
Method Article
Modélisation simple, abordable et modulaire des cellules à l’aide de l’ADN
Dans cet article
Résumé
Nous présentons ici un protocole pour les cellules micromotérinaires à résolution unicellulaire en utilisant l’adhésion programmée par l’ADN. Ce protocole utilise une plate-forme de photolithographie de paillasse pour créer des motifs d’oligonucléotides d’ADN sur une lame de verre, puis marque les membranes cellulaires avec des oligonucléotides complémentaires disponibles dans le commerce. L’hybridation des oligos entraîne une adhésion cellulaireprogrammée.
Résumé
Le positionnement relatif des cellules est une caractéristique clé du microenvironnement qui organise les interactions cellule-cellule. Pour étudier les interactions entre des cellules du même type ou d’un type différent, les techniques de micromotrage se sont avérées utiles. L’assemblage programmé de cellules par ADN (DPAC) est une technique de micromotif qui cible l’adhésion des cellules à un substrat ou à d’autres cellules en utilisant l’hybridation de l’ADN. Les opérations les plus élémentaires dans le DPAC commencent par décorer les membranes cellulaires avec des oligonucléotides modifiés par les lipides, puis les faire circuler sur un substrat qui a été modelé avec des séquences d’ADN complémentaires. Les cellules adhèrent sélectivement au substrat uniquement lorsqu’elles trouvent une séquence d’ADN complémentaire. Les cellules non adhérentes sont emportées, révélant un modèle de cellules adhérentes. D’autres opérations comprennent d’autres cycles d’adhésion cellule-substrat ou cellule-cellule, ainsi que le transfert des motifs formés par le DPAC dans un hydrogel d’incorporation pour une culture à long terme. Auparavant, les méthodes de modelage des oligonucléotides sur les surfaces et de décoration des cellules avec des séquences d’ADN nécessitaient un équipement spécialisé et une synthèse personnalisée de l’ADN, respectivement. Nous rapportons une version mise à jour du protocole, utilisant une configuration de photolithographie de paillasse peu coûteuse et des oligonucléotides modifiés par le cholestérol (CMO) disponibles dans le commerce déployés à l’aide d’un format modulaire. Les cellules étiquetées CMO adhèrent avec une grande efficacité aux substrats à motifs d’ADN. Cette approche peut être utilisée pour modéliser plusieurs types de cellules à la fois avec une grande précision et pour créer des réseaux de microtissuons intégrés dans une matrice extracellulaire. Les avantages de cette méthode comprennent sa haute résolution, sa capacité à intégrer des cellules dans un microenvironnement tridimensionnel sans perturber le micromot et sa flexibilité dans la modélisation de tout type de cellule.
Introduction
Le positionnement des cellules les unes par rapport aux autres dans un tissu est une caractéristique importante du microenvironnement1,2,3,4. Les techniques utilisées pour modeler les cellules vivantes en arrangements contrôlés spatialement sont des outils expérimentaux précieux pour étudier la différenciation4,5,6,7,8, la motilité cellulaire9, la morphogenèse10,11,12, le métabolisme13et les interactions cellule-cellule7,14 . Il existe une variété de méthodes pour modéliser les cellules, chacune avec ses propres avantages et inconvénients3,4. Les méthodes qui créent des îlots adhésifs de protéines de matrice extracellulaire (ECM), telles que l’impression par microcontact et les pochoirs découpés au laser, sont simples et évolutives. Cependant, il est difficile de modeler plus d’un ou deux types de cellules à la fois car les propriétés adhésives de différents types de cellules à différentes molécules ECM sont souvent similaires15,16,17. Des micromotèges plus complexes peuvent être créés avec l’adsorption moléculaire induite par la lumière (LIMAP), une technique qui utilise la lumière UV pour ablater les régions enduites de PEG et permettre une adsorption ultérieure des protéines18,19. Ce processus peut être répété pour créer des micromots à haute résolution avec plusieurs types de cellules. Cependant, une liaison croisée des cellules aux différents patchs protéiques peut se produire, entraînant une mauvaise spécificité du modèle19. Les méthodes physiques telles que l’ensemencement de cellules sur des dispositifs de culture micromécaniques reconfigurables peuvent créer des co-cultures structurées avec contrôle dynamique, mais sans la flexibilité dans la conception de motifs d’impression par microcontact ou LIMAP14,8. Contrairement aux autres techniques, la bio-impression peut créer des arrangements tridimensionnels de cellules dans les hydrogels20,21. Cependant, les constructions bioimprimées ont une résolution beaucoup plus faible que les autres techniques de micromotériste, avec une taille moyenne de caractéristiques de l’ordre de centaines de microns22. Une méthode idéale de modélisation cellulaire aurait une haute résolution, des modèles multiples types de cellules, utiliserait des équipements et des réactifs facilement accessibles et aurait la capacité d’intégrer des motifs réussis dans un hydrogel pour la culture cellulaire tridimensionnelle (3D). Dans cet article, nous présentons le CMO-DPAC, une technique de micromotif cellulaire qui utilise la flexibilité et la vitesse de l’hybridation de l’ADN pour cibler l’adhésion cellulaire à un substrat. Cette méthode a été adaptée de nos protocoles précédents23,24 pour la rendre plus abordable, modulaire et accessible. En utilisant le protocole actuel, tout laboratoire devrait être en mesure de mettre en place un système entièrement fonctionnel sans équipement ou expertise spécialisé.
L’assemblage programmé de cellules par ADN (DPAC) est une puissante technique d’ingénierie tissulaire qui modèle les cellules à une résolution unicellulaire avec un contrôle précis de l’espacement cellule-cellule et de la géométrie tissulaire. Dans le DPAC, les membranes cellulaires sont décorées d’oligonucléotides d’ADN (oligos) à l’aide de deux oligos modifiés par les lipides conçus pour s’hybrider sur la membrane cellulaire. Parce que les oligos sont conjugués à des lipides hydrophobes, ils se partitionnent rapidement avec la membrane cellulaire25 où ils s’hybrident, augmentant l’hydrophobicité nette des molécules non liées par covalent, et améliorant ainsi leur durée de vie à la surface de la cellule26. Les oligos sont présentés à la surface de la cellule d’une manière où ils peuvent s’hybrider avec des oligos complémentaires sur d’autres cellules ou des lames de verre fonctionnalisées par l’ADN pour créer des modèles cellulaires 2D ou 3D définis avec la composition prescrite, l’espacement cellule-cellule et la géométrie23,24. Les microtissuons à motifs peuvent être clivés de la surface de manière enzymatique et incorporés dans un hydrogel pour une culture 3D prolongée. Lorsqu’elles sont utilisées en combinaison avec des cellules primaires ou des cellules souches, les collections de cellules résultantes peuvent subir une morphogenèse et se former en organoïdes23,27,28. Le DPAC a été appliqué pour étudier la dynamique du devenir des cellules souches neurales adultes en réponse à des signauxconcurrents 6,29,pour étudier l’auto-organisation des cellules épithéliales mammaires23,28et pour générer de l’origami tissulaire par condensation mésenchymateuse27.
DPAC permet le placement précis de plusieurs populations cellulaires et a une résolution nettement meilleure que les bioimprimantes à base d’extrusion (de l’ordre du micron)22,23. De plus, contrairement aux méthodes de modelage basées sur l’ECM telles que l’impression par microcontact, le DPAC ne nécessite pas d’adhésion différentielle des différents types de cellules à une surface revêtue d’ECM15,23. Il est idéal pour répondre à des questions sur la façon dont la composition d’un tissu affecte son comportement, comment les cellules intègrent de multiples indices cellulaires et microenvironnementaux lors de la prise de décisions6,29et comment des paires de cellules interagissent les unes avec les autres. Un avantage de cette méthode par rapport à d’autres méthodes de micromotbrement est qu’elle peut être utilisée pour la culture cellulaire 3D dans un seul plan d’imagerie, facilitant les études en accéléré de l’auto-organisation tissulaire et de la morphogenèse organoïde23,27,30.
Malgré ces avantages, la mise en œuvre réussie du DPAC a nécessité la synthèse de réactifs oligonucléotidiques personnalisés et l’accès à un équipement spécialisé pour le modelage de l’ADN23,24,limitant ainsi l’adoption généralisée. Par exemple, les oligos modifiés par les lipides (LMO) optimaux utilisés dans le protocole original doivent être synthétisés sur mesure, modifiés avec de l’acide lignocérique ou de l’acide palmitique, et purifiés26. Ce processus nécessite l’utilisation d’un synthétiseur d’ADN et d’un instrument de chromatographie liquide à haute performance, ainsi que l’achat des réactifs associés tels que la méthylamine, une substance contrôlée soumise à la réglementation institutionnelle et fédérale. Comme alternative, les LMO peuvent être achetés sur mesure en vrac, mais cela nécessite un investissement préalable important dans la technologie.
Pour surmonter ces limitations, nous avons développé une version révisée du DPAC qui utilise des oligos modifiés par le cholestérol (CMO) disponibles dans le commerce à la place des LMO synthétisés sur mesure. Pour réduire davantage les coûts et augmenter la flexibilité de la plate-forme, nous sommes passés à un système modulaire à trois oligo. Au lieu de commander un nouvel oligo modifié par le cholestérol pour chaque population cellulaire unique, un utilisateur de ce protocole peut plutôt utiliser les mêmes oligos modifiés par le cholestérol (« Ancre universelle » et « Co-ancre universelle ») pour chaque population cellulaire, puis utiliser un oligo peu coûteux et non modifié (« Brin adaptateur ») qui s’hybride à la fois avec l’ancre universelle et l’ADN fonctionnalisé par les amines à la surface ou le brin adaptateur d’un autre type de cellule.
Une autre limitation du protocole DPAC original était qu’il créait les diapositives à motifs d’ADN en utilisant une imprimante liquide haute résolution (par exemple, Nano eNabler, BioForce Nanosciences)23,24. Bien que cet instrument offre une résolution extraordinaire et de faibles exigences en réactifs, il n’est pas disponible pour la plupart des institutions et a un taux d’impression relativement faible (environ 1 caractéristique par seconde). Récemment, deux méthodes photolithographiques ont été développées pour modeler les caractéristiques de l’ADN sur les surfaces. Viola et ses collègues ont utilisé un revêtement de polyacrylamide et de benzophénone qui liait de manière covalente des oligos d’ADN simple brin lors de l’exposition à la lumière UV30. En utilisant cette méthode, ils ont pu créer des échafaudages tissulaires qui ont subi des changements de forme programmés à grande échelle en raison de la contractilité cellulaire et de l’auto-organisation. Scheideler et al. ont développé une méthode qui utilise l’exposition aux UV d’une photorésiste positive pour exposer sélectivement des oligos d’ADN modifiés par les amines à une lame fonctionnalisée par l’aldéhyde29. Après la cuisson et l’amination réductrice, l’ADN modifié par les amines est lié de manière covalente à la surface. Cette méthode a été utilisée pour étudier la réponse des cellules souches neurales adultes aux signaux d’auto-renouvellement et de différenciation présentés spatialement. Cet article adapte le protocole de Scheideler et al. pour créer les modèles d’ADN qui captureront les cellules étiquetées CMO. Ce protocole de photomodèlement peut être effectué sans utiliser de salle blanche. Il utilise de l’équipement peu coûteux et disponible dans le commerce qui est facilement déployé sur une paillasse ou une hotte aspirante. L’utilisation d’équipements de photolithographie peu coûteux ou de bricolage augmente l’accessibilité aux chercheurs sans accès aux installations de salle blanche et permet aux chercheurs d’essayer la technique sans un investissement important de temps ou de ressources31,32. Cependant, une meilleure résolution et l’alignement de plusieurs caractéristiques de l’ADN peuvent être obtenus en utilisant le spin coater commercial et l’aligneur de masque que l’on trouve couramment dans les salles blanches.
Ici, nous décrivons une méthode pour modéliser des cellules à une résolution unicellulaire en utilisant l’adhésion basée sur l’ADN. Tout d’abord, le photomodèlement avec une photorésine positive est utilisé pour créer des motifs à haute résolution d’ADN modifié par les amines sur un substrat de verre modifié par l’aldéhyde. Ensuite, la diapositive est traitée pour réduire l’attachement cellulaire non spécifique et des cellules de flux PDMS sont créées pour confiner les cellules sur des régions à motifs. Les cellules sont ensuite étiquetées avec de courts oligonucléotides d’ADN qui sont fonctionnalisés avec du cholestérol et, par conséquent, insérés dans la membrane cellulaire. Les cellules sont ensuite acheminées sur les micromots d’ADN. L’hybridation entre l’ADN de la surface cellulaire et l’ADN sur la surface du verre entraîne une adhésion spécifique des cellules au modèle d’ADN. Les cellules non adhérentes sont emportées, révélant le motif cellulaire adhérent. Ce processus peut être répété pour modéliser plusieurs types de cellules ou pour créer des structures multicouches. Si vous le souhaitez, les cellules peuvent être entièrement intégrées dans un ECM pour la culture cellulaire 3D.
Protocole
1. Expérience de conception
- Planifiez l’expérience souhaitée, en tenant compte de la taille des entités, de l’espacement des entités, du nombre de types de cellules impliqués et de la disposition des cellules les unes par rapport aux autres. Reportez-vous au fichier supplémentaire 1, un guide pour la conception expérimentale, et au fichier supplémentaire 2, qui contient des exemples de séquences oligo.
- Concevoir un photomasque à l’aide d’un logiciel de conception assistée par ordinateur. Un exemple de photomasque est fourni dans le fichier supplémentaire 3.
- Dessinez un rectangle des dimensions d’une lame de microscope standard (25 mm x 75 mm).
- Dessinez quatre régions rectangulaires de 10 mm de large et 10 mm de long, réparties uniformément sur la diapositive.
- Dans chaque région, dessinez des entités dont la taille, la forme et l’espacement souhaités pour l’expérience sont souhaitées. Les cellules n’adhéreront qu’à ces caractéristiques dans l’expérience.
- Pour créer des photomasques alignés pour plusieurs types de cellules, créez un dessin principal avec tous les ensembles de fonctions, puis enregistrez les versions correspondant à chaque type de cellule.
- Commandez un photomasque transparent haute résolution (au moins 20 000 points par pouce) à partir de ce dessin CAO avec les caractéristiques dessinées en transparent 1.2.3 et les grandes régions en noir.
2. ADN photomotérien sur lames fonctionnalisées par l’aldéhyde (protocole adapté de Scheideler et al.29)
- Si vous modélisez plusieurs types de cellules, fabriquez des marqueurs fiduciaux sur la lame fonctionnalisée à base d’aldéhyde avant tout motif d’ADN pour faciliter l’alignement des caractéristiques. D’autres méthodes de création de marqueurs fiduciaires sont suggérées dans le fichier supplémentaire 1.
- Pour créer des marqueurs fiducial métalliques, appliquez la photorése positive S1813 comme décrit aux étapes 2.3 à 2.11. Utilisez un photomasque qui contient de grandes caractéristiques qui seront faciles à aligner plus tard. Intégrez ces caractéristiques dans la conception des photomasques qui seront utilisés pour le modelage de l’ADN.
- Déposer un mince film (100 Angstroms) de titane sur la lame en utilisant l’évaporation du pistolet àélectrons 29. Enlevez l’excès de métal et de photorésine à l’aide d’acétone, puis passez au photomodèlement de l’ADN.
- Préparer une solution de 20 μM d’un oligo modifié par 5'-amine dans un tampon d’ADN (50 mM de phosphate de sodium dans l’eau, pH = 8,5). Voir le fichier supplémentaire 2 pour les séquences oligo suggérées.
REMARQUE: Il est possible d’utiliser aussi peu que 5 μM d’oligo modifié par les amines pour certains modèles et applications, de sorte que la concentration d’ADN de surface peut devoir être optimisée. - Préchauffer une plaque chauffante à 100 °C.
- Utilisez du ruban adhésif double face ou un aspirateur pour fixer une lame de verre fonctionnalisée à l’aldéhyde au rotor d’un enrobeur de spin.
ATTENTION : Le détachement des glissières pendant le revêtement par essorage constitue un risque pour la sécurité. Utilisez toujours le revêtement de spin dans un récipient fermé avec un couvercle, tel qu’une boîte en acrylique.
REMARQUE: Étiquetez un coin de la diapositive à l’aide d’un scribe en diamant ou d’un outil similaire pour rayer le verre. Cela aide à l’identification et à l’orientation des glissières après que la photorésiste a été emportée. - Utilisez une pipette jetable pour déposer la photorésiste positive sur la lame d’aldéhyde. Pour des revêtements uniformes, ajoutez de petites gouttes de la photorése sur la diapositive, au lieu d’une grosse goutte au milieu(Figure supplémentaire 1A).
- À l’aide de l’enrobeur de rotation, faites tourner la glissière à 3000 tr/min pendant 30 s.
- Placez la lame sur une plaque chauffante à 100 °C pendant 1,5 min (cuisson douce) pour réticuler la résine photoserésure.
- Retirez la glissière de la plaque chauffante. Placez un photomasque avec les caractéristiques souhaitées pour cette expérience sur le dessus de la diapositive et alourdez le photomasque avec un morceau de verre (Figure supplémentaire 1B, C). Couvrez l’ensemble de la configuration dans une boîte opaque (Figure supplémentaire 1D). Exposer avec une lampe UV (longueur d’onde 365 nm, 360 mW, 5 poucesde glissière, densité d’énergie rayonnante totale 100 mJ/cm2) pendant 2 min.
REMARQUE: La lumière UV brisera les liaisons polymères dans la résine photoserrière les régions transparentes du photomasque, créant des régions où l’ADN pourra plus tard adhérer. - Développez la diapositive en l’immergeant dans la solution de développement pendant 3 à 5 minutes(Figure supplémentaire 1E).
- Rincez l’excès de solution de développement avec de l’eau. Sécher sous un courant d’air ou d’azote. (Figure supplémentaire 1F).
- Confirmez que la photolithographie a réussi en regardant la lame au microscope. Étant donné que la photorésiste est sensible aux rayons UV, effectuez cette étape rapidement, puis stockez la diapositive dans l’obscurité tout en préparant d’autres diapositives (le cas échéant).
REMARQUE : une diapositive à motifs réussis doit avoir des arêtes bien définies pour chaque fonction, aucune fissure et aucune distorsion de la fonction sur les bords. Des exemples de photolithographie correcte et incorrecte sont fournis dans la figure supplémentaire 2A. Reportez-vous au tableau 1 pour obtenir des suggestions de dépannage si la photolithographie ne fournit pas la qualité de fonctionnalité souhaitée. - Ajouter une gouttelette de la solution oligo modifiée par amine de 20 μM (étape 2.1) sur chaque région photomotée de la lame. Utilisez une pointe de pipette pour étaler doucement la gouttelette sur toute la région, en prenant soin de ne pas rayer la glissière. (Figure supplémentaire 1G).
- Cuire la lame dans un four à 65-70 °C jusqu’à ce que la solution d’ADN ait complètement séché sur la surface de la lame (environ 1 h).
- Effectuez une amination réductrice en plaçant les lames à motifs cuites dans un plat de culture cellulaire de 15 cm et placez-les dans une hotte sur un agitateur. Peser 100 mg de borohydrure de sodium. Dans une hotte, ajouter 40 mL de solution saline tamponnée au phosphate (PBS), mélanger doucement et ajouter au plat contenant les lames à motifs. Laissez la réaction se poursuivre pendant 15 min en secouant doucement.
REMARQUE: L’amine sur l’oligo forme d’abord une base de Schiff avec les aldéhydes sur la surface de la lame. Il s’agit d’une liaison covalente réversible qui doit être convertie en une liaison irréversible avant d’être utilisée dans DPAC. L’ajout d’un agent réducteur (borohydrure de sodium) convertit la base de Schiff en amine secondaire par amination réductrice.
ATTENTION: La réaction du borohydrure de sodium avec l’eau crée de l’hydrogène gazeux et continuera à le faire pendant des heures ou des jours après le début de la réaction. Effectuez l’étape d’amination réductrice dans une hotte et conservez tous les déchets de solution de borohydrure de sodium dans un récipient ouvert ou mal bouché dans la hotte pendant au moins 24 h. - Éliminer l’ADN non réagi en se lavant deux fois avec du dodécylsulfate de sodium (SDS) à 0,1% dans de l’eau, puis trois fois avec de l’eau distillée. Séchez la lame sous un jet d’azote ou d’air.
- Rincez la lame avec de l’acétone pour enlever la photorésine restante.
REMARQUE: À ce stade, l’ADN a été attaché de manière irréversible et covalente à la lame et tous les groupes fonctionnels aldéhydes non réagis ont été convertis en alcools. La photorésistante n’est plus nécessaire. - Si plusieurs oligos doivent être modelés, revenez à l’étape 2.4, alignez le photomasque avec des marques fiduciaires et répétez.
REMARQUE: L’expérience peut être mise en pause ici. Rangez les glissières dans un dessiccateur sous vide. Dans des conditions sèches, les lames peuvent être conservées jusqu’à 3 mois sans perte de qualité.
3. Rendre la diapositive hydrophobe (facultatif) (protocole adapté de Todhunter et al.24)
REMARQUE: Il est avantageux, mais pas obligatoire, de modifier la chimie de surface de la lame pour la rendre plus inerte et hydrophobe. La fixation cellulaire non spécifique est réduite sur ces surfaces33, ce qui atténue la liaison non spécifique des cellules aux zones non modelées de la diapositive. De plus, si les cellules à motifs sont finalement incorporées dans un hydrogel et transférées hors de la lame, le traitement de surface est essentiel pour un mouvement fiable de l’hydrogel chargé de cellules à travers la lame sans distorsion ni déchirure. La silanisation avec du diméthylchlorosilane (tridécafluoro-1,1,2,2-tétrahydrooctyle) entraîne la présence de groupes fluoroalkyles hydrophobes sur la surface de la lame.
ATTENTION : Effectuez toutes les étapes à partir de l’article 3.1 dans une hotte chimique pour éviter l’exposition aux vapeurs d’acide acétique et de chlorure de méthylène.
- Rincez la lame avec 10% d’acide acétique, puis séchez sous un flux d’air.
- Dans un bocal Coplin en verre, préparer une solution de 60 mL de chlorure de méthylène (dichlorométhane), de 0,6 mL de triéthylamine et de 0,6 mL de diméthylchlorosilane (tridécafluoro-1,1,2,2-tétrahydrooctyle). Remuer avec une spatule métallique pour mélanger.
REMARQUE: Ces réactifs sont sensibles à l’eau. Ils doivent être conservés dans des conditions sèches et utilisés aussi frais que possible. - Ajouter la lame au pot Coplin contenant la solution de silane. Placez le pot Coplin sur un agitateur orbital (réglé sur 60-80 tr/min) et laissez la réaction du silane et la glissière progresser pendant 15 min.
- Utilisez des pinces métalliques pour retirer la glissière de la solution de silane. Immerger la lame dans un pot Coplin contenant du chlorure de méthylène pendant 1 minute pour éliminer l’excès de silane de la lame.
- Immerger la lame dans un tube conique de 50 mL contenant de l’éthanol. Agiter. Immerger la lame dans un tube conique de 50 mL contenant de l’eau distillée. Agiter.
REMARQUE: Le chlorure de méthylène et l’eau ne sont pas miscibles, de sorte qu’un rinçage à l’éthanol est nécessaire pour éliminer l’excès de chlorure de méthylène avant le rinçage final à l’eau. - Retirez la glissière de l’eau et inspectez-la. La lame doit être assez sèche, toutes les gouttelettes d’eau ayant un angle de contact supérieur à 90 °. Laisser sécher complètement les lames et les conserver dans un dessiccateur sous vide jusqu’à leur utilisation.
REMARQUE: L’expérience peut être mise en pause ici. Conservez la lame dans des conditions sèches.
4. Préparer les cellules d’écoulement PDMS et la diapositive pour l’expérience
REMARQUE : Les cellules d’écoulement PDMS rectangulaires sont utilisées pour concentrer les cellules sur les régions à motifs de la diapositive. Pour les expériences cultivées en 3D, les cellules d’écoulement forment un moule pour l’hydrogel.
- Fabriquez un maître SU-8 à utiliser comme moule pour les cellules de flux PDMS.
- Préchauffer la plaque chauffante à 95 °C.
- Ajouter 5 mL de SU-8 2075 à une plaquette de silicium.
- Enduire le SU-8 sur la plaquette à 500 tr/min pendant 10 s, suivi de 1 000 tr/min pendant 30 s. Cela devrait créer des caractéristiques allant jusqu’à 240 μm de hauteur34.
- Cuire doucement la plaquette sur la plaque chauffante pendant au moins 45 min.
- Retirez la plaquette de la plaque chauffante. Placez le photomasque (voir fichier supplémentaire 4)(côté émulsion vers le bas) sur le dessus de la plaquette et alourdissez-le avec un disque en verre pour assurer le contact entre le photomasque et la glissière.
- Exposer à la lumière UV (365 nm) pour une densitéd’énergie rayonnante de 350 mJ/cm2.
- Cuire la plaquette sur la plaque chauffante pendant 12-15 min.
- Placez la plaquette dans un large récipient en verre. Couvrez la plaquette avec la solution de développement SU-8. Placer sur un shaker et développer tout en agitant pendant au moins 15 min.
- Utilisez des pinces pour retirer la plaquette de la solution de développement. Rincez pendant 5 s en pulvérisant plus de solution de développement à partir d’un flacon de gicleur. Vaporiser avec de l’alcool isopropylique pour rincer. Si un précipité blanc apparaît, retournez la plaquette à la solution de développement et développez-la plus longtemps.
- Plaquette sèche sous un courant d’air ou d’azote.
- Faire cuire la diapositive pendant 5 min.
REMARQUE: Une fois la plaquette principale créée, elle peut être réutilisée indéfiniment tant que les fonctionnalités restent intactes.
- Préparez pdMS.
- Dans un bateau de pesage, ajoutez de l’élastomère de polydiméthylsiloxane et du réticulateur dans un rapport de 10:1 (en masse). Remuer vigoureusement pour assurer un mélange uniforme.
- Dégazez le PDMS dans un dessiccateur sous vide pendant 15 à 30 minutes jusqu’à ce qu’il n’y ait plus de bulles visibles.
- Placez la plaquette maîtresse dans un plat de culture tissulaire de 15 cm. Versez PDMS sur la plaquette. Si des bulles apparaissent, dégazez dans un dessiccateur sous vide pendant quelques minutes.
- Cuire au four à 60 °C pendant 3 h.
REMARQUE: Après la cuisson, les cellules d’écoulement PDMS peuvent être stockées sur la paillasse indéfiniment.
- Préparez des cellules de flux PDMS pour l’expérience.
- Peu de temps avant de commencer une expérience CMO-DPAC, découpez le nombre requis de cellules de flux PDMS de la plaquette maîtresse. Le plasma s’oxyde avec 10 cc/min d’air ambiant pendant 90 s pour rendre la surface hydrophile.
- Découpez chaque cellule d’écoulement individuelle de sorte qu’il reste 1 à 2 mm de PDMS de chaque côté, puis ouvrez le haut et le bas de la cellule d’écoulement pour créer une entrée et une sortie.
- Récupérez la diapositive à motifs créée aux étapes 2 et 3. Aligner sur le dessus du masque photo.
- En utilisant le photomasque comme référence, placez les cellules de flux PDMS sur la diapositive à l’emplacement de chaque région à motifs.
- Ajouter 50 μL de solution saline tamponnée au phosphate (PBS) + 1 % d’albumine sérique bovine (BSA) à l’entrée de chaque cellule d’écoulement, comme le montre la figure supplémentaire 1H. Confirmez que la cellule d’écoulement est complètement remplie par le PBS + 1% BSA et qu’il n’y a pas de grosses bulles. Passez immédiatement aux étapes 5 et 6.
REMARQUE: Le blocage avec BSA minimise l’adhérence des cellules non spécifiques à la surface de la glissière.
5. Soulevez et étiquetez les cellules avec de l’ADN modifié par le cholestérol
- Préparez les solutions d’ADN modifiées par le cholestérol.
- Pour chaque ensemble de cellules de l’expérience, mélanger ensemble 3 μL d’une solution mère de 100 μM du brin d’ancrage universel modifié par le cholestérol avec 3 μL d’une solution mère de 100 μM d’un brin adaptateur. Incuber pendant 1 minute. Cela pré-hybridera les oligos. Ajouter 69 μL de solution saline tamponnée au phosphate (PBS) pour créer une solution Universal Anchor + Adapter de 4 μM.
- Pour chaque ensemble de cellules de l’expérience, ajoutez 3 μL d’une solution mère de 100 μM de Co-Anchor Strand modifiée par cholestérol universel à 12 μL de PBS, créant ainsi une solution de 20 μM.
- Préparez la ou les suspensions unicellulaires.
- Pour les cellules adhérentes, utilisez la trypsine ou un autre agent de dissociation pour retirer les cellules de la fiole de culture. Ajouter un milieu de culture pour neutraliser la trypsine et centrifuger pour granuler les cellules. Pour les cellules non adhérentes, prélever la suspension cellulaire et centrifuger pour granuler les cellules.
- Resuspendez la pastille cellulaire dans 1 mL de PBS glacé ou de milieu sans sérum. Transférer 1 à 3 millions de cellules dans un tube microcentrifuge de 1,5 mL. Centrifuger à 160 x g pendant 4 min.
REMARQUE: Si le type de cellule utilisé est sujet à l’agglutination / agrégation, utilisez PBS sans ions calcium et magnésium pour toutes les étapes de lavage afin de réduire l’agrégation cellulaire indésirable. Si la viabilité est une préoccupation particulière pour le type de cellule utilisé, utilisez un milieu sans sérum au lieu de PBS. Les milieux contenant du sérum bovin fœtal ne sont pas recommandés pour le profilage cellulaire car ils peuvent entraver l’incorporation d’oligos modifiés par les lipides. 35
- Étiquetez les cellules avec des oligos modifiés par le cholestérol.
- Resuspendez la pastille cellulaire dans 75 μL de PBS glacé ou de milieu sans sérum. Conservez les cellules dans un seau à glace tout au long du processus d’étiquetage et de lavage afin de maximiser la viabilité cellulaire et de minimiser la perte des oligos modifiés par le cholestérol de la surface cellulaire.
REMARQUE: La réutilisation des cellules avant d’ajouter l’ADN garantit que la distribution de l’ADN est uniforme dans la population cellulaire. - Ajouter les 75 μL de la solution Universal Anchor + Adapter de 4 μM créée à l’étape 5.1.1 au tube de microcentrifugation contenant la suspension cellulaire. Bien mélanger par pipetage. Incuber pendant 5 min sur de la glace.
- Ajouter 15 μL de la solution universelle de co-ancrage au tube de microcentrifugation. Bien mélanger par pipetage. Incuber pendant 5 min sur de la glace.
- Éliminer l’excès d’oligos de la suspension cellulaire. Ajouter 1 mL de PBS glacé ou de milieu sans sérum au tube de microcentrifugation. Mélanger avec une pipette P1000. Centrifuger à 160 x g pendant 4 min à 4 °C. Jetez le surnageant. Répétez deux fois de plus.
REMARQUE: Si les cellules sont sujettes à l’agglutination, passez la suspension cellulaire à travers un filtre de 40 μm avant le lavage final. Si les cellules sont sujettes à l’adsorption sur le côté du tube de microcentrifugation, envisagez de préblocer le tube avec de la caséine.
- Resuspendez la pastille cellulaire dans 75 μL de PBS glacé ou de milieu sans sérum. Conservez les cellules dans un seau à glace tout au long du processus d’étiquetage et de lavage afin de maximiser la viabilité cellulaire et de minimiser la perte des oligos modifiés par le cholestérol de la surface cellulaire.
6. Modéliser les cellules étiquetées par l’ADN
- Resuspendez les cellules dans des PBS glacés ou des milieux sans sérum pour créer une solution dense en cellules d’au moins 25 millions de cellules /mL.
REMARQUE: Pour une lame utilisant quatre des cellules d’écoulement PDMS de 10 mm x 15 mm x 200 μm décrites à l’étape 4, environ 100 μL de cette suspension de cellules denses sont nécessaires. Bien que la plupart de ces cellules n’adhèrent pas au modèle et seront finalement jetées, le fait d’avoir une solution extrêmement concentrée de cellules sur le motif améliore considérablement l’efficacité du motif cellulaire. - Prenez la glissière et inclinez-la légèrement. Ajouter 25 μL de suspension cellulaire à l’entrée de chaque cellule d’écoulement sur la glissière à motifs. Retirez la solution PBS + 1% BSA de la sortie, ce qui permet à la suspension de la cellule de remplir la cellule d’écoulement PDMS. Incuber sur de la glace ou à température ambiante pendant 30 s.
REMARQUE: À ce stade, l’examen de la cellule d’écoulement au microscope devrait montrer des cellules densément emballées avec peu ou pas d’espaces visibles entre les cellules. Voir la figure supplémentaire 2B. - Aspirer 5 μL de suspension cellulaire à partir de la sortie de la glissière et la rajouter dans l’entrée. Répétez 10 fois par cellule d’écoulement.
REMARQUE: L’adhésion des cellules étiquetées CMO à la lame à motif d’ADN est presque instantanée. Faire circuler les cellules sur le motif plusieurs fois augmente la probabilité qu’une cellule circule sur un point d’ADN donné et soit capturée. - Pipettez doucement le PBS ou un milieu sans sérum dans l’entrée de chaque cellule d’écoulement pour éliminer les cellules en excès. Prélever la suspension cellulaire de la sortie. Répétez 2 à 4 fois ou jusqu’à ce qu’une inspection visuelle de la lame au microscope confirme qu’il ne reste plus de cellules en excès.
REMARQUE: Il peut être avantageux de sauver les cellules en excès du premier lavage. Si l’efficacité du motif n’est pas satisfaisante, les cellules excédentaires peuvent être centrifugées et remises en service dans un volume inférieur de PBS pour créer une solution plus dense en cellules, puis le processus peut être répété à partir de l’étape 6.2. - Répétez les étapes 6.1 à 6.4 pour chaque ensemble de cellules du modèle. Pour les modèles dans lesquels plusieurs types de cellules sont directement modelés par le modèle de surface, commencez par le type de cellule le moins abondant du motif et terminez par le type de cellule le plus abondant.
REMARQUE: Il est conseillé de faire chaque tour d’assemblage cellulaire séquentiellement au lieu de regrouper les cellules, même dans des conditions où les cellules sont toutes étiquetées avec des séquences d’ADN orthogonales. La mise en commun des cellules dilue efficacement chaque population cellulaire et réduit l’efficacité de la modélisation. - Une fois la dernière série d’assemblage de cellules terminée, les étapes suivantes varient en fonction de l’expérience spécifique. Si les cellules sont destinées à rester sur le verre, ajoutez un support à une boîte de Petri contenant la lame, puis utilisez doucement une pince pour pousser les cellules de flux PDMS hors de la lame. Si les cellules doivent être incorporées dans un hydrogel et cultivées en 3D, passez à l’étape 7.
7. Transfert en hydrogel pour culture 3D (facultatif)
- Préparer une solution de précurseur d’hydrogel contenant 2% de DNase.
REMARQUE: La composition de la solution varie en fonction de la configuration expérimentale. Matrigel et mélanges de Matrigel et de collagène Je fonctionne bien dans ce protocole, mais d’autres hydrogels sont également possibles. - Ajouter 50 μL de solution d’hydrogel contenant 2% de DNase à l’entrée de chaque cellule d’écoulement. Aspirer l’excès de liquide de la sortie, en conduisant la solution d’hydrogel dans la cellule d’écoulement. Pour les précurseurs d’hydrogel visqueux, il peut être nécessaire d’incliner légèrement la glissière pour aider l’hydrogel à s’écouler dans la cellule d’écoulement.
- Incuber la lame à 37 °C pendant 30 à 45 min (selon la cinétique de gélification de l’hydrogel) pour permettre à l’hydrogel de se fixer et de cliver l’adhérence à base d’ADN entre les cellules et la surface.
- Retirez chaque cellule d’écoulement de la lame et placez-la sur la solution de précurseur d’hydrogel.
- Ajouter 50 μL de précurseur d’hydrogel à un puits d’une lame de chambre à 2 puits ou d’une plaque de 6 puits.
- Pipette 10 μL de PBS de chaque côté de chaque cellule d’écoulement.
- Utilisez une lame de rasoir ou une pince à épiler à pointe fine pour répartir le PBS sur toute la longueur de la cellule d’écoulement, puis soulevez doucement les côtés de la cellule d’écoulement afin que le PBS se précipite sous l’hydrogel.
REMARQUE: Cela « flottera » l’hydrogel sur la glissière, permettant le transfert sans distorsion ni déchirure. - Utilisez une lame de rasoir pour déplacer doucement la cellule d’écoulement vers le bord de la glissière en verre.
- Inverser la diapositive. Avec la lame de rasoir, poussez la cellule d’écoulement hors de la glissière afin qu’elle atterrisse sur le dessus de la lame de rasoir.
- Choisissez la cellule d’écoulement de la lame de rasoir à l’aide de pinces incurvées. Inverser la cellule d’écoulement de sorte que les cellules soient sur le fond, puis placer sur le dessus de la gouttelette de solution de précurseur d’hydrogel.
- Répétez les étapes 7.4.1 à 7.4.6 pour chaque cellule d’écoulement.
- Incuber pendant au moins 30 minutes afin que l’hydrogel contenant les cellules à motifs puisse se lier à la sous-couche d’hydrogel, ce qui entraîne l’intégration complète des cellules à motifs.
- Retirez la cellule de flux PDMS.
- Ajoutez suffisamment de média pour immerger la cellule de flux PDMS.
REMARQUE: L’afflux de milieux desserre l’adhérence entre l’hydrogel et la cellule d’écoulement PDMS. - Utilisez des pinces incurvées, orientées le long de l’axe long de la cellule d’écoulement, pour pousser doucement la cellule d’écoulement jusqu’à ce qu’elle se détache et flotte dans le média. Collectez la cellule d’écoulement avec des pinces et jetez-la.
REMARQUE: Pour des résultats optimaux, étalez les pinces incurvées et appliquez une légère pression sur les parois de la cellule d’écoulement PDMS. Appliquez la force dans la direction de l’axe long de la cellule d’écoulement.
- Ajoutez suffisamment de média pour immerger la cellule de flux PDMS.
8. Confirmer l’étiquetage réussi des cellules avec CMO (facultatif, pour le dépannage)
- Commandez un oligonucléotide modifié par fluorescence (FAM ou AF647) qui est complémentaire à la séquence d’adhésion de surface du brin adaptateur utilisé dans l’expérience.
- Étiquetez les cellules avec de l’ADN CMO et lavez l’excès d’ADN comme décrit à l’étape 5. Resuspendez dans 200 μL de PBS glacé.
- Constituer une solution de 4 μM de l’oligonucléotide complémentaire marqué par fluorescence dans le PBS. Ajouter 200 μL de cette solution à la suspension cellulaire. Incuber sur de la glace pendant 5 min.
- Ajouter 1 mL de PBS glacé. Mélanger. Centrifuger les cellules pour les granuler. Retirer le surnageant. Répétez ce processus deux fois de plus pour laver tout ADN qui ne s’est pas hybridé.
- Effectuer une cytométrie analytique en flux pour quantifier la présence d’ADN à la surface de la cellule.
- Sur un cytomètre en flux, analysez les cellules témoins qui n’ont pas été étiquetées avec de l’ADN. Mettez en place des portes en fonction de cette population.
- Analyser les cellules étiquetées CMO qui ont été traitées avec un oligonucléotide complémentaire marqué par fluorescence.
- Calculer l’intensité moyenne de fluorescence.
Résultats
Ce protocole permet de modeler des cellules en 2D et 3D avec une grande précision et sans l’utilisation de réactifs personnalisés ou d’équipements de salle blanche coûteux. La figure 1 montre une vue d’ensemble du protocole. Tout d’abord, les lames fonctionnalisées par l’ADN sont créées par photolithographie. Ensuite, les cellules sont étiquetées avec des CMO. Les cellules sont ensuite acheminées sur la lame, où elles ne se fixent qu’aux régions fonctionnalisées par l’ADN de la lame. Une fois que les cellules en excès sont emportées, le modèle souhaité de cellules est révélé. Ces cellules peuvent être cultivées sur la lame ou incorporées dans un hydrogel contenant de la DNase et transférées hors de la lame pour la culture cellulaire 3D.
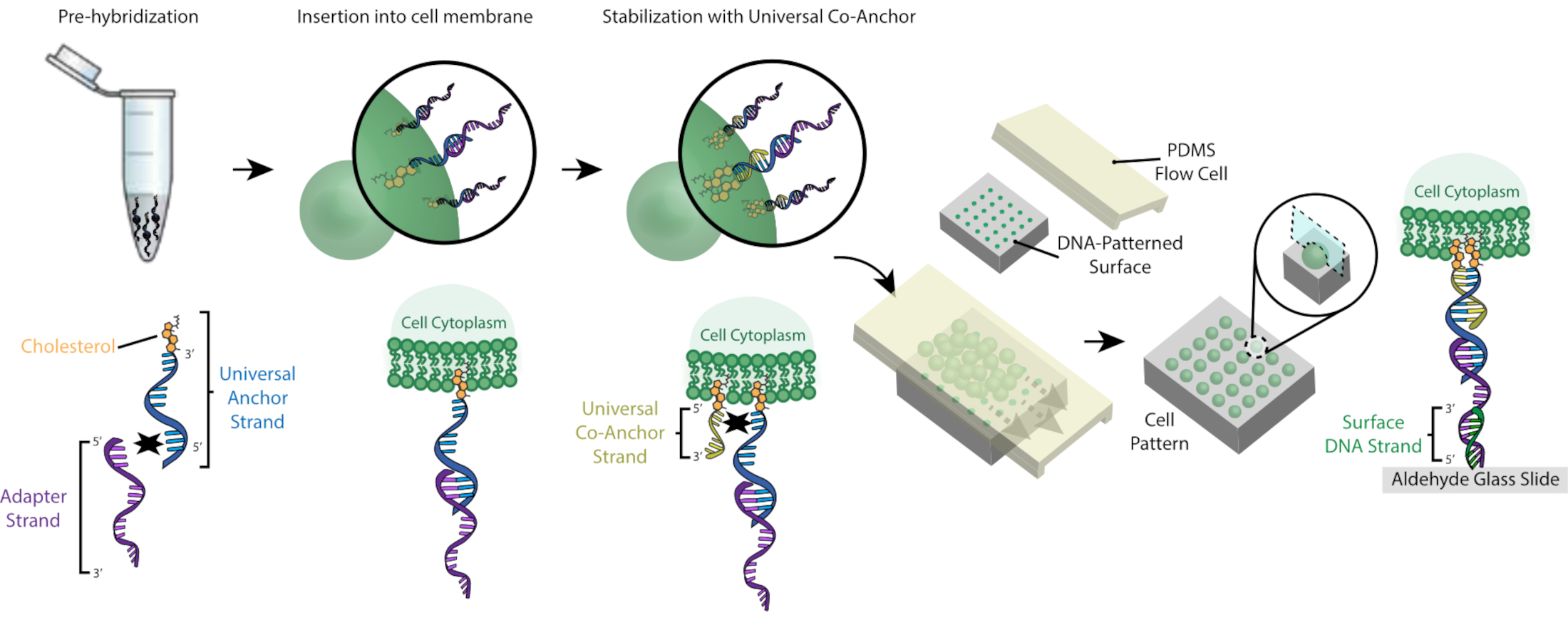
L’étiquetage des cellules avec des ocM permet leur fixation à la lame à motifs d’ADN (Figure 2). Tout d’abord, le brin d’ancrage universel modifié par le cholestérol est pré-hybridé avec le brin adaptateur. Ensuite, la solution Universal Anchor + Adapter est mélangée 1:1 avec la suspension de cellule. Le cholestérol du complexe Universal Anchor + Adapter s’insère dans la membrane cellulaire. L’ajout du brin de co-ancrage universel modifié par le cholestérol, qui s’hybride avec le brin d’ancrage universel, améliore la stabilité du complexe CMO dans la membrane cellulaire en augmentant l’hydrophobicité nette du complexe26. Après avoir éliminé l’excès d’ADN de la suspension cellulaire, les cellules sont acheminées sur la lame. L’hybridation entre le brin adaptateur et le brin d’ADN de surface entraîne la fixation des cellules aux régions à motifs d’ADN de la diapositive.
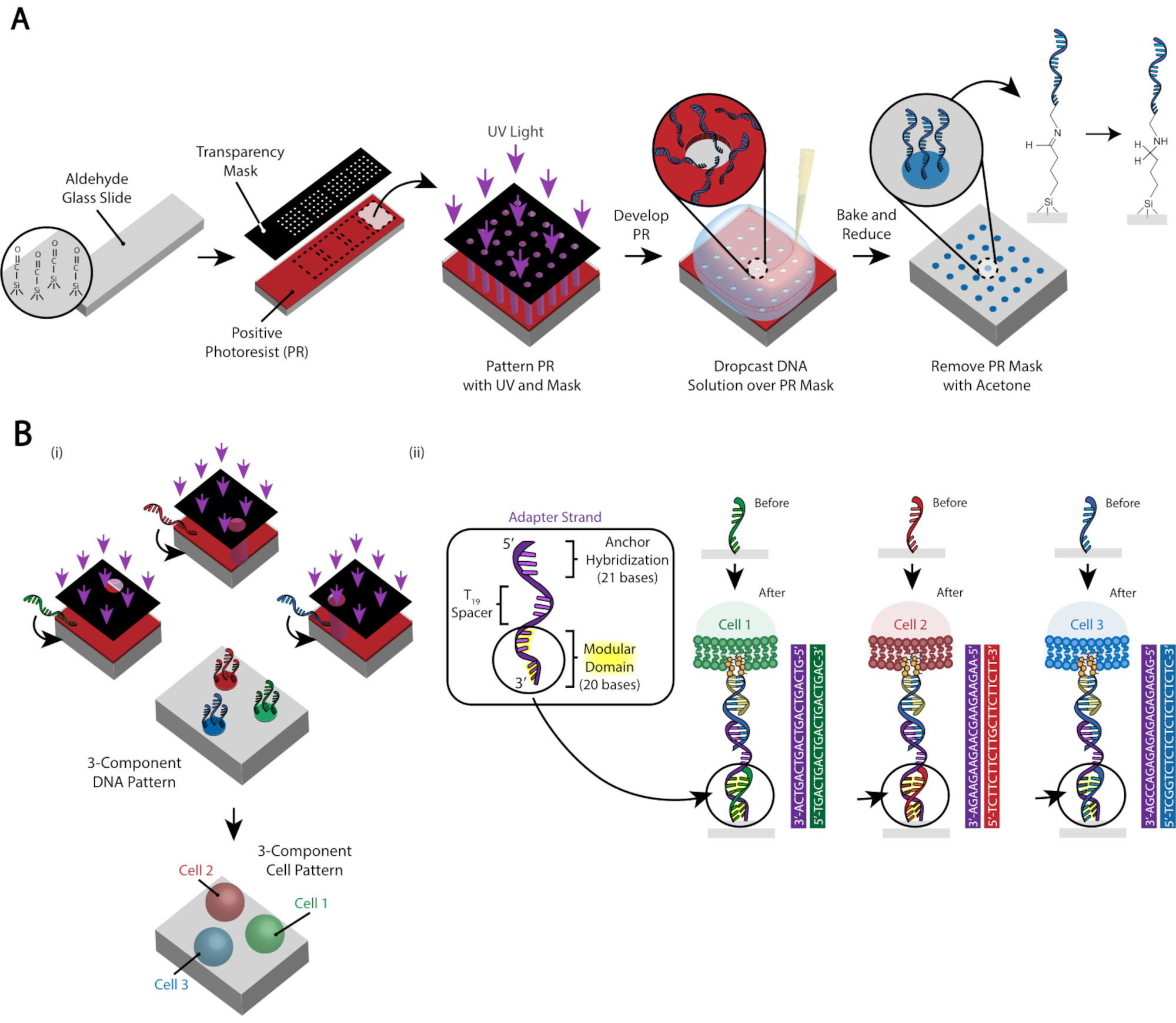
Le motif des cellules est créé en utilisant la photolithographie pour restreindre la fixation d’oligos d’ADN modifiés par les amines à des régions spécifiques d’une lame de verre modifiée par l’aldéhyde29 (Figure 3A). La photorésiste positive est recouverte d’une lame fonctionnalisée par l’aldéhyde. Un photomasque transparent est ensuite placé sur le dessus de la diapositive et la diapositive est exposée à la lumière UV. Après le développement, les régions de la lame qui ont été exposées à la lumière UV ne sont plus recouvertes de photorésistante et ont donc exposé des groupes aldéhydes. Une solution de 20 μM d’oligos d’ADN modifiés par les amines est ensuite déposée sur la lame et étalée pour couvrir les régions à motifs. La cuisson suivie d’une amination réductrice entraîne une liaison covalente entre l’ADN modifié par les amines et la lame. Remarquablement, ce processus peut être répété pour modeler plusieurs oligos sans aucune perte de fonctionnalité des oligos précédemment modelés (Figure 3B). Cependant, il faut veiller à éviter les chevauchements de modèles, ce qui entraîne la présence des deux oligos à une concentration réduite (Figure supplémentaire 3). Plusieurs populations de cellules peuvent être modelées séquentiellement à l’aide de brins d’adaptateur qui diffèrent par leur domaine modulaire (les 20 bases les plus proches de l’extrémité 3').
Bien que ce protocole de photomodèlement ait été développé par Scheideler et al. dans le contexte d’une salle blanche, nous avons démontré qu’il est possible d’obtenir des résultats similaires avec une configuration de photolithographie peu coûteuse et « brassée à la maison » qui s’intègre facilement dans une hotte chimique. La configuration comprend un enrobeur de spin de 400 $ composé d’un moteur à courant continu, d’un contrôleur numérique et d’une boîte à gâteaux CD, ainsi qu’une lampe UV assemblée à partir de composants individuels et logée dans un récipient d’objets tranchants réutilisés(figure supplémentaire 1). Le principal avantage de la configuration de photolithographie maison est qu’elle est très abordable (< 1000 $ pour tout l’équipement) tout en étant capable de créer des fonctionnalités de taille unique. Cependant, l’utilisation d’équipements peu coûteux a ses limites - par exemple, il est plus difficile d’aligner avec précision les marqueurs fiducial pour modeler plusieurs oligos d’ADN sans utiliser d’aligneur de masque. Nous recommandons cette configuration de photolithographie peu coûteuse pour les laboratoires qui n’ont pas un accès pratique à une salle blanche ou qui veulent essayer cette méthode sans un investissement important.
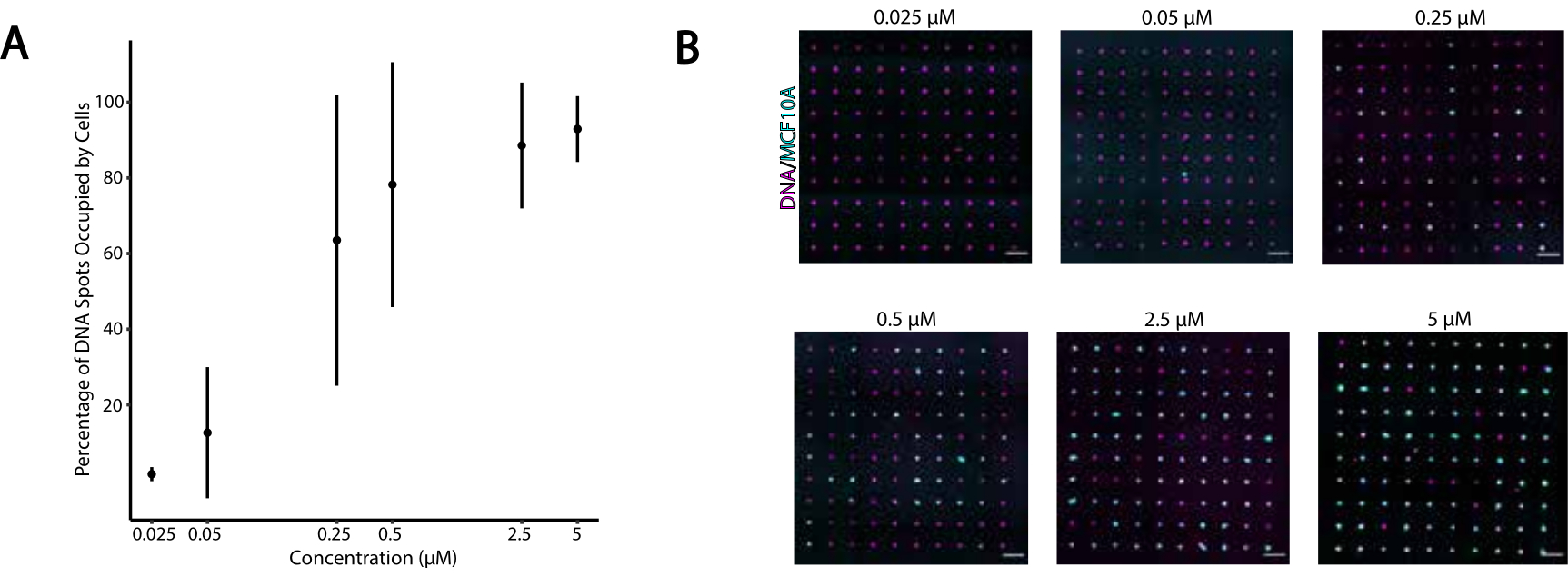
Pour identifier les conditions optimales pour l’adhésion cellulaire programmée par l’ADN, nous avons systématiquement fait varier les concentrations de brins d’ADN sur les surfaces cellulaires et mesuré l’efficacité de l’adhésion cellulaire aux surfaces en verre modifiées par l’ADN. La concentration d’Universal Anchor + Adapter Strand et d’Universal Co-Anchor dans les solutions de marquage a varié selon plusieurs ordres de grandeur (Figure 4A, B), ce qui a donné 104 -10 6 complexes d’ADN par cellule (Figure supplémentaire 4). L’adhésion cellulaire dépendait de la dose, avec une adhérence cellulaire minimale au modèle d’ADN lorsque les cellules étaient étiquetées avec des OCM à une concentration de 0,05 μM ou moins, et une occupation élevée à une concentration de 2,5 μM et plus. Nous avons donc utilisé une solution de 2 μM d’universal anchor + adapter Strand et une solution de 2 μM de Universal Co-Anchor dans la plupart des expériences. On s’attendrait également à ce que l’adhérence des cellules diminue si la quantité d’ADN utilisée sur la surface du verre diminuait de29 ou si les incohérences entre le brin adaptateur et le brin de surface augmentaient. Pour plus d’informations sur la conception de séquence de brins d’adaptateur, consultez le fichier supplémentaire 2. L’étiquetage CMO à l’aide de brins d’adaptateur sans répétitions CpG n’a pas stimulé TLR9 dans les cellules HEK exprimant TLR9 de souris ( Figure supplémentaire5).
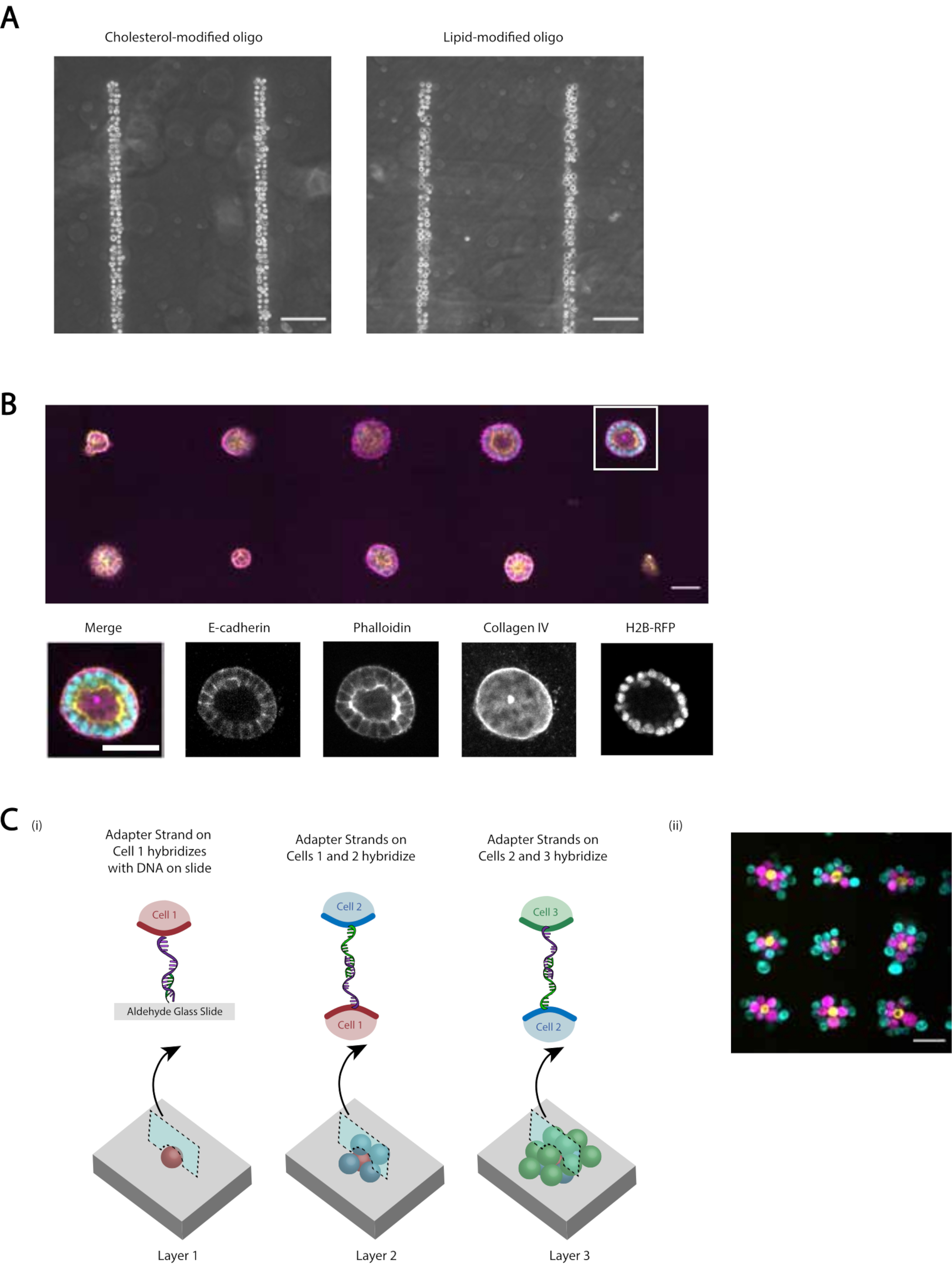
Nous fournissons plusieurs démonstrations que le protocole révisé fournit une adhésion cellulaire reproductible et efficace programmée par l’ADN. Par exemple, les cellules endothéliales de la veine ombilicale humaine (HUVECs) étiquetées avec des CMO ont adhéré à des modèles d’ADN avec une grande efficacité. Les HUVÉCs étiquetés CMO adhéraient ainsi que les HUVÉC étiquetés LMO (Figure 5A). Les cellules modélisées à l’aide de CMO-DPAC ont conservé leur viabilité et leur fonctionnalité. Les cellules étiquetées avec des OCM ont été colorées par la calcéine AM et l’homodimère d’éthidium pour évaluer la viabilité (Figure supplémentaire 6). Les différences de viabilité par rapport aux cellules témoins non marquées étaient faibles (94 % contre 97 %). Les CDK simples modelés via CMO-DPAC et transférés dans Matrigel ont pu proliférer et se polariser correctement après 5 jours de culture(Figure 5B). DPAC fournit également un moyen d’élaborer des modèles de cellules dans la troisième dimension (Figure 5C). Par exemple, des agrégats multicouches et multicellulaires peuvent être créés en alternant des couches de cellules étiquetées avec des OCM complémentaires(Figure 5C). Ces expériences démontrent que le protocole est reproductible, n’affecte pas négativement la viabilité ou la fonctionnalité des cellules et produit des modèles cellulaires qui peuvent être cultivés avec succès dans un seul plan d’imagerie dans un ECM 3D.
En fournissant des séquences d’ADN orthogonales pour diriger l’adhésion cellulaire, DPAC fournit un moyen de modéliser plusieurs types de cellules sur une seule surface. Pour mettre en œuvre cette caractéristique du DPAC, les modèles d’ADN générés par photolithographie doivent être alignés les uns par rapport aux autres. Les marqueurs fiduciaires métalliques déposés sur la lame ont permis l’alignement de plusieurs photomasques et donc la modelage de plusieurs types de cellules à la fois. Les MCF10A colorés avec différents colorants uniques ont été étiquetés avec des CMO orthogonaux et modelés pour créer une visualisation des logos UC Berkeley et UCSF (Figure 6). Cette expérience démontre que plusieurs populations cellulaires uniques peuvent être modelées ensemble avec une grande précision et sans contamination croisée.
La modélisation réussie des cellules à l’aide de CMO-DPAC nécessite une photolithographie de haute qualité, une concentration suffisante d’oligo à la surface de la cellule, une densité élevée de cellules sur le motif et un lavage suffisant. L’échec de l’une de ces étapes affecte le résultat final. La figure supplémentaire 2 comprend des exemples d’images de photolithographie correcte et incorrecte (Figure supplémentaire 2A), la densité cellulaire souhaitée sur le motif pour créer des motifs entièrement occupés (Figure supplémentaire 2B), la perte de cellules à motifs due à un pipetage trop vigoureux au cours des étapes ultérieures du DPAC (Figure supplémentaire 2C), et l’agglutination indésirable des cellules (Figure supplémentaire 2D). Le tableau 1 inclut une liste des points de défaillance courants et le dépannage suggéré. L’utilisation d’oligos complémentaires fluorescents est recommandée comme outil de dépannage pour confirmer la présence d’ADN à motifs sur la lame et la présence d’OCM à la surface de la cellule par cytométrie en flux (voir l’étape 8 du protocole).

Figure 1: Vue d’ensemble du protocole CMO-DPAC. Tout d’abord, une lame à motifs d’ADN est créée en recouvrant une lame de verre fonctionnalisée à l’aldéhyde d’une photorésiste positive, en la recouvrant d’un masque transparent dans le motif souhaité et en l’exposant à la lumière UV. La photorésisse exposée aux UV est emportée par le révélateur, laissant les régions exposées de la lame d’aldéhyde et permettant la liaison de l’ADN fonctionnalisé par les amines à la surface. Les cellules sont ensuite étiquetées avec des CMO et circulent sur la surface. L’ADN sur la membrane cellulaire s’hybride à l’ADN à la surface, ce qui entraîne une adhésion. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2: Les cellules sont étiquetées avec des CMO dans un processus par étapes. Tout d’abord, le brin d’ancrage universel modifié par le cholestérol est pré-hybridé avec le brin adaptateur. Ensuite, la solution Universal Anchor + Adapter est mélangée à la suspension de cellule. Le cholestérol du complexe Universal Anchor + Adapter s’insère dans la membrane cellulaire. Après l’incubation, le brin de co-ancrage universel modifié par le cholestérol est ajouté à la suspension cellulaire, où il s’hybride avec le brin d’ancrage universel et s’insère dans la membrane cellulaire. L’ajout de la deuxième molécule de cholestérol augmente l’hydrophobicité nette du complexe ADN et le stabilise au sein de la membrane26. Après avoir éliminé l’excès d’ADN, les cellules sont concentrées et ajoutées à une cellule d’écoulement PDMS au-dessus de la surface à motifs. L’extrémité 3' du brin adaptateur s’hybride avec le brin d’ADN de surface sur la lame de verre, ce qui entraîne une adhérence à la lame spécifiquement dans les régions fonctionnalisées avec de l’ADN complémentaire. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3: La photolithographie est utilisée pour créer les lames à motifs d’ADN qui dicteront finalement le placement des cellules. (A) Vue d’ensemble du procédé de photolithographie. Une lame fonctionnalisée à l’aldéhyde est recouverte d’une photorésiste positive. La lumière UV brille sur la glissière à travers un photomasque transparent transparent où l’adhésion cellulaire est souhaitée. Une fois la lame développée, les régions qui étaient auparavant exposées à la lumière UV ont maintenant exposé des groupes aldéhydes. Une solution de 20 μM d’un oligo d’ADN fonctionnalisé par les amines est ensuite déposée sur la lame et étalée sur les régions à motifs. La lame est ensuite cuite pour induire la formation de liaisons de Schiff (C=N) entre les groupes amine et aldéhyde, une liaison covalente réversible29. L’amination réductrice ultérieure avec 0,25% de borohydrure de sodium dans le PBS convertit la base de Schiff en amine secondaire par amination réductrice, ce qui entraîne une liaison irréversible entre l’ADN et la lame. La photorésine restante peut ensuite être enlevée par rinçage à l’acétone. (B) Ce processus peut être répété pour créer des modèles d’ADN multi-composants et donc effectuer des expériences avec plusieurs populations cellulaires. (i) Une fois le premier oligo modelé, la lame est à nouveau recouverte de photorésistante et le protocole se déroule comme auparavant. L’alignement des photomasques à l’aide de marqueurs fiduciaires est nécessaire pour modéliser plusieurs brins d’ADN. (ii) Chaque type de cellule modelé diffère dans le domaine modulaire à 20 bases du brin adaptateur. En utilisant des ensembles orthogonaux d’oligos complémentaires, plusieurs types de cellules peuvent être modelés sans adhésion croisée. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4: L’adhésion des cellules étiquetées CMO aux profils d’ADN augmente en fonction de la concentration de CMO pendant le profilage. Dans cette expérience, l’Universal Anchor + Adapter Strand (pré-hybridé) et le Universal Co-Anchor ont été utilisés à des concentrations égales. La concentration fait référence à la concentration de CMO dans la suspension cellulaire pendant le profilage CMO des cellules. (A) Quantification du pourcentage de taches d’ADN de 15 μm de diamètre qui étaient occupées par des cellules MCF10A étiquetées CMO en fonction de la concentration de CMO lors du profilage cellulaire. Données représentées comme la moyenne ± écart-type par rapport à trois expériences. (B) Images représentatives des profils d’ADN (magenta) et des MCF10A adhérés (cyan) à différentes concentrations de CMO. Barre d’échelle = 100 μm. Veuillez cliquer ici pour afficher une version agrandie de cette figure.
{kind=link}

Figure 5: CMO-DPAC peut être utilisé pour créer des motifs cellulaires bidimensionnels qui peuvent ensuite être incorporés dans un hydrogel tridimensionnel pour la culture et / ou stratifiés pour créer des structures multicouches. (A) La comparaison directe entre les cellules endothéliales de la veine ombilicale humaine marqués par CMO (HUVEC) et les HUVEC marqués LMO ont adhéré à un modèle d’ADN linéaire. Les deux méthodes de profilage cellulaire entraînent une occupation de près de 100% du motif ADN. (B) Des cellules simples du rein canin Madin-Darby (CMDK) exprimant H2B-RFP ont été modelées sur des taches de 15 μm de diamètre espacées de 200 μm et ensuite incorporées dans Matrigel. Après 120 h de culture, les kystes épithéliaux résultants ont été fixés et colorés pour l’E-cadhérine, l’actine et le collagène IV. Sphéroïde dans la boîte blanche est montré en détail. Barre d’échelle = 50 μm. (C) Des structures cellulaires multicouches peuvent être créées en étiquetant des populations cellulaires séparées avec des brins adaptateurs complémentaires et en modelant séquentiellement de sorte que chaque nouvel ajout de cellules adhère à la couche cellulaire qui la précède. (i) Schéma de la modélisation séquentielle des populations cellulaires pour créer des structures multicouches. ii) Des agrégats cellulaires à trois couches de MCF10A (visualisés à l’aide de colorants) ont été créés à l’aide de ce processus. Barre d’échelle = 50 μm. Veuillez cliquer ici pour afficher une version agrandie de cette figure.
{kind=link}

Figure 6: Plusieurs types de cellules peuvent être modelés sans contamination croisée ni perte d’adhérence. De multiples oligos d’ADN modifiés par les amines ont été modelés séquentiellement sur une lame d’aldéhyde et alignés à l’utilisation de marqueurs fiduciaires métalliques. Trois populations de MCF10A (cyan, magenta, jaune) ont été colorées avec des colorants uniques étiquetés avec des CMO complémentaires, et modelées sur la diapositive, ce qui a donné une image des logos UC Berkeley et UCSF. Barre d’échelle 1 mm. Veuillez cliquer ici pour agrandir cette figure.
{kind=link}
Figure supplémentaire 1 : Exemples d’images de la configuration de la photolithographie de paillasse. (A) Glissière sur enduit de spin, recouvert de photorésistant positif, avant le revêtement de spin. (B) Image du photomasque transparent. (C) Pendant l’exposition, le photomasque est pris en sandwich entre la lame revêtue de photorésistante et un disque en verre. (D) Le boîtier de la lampe UV a été fabriqué à partir d’un récipient d’objets tranchants réutilisé. (E) Diapositive immergée dans la solution de développement. (F) Diapositive développée. (G) Solution d’ADN modifiée par les amines étalée sur des régions à motifs de la lame. (H) Cellules d’écoulement PDMS placées au-dessus des régions à motifs de la diapositive. Veuillez cliquer ici pour télécharger ce fichier.
{kind=link}
Figure supplémentaire 2 : Quelques exemples d’échecs courants de ce protocole. (A) (i) Une sous-cuisson avant l’exposition aux UV ou des caractéristiques de surdémisement après l’exposition peut entraîner des caractéristiques qui ont des bords déchiquetés et peuvent être de taille irrégulière. (ii) Exemple d’une diapositive correctement photomotée qui présente des bords propres autour des entités, une taille de caractéristique uniforme et aucune fissure évidente dans le motif. Barre d’échelle = 50 μm. (B) La densité des cellules est essentielle à l’efficacité de la modélisation. Lors de l’observation des cellules au-dessus du motif au microscope, peu d’espaces doivent exister entre les cellules, comme en témoigne l’exemple d’image à gauche. Barre d’échelle = 50 μm. (C) Les cellules à motifs peuvent être sensibles aux forces du fluide résultant d’un pipetage trop vigoureux, ce qui peut endommager et déloger les cellules à motifs. Les agrégats cellulaires multicouches sont particulièrement vulnérables, car une cellule au fond supporte une structure de plusieurs cellules. (i) Un tableau d’agrégats cellulaires intégrés avec succès dans Matrigel. ii)Grille d’agrégats cellulaires qui se sont délogés à la suite d’un pipetage trop vigoureux de Matrigel visqueux. (D) L’agglutination des cellules peut se produire, en particulier avec les cellules épithéliales. Ces amas sont généralement homotypiques mais peuvent être hétérotypiques (cellules adhérant à des cellules déjà modelées d’un type différent) si les cellules sont particulièrement collantes. L’image montre que trois populations différentes de MCF10A ont été modelées sur un réseau composé de trois taches d’ADN différentes de taille unicellulaire (15 μm). La plupart des taches d’ADN ont 2-4 cellules attachées. L’agglutination peut être résolue par un traitement EDTA ou en filtrant les touffes avant le modelage. Barre d’échelle = 100 μm. Veuillez cliquer ici pour télécharger ce fichier.
{kind=link}
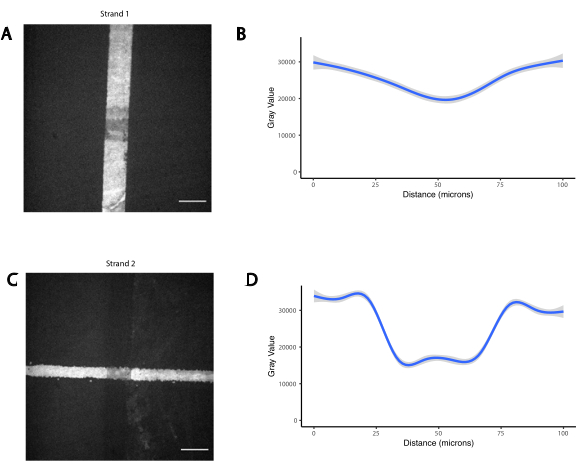
Figure supplémentaire 3 : Le chevauchement des photomots entraîne la présence des deux oligos à une concentration réduite. Deux oligos orthogonaux modifiés par les amines ont été photomoptérisés séquentiellement, d’abord une ligne verticale (brin 1), suivie d’une ligne horizontale qui la chevauchait (brin 2). Les oligos ont ensuite été visualisés par hybridation avec des oligos complémentaires fluorescents. (A) Image de fluorescence du brin 1. (B) Quantification du profil de fluorescence du brin 1 sur une ligne verticale de 100 μm couvrant le chevauchement. (C) Image de fluorescence du brin 2. (D) Quantification du profil de fluorescence du brin 2 sur une ligne horizontale de 100 μm couvrant le chevauchement. Barre d’échelle = 50 μm. Veuillez cliquer ici pour télécharger ce fichier.
{kind=link}
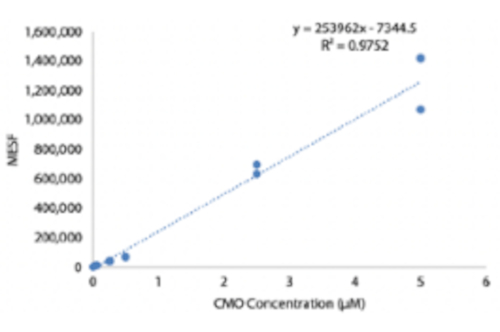
Figure supplémentaire 4 : Quantification des complexes d’ADN à la surface de la cellule en fonction de la concentration de marquage CMO. Les HUVECs ont été marqués avec différentes concentrations de solution de CMO, lavés, puis incubés avec un brin complémentaire fluorescent. Un kit de microsphère MESF (Molecules of Equivalent Soluble Fluorochrome) a été utilisé pour effectuer une cytométrie quantitative en flux et estimer le nombre de complexes d’ADN à la surface de la cellule en fonction de la concentration de CMO lors du profilage. Veuillez cliquer ici pour télécharger ce fichier.
{kind=link}
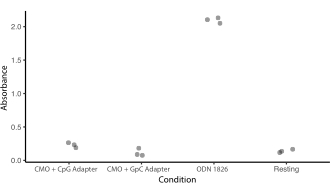
Figure supplémentaire 5 : L’étiquetage CMO ne stimule pas la réponse TLR9. Une expérience a été menée pour voir si le label CMO déclencherait le mécanisme de détection de l’ADN de TLR9 et si cela serait affecté par les CpG dans la séquence Adapter Strand. Les cellules HEK exprimant le TLR9 de souris ont été incubées pendant la nuit avec 0,2 μM d’ODN 1826 (un agoniste TLR9 contenant du CpG), d’ancre universelle CMO + co-ancre universelle + brin d’adaptateur contenant la même séquence que ODN 1826 (CMO-CpG), ou d’ancre universelle CMO + co-ancre universelle + brin d’adaptateur contenant une séquence similaire mais avec remplacement des CpG par des GpC (CMO-GpC). La stimulation TLR9 entraînerait la production de PAED (phosphatase alcaline embryonnaire sécrétée). La sécrétion de PAED a été quantifiée par un test colorimétrique (absorbance). Les conditions de traitement ont été comparées aux cellules au repos qui n’ont été traitées qu’avec du PBS. L’incubation avec CMO-GPC n’a pas stimulé l’expression de TLR9. L’incubation avec CMO-CpG était légèrement supérieure à celle des cellules au repos, mais beaucoup plus faible que l’ODN-1826. Veuillez cliquer ici pour télécharger ce fichier.
{kind=link}
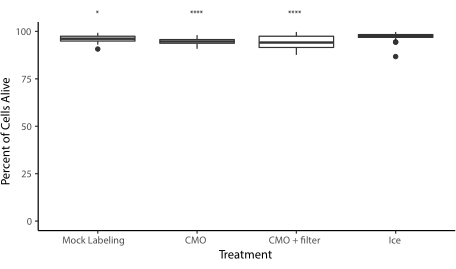
Figure supplémentaire 6 : Viabilité des cellules après le processus de marquage CMO. Pour évaluer l’impact du protocole sur la viabilité, les HUVEC ont été divisés en quatre populations : une est restée sur la glace pendant 1 h, une a été étiquetée avec du PBS mais a été prise en compte à travers toutes les étapes de centrifugation et de lavage, une a été étiquetée avec des CMO et une a été étiquetée avec des CMO et filtrée à travers un filtre de 40 μm pour éliminer les amas. Les cellules ont ensuite été colorées avec de la calcéine AM et de l’homodimère d’éthidium pour évaluer le nombre de cellules vivantes et mortes. Tous les traitements ont entraîné une diminution significative de la viabilité par rapport au contrôle de la glace (ANOVA unidirectionnelle avec analyse post-hoc de Tukey), mais la viabilité médiane pour l’étiquetage CMO (avec ou sans filtrage) était d’environ 94%. Données recueillies à partir de trois expériences indépendantes. * = p < 0,05. = p < 0,0001 Veuillez cliquer ici pour télécharger ce fichier.
{kind=link}
| Résultat | Cause(s) possible(s) | Correctifs suggérés |
| Photolithographie – les caractéristiques sont fissurées | Cuisson molle incohérente ou inadéquate | Augmenter le temps de cuisson molle jusqu’à 3 minutes; vérifier la température réelle de la plaque chauffante et augmenter la température au besoin |
| Photolithographie – les caractéristiques ne sont pas nettes ou ont une photorésine restante à l’intérieur | Sous-développement | Augmentez le temps que la diapositive passe dans la solution de développement; incorporer une agitation douce |
| Photolithographie – caractéristiques incohérentes sur la diapositive | La lumière UV peut ne pas être centrée ou mal focalée | Ajustez la configuration de la lumière UV pour assurer une lumière collimée d’intensité uniforme |
| Les cellules n’adhèrent pas aux taches à motifs avec une grande efficacité | Pas assez d’ADN à la surface | Confirmer que l’ADN est présent en surface en hybridant la lame avec des oligos complémentaires fluorescents, puis en imageant au microscope |
| Les cellules ne sont pas correctement étiquetées avec CMO | Ajouter des oligos complémentaires fluorescents à la suspension cellulaire et confirmer la fluorescence par cytométrie en flux | |
| Pas assez de cellules sur le motif | Prélever les cellules en les lavant de la cellule d’écoulement PDMS, centrifuger et re-suspendre dans un volume inférieur pour concentrer les cellules | |
| Trop de CMO restant en suspension cellulaire, hybridation avec l’ADN sur lame | Ajoutez une autre étape de lavage. Assurez-vous d’enlever autant de surnageant que possible à chaque lavage. | |
| Trop d’internalisation du CMO en raison du temps et de la température | Travailler rapidement après avoir marqué les cellules avec CMO; garder les cellules et les glisser sur la glace et utiliser des réactifs glacés | |
| Amas de cellules | Les cellules n’ont pas été séparées adéquatement pendant la trypsinisation | Utilisez PBS + 0,04% EDTA pendant le lavage des cellules; passer la suspension cellulaire à travers un filtre de 35 μm avant le lavage final |
| Les cellules adhèrent de manière non spécifique | Si dans une zone spécifique – pourrait être dû à des rayures sur la diapositive, un désalignement des cellules de flux PDMS ou un déversement d’ADN en dehors de la région du motif | Évitez les rayures, veillez à aligner les cellules de flux PDMS sur la région du motif |
| Si les cellules adhèrent partout – blocage ou lavage inadéquat | Ajoutez plus de lavages après avoir modelé les cellules; pipet plus vigoureusement pendant les lavages; bloc avec 1% de BSA pendant plus longtemps avant de commencer la modélisation cellulaire; silaniser la lame (étape 3 facultative) ou confirmer que la silanisation a réussi en mesurant l’angle de contact des gouttelettes d’eau | |
| Des bulles se forment dans la cellule d’écoulement | Erreurs de pipetage, surface hydrophile inégale créée lors de l’oxydation au plasma | Si les bulles sont petites, ajoutez pbS à l’entrée de la cellule d’écoulement et elles peuvent être lavées. Si les bulles sont plus grandes, appliquez une légère pression sur la cellule d’écoulement PDMS, en faisant pousser les bulles vers l’entrée ou la sortie. |
| Les cellules adhèrent initialement au motif, mais sont enlevées lors des lavages, de la modelage d’autres types de cellules ou de l’ajout du précurseur de l’hydrogel. | Les forces de cisaillement dues au pipetage trop vigoureusement peuvent provoquer le détachement des cellules de la surface | Pipet plus doucement lors des lavages ultérieurs, des rondes de motifs cellulaires ou de l’ajout de précurseurs d’hydrogel. Parce que les précurseurs de l’hydrogel sont visqueux, ils sont plus susceptibles de provoquer le délogement du motif, alors prenez des précautions supplémentaires. Les structures multicouches ont tendance à être lourdes et sont plus susceptibles d’être délogées. |
| Les tissus se déforment lors du transfert 3D | L’hydrogel colle à glisser | Confirmer l’hydrophobicité de la glissière à l’aide de mesures d’angle de contact |
| Utilisez une lame de rasoir pour soulever complètement le PDMS sur les deux bords, permettant au PBS de flotter sous le tissu | ||
| Cela peut se produire avec des hydrogels de collagène pur – envisagez d’ajuster la concentration en protéines ou la composition de l’hydrogel | ||
| Les cellules ne sont pas transférées avec l’hydrogel et restent sur la lame | Augmenter la concentration de Turbo DNAse ou augmenter le temps d’incubation | |
| L’hydrogel n’est pas assez solide | Augmenter le temps d’incubation et/ou le mécanisme de gélification de l’hydrogel en question (par exemple pour le collagène, assurez-vous que le pH est correct) | |
| L’hydrogel se déchire lors de l’élimination du PDMS | Rendre les cellules d’écoulement PDMS hydrophiles en utilisant l’oxydation plasma avant de commencer l’expérience afin qu’elles se détachent facilement lors de l’ajout de milieu. Utilisez des pinces très doucement pour détacher le PDMS. |
Tableau 1 : Guide de dépannage pour identifier et résoudre les défaillances potentielles pouvant découler de ce protocole. En particulier, une mauvaise adhésion des cellules au modèle peut avoir de nombreuses causes profondes et ce guide devrait aider à identifier et à résoudre ces problèmes.
Dossier supplémentaire 1. Veuillez cliquer ici pour télécharger ce fichier.
Dossier supplémentaire 2. Veuillez cliquer ici pour télécharger ce fichier.
Dossier supplémentaire 3. Veuillez cliquer ici pour télécharger ce fichier.
Dossier supplémentaire 4. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Dans cet article, nous présentons un protocole détaillé pour le modelage à haute résolution de cellules en 2D et 3D pour des expériences de culture cellulaire in vitro. Contrairement aux versions précédemment publiées de cette méthode, le protocole présenté ici se concentre sur la convivialité: il ne nécessite pas d’équipement hautement spécialisé et tous les réactifs peuvent être achetés auprès de fournisseurs au lieu de nécessiter une synthèse personnalisée. Contrairement à d’autres méthodes de micromotriction cellulaire, cette méthode est rapide et agnostique de type cellulaire: elle ne nécessite pas d’adhésion spécifique aux protéines de la matrice extracellulaire15. Les cellules modelées par CMO-DPAC peuvent être incorporées dans une matrice extracellulaire telle que le Matrigel ou le collagène, ce qui donne des cultures 3D avec une résolution spatiale beaucoup plus élevée que ce qui est actuellement possible avec les méthodes basées sur l’impression par extrusion22. CMO-DPAC peut être utilisé pour créer des centaines à des milliers de caractéristiques microscopiques par diapositive, ce qui permet d’effectuer de nombreuses réplications en même temps.
L’un des paramètres les plus importants dans le succès de ce protocole est la densité des cellules ajoutées aux cellules d’écoulement au-dessus de la diapositive à motifs. Idéalement, la densité devrait être d’au moins 25 millions de cellules/mL. Lorsqu’elle est chargée dans les cellules d’écoulement, cette densité de cellules se traduit par une monocouche de cellules presque serrée au-dessus du motif (Figure supplémentaire 2B). Ces densités cellulaires élevées maximisent la probabilité qu’une cellule s’installe directement sur une tache d’ADN et adhère. La réduction de la densité cellulaire diminuera l’efficacité globale de la modélisation. Une autre étape critique de ce protocole consiste à suspendre complètement les cellules dans un PBS ou un milieu sans sérum avant d’ajouter la solution CMO. Les CMO se divisent très rapidement en membranes cellulaires et l’ajout de la solution CMO directement à une pastille cellulaire entraînera un étiquetage hétérogène des cellules. Après avoir ajouté la solution CMO à la suspension cellulaire, il est important de bien mélanger par pipetage afin que les cellules soient uniformément étiquetées avec les CMO. Après les incubations, il est nécessaire de laver soigneusement les CMO en excès par de multiples étapes de centrifugation et de lavage. L’excès de CMO libre présent dans la suspension cellulaire se liera à l’ADN modifié par les amines sur la lame de verre, bloquant l’hybridation et l’adhésion des cellules modifiées par CMO en suspension. Le temps est également une considération clé pour ce protocole. Il est important de travailler le plus rapidement possible lors de l’utilisation d’OCM et de garder les cellules sur la glace afin de minimiser l’internalisation des OCM et de maximiser la viabilité cellulaire. Des expériences de cytométrie en flux ont montré que les CMO ne persistent pas aussi longtemps à la surface des cellules que les LMO, avec une perte de 25% des complexes CMO sur deux heures d’incubation sur glace36. De plus, la viabilité des cellules diminuera à mesure que le temps de manipulation des cellules augmentera. La viabilité peut être maximisée en travaillant rapidement, en gardant les cellules sur la glace, en utilisant des réactifs glacés et en utilisant des milieux sans sérum pour fournir certains nutriments.
Bien que le CMO-DPAC puisse être un moyen puissant d’étudier la biologie cellulaire en modélisant les cellules avec une grande précision, il a ses limites. Les expériences CMO-DPAC peuvent être difficiles, d’autant plus que la complexité expérimentale est ajoutée avec plusieurs types de cellules, couches ou culture cellulaire 3D (Fichier supplémentaire 1). Les échecs expérimentaux peuvent être fréquents lors du démarrage de ce protocole, comme décrit dans le tableau 1. Par conséquent, nous recommandons aux utilisateurs d’instituer des contrôles de qualité (confirmation de la présence d’ADN sur la lame, confirmation que les cellules sont suffisamment étiquetées avec de l’ADN (étape 8), confirmation que les cellules excédentaires ont été soigneusement lavées, etc.) pour s’assurer que l’expérience réussit et pour identifier les étapes qui peuvent nécessiter une optimisation supplémentaire. Nous espérons que les informations fournies dans ce manuscrit et ses fichiers supplémentaires aideront à faciliter tout dépannage requis.
Le cholestérol est une molécule bioactive dont l’internalisation peut influencer le métabolisme cellulaire, l’expression des gènes et la fluidité membranaire37,38. Une étude précédente a comparé les effets sur l’expression génique des cellules étiquetées CMO et LMO en utilisant le séquençage de l’ARN unicellulaire. Les cellules HEK étiquetées CMO avaient une expression génique altérée par rapport aux cellules non étiquetées et étiquetées LMO36. Le profilage des cellules avec des OCM a entraîné l’expression différentielle (> 1,5 fois) de huit gènes par rapport à des témoins non marqués, dont AP2B1, qui a été lié au transport du cholestérol et des sphingolipides (GeneCards), et MALAT1, un long ARN non codant qui régule l’accumulation de cholestérol39. Bien que mineures, ces réponses transcriptionnelles peuvent néanmoins être préoccupantes si l’expérience en question étudie le métabolisme, la dynamique membranaire ou d’autres voies associées au cholestérol dans les cellules.
Ce protocole est flexible et peut être ajusté pour répondre aux besoins de chaque expérience. Parce que le CMO s’insère dans la membrane lipidique au lieu d’utiliser un récepteur spécifique, la méthode est indépendante du type cellulaire (les HUVECs, MCF10As, HEKs et MTDC ont été démontrés ici). Bien que le cholestérol soit un ancrage hydrophobe différent de nos LMO précédemment publiés, nous avons jusqu’à présent constaté qu’ils se comportaient de manière similaire. Ainsi, nous nous attendons à ce que les OCM travaillent avec l’un des types de cellules que nous avons précédemment publiés avec les OVM, y compris, mais sans s’y limiter, les cellules souches neurales, les fibroblastes, les cellules mononucléaires du sang périphérique, les cellules tumorales et les cellules épithéliales mammaires primaires6,23,27,29,36 . Le label CMO ne stimule pas TLR9, ce qui suggère que le protocole est compatible avec les cellules immunitaires. L’incorporation membranaire du CMO est fonction de la taille totale de la cellule et du degré de charge négative dans le glycocalyxcellulaire 35. Ainsi, nous avons inclus un protocole (étape 8) pour tester l’étendue de l’incorporation de la membrane qui se prête à une optimisation rapide. Les caractéristiques spécifiques de chaque modèle de cellule varieront inévitablement en fonction de la conception expérimentale (voir le fichier supplémentaire 1 pour plus de conseils). Bien que le protocole de photomodèlement décrit ci-dessus pour modeler l’ADN soit recommandé, toute méthode de confinement spatial des gouttelettes de solution amine-ADN devrait fonctionner, comme l’utilisation d’imprimantes à gouttelettes à haute résolution. La résolution du motif et l’espacement minimal des fonctions varient en fonction de la méthode utilisée. Il est également théoriquement possible de combiner les sections de photomotrage d’ADN de ce protocole avec d’autres méthodes qui ont été utilisées pour étiqueter les cellules avec de l’ADN, comme avec de l’ADN hybridé à des doigts de zinc exprimés par membrane40, en utilisant de l’ADN conjugué NHS41, et en réagissant des résidus d’acide azido sialique à la surface cellulaire avec de l’ADN conjugué à la phosphine42 . CMO-DPAC peut être appliqué à une variété d’expériences qui nécessitent un contrôle étroit sur l’espacement cellule-cellule, y compris des études des interactions entre les paires de cellules, des expériences de co-culture portant sur le transfert de signaux des cellules « émettreuses » aux cellules « réceptrices » et des études sur l’effet des signaux extracellulaires voisins sur la différenciation des cellules souches6,29 . La méthode peut également être utilisée pour créer des microtissus qui peuvent être utilisés pour étudier la migration cellulaire en trois dimensions, l’auto-organisation des cellules dans les tissus23,27, et l’interaction dynamique entre les cellules et l’ECM27. Nous espérons que ce protocole fournira aux chercheurs une plate-forme accessible pour explorer de nouvelles applications de la modélisation cellulaire à haute résolution à base d’ADN dans leurs propres laboratoires.
Déclarations de divulgation
Z.J.G. est conseiller et actionnaire de Provenance Biosciences.
Remerciements
Les auteurs tiennent à remercier Jeremy Garcia d’avoir testé ce protocole et Bhushan Kharbikar d’avoir fourni une formation sur l’équipement de l’UCSF Biomedical Micro and Nanotechnology Core. Cette recherche a été soutenue en partie par des subventions du programme de recherche sur le cancer du sein du ministère de la Défense (W81XWH-10-1-1023 et W81XWH-13-1-0221), NIH (U01CA199315, DP2 HD080351-01, 1R01CA190843-01, 1R21EB019181-01A et 1R21CA182375-01A1), du NSF (MCB1330864) et du UCSF Center for Cellular Construction (DBI-1548297), un centre scientifique et technologique de la NSF. O.J.S a été financé par une bourse de recherche aux cycles supérieurs de la NSF, une bourse Siebel et une bourse P.E.O. Z.J.G et A.R.A. sont des enquêteurs Chan-Zuckerberg BioHub.
matériels
| Name | Company | Catalog Number | Comments |
| 2-well Chambered Coverglass w/ non-removable wells | Thermo Fisher Scientific | 155379 | |
| Acetic Acid | Sigma-Aldrich | A6283 | |
| Adapter with External SM1 Threads and Internal SM3 Thread | ThorLabs | SM3A1 | |
| Aldehyde Functionalized Slides | Schott | Nexterion Slide AL | Store under dry conditions after opening. |
| All Plastic Syringes, 1 mL | Fisher Scientific | 14-817-25 | |
| Amine-Modified DNA Oligo | IDT | n/a | See Supplemental File 1 for suggested sequences. |
| Aspheric Condenser Lens | ThorLabs | ACL7560 | |
| Borosilicate Disc, 6in Diameter X 1/2in Thick | Chemglass | CG-1906-23 | |
| Cell Culture Dishes 60x15 mm style | Corning | 353002 | |
| Cholesterol-Modified Oligo | IDT | n/a | See Supplemental File 1 for suggested sequences. |
| Diamond Scribe | Excelta | 475B | |
| DNA Oligonucleotide | IDT | n/a | See Supplemental File 1 for suggested sequences. |
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | 14190250 | |
| Isopropyl Alcohol | Sigma-Aldrich | 278475 | |
| Matrigel Matrix, Growth Factor Reduced | Corning | 354230 | |
| Methylene Chloride (Stabilized/Certified ACS) | Fisher Scientific | D37-4 | |
| MF-321 Developer | Kayaku Advanced Materials | n/a | |
| Microposit S1813 Positive Photoresist | Kayaku Advanced Materials | n/a | |
| Ø3" Adjustable Lens Tube, 0.81" Travel | ThorLabs | SM3V10 | |
| Oven | Thermo Scientific | 51-028-112H | |
| PE-50 Compact Benchtop Plasma Cleaning System | Plasma Etch | PE-50 | |
| Photomask (custom) | CAD/Art Services | n/a | Minimum feature size guaranteed by CAD/Art Services is 10 microns. |
| Razor Blades | Fisher Scientific | 12-640 | |
| RCT Basic Hot Plate | IKA | 3810001 | |
| Silicon Wafer (100 mm) | University Wafer | 590 | |
| Sodium Borohydride, 98%, granules | Acros Organics | 419471000 | |
| Spin Coater Kit | Instras | SCK-200 | This is a low cost option, but any spin coater that can maintain a speed of 3000 rpm will suffice. |
| SU-8 2075 | Microchem | Y111074 0500L1GL | |
| SU-8 Developer | Microchem | Y020100 4000L1PE | |
| Sylgard 184 Silicone Elastomer Kit | Dow | 2646340 | |
| Syringe Needles | Sigma-Aldrich | Z192341 | |
| T-Cube LED Driver, 1200 mA Max Drive Current | ThorLabs | LEDD1B | |
| Tridecafluoro-1,1,2,2-tetrahydrooctyl dimethylchlorosilane | Gelest | SIT8170.0 | |
| Triethylamine | Sigma-Aldrich | 90335 | |
| Turbo DNase | Thermo Fisher Scientific | AM2238 | |
| Tweezers Style N7 | VWR | 100488-324 | The curved shape of these tweezers is essential for delicately picking up the PDMS flow cells containing patterned tissues. |
| UV LED (365 nm, 190 mW (Min) Mounted LED, 700 mA) | ThorLabs | M365L2 | |
| Wafer Tweezers | Agar Scientific | T5063 | |
| WHEATON Dry-Seal vacuum desiccator | Millipore Sigma | W365885 |
Références
- Kreeger, P. K., Strong, L. E., Masters, K. S. Engineering approaches to study cellular decision-making. Annual Review of Biomedical Engineering. , 49-72 (2018).
- Goubko, C. a., Cao, X. Patterning multiple cell types in co-cultures: A review. Materials Science and Engineering C. 29 (6), 1855 (2009).
- Sun, W., et al. The bioprinting roadmap. Biofabrication. 12 (2), 022002 (2020).
- Liu, W. F., Chen, C. S. Cellular and multicellular form and function. Advanced Drug Delivery Reviews. 59 (13), 1319-1328 (2007).
- Duffy, R. M., Sun, Y., Feinberg, A. W. Understanding the role of ECM protein composition and geometric micropatterning for engineering human skeletal muscle. Annals of Biomedical Engineering. 44 (6), 2076-2089 (2016).
- Chen, S., et al. Interrogating cellular fate decisions with high-throughput arrays of multiplexed cellular communities. Nature Communications. 7, 10309 (2016).
- Shaya, O., et al. Cell-cell contact area affects notch signaling and notch-dependent patterning. Developmental Cell. 40 (5), 505-511 (2017).
- Rao, N., et al. A co-culture device with a tunable stiffness to understand combinatorial cell-cell and cell-matrix interactions. Integrative Biology. 5 (11), 1344 (2013).
- Sriraghavan, V., Desai, R. A., Kwon, Y., Mrksich, M., Chen, C. S. Micropatterned dynamically adhesive substrates for cell migration. Langmuir. 26 (22), 17733-17738 (2010).
- Wong, L., Pegan, J. D., Gabela-Zuniga, B., Khine, M., McCloskey, K. E. Leaf-inspired microcontact printing vascular patterns. Biofabrication. 9 (2), 021001 (2017).
- Chen, T. H., et al. Directing tissue morphogenesis via self-assembly of vascular mesenchymal cells. Biomaterials. 33 (35), 9019-9026 (2012).
- Laurent, J., et al. Convergence of microengineering and cellular self-organization towards functional tissue manufacturing. Nature Biomedical Engineering. 1 (12), 939-956 (2017).
- Lin, C., Khetani, S. R. Micropatterned co-cultures of human hepatocytes and stromal cells for the assessment of drug clearance and drug-drug interactions. Current Protocols in Toxicology. 2017, 1-23 (2017).
- Hui, E. E., Bhatia, S. N. Micromechanical control of cell-cell interactions. Proceedings of the National Academy of Sciences of the United States of America. 104 (14), 5722-5726 (2007).
- D'Arcangelo, E., McGuigan, A. P. Micropatterning strategies to engineer controlled cell and tissue architecture in vitro. BioTechniques. 58 (1), 13-23 (2015).
- Martinez-Rivas, A., González-Quijano, G. K., Proa-Coronado, S., Séverac, C., Dague, E. Methods of micropatterning and manipulation of cells for biomedical applications. Micromachines. 8 (12), (2017).
- Lee, S., et al. Simple lithography-free single cell micropatterning using laser-cut stencils. Journal of Visualized Experiments. (158), e60888 (2020).
- Strale, P. O., et al. Multiprotein printing by light-induced molecular adsorption. Advanced Materials. 28 (10), 2024-2029 (2016).
- Melero, C., et al. Light-induced molecular adsorption of proteins using the primo system for micro-patterning to study cell responses to extracellular matrix proteins. Journal of Visualized Experiments. (152), e60092 (2019).
- Reid, J. A., Mollica, P. M., Bruno, R. D., Sachs, P. C. Consistent and reproducible cultures of large-scale 3D mammary epithelial structures using an accessible bioprinting platform. Breast Cancer Research. , 1-13 (2018).
- Wang, Z., Lee, S. J., Cheng, H. -. J., Yoo, J. J., Atala, A. 3D bioprinted functional and contractile cardiac tissue constructs. Acta Biomaterialia. 70, 48-56 (2018).
- Miri, A. K., et al. Effective bioprinting resolution in tissue model fabrication. Lab on a Chip. 19 (11), 2019-2037 (2019).
- Todhunter, M. E., et al. Programmed synthesis of three-dimensional tissues. Nature Methods. 12 (10), 975-981 (2015).
- Todhunter, M. E., Weber, R. J., Farlow, J., Jee, N. Y., Gartner, Z. J. Fabrication of 3D microtissue arrays by DNA programmed assembly of cells. Current Protocols in Chemical Biology. 8 (3), 147-178 (2016).
- Csizmar, C. M., Petersburg, J. R., Wagner, C. R. Programming cell-cell interactions through non-genetic membrane engineering. Cell Chemical Biology. 25 (8), 931-940 (2018).
- Weber, R. J., Liang, S. I., Selden, N. S., Desai, T. A., Gartner, Z. J. Efficient targeting of fatty-acid modified oligonucleotides to live cell membranes through stepwise assembly. Biomacromolecules. 15 (12), 4621-4626 (2014).
- Hughes, A. J., et al. Engineered tissue folding by mechanical compaction of the mesenchyme. Developmental Cell. 44 (2), 165-178 (2018).
- Weber, R. J., et al. Rapid organoid reconstitution by chemical micromolding. ACS Biomaterials Science & Engineering. 2 (11), 1851-1855 (2016).
- Scheideler, O. J., et al. Recapitulating complex biological signaling environments using a multiplexed, DNA-patterning approach. Science Advances. 6 (12), (2020).
- Viola, J. M., et al. Guiding cell network assembly using shape-morphing hydrogels. Advanced materials (Deerfield Beach, Fla.). , 2002195 (2020).
- Mohammad, A., Davis, M., Aprelev, A., Ferrone, F. A. Note: Professional grade microfluidics fabricated simply. Review of Scientific Instruments. 87 (10), 1-4 (2016).
- Lee, O. J., Chuah, H. S., Umar, R., Chen, S. K., Yusra, A. F. I. Construction of cost effective homebuilt spin coater for coating amylose-amylopectin thin films. Journal of Fundamental and Applied Sciences. 9 (2), 279 (2018).
- Webb, K., Hlady, V., Tresco, P. A. Relative importance of surface wettability and charged functional groups on NIH 3T3 fibroblast attachment, spreading, and cytoskeletal organization. Journal of Biomedical Materials Research. 41 (3), 422-430 (1998).
- Processing Guidelines for: SU-8 2025, SU-8 2035, SU-8 2050, SU-8 2075. Microchem SU-8 2000 Permanent Expoxy Negative Photoresist Available from: https://kayakuam.com/wp-content/uploads/2019/09/SU-82000DataSheet2025thru2075Ver4.pdf (2019)
- Palte, M. J., Raines, R. T. Interaction of nucleic acids with the glycocalyx. Journal of the American Chemical Society. 134 (14), 6218-6223 (2012).
- McGinnis, C. S., et al. MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices. Nature Methods. 16 (7), 619-626 (2019).
- Maxfield, F. R., van Meer, G. Cholesterol, the central lipid of mammalian cells. Current Opinion in Cell Biology. 22 (4), 422-429 (2010).
- Luo, J., Yang, H., Song, B. L. Mechanisms and regulation of cholesterol homeostasis. Nature Reviews Molecular Cell Biology. 21 (4), 225-245 (2020).
- Liu, L., Tan, L., Yao, J., Yang, L. Long non-coding RNA MALAT1 regulates cholesterol accumulation in ox-LDL-induced macrophages via the microRNA-17-5p/ABCA1 axis. Molecular Medicine Reports. 21 (4), 1761-1770 (2020).
- Mali, P., Aach, J., Lee, J. H., Levner, D., Nip, L., Church, G. M. Barcoding cells using cell-surface programmable DNA-binding domains. Nature Methods. 10 (5), 403-406 (2013).
- Hsiao, S. C., et al. Direct cell surface modification with DNA for the capture of primary cells and the investigation of myotube formation on defined patterns. Langmuir. 25 (12), 6985-6991 (2009).
- Gartner, Z. J., Bertozzi, C. R. Programmed assembly of 3-dimensional microtissues with defined cellular conductivity. Proceedings of the National Academy of Sciences. (17), 1-5 (2009).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.