитываться при анализе статистических свойств жидкости.
- Если MD моделирование проводилось в VASP, то в командной строке введите:
- Преобразование платформы . umd файлы в . файлы xyz для облегчения визуализации на различных других пакетах, таких как VMD4 или Vesta5. В командной строке введите:
umd2xyz.py -f -i -s
где –f определяет имя платформы . umd файл, –i определяет период термализации, который должен быть отброшен, и –s частоту выборки траектории, хранящейся в . umd файл. Значениями по умолчанию являются –i 0 –s 1, т.е. с учетом всех этапов моделирования, без отбрасывания. - Переверните файл umd в файлы POSCAR типа VASP с помощью скрипта umd2poscar.py; снимки симуляций могут быть выбраны с заданной частотой. В командной строке введите:
umd2poscar.py -f -i -l -s
где –l представляет собой последний шаг, преобразуемый в файл POSCAR. Значения по умолчанию : -i 0 -l 10000000 -s 1. Это значение –l достаточно велико, чтобы покрыть типичную всю траекторию.
2. Выполните расчет конструкций
- Запустите сценарий gofrs_umd.py для вычисления функции распределения пар (PDF) gᴀʙ(r) для всех пар атомарных типов A и B (рисунок 3). Выходные данные записываются в один ASCII-файл, разделенный табуляцией, с расширением gofrs.dat. В командной строке введите:
gofrs_umd.py -f -s < Sampling_Frequency > -d <ДискретизацияInterval> -i
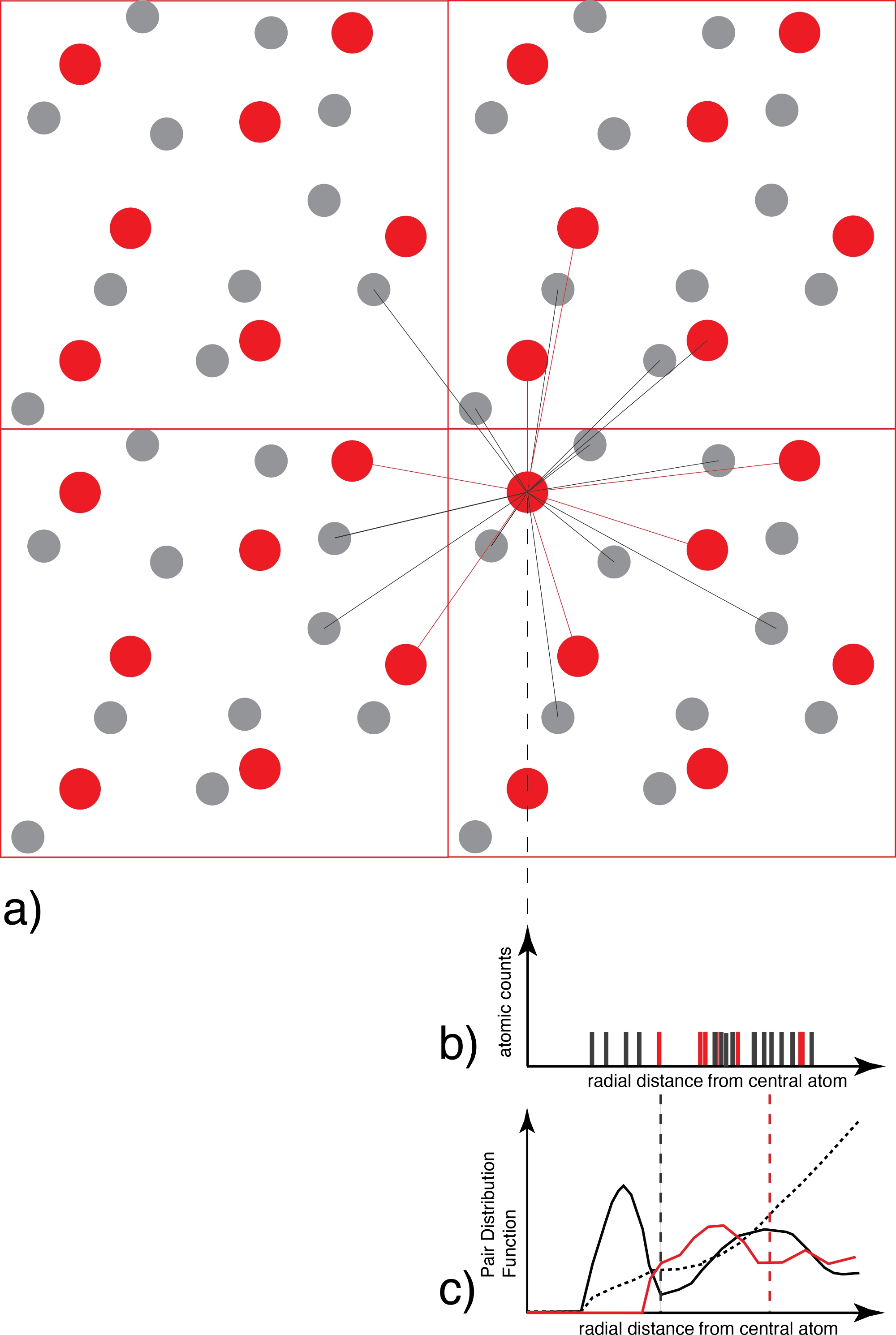
ПРИМЕЧАНИЕ: Значениями по умолчанию являются Sampling_Frequency (частота выборки траектории) = 1 шаг; ДискретизацияИнтервал (для построения графика g(r)) = 0,01 Å; InitialStep (количество шагов в начале траектории, которые отбрасываются) = 0. Радиальный PDF, gᴀʙ(r) — среднее число атомов типа B на расстоянии d_ᴀʙ в пределах сферической оболочки радиусом r и толщиной dr, центрированной на атомах типа A (рисунок 3):
с ρ атомная плотность, NA и NB — число атомов типов A и B, а δ (r−rᴀʙ) дельта-функция, равная 1, если атомы A и B лежат на расстоянии между r и r+dr. Абсцисса первого максимума gᴀʙ(r) дает наибольшую вероятность длины связи между атомами типа A и B, которая является ближайшей к среднему расстоянию связи, которое мы можем определить. Первый минимум разграничивает масштаб первой координационной сферы. Следовательно, интеграл над PDF до первого минимума дает среднее координационное число. Сумма преобразований Фурье gᴀʙ(r) для всех пар атомных типов A и B дает дифракционную картину жидкости, полученную экспериментально с помощью дифрактометра. Однако в действительности, поскольку часто координационные сферы высокого порядка отсутствуют в gᴀʙ(r), дифракционная картина не может быть получена во всей ее полноте.

Рисунок 3: Определение функции распределения пар.
а) Для каждого атома одного вида (например, красного) все атомы координирующего вида (например, серого и/или красного) учитываются как функция расстояния. b) Результирующий график распределения расстояний для каждого снимка, который на данном этапе представляет собой лишь набор дельта-функций, затем усредняется по всем атомам и всем моментальным снимкам и взвешивается распределением идеального газа для получения c) функции парного распределения, которая является непрерывной. Первым минимумом g(r) является радиус первой координационной сферы, используемый позже в анализе видообразования. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
- Извлеките средние расстояния межатомных связей в виде радиусов первых координационных сфер. Для этого определите положение первого максимума функций gᴀʙ(r): постройте файл gofrs.dat в приложении для работы с электронными таблицами и выполните поиск максимумов и минимумов для каждой пары атомов.
- Определите радиус первой координационной сферы, как первый минимум PDF, gᴀʙ(r), с помощью программного обеспечения для работы с электронными таблицами. Это является основой для всего структурного анализа жидкости; PDF дает среднее состояние связывания атомов в жидкости.
- Извлеките расстояния первых минимумов, т.е. абсциссы, и запишите их в отдельный файл, называемый, например, bonds.input. Кроме того, можно запустить один из analyze_gofr сценариев пакета UMD, чтобы определить максимумы и минимумы функций gᴀʙ(r). В командной строке введите:
analyze_gofr_semi_automatic.py - Нажмите на положение максимума и минимума функции gᴀʙ(r), отображаемых на графике, который открывается программой. Скрипт автоматически сканирует текущую папку, идентифицирует все файлы gofrs.dat и выполняет анализ для каждого из них. Нажимайте еще раз на максимум и минимум в окне каждый раз, когда скрипт нуждается в обоснованном первоначальном предположении.
- Откройте и просмотрите автоматически сгенерированный файл bonds.input , содержащий расстояния междуатомных связей.
3. Выполните анализ видообразования
- Вычислите топологию связи между атомами, используя концепцию связности в теории графов: атомы — это узлы, а межатомные связи — это пути. Сценарию speciation_umd.py требуются расстояния межатомной связи, определенные в файле bonds.input .
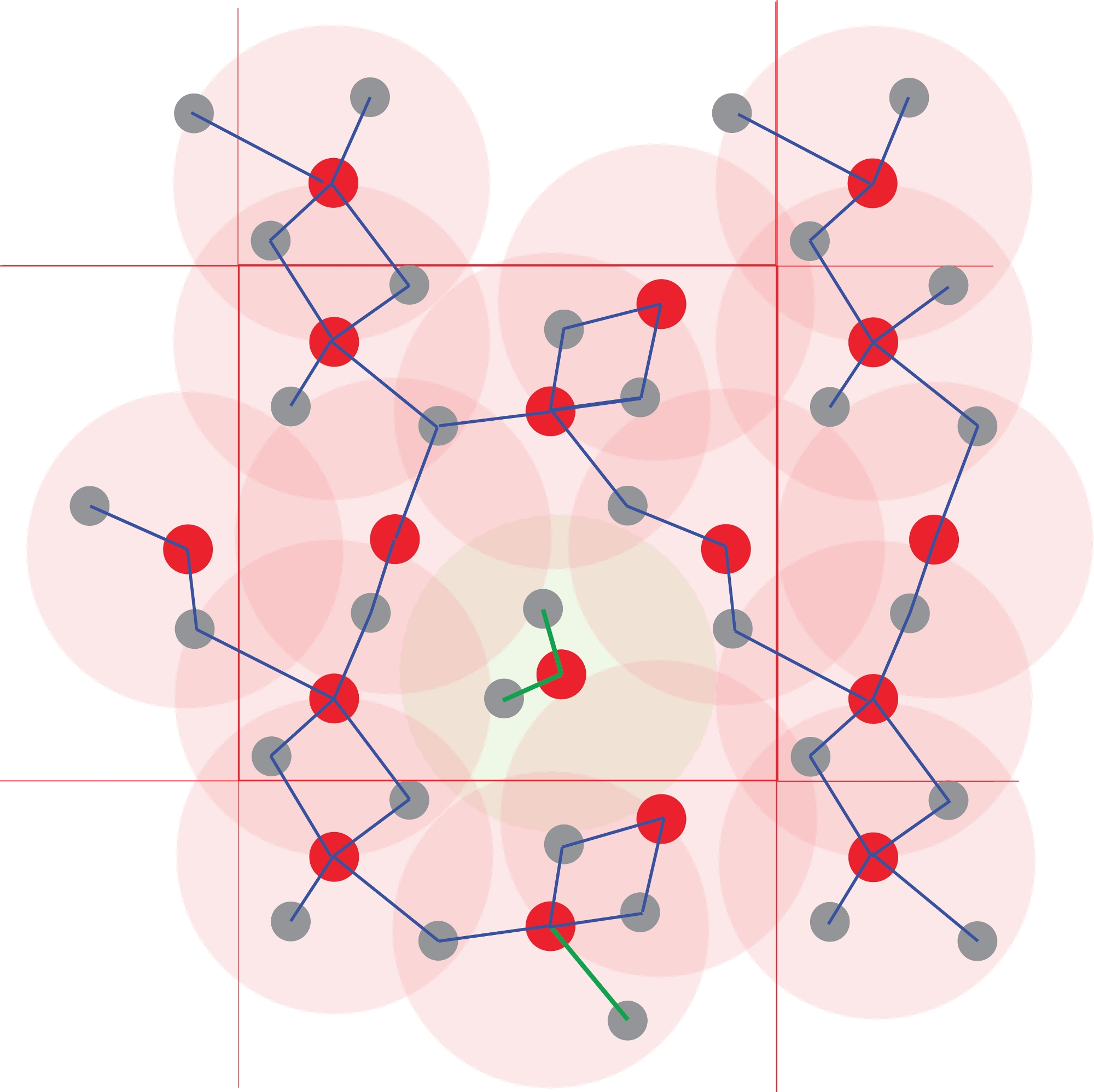
ПРИМЕЧАНИЕ: Матрица связности строится на каждом временном шаге: два атома, лежащие на расстоянии, меньшем радиуса соответствующей им первой координационной сферы, считаются связанными, т.е. связанными. Различные атомные сети строятся путем рассмотрения атомов как узлов в графе, связи которых определяются этим геометрическим критерием. Эти сети являются атомными видами, и их ансамбль определяет атомное видообразование в этой конкретной жидкости (рисунок 4).

Рисунок 4: Идентификация атомных кластеров.
Координационные многогранники определяются с помощью межатомных расстояний. Все атомы на расстоянии, меньшем заданного радиуса, считаются связанными. Здесь порог соответствует первой координационной сфере (светло-красным кругам), определенной на рисунке 1. Полимеризация и, таким образом, химические виды получают из сетей связанных атомов. Обратите внимание на центральный кластер Red1Grey2, который изолирован от других атомов, которые образуют бесконечный полимер. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
- Запустите сценарий видообразования для получения матрицы связности и получения координационных многогранников или полимеризации. В командной строке введите:
speciation_umd.py -f -s -i -l -c -a -m -r
где флаг -i дает файл с расстояниями межатомной связи, который был получен, например, на предыдущем шаге. Кроме того, можно запустить сценарий с одной единственной длиной для всех связей, определенных флагом -l.
ПРИМЕЧАНИЕ: Флаг -c указывает центральные атомы, а флаг -a — лиганды. Как центральные атомы, так и лиганды могут быть разных типов; в этом случае они должны быть разделены запятыми. Флаг -m дает минимальное время, в течение которого вид должен жить, чтобы быть рассмотренным в анализе. По умолчанию это минимальное время равно нулю, все вхождения подсчитываются в конечном счете.- Запустите сценарий speciation_umd.py с флагом –r 0, который выбирает граф связности на первом уровне для идентификации координационных многогранников. Например, центральный атом, обозначаемый катионом , может быть окружен одним или несколькими анионами (фиг.4). Сценарий видообразования идентифицирует каждый из координационных многогранников. Средневзвешенное значение всех координационных многогранников дает координационное число, идентичное тому, которое получено при интеграции PDF. В командной строке введите:
speciation_umd.py -f -i -c <Катионы> -a -r 0
ПРИМЕЧАНИЕ: Средние координационные числа в жидкостях являются дробными числами. Эта дробность исходит из средней характеристики координации. Определение, основанное на видообразовании, дает более интуитивное и информативное представление структуры жидкости, где количественные пропорции различных видов, то есть координации. - Запустите сценарий speciation_umd.py с флагом –r1, который сэмплирует график связности на всех уровнях глубины для получения полимеризации. Сеть через атомный граф имеет определенную глубину, так как атомы связаны дальше с другими связями (например, в последовательностях чередующихся катионов и анионов) (рисунок 4).
- Запустите сценарий speciation_umd.py с флагом –r 0, который выбирает граф связности на первом уровне для идентификации координационных многогранников. Например, центральный атом, обозначаемый катионом , может быть окружен одним или несколькими анионами (фиг.4). Сценарий видообразования идентифицирует каждый из координационных многогранников. Средневзвешенное значение всех координационных многогранников дает координационное число, идентичное тому, которое получено при интеграции PDF. В командной строке введите:
- Откройте два файла . попул.dat и . стат.dat последовательно; они являются выходными данными сценария видообразования. Каждый кластер записывается на одной строке, указывая его химическую формулу, время, в которое он сформировался, время, в которое он умер, его время жизни, матрицу со списком атомов, образующих этот кластер. График времени жизни каждого атомного кластера всех химических видов, обнаруженных в моделировании, как найдено в файле .popul.dat (рисунок 5).
- График анализа популяции с обилием каждого вида, как найдено в . stat.dat файл. Этот анализ, как абсолютный, так и относительный, соответствует фактической статистике координационных многогранников для случая -r 0; в случае полимеризации с -r1 это необходимо тщательно рассматривать, поскольку может потребоваться некоторая нормализация относительного числа атомов. Изобилие соответствует интегралу на протяжении всех жизней. Тем. stat.dat файл также перечисляет размер каждого кластера, то есть сколько атомов его образует.
4. Вычисление коэффициентов диффузии
- Извлеките средние квадратные смещения (MSD) атомов в зависимости от времени для получения самодиффузионной способности. Стандартная формула MSD:

где префакторами являются перенормировки. С помощью инструмента MSD существуют различные способы анализа динамических аспектов жидкостей.
ПРИМЕЧАНИЕ: T - общее время моделирования, а N α - количество атомов типа α. Начальное время t0 является произвольным и охватывает первую половину моделирования. Нинит — это количество начальных раз. τ — ширина временного интервала, за который вычисляются MSD; его максимальное значение составляет половину временной продолжительности моделирования. В типичных реализациях MSD каждое окно начинается в конце предыдущего. Но более разреженная выборка может ускорить вычисление MSD, не изменяя наклон MSD. Для этого i-е окно начинается в момент времени t0(i), но (i+1)-е окно начинается в момент t0(i) + τ + v, где значение v определяется пользователем. Аналогично, ширина окна увеличивается дискретными шагами, определенными пользователем, как таковые: τ(i) = τ(i-1) + z. Значения z («горизонтальный шаг») и v («вертикальный шаг») являются положительными или нулевыми; значение по умолчанию для обоих значений равно 20. - Вычислите MSD с помощью серии msd_umd сценариев. Их выходные данные печатаются в формате . msd.dat файл, где MSD каждого атома, атома или кластера печатается на одном столбце в зависимости от времени.
- Вычислите средний MSD каждого атомарного типа. MSD вычисляются для каждого атома, а затем усредняются для каждого типа атома. Выходной файл содержит по одному столбцу для каждого атомарного типа. В командной строке введите:
msd_umd.py -f -z -v -b - Вычислите MSD каждого атома. MSD вычисляются для каждого атома, а затем усредняются для каждого типа атома. Выходной файл содержит один столбец для каждого атома в моделировании, а затем один столбец для каждого типа атома. Эта особенность позволяет идентифицировать атомы, которые диффундируют в двух разных средах, таких как жидкость и газ, или две жидкости. В командной строке введите:
msd_all_umd.py -f -z -v -b - Вычислите MSD химических веществ. Используйте совокупность кластеров, идентифицированных с помощью сценария видообразования и напечатанных в . popul.dat файл. MSD вычисляются для каждого отдельного кластера. Выходной файл содержит по одному столбцу для каждого кластера. Чтобы избежать рассмотрения крупномасштабных полимеров, установите ограничение на размер кластера; по умолчанию — 20 атомов. В командной строке введите:
msd_cluster_umd.py -f -p -s -b -c
ПРИМЕЧАНИЕ: Значения по умолчанию: –b 100 –s 1 –c 20.
- Вычислите средний MSD каждого атомарного типа. MSD вычисляются для каждого атома, а затем усредняются для каждого типа атома. Выходной файл содержит по одному столбцу для каждого атомарного типа. В командной строке введите:
- Построение MSD с помощью программного обеспечения на основе электронных таблиц (рисунок 6). В логарифмическом представлении MSD по времени определите изменение уклона. Отделите первую часть, обычно короткую, которая представляет собой баллистический режим, т.е. сохранение скорости атомов после столкновений. Вторая более длинная часть представляет собой диффузный режим, т.е. рассеяние скорости атомов после столкновений.
- Вычислите коэффициенты диффузии из наклона MSD следующим образом:

где Z — число степеней свободы (Z = 2 для диффузии в плоскости, Z = 3 для диффузии в пространстве), а t — шаг времени.
5. Функции корреляции времени
- Вычислить функции временной корреляции как меру инерции системы по общей формуле:

A может быть множеством зависящих от времени переменных, таких как атомные положения, атомные скорости, напряжения, поляризация и т. д., каждая из которых дает — через отношения Грина-Кубо12,13 — различные физические свойства, иногда после дальнейшего преобразования. - Анализ атомных скоростей для получения колебательного спектра жидкости и альтернативного выражения атомных коэффициентов самодиффузии.
- Запустите сценарий vibr_spectrum_umd.py для вычисления функции автокорреляции атомная скорость-скорость (VAC) для каждого типа атома и выполните его быстрое преобразование Фурье. В командной строке введите:
vibr_spectrum_umd.py -f -t
где –t — температура, которая должна быть определена пользователем. Сценарий печатает два файла: . файл vels.scf.dat с функцией VAC для каждого атомарного типа и . vibr.dat файл с колебательным спектром, разложенным по каждому атомному виду и общим значением. - Откройте и прочитайте файл vels.scf.dat. Настройте функцию VAC из файла vels.scf.dat с помощью программного обеспечения, похожего на электронную таблицу.
- Сохраните реальную часть Fourier VAC. Вот что дает вибрационный спектр, как функцию частоты:

где m — атомные массы. - График вибрационного спектра из файла vibr.dat с помощью программного обеспечения, похожего на электронную таблицу (рисунок 7). Определите конечное значение при ω=0, соответствующее диффузионному характеру жидкости и различным пикам спектра на конечной частоте. Определите участие каждого атомного типа в вибрационном спектре.
ПРИМЕЧАНИЕ: Разложение на атомные типы показывает, что разные атомы имеют разные вклады ω=0, соответствующие их коэффициентам диффузии. Общая форма спектра намного более гладкая с меньшим количеством признаков, чем для соответствующего твердого тела. - На оболочке считывают интеграл по колебательному спектру, который дает коэффициенты диффузии для каждого атомного вида.
ПРИМЕЧАНИЕ: Термодинамические свойства могут быть получены путем интегрирования из колебательного спектра, но результаты следует использовать с осторожностью из-за двух приближений: интеграция действительна в рамках квазигармонического приближения, которое не обязательно держится при высоких температурах; и газообразная часть спектра, соответствующая диффузии, должна быть отброшена. Затем интеграция должна осуществляться только над решетчатой частью спектра. Но это разделение обычно требует нескольких дополнительных этапов постобработки и вычислений14, которые не охватываются настоящим пакетом UMD.
- Запустите сценарий vibr_spectrum_umd.py для вычисления функции автокорреляции атомная скорость-скорость (VAC) для каждого типа атома и выполните его быстрое преобразование Фурье. В командной строке введите:
- Запустите сценарий viscosity_umd.py для анализа самокорреляции тензора напряжений компонентов для оценки вязкости расплава. В командной строке введите:
viscosity_umd.py -f -i -s -o -l
ПРИМЕЧАНИЕ: Эта функция является исследовательской, и любые результаты должны восприниматься с осторожностью. Во-первых, тщательно проверьте сходимость вязкости относительно длины моделирования.- Выведите вязкость жидкости из самокорреляции тензора напряжений15 следующим образом:

где V и T — объём и температура соответственно, κB — постоянная Больцмана, а σ ij — внедиагональная составляющая ij тензора напряжений, выраженная в декартовых координатах. - Используйте более адекватное соответствие для получения более надежной оценки вязкости15,16 и избегайте шума функции автокорреляции напряжений-тензоров, которые могут возникнуть из-за конечного размера и конечной длительности моделирования. Для автокорреляционной функции тензора напряжений используйте следующую функциональную форму15,16, которая дает хорошие результаты:

где A, B, τ1, τ2 и ω — параметры соответствия. После интегрирования выражение для вязкости становится:
- Выведите вязкость жидкости из самокорреляции тензора напряжений15 следующим образом:
6. Термодинамические параметры, вытекающие из моделирования.
- Выполните averages.py , чтобы извлечь средние значения и разброс (в качестве стандартного отклонения) для давления, температуры, плотности и внутренней энергии из файлов umd . В командной строке введите:
averages.py -f -s
с –s 0 по умолчанию. - Вычислите статистическую погрешность среднего значения, используя методы блокировки.
ПРИМЕЧАНИЕ: Существуют различные разновидности этого метода. Следуя работе Аллена и Тилдесли2, принято усреднять последовательности временных блоков все большей длины и оценивать стандартное отклонение по отношению к среднему арифметическому17. Сходимость может быть достигнута в пределе многих и достаточно длинных размеров блоков, когда выборка некоррелирована. Хотя фактическое пороговое значение для сходимости обычно нужно выбирать вручную.- Используйте метод половинки18: начиная с исходной выборки данных, на каждом шаге κ уменьшайте вдвое число выборок путем усреднения по каждому двум соответствующим последовательным выборкам из предыдущего шага κ−1:

- Запустите сценарий fullaverages.py для выполнения полного статистического анализа, включая погрешность среднего значения. В командной строке введите:
fullaverages.py -s -u <ъединицы>
ПРИМЕЧАНИЕ: Скрипт автоматизирован до такой степени, что поиск всех файлов .umd.dat в текущем каталоге и выполнение анализа для всех из них. Значения по умолчанию — –s 0 –u 0. Для -u 0 выход минимален, а для -u 1 выход является полным, с несколькими распечатанными альтернативными единицами. Этот скрипт требует графической поддержки, так как создает графическое изображение для проверки сходимости для оценки ошибки по среднему значению.
- Используйте метод половинки18: начиная с исходной выборки данных, на каждом шаге κ уменьшайте вдвое число выборок путем усреднения по каждому двум соответствующим последовательным выборкам из предыдущего шага κ−1:
Результаты
Пиролит представляет собой модель многокомпонентного силикатного расплава (0,5Na2O 2CaO 1,5Al2O3 4FeO 30MgO 24SiO2), который лучше всего приближается к составу объемного силиката Земли — геохимическому среднему или всей нашей планете, за исключением ее ядра на основе железа19. На ранней Земле доминировала серия крупномасштабных событий плавления20, последнее из которых могло охватить всю планету после ее конденсации для протолунного диска21. Пиролит представляет собой лучший аппроксимант к составу таких океанов магмы планетарного масштаба. Следовательно, мы тщательно изучили физические свойства расплава пиролита в диапазоне температур 3000\u20125,000 K и диапазоне давлений 0\u2012150 ГПа из моделирования молекулярной динамики ab initio в реализации VASP. Эти термодинамические условия полностью характеризуют самые экстремальные условия океана магмы на Земле. Наше исследование является отличным примером успешного использования пакета UMD для всего углубленного анализа расплавов22. Мы рассчитали распределение и среднюю длину связи, проследили изменения катионно-кислородной координации и сравнили наши результаты с предыдущими экспериментальными и вычислительными исследованиями аморфных силикатов различного состава. Наш углубленный анализ помог разложить стандартные координационные числа на их основные составляющие, обозначить наличие экзотических координационных многогранников в расплаве и извлечь сроки жизни для всех координационных многогранников. Он также подчеркнул важность выборки в моделировании с точки зрения как длины траектории, так и количества атомов, присутствующих в моделируемой системе. Что касается постобработки, то анализ UMD не зависит от этих факторов, однако их следует учитывать при интерпретации результатов, предоставляемых пакетом UMD. Здесь мы покажем несколько примеров того, как пакет UMD может быть использован для извлечения нескольких характерных особенностей расплавов с применением к расплавленному пиролиту.
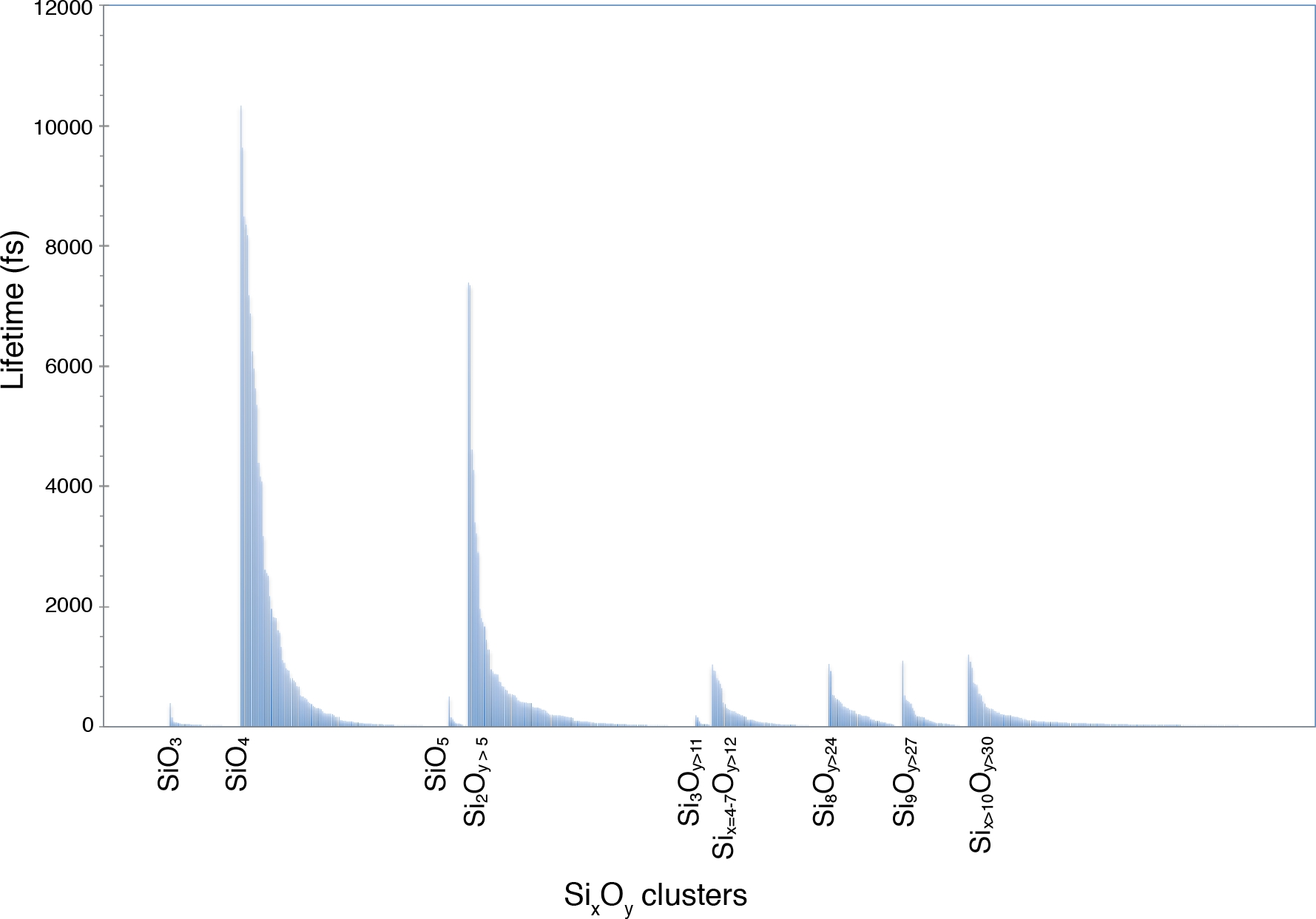
Функция распределения пары Si-O, полученная из сценария gofrs_umd.py, показывает, что радиус первой координационной сферы, которая является первым минимумом функции g(r), лежит около 2,5 ангстрем при T = 3000 K и P = 4,6 ГПа. Максимум g(r) лежит на 1,635 Å — это лучшее приближение к длине изгиба. Длинный хвост обусловлен температурой. Используя этот предел в качестве расстояния связи Si-O, анализ видообразования показывает, что единицы SiO4, которые могут длиться до нескольких пикосекунд, доминируют в расплаве (рисунок 5). Существует важная часть расплава, которая показывает частичную полимеризацию, что отражается присутствием димеров, таких как Si2O7, и тримеров, таких как единицы Si3Ox. Их соответствующее время жизни находится в порядке пикосекунды. Все полимеры более высокого порядка имеют значительно более короткий срок службы.

Рисунок 5: Время жизни химических веществ Si-O.
Видообразование определяется в многокомпонентном расплаве при 4,6 ГПа и 3000 К. На этикетках обозначены мономеры SiO3, SiO4 и SiO5 и различные полимеры SixOy. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
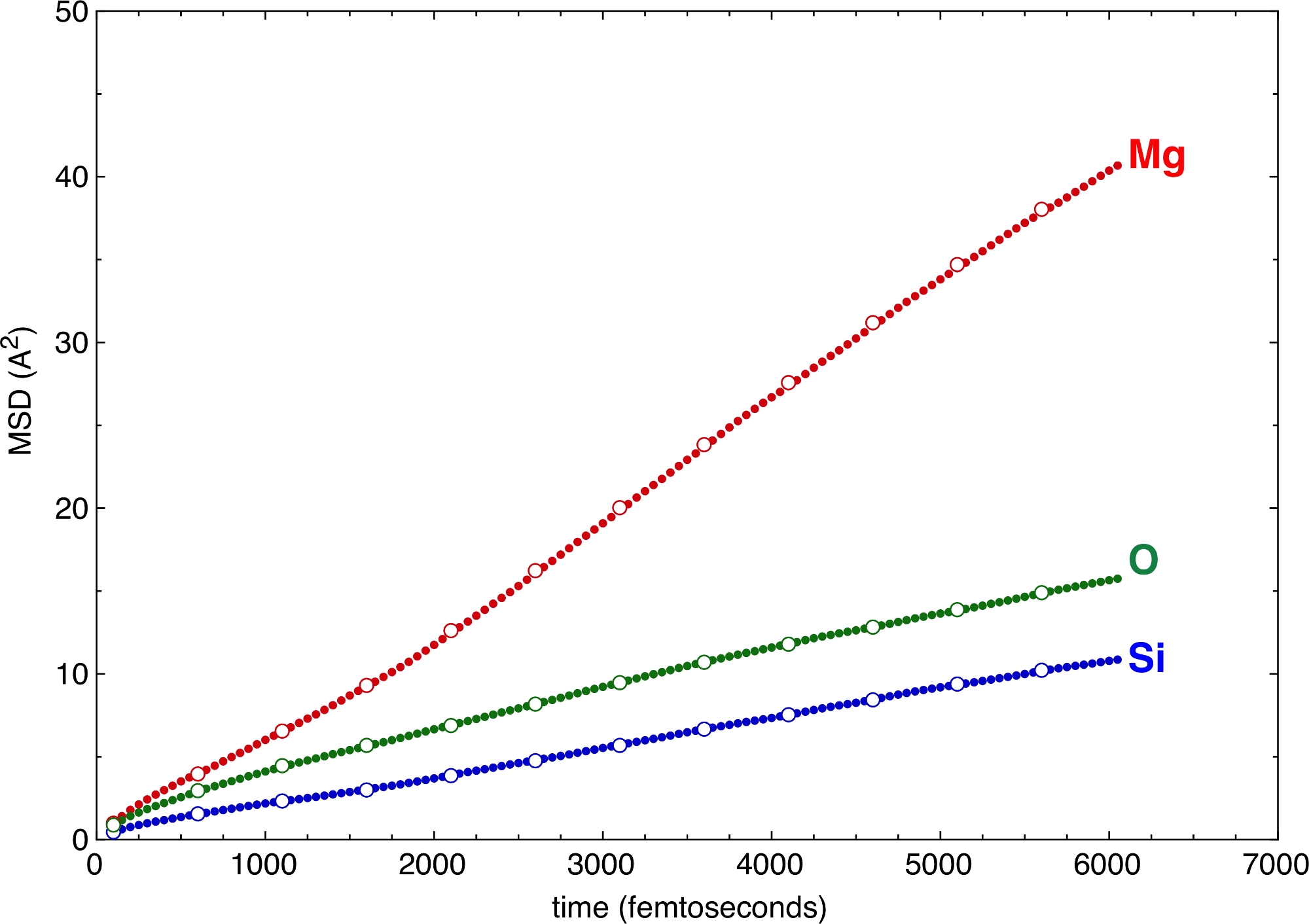
Различные значения вертикального и горизонтального шагов, определенные флагами –z и –v выше, дают различные выборки MSD (рисунок 6). Даже больших значений z и v достаточно, чтобы определить наклоны и, следовательно, коэффициенты диффузии различных атомов. Выигрыш во времени для постобработки примечателен при переходе к большим значениям z и v. MSD предлагает очень сильный критерий проверки качества моделирования. Если диффузионная часть MSD недостаточно длинная, это признак того, что моделирование слишком короткое и не достигает состояния жидкости в статистическом смысле. Минимальные требования к диффузной части MSD сильно зависят от системы. Можно потребовать, чтобы все атомы хотя бы раз изменили свое место в структуре расплава, чтобы его можно было рассматривать как жидкость10. Отличным примером применения в планетарных науках являются сложные силикатные расплавы при высоких давлениях, близких или даже ниже их жидкостной линии11. Атомы Si, основные сетеобразующие катионы, переключаются между участками через более чем два десятка пикосекунд. Моделирование, более короткое, чем это пороговое значение, будет значительно занижать выборку возможного конфигурационного пространства. Однако, поскольку координирующие анионы, а именно атомы O, движутся быстрее, чем центральные атомы Si, они могут компенсировать часть медленной подвижности Si. Таким образом, вся система действительно могла бы покрыть лучшую выборку конфигурационного пространства, чем предполагалось только из смещений Si.

Рисунок 6: Среднеквадратичные смещения (MSD).
MSD проиллюстрированы для нескольких атомных типов многокомпонентного силикатного расплава. Отбор проб с различными горизонтальными и вертикальными шагами, z и v, дает последовательные результаты. Сплошные круги: -z 50 –v 50. Открытые круги: -z 250 –v 500. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
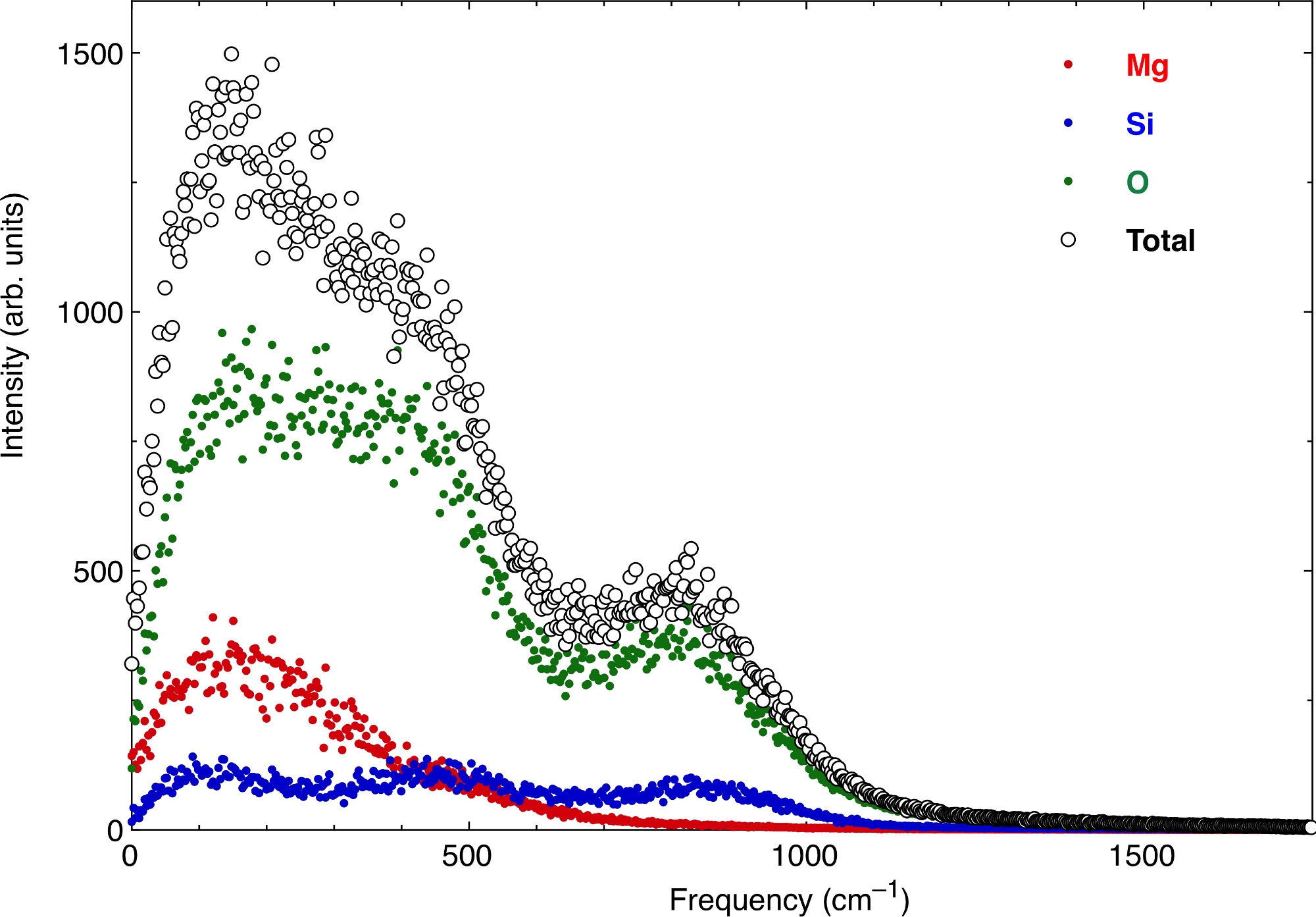
Наконец, атомные функции VAC дают вибрационный спектр расплава. На рисунке 7 показан спектр при тех же условиях давления и температуры, что и выше. Мы представляем вклад атомов Mg, Si и O, а также общее значение. При нулевой частоте существует конечное значение спектра, которое соответствует диффузионному характеру расплава. Извлечение термодинамических свойств из колебательного спектра необходимо убрать этот газообразный диффузионный характер с нуля, а также правильно учесть его распад на более высоких частотах.

Рисунок 7: Колебательный спектр расплава пиролита.
Действительная часть преобразования Фурье функции самокорреляции атомная скорость-скорость дает вибрационный спектр. Здесь вычисляется спектр для многокомпонентного силикатного расплава. Жидкости имеют ненулевой газоподобный диффузионный характер на нулевой частоте. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Обсуждение
Пакет UMD был разработан для лучшей работы с моделированием ab initio, где количество снимков обычно ограничивается десятками и сотнями тысяч снимков, с несколькими сотнями атомов на единицу ячейки. Более крупные симуляции также поддаются обработке при условии, что машина, на которой выполняется постобработка, имеет достаточно активных ресурсов памяти. Код отличается разнообразием свойств, которые он может вычислять, и лицензией с открытым исходным кодом.
Файлы umd.dat соответствуют ансамблям, которые сохраняют количество частиц неизменным на протяжении всего моделирования. Пакет UMD может считывать файлы, полученные в результате вычислений, где форма и объем блока моделирования различаются. Они охватывают наиболее распространенные расчеты, такие как NVT и NPT, где количество частиц N, температура T, объем, V и / или давление, P, остаются постоянными.
Для начала работы функция распределения пар, а также все скрипты, необходимые для оценки межатомных расстояний, как и скрипты видообразования, работают только для ортогональных единичных ячеек, то есть для кубических, тетрагональных и орторомбических ячеек, где углы между осями составляют 90°.
Основными направлениями развития версии 2.0 являются снятие ограничения ортогональности для расстояний и добавление дополнительных возможностей для скриптов видообразования: анализ отдельных химических связей, анализ межатомных углов и реализация второй координационной сферы. С помощью внешнего сотрудничества мы работаем над переносом кода на графический процессор для более быстрого анализа в больших системах.
Раскрытие информации
Авторам нечего раскрывать.
Благодарности
Эта работа была поддержана Европейским исследовательским советом (ERC) в рамках исследовательской и инновационной программы Европейского союза Horizon 2020 (номер грантового соглашения 681818 IMPACT to RC), Директоратом по экстремальной физике и химии Обсерватории глубокого углерода и Исследовательским советом Норвегии через свою схему финансирования Центров передового опыта, номер проекта 223272. Мы признаем доступ к суперкомпьютерам GENCI через серию вычислительных грантов eDARI stl2816, к суперкомпьютеру Irene AMD через проект PRACE RA4947 и к суперкомпьютеру Fram через UNINETT Sigma2 NN9697K. FS была поддержана проектом Марии Склодовской-Кюри (грантовое соглашение ABISSE No 750901).
Материалы
| Name | Company | Catalog Number | Comments |
| getopt library | open-source | ||
| glob library | open-source | ||
| matplotlib library | open-source | ||
| numpy library | open-source | ||
| os library | open-source | ||
| Python software | The Python Software Foundation | Version 2 and 3 | open-source |
| random library | open-source | ||
| re library | open-source | ||
| scipy library | open-source | ||
| subprocess library | open-source | ||
| sys library | open-source |
Ссылки
- Frenkel, D., Smit, B. . Understanding Molecular Simulation. From Algorithms to Applications. , (2001).
- Allen, M. P., Tildesley, D. J., Allen, T. . Computer Simulation of Liquids. , (1989).
- Zepeda-Ruiz, L. A., Stukowski, A., Oppelstrup, T., Bulatov, V. V. Probing the limits of metal plasticity with molecular-dynamics simulations. Nature Publishing Group. 550 (7677), 492-495 (2017).
- Humphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics & Modeling. 14 (1), 33-38 (1996).
- Momma, K., Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. Journal of Applied Crystallography. 44 (6), 1272-1276 (2011).
- Brehm, M., Kirchner, B. TRAVIS - A free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. Journal of Chemical Information and Modeling. 51 (8), 2007-2023 (2011).
- Stixrude, L. Visualization-based analysis of structural and dynamical properties of simulated hydrous silicate melt. Physics and Chemistry of Minerals. 37 (2), 103-117 (2009).
- Kresse, G., Hafner, J. Ab initio Molecular-Dynamics for Liquid-Metals. Physical Review B. 47 (1), 558-561 (1993).
- Gygi, F. Architecture of Qbox: A scalable first-principles molecular dynamics code. IBM Journal of Research and Development. 52 (1-2), 137-144 (2008).
- Harvey, J. P., Asimow, P. D. Current limitations of molecular dynamic simulations as probes of thermo-physical behavior of silicate melts. American Mineralogist. 100 (8-9), 1866-1882 (2015).
- Caracas, R., Hirose, K., Nomura, R., Ballmer, M. D. Melt-crystal density crossover in a deep magma ocean. Earth and Planetary Science Letters. 516, 202-211 (2019).
- Green, M. S. Markoff Random Processes and the Statistical Mechanics of Time-Dependent Phenomena. II. Irreversible Processes in Fluids. The Journal of Chemical Physics. 22 (3), 398-413 (1954).
- Kubo, R. Statistical-Mechanical Theory of Irreversible Processes. I. General Theory and Simple Applications to Magnetic and Conduction Problems. Journal of the Physical Society of Japan. 12 (6), 570-586 (1957).
- Lin, S. T., Blanco, M., Goddard, W. A. The two-phase model for calculating thermodynamic properties of liquids from molecular dynamics: Validation for the phase diagram of Lennard-Jones fluids. The Journal of Chemical Physics. 119 (22), 11792-11805 (2003).
- Meyer, E. R., Kress, J. D., Collins, L. A., Ticknor, C. Effect of correlation on viscosity and diffusion in molecular-dynamics simulations. Physical Review E. 90 (4), 1198-1212 (2014).
- Soubiran, F., Militzer, B., Driver, K. P., Zhang, S. Properties of hydrogen, helium, and silicon dioxide mixtures in giant planet interiors. Physics of Plasmas. 24 (4), 041401-041407 (2017).
- Flyvbjerg, H., Petersen, H. G. Error estimates on averages of correlated data. The Journal of Chemical Physics. 91 (1), 461-466 (1989).
- Tuckerman, M. E. . Statistical mechanics: theory and molecular simulation. , (2010).
- McDonough, W. F., Sun, S. S. The composition of the Earth. Chemical Geology. 120, 223-253 (1995).
- Elkins-Taton, L. T. Magma oceans in the inner solar system. Annual Review of Earth and Planetary Sciences. 40, 113-139 (2012).
- Lock, S. J., et al. The origin of the Moon within a terrestrial synestia. J. Geophysical Research: Planets. 123, 910-951 (2018).
- Solomatova, N. V., Caracas, R. Pressure-induced coordination changes in a pyrolitic silicate melt from ab initio molecular dynamics simulations. Journal of Geophysical Research: Solid Earth. 124, 11232-11250 (2019).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены