
Method Article
Isolation of Uterine Innate Lymphoid Cells for Analysis by Flow Cytometry
* These authors contributed equally
In This Article
Summary
This is a method to isolate uterine lymphoid cells from both pregnant and non-pregnant mice. This method can be used for multiple downstream applications such as FACS phenotyping, cell sorting, functional assays, RNA-seq, and proteomics. The protocol here demonstrates how to phenotype group 1 uterine innate lymphoid cells by flow cytometry.
Abstract
Described here is a simple method to isolate and phenotype mouse group 1 uterine innate lymphoid cells (g1 uILCs) from individual pregnant uterus by flow cytometry. The protocol describes how to set up time mating to obtain multiple synchronous dams, the mechanical and enzymatic digestion of the pregnant uterus, the staining of single-cell suspensions, and a FACS strategy to phenotype and discriminate g1 uILCs. Although this method inevitably loses the spatial information of cellular distribution within the tissue, the protocol has been successfully applied to determine uILC heterogeneity, their response to maternal and foetal factors affecting pregnancy, their gene expression profile, and their functions.
Introduction
Described here is a simple method to obtain a high yield of uterine innate lymphocytes from individual pregnant uterus. This method preserves protein surface expression and functionality of uterine innate lymphocytes and it is suitable for subsequent applications such as FACS phenotyping, RNAseq, proteomics or functional assays. Here, the focus is on the phenotyping of group 1 uILCs by flow cytometry.
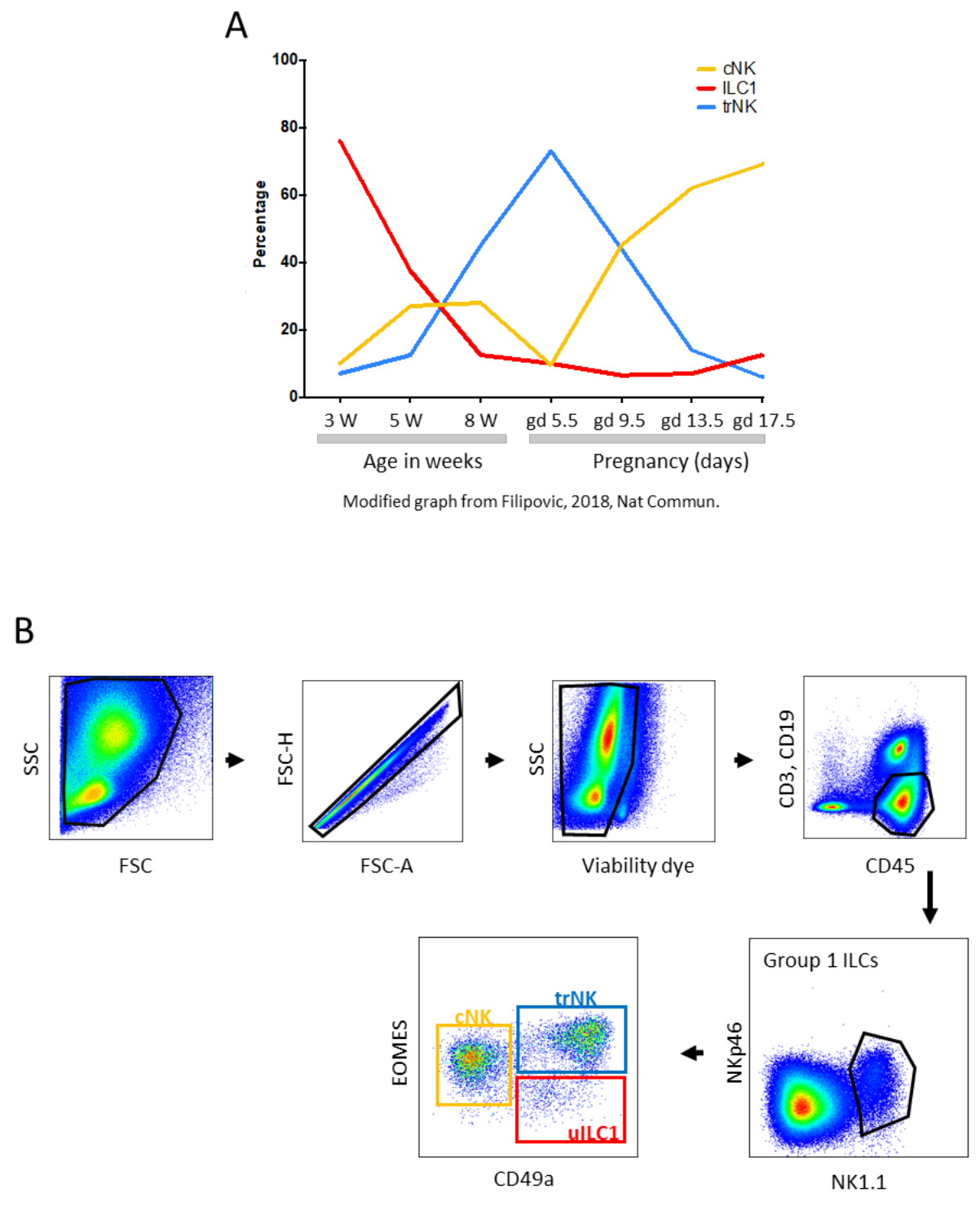
The uterus is composed of three layers: the endometrium, myometrium, and perimetrium (Figure 1). The endometrium is the mucosa, lining the lumen of the uterus. Progesterone, produced by the corpus luteum, converts the endometrium into decidua. The myometrium is composed of two layers of smooth muscle that make up the uterine wall. The perimetrium is the serosa that wraps the uterus and connects it to the peritoneum through the broad ligament called mesometrium. In a cross-section of the uterus, the part opposite to the lumen is called the mesometrial side, while the part close to the lumen is called the anti-mesometrial side. A variety of maternal leukocytes populate the endometrium and the decidua, including several types of cells, where the innate immune cells represent the vast majority of cells. Innate lymphoid cells (ILCs), macrophages, dendritic cells (DC), as well as CD4+ and CD8+ T lymphocytes, regulatory T cells (Tregs), and rare B cells, may all play important roles in the regulation of the uterine environment throughout pregnancy1,2. ILCs in the uterus are found not only in the mucosa, but also in the myometrium in mice. Including all three groups of ILCs, the uterus is indeed the organ most densely populated by group 1 ILCs. With the structural transformation of uterine tissues throughout gestation, the number and proportion of uterine leukocytes also change (see Figure 2A for an example of variations in the percentage of group 1 uILC subsets)3,4.
When mice are referred to in this paper, the C57BL/6 strain of inbred laboratory mice is meant. Outbred mice (e.g., NMRI mice) are often used in reproductive research because of their high reproductive rate. However, the use of inbred strains is necessary to generate consistent results, and the immunologist's favorite genetic background is C57BL/6, also known as B6.
Approximately 30% of uterine leukocytes in B6 dams at mid-gestation are g1 uILCs, which are defined by flow cytometry as viable CD45+CD3-CD19-NK1.1+NKp46+ cells (Figure 2B): pro-angiogenic tissue-resident NK (trNK), IFN-g producing conventional NK (cNK), and uILC14,5. The percentage of uNK cells is even higher in humans, reaching about 70% in the first trimester6. There are more similarities than differences between human and mouse uNK and uILC7,8. Although it is important to keep the differences in mind, it is useful to integrate information available on the two species. When one combines information obtained from investigating uILC in humans and laboratory rodents, it is clear that NK cells assist in homeostatic changes essential for the biology of the uterus, including maintenance of arterial integrity9 and spiral artery remodeling10, as well as trophoblast invasion11,12. They also play specific roles in the defense against pathogens13,14. In mice and rats, besides filling the decidua around the implantation site, NK cells accumulate between the two muscle layers of the myometrium of dams in a transient structure known as the Mesometrial Lymphoid Aggregate of pregnancy (MLAp)15 (Figure 1B), also known in the past as the metrial gland, the function of which is yet to be discovered.
Described here is a detailed protocol of the method used in the laboratory to isolate lymphocytes from the uterus of pregnant mice using a combination of mechanical disaggregation and enzymatic digestion. As the whole uterus is used in the method, lymphocytes isolated from the uterus during gestation are a mixture of decidual and myometrial cells. Further dissection of the decidua from the uterine wall and its MLAp is possible, and it has been described before16. The method described here was developed to obtain uterine lymphocytes while preserving protein surface expression, cellular functionality, and viability. The outcome is a single cell suspension with minimal residual cellular debris and a yield typically ranging from 1-5 million cells at mid-gestation (10.5 days) for a pregnant uterus. The applications of this method encompass phenotyping by flow cytometry, cell sorting for subsequent transcriptomic or proteomic studies, functional studies such as intracellular cytokine production, degranulation, ELISPOT or cytotoxic assays. The protocol presented here focuses on identifying group 1 ILCs but can be adapted for other cell types such as other ILCs, T cells, B cells, DC, or macrophages with minor modifications of the antibody panel used for FACS analysis. The protocol can also be used to isolate cells from other tissues and for pooled non-pregnant uteri.
Protocol
All animal experiments described in this paper were conducted according to the Animals (Scientific Procedures) Act 1986 under PP2363781 issued by the UK Home Office. The protocol below consists of several sections starting from mice husbandry and finishing with staining for FACS analysis. Figure 3 reflects the main steps of the protocol. The materials used in the protocol are listed in the Table of Materials.
1. General mice husbandry, mating, and dissection
- Keep 7-14 week old female mice under specific pathogen-free (SPF) conditions and group-housed (typically 4-6 females according to cage size and animal weight), for 10-14 days to trigger the Lee-Boot effect, which results in estrus synchronization17.
- Keep the stud males in SPF conditions, single-housed, and rested for at least 48 h between each mating (time for sperm regeneration). It is preferable to use experienced proven 3-4 month old stud males as they are typically more performant than the young ones.
- To increase the likelihood of females becoming pregnant, introduce soiled bedding from a male's cage in the female cage 3 days prior to mating. This triggers the Whitten effect18 by exposure to male urine pheromones, and results in synchronized estrus as well as enhanced receptivity for mating.
- On Day 0 (D0), set up the mice for mating using one stud male per two females; consider a plug rate of approximately 20%-25%.
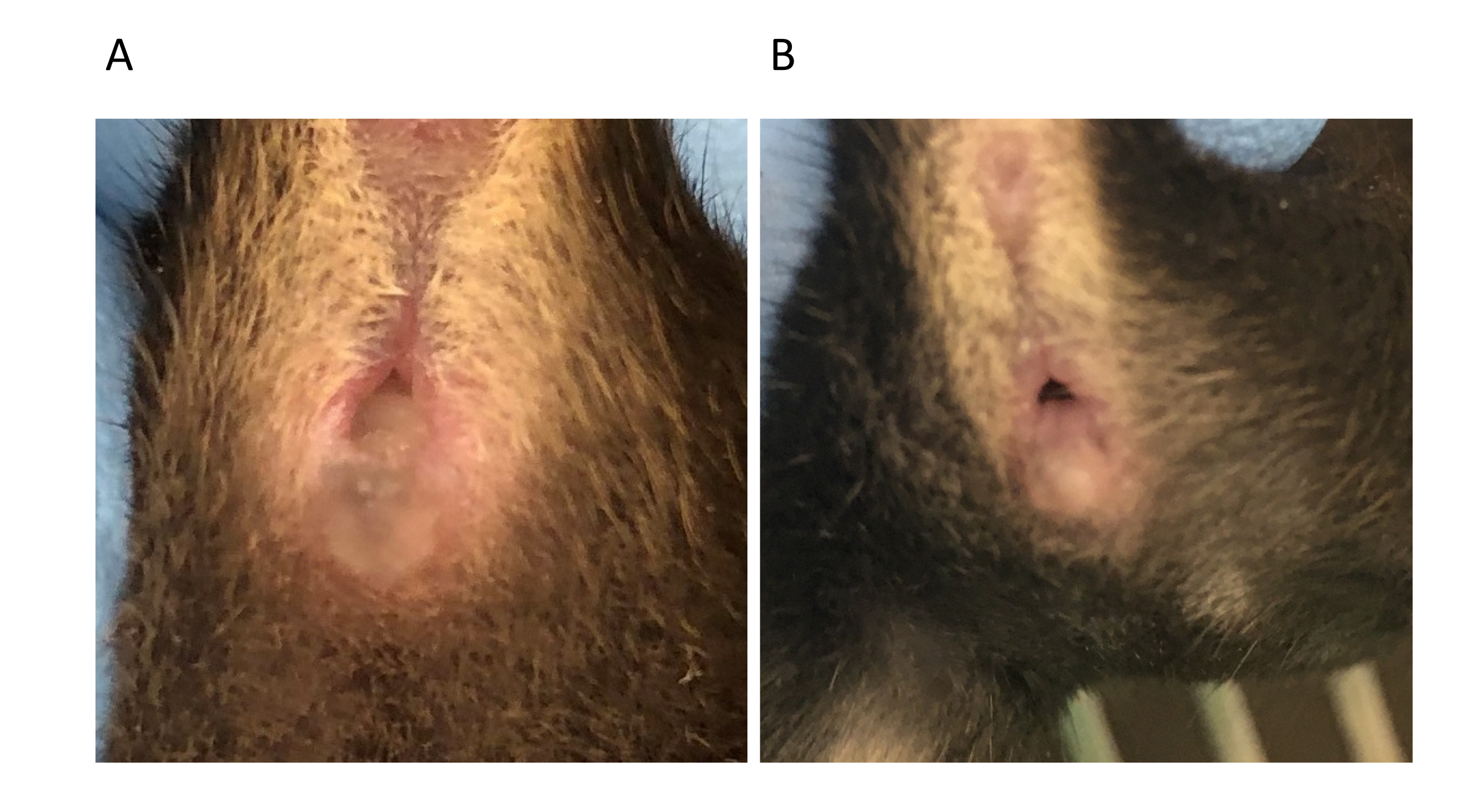
NOTE: Mating will likely occur at night since mice are nocturnal animals. - In the morning following mating (D0.5), check for the presence of a vaginal plug, which is an indicator of copulation (Figure 4). The vaginal plug is an aggregate of the male ejaculate and typically persists for up to 8-24 h after mating. Check the plugs early morning.
- Consolidate the plugged females in a new cage and earmark them. Return the males to their cages for resting.
- At D9.5 or 10.5-after mating, prepare 5 mL tubes with 1 mL sterile HBSS 1x (with Mg2+ and Ca2+) for tissue collection and place them on ice.
- Proceed to animal euthanasia by cervical dislocation, followed by exsanguination to confirm death.
- Work in a sterile environment if the downstream application requires to do so. Directly after euthanasia, wipe the mouse body with 70% ethanol and proceed to dissection under a laminar flux cabinet with sterile instruments.
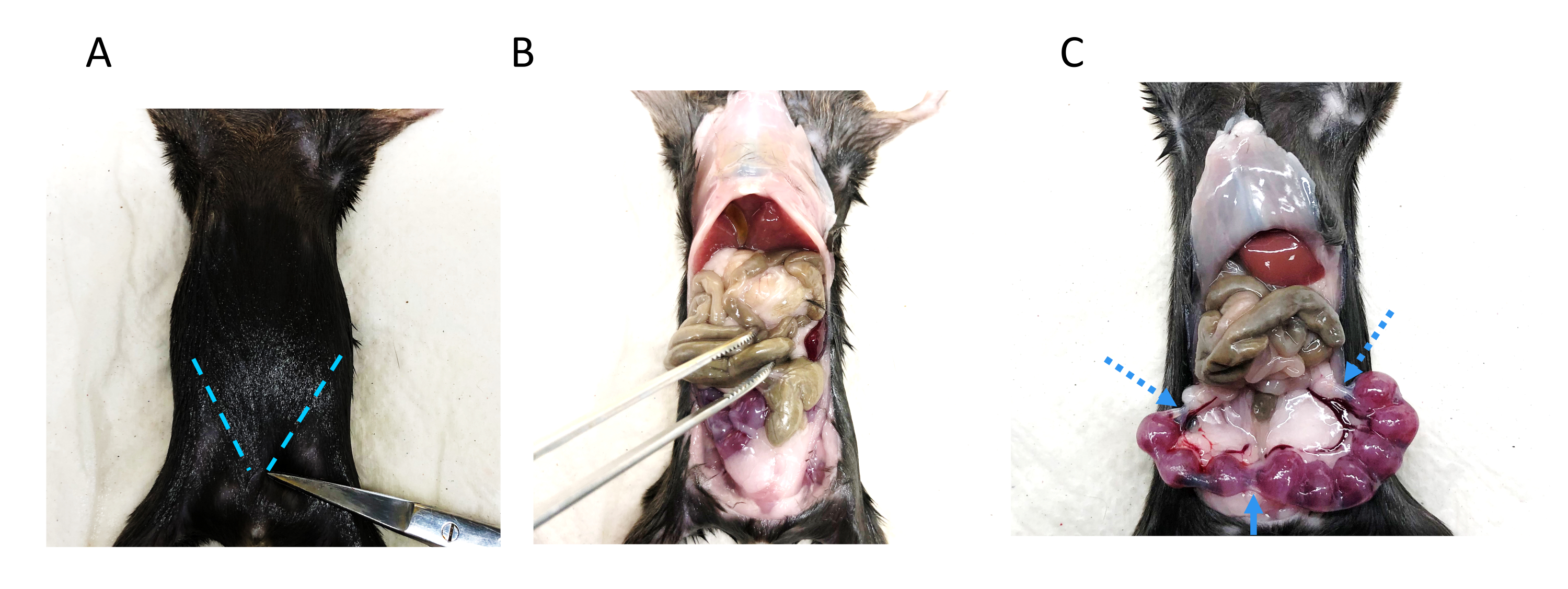
- Dissect the pregnant uterus free of mesometrial fat (Figure 5) and place the whole uterus in a prepared and appropriately labeled 5 mL tube. Keep the tubes on ice.
2. Mechanical and enzymatic digestion of the uterus
- To prepare the enzymatic digestion solution, mix 3 mL per uterus of sterile HBSS 1x with 30 µg/mL of DNAse and 0.1 Wünsch unit (WU)/mL of Liberase DH or 0.52 WU/mL of Liberase TM. Put the solution in a water bath at 37 °C.
NOTE: Both Liberase TM and Liberase DH can be used. The choice of one over the other must be guided by their potential effect on epitopes recognized by antibodies used for subsequent flow cytometry analysis.
CAUTION: If using lyophilized enzymes, work under the hood. - Prepare 20 mL of 5 mM EDTA in PBS (no Ca2+/Mg2+). Place half the solution at 37 °C in a water bath and the other half on ice.
- Under a laminar flux cabinet, gently remove the fat surrounding the pregnant uterus with sterile instruments in a sterile Petri dish. Do not allow the tissue to dry.
- Dissect each implantation site with sterile instruments to remove the fetuses (seahorse-shaped translucid structure, around 1 mm in length) (Figure 6A). Discard the fetuses.
- Return the uterus to their original collection 5 mL tube and mince the tissue using scissors directly in the 5 mL tube and collection medium. Keep the tubes on ice all the time between the procedures.
- Place the 5 mL tubes containing the minced tissue in a water bath at 37 °C.
- Add 3 mL of warm enzymatic digestion mix to each sample so that the total volume of liquid in the tube is 4 mL (1 mL of collection medium with the minced uterus and 3 mL of enzymatic digestion solution). Incubate the 5 mL tubes for 30 min at 37 °C with agitation to enhance enzymatic digestion activity.
- Vortex the 5 mL tubes and place them on ice to inhibit the action of enzymes. Then, subsequently, transfer the contents into properly labeled 15 mL centrifuge tubes.
- Flush everything out of the 5 mL tubes into the 15 mL centrifuge tubes using 10 mL of ice-cold 5 mM EDTA PBS solution.
- Centrifuge the 15 mL centrifuge tubes containing the digested tissues for 10 min at 400 x g.
- Discard the supernatant, gently flick the pellet, and then resuspend it in 10 mL of warm (37 °C) 5 mM EDTA PBS solution.
- Incubate the samples in the 15 mL centrifuge tubes at 37 °C with agitation for 15 min to remove the left-over digestion medium and reduce the cell clumping.
- Vortex the samples on high for 10 s to further facilitate tissue dissociation.
3. Processing of the uterus into a single cell suspension
- Using the plunger of a sterile 1 mL syringe, force the digested tissue through a 70 µm strainer onto a properly labeled and sterile 50 mL centrifuge tube to remove the cell clumps and the undissociated tissue.
- Wash the strainer several times with a total of 10 mL of cold PBS to collect all the cells.
- Spin the 50 mL centrifuge tube for 10 min at 400 x g.
NOTE: Perform the further steps using Option A or Option B. Option A allows better enrichment of the lymphocytes with less debris and stromal cell contamination than Option B. However, Option B gives a higher immune cell yield due to less cell loss and less variability of cell yield between samples. Option B is also easier to perform technically. Therefore, depending on preference, proceed with Option A or Option B.- The steps for Option A are as follows.
- Label one sterile 15 mL centrifuge tube per sample containing 5 mL of 80% (v/v) isotonic Percoll diluted in PBS.
- After the spin, discard the supernatant from the 50 mL centrifuge tube. Use a pipet boy to resuspend each pellet in 8 mL of 40% (v/v) isotonic Percoll in PBS.
- Use a pipet boy on slow speed to carefully overlay the pellet resuspended in the 40% Percoll solution onto 80% Percoll solution. Pipette slowly and continuously; hold the 15 mL tube at an angle of 45° (Figure 6B).
- Without disturbing the overlay, centrifuge the 15 mL centrifuge tubes for 20 min at 850 x g, at room temperature (medium acceleration and minimum break).
- Carefully remove the tubes from the centrifuge without disturbing the Percoll layers (Figure 6C).
- Without disturbing the ring of leukocytes at the interface of the two Percoll solutions, use a sterile Pasteur Pipette to discard all except for approximately 0.5-1 mL of the top Percoll layer.
- While trying to suck a minimum amount of Percoll solution (up to 4-5 mL total), carefully collect the ring of leukocytes and transfer the cells into a new labeled 15 mL centrifuge tube.
- Top up each sample with 10 mL of sterile RMPI-1640 medium supplemented with 10% of heat-inactivated FBS.
- Centrifuge for 5 min at 500 x g at 4 °C.
- Discard the supernatant and proceed to RBC lysis.
- The steps for Option B are as follows.
- Label one sterile 15 mL centrifuge tube per sample.
- After the spin, discard the supernatant from the 50 mL centrifuge tube. Use a pipet boy to resuspend each pellet with 8 mL of 35% (v/v) isotonic Percoll in RPMI-1640 medium.
- Transfer the samples into 15 mL centrifuge tubes.
- Centrifuge the samples at 940 x g for 10 min at room temperature with medium acceleration and minimum break.
- Aspirate the supernatant carefully using an aspirator or pipette boy (not by inverting the tube).
- Resuspend the pellet in 14 mL of RPMI-1640 medium supplemented with 10% heat-inactivated FBS and then centrifuge the sample at 500 x g for 5 min at 4 °C.
- Discard the supernatant by aspiration and proceed to RBC lysis.
- The steps for Option A are as follows.
4.RBC lysis
- To lyse the RBCs, resuspend the samples in 3 mL of 1x RBC lysing solution and incubate for 3 min at room temperature.
- Add 10 mL of PBS into the samples to stop the reaction.
- Centrifuge the tubes at 400 x g for 5 min and discard the supernatant.
- Add 10 mL of PBS and repeat step 4.3.
- Resuspend each pellet in 1 mL of RPMI-1640 medium supplemented with 10% of heat-inactivated FBS.
- Pass the samples through sterile 70 µm cell strainers.
- Perform the cell count using trypan blue and a Neubauer Chamber as per the manufacturer's instructions.
- Adjust the concentration of the cell suspension to 1-2 million cells in 100 µL of PBS or medium.
5. Panel design strategy and controls
NOTE: The panel described in this paper is suitable for the discrimination of uILC1, trNK, and cNK cells and was designed to be used on a 5-laser BD LSRFortessa. Minor modifications can be made to study different cell populations and use alternative fluorochromes. It is recommended to check the configuration of the instrument, using titrated antibodies for optimal separation, consulting the manufacturer's brightness index and using the brightest dyes for low expressing antigens such as NKp46, and following general guidelines19. It is recommended to include a Fluorescence Minus One (FMO) control for NKp46.
6. Innate lymphoid cell staining for FACS phenotyping
- Transfer 1-2 million cells per well into a round-bottom 96-well plate.
- Spin the plate at 400 x g for 3 min at 4 °C and discard the supernatant by flicking it into a sink.
- Resuspend the cell pellets into 100 µL of PBS (protein and azide-free) using a multi-channel pipette.
NOTE: Ensure that the PBS contains no sodium azide, no Tris, or any proteins for the subsequent step. - Repeat step 6.2.
- Resuspend cells in 50 µL of fixable viability dye diluted in PBS (protein and azide-free) (1:1,000). Incubate the cells at room temperature for 30 min in the dark.
NOTE: Ensure that the PBS contains no sodium azide, no Tris, or any proteins such as FBS or BSA as this may result in decreased staining intensity of dead cells and/or increased background staining for the live cells.
CAUTION: If viability dye is powdered, use under the hood. - Add 150 µL of PBS, resuspend the cells with a multi-channel pipette, and then repeat step 6.2.
- Resuspend the cells in 25 µL of FACS Buffer (PBS supplemented with 1% BSA or 2% FBS) containing Fc receptor blocking reagent. Incubate the cells for 5 min at 4 °C.
- Add 25 µL of a surface antibody cocktail.
NOTE: Always titrate antibodies and optimize the antibody panel prior to the experiment. - Incubate the samples at room temperature for 20 min in the dark.
- Add 150 µL of FACS Buffer to each well, mix thoroughly, and then repeat step 6.2.
- Repeat step 6.10.
NOTE: Perform further steps using Option A or Option B. Use option A to stain the cells with surface markers. Use option B to study the intracellular markers by flow cytometry.- The steps for Option A are as follows.
- Resuspend the samples in 100 µL of 4% paraformaldehyde (PFA) per well and incubate for 20 min at room temperature.
CAUTION: Use PFA under the hood. Please refer to the safety sheet for discarding PFA waste/objects that have come into contact with PFA (e.g., pipettes) safely. - Repeat step 6.2, twice.
CAUTION: Do not discard by flicking into the sink here as it contains PFA. Aspirate with a pipette and discard the waste as per the safety sheet. - Resuspend the samples in 200 µL of PBS.
- Transfer the samples into labeled FACS tubes and top up with 100 µL of PBS. Keep the tubes on ice or in a fridge until processing with FACS analysis. Acquire the samples on a flow cytometer within 24 h.
- Resuspend the samples in 100 µL of 4% paraformaldehyde (PFA) per well and incubate for 20 min at room temperature.
- The steps for Option B are as follows.
- Resuspend the samples in 100 µL of fixation / permeabilization solution per well (containing paraformaldehyde) and incubate for 20 min at 4 °C.
CAUTION: Use PFA under the hood. Please refer to the safety sheet for discarding PFA waste/objects that have come into contact with PFA (e.g., pipettes) safely. - Repeat step 6.2.
CAUTION: Do not discard by flicking into the sink here as it contains PFA. Aspirate with a pipette and discard the waste as per the safety sheet. - Add 200 µL of 1x permeabilization / washing buffer, mix well, and then repeat step 6.2.
- Repeat step 6.11.2.3.
- Resuspend the fixed and permeabilized cells in 50 µL of 1x permeabilization/washing buffer containing antibodies mix for intracellular staining.
- Incubate the samples at 4 °C for 30 min in the dark.
- Add 200 µL of 1x permeabilization / washing solution, mix well, and then repeat step 6.2.
- Repeat step 6.11.2.7.
- Resuspend the samples in 200 µL of PBS.
- Transfer the samples into labeled FACS tubes and top up with 100 µL of PBS. Keep the tubes on ice or in a fridge until processing with FACS analysis. Acquire the samples on a flow cytometer within 24 h.
NOTE: After performing this protocol, the decidual cell suspension is ready for FACS analysis. It is recommended to record as many events as possible per sample; at least 1,000-3,000 events of a parental population have to be obtained to achieve reliable results.
- Resuspend the samples in 100 µL of fixation / permeabilization solution per well (containing paraformaldehyde) and incubate for 20 min at 4 °C.
- The steps for Option A are as follows.
Results
The main steps of the method that are described to obtain a single cell suspension of uterine leukocytes are summarized in Figure 3. Demonstrated in Figure 2B are the basic FACS gating strategy used for the identification of three subsets of g1 ILCs in B6 mice: uILC1 (CD49a+Eomes-), trNK (CD49a+Eomes+), and cNK (CD49a-Eomes+) cells. Further analysis of these populations can be performed to study various surface and intracellular markers of g1 ILCs. As an example, the co-expression of IFN-ɣ and self-MHC receptors can be assessed in uILC1, trNK, and cNK cells after stimulation with anti-NK1.1 antibody (Figure 7).
Depending on the research question, both the protocol (Figure 3) and the antibody panel can be adapted. Importantly, it is recommended to use both anti-NK1.1 and anti-NKp46 antibodies in one FACS panel for g1 ILC gating (Figure 2B and Table 1). It should be noted that g1 ILCs obtained from blood, spleen, or liver have a higher expression of NKp46 on their surface than the uterine counterpart (Figure 8). Surface staining for NK1.1 gives a better separation and enables uterine g1 ILCs to be gated easily (Figure 8). While NKp46 is expressed by all mouse strains, the NKR-P1C antigen recognized by the anti-NK1.1 antibody PK136 is only expressed by some mouse strains, including C57BL/6 (i.e., B6), FVB/N, and NZB, but not in AKR, BALB/c, CBA/J, C3H, DBA/1, DBA/2, NOD, SJL, or 129. In addition, if the investigator intends to study crucial NK cell receptors such as the MHC receptors Ly49, it is important to be aware of allelic variations in laboratory mouse strains, which recapitulate the high variability of human killer-cell immunoglobulin-like receptors (KIR). Moreover, if the cells are to be stimulated with NK1.1 for a functional assay, as described in Kim, S. et al.20, it might be desirable to stain the cells with anti-NKp46 rather than anti-NK1.1, as the NKR-P1C antigen may be occupied by the crosslinking anti-NK1.1 or a receptor downregulation may follow the stimulation. Either receptor occupancy or downregulation can impede staining with the same antibody used to stimulate.
A common problem with enzymatic tissue dissociation is the alteration of surface epitopes on cells by enzymes used for a digestion medium. For example, staining for the MHC CD94:NKG2A receptor is poor if Liberase TM is used. However, digestion with Liberase DH preserves NKG2A recognition by 16A11 antibody clone (Figure 9). It is recommended to check the influence of enzymes on all epitopes in one's FACS panel. For this purpose, use the suspension of mouse splenocytes obtained by mechanical dissociation (passing the whole spleen through a 70 µm strainer). The sample is then divided into two or more parts followed by incubation with a medium with or without enzyme(s).
As mentioned before, blood-derived cells are present in tissue dissociated samples. If required, blood contaminants can be excluded using an intravascular staining method as developed in Masopust laboratory21. Figure 10 demonstrates that around 6.5% of g1 ILCs present in uterine tissue samples at gestation day 8.5 are blood-derived. Anti-CD45 antibodies used for intravascular staining can be conjugated with a fluorochrome used for a dump-channel; this will exclude blood contaminants without using an extra fluorescence channel. The most common problems and their solutions are presented in Table 2.

Figure 1: Cross-section of a mouse uterus. (A) Mouse uterus cross-section (non-pregnant) indicating a variety of maternal leukocytes which populate the uterus. (B) Mouse uterus cross-section (gestation day 8.5). (C) Mouse uterus cross-section (gestation day 13.5). (D) Comparison of mouse versus human placenta formation from blastocyst stage onward. Images created with BioRender.com. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Subpopulations of uterine g1 ILC1. (A) Percentages of uterine cNK, ILC1, and trNK in mice during early life and pregnancy. W - weeks, gd - gestation day. Modified graph from Filipovic, I. et al.4. (B) Gating strategy to analyze uterine group 1 ILC subsets by flow cytometry. Lymphocytes were isolated from the uterine tissues at gestation day 10.5. Tissue digestion was performed using a digestion medium containing Liberase TM. Cells were gated based on their ability to scatter light. Doublets were excluded using a FSC-A versus FSC-H plot, and only CD45+CD3-CD19- viable cells were analyzed further. Within CD45+CD3-CD19- viable cells, the group 1 ILC gate was identified as NK1.1+ NKp46+ cells. Within group 1 ILCs, three subsets can be identified: CD49a-Eomes+ conventional NK cells (cNK), CD49a+Eomes+ tissue-resident NK cells (trNK), and CD49a+Eomes- uILC1. Please click here to view a larger version of this figure.
{kind=link}

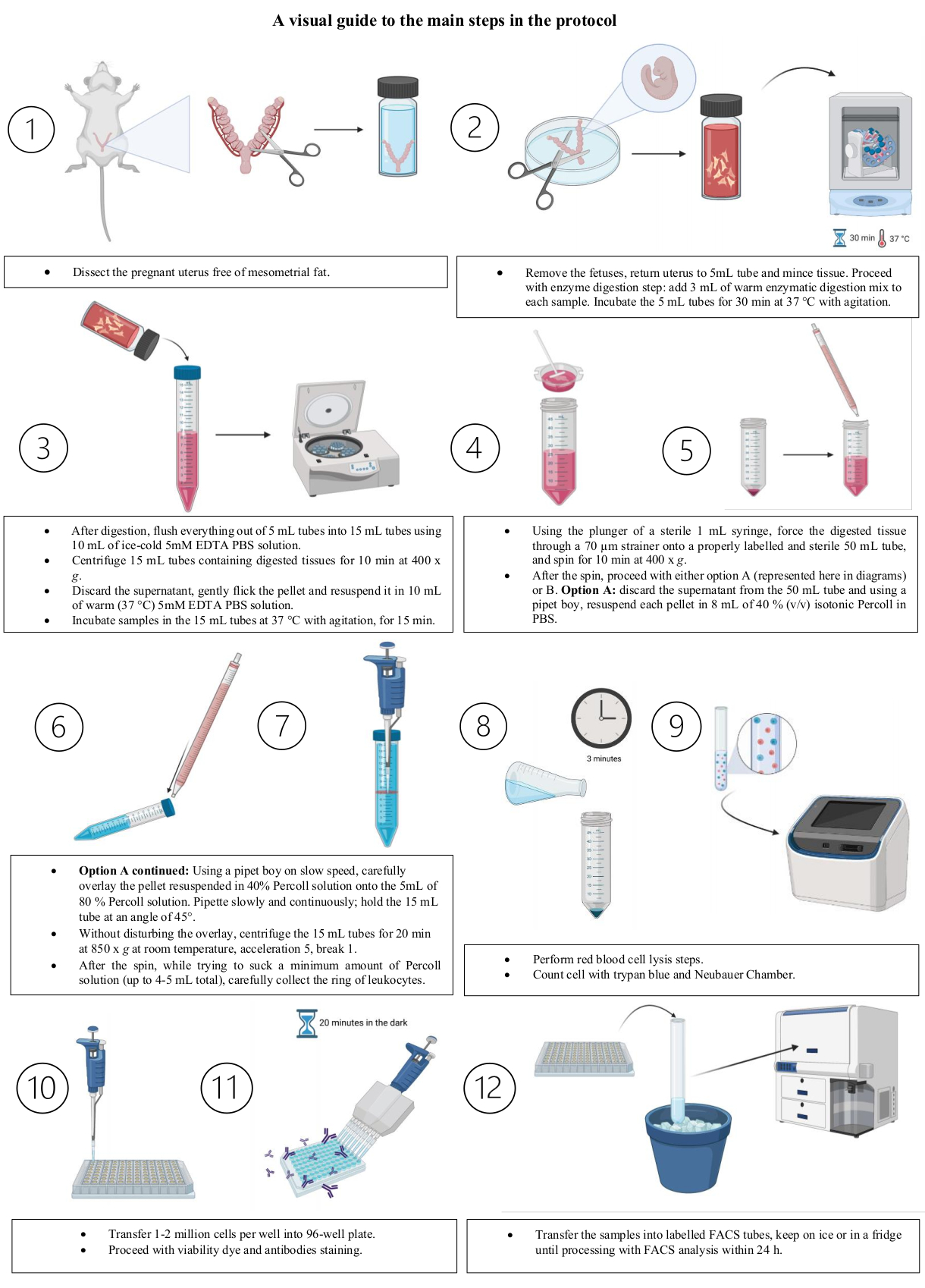
Figure 3: A visual guide for the main steps of the protocol. (1) Dissect the pregnant uterus free of mesometrial fat. (2) Remove the fetuses; return the uterus to the 5 mL tube and mince the tissue. Proceed with the enzyme digestion step: add 3 mL of warm enzymatic digestion mix to each sample. Incubate the 5 mL tubes for 30 min at 37 °C with agitation. (3) (i) After digestion, flush everything out of 5 mL tubes into 15 mL tubes using 10 mL of ice-cold 5 mM EDTA PBS solution. (ii) Centrifuge 15 mL tubes containing digested tissues for 10 min at 400 x g. (iii) Discard the supernatant; gently flick the pellet and resuspend it in 10 mL of warm (37 °C) 5 mM EDTA PBS solution. (iv) Incubate samples in the 15 mL tubes at 37 °C with agitation, for 15 min. (4) Using the plunger of a sterile 1 mL syringe, force the digested tissue through a 70 µm strainer onto a properly labeled and sterile 50 mL tube, and spin for 10 min at 400 x g. (5) After the spin, proceed with either option A (represented here in diagrams) or B. Option A: discard the supernatant from the 50 mL tube and, using a pipet boy, resuspend each pellet in 8 mL of 40% (v/v) isotonic Percoll in PBS. (6) (i) Option A continued: Using a pipet boy on slow speed, carefully overlay the pellet resuspended in 40% Percoll solution onto the 5 mL of 80% Percoll solution. Pipette slowly and continuously; hold the 15 mL tube at an angle of 45°. (ii) Without disturbing the overlay, centrifuge the 15 mL tubes at 850 x g for 20 min at room temperature, with medium acceleration and slow break. (7) After the spin, while trying to suck a minimum amount of Percoll solution (up to 4-5 mL total), carefully collect the ring of leukocytes. (8) Perform red blood cell lysis steps. (9) Count cell using trypan blue and a Neubauer Chamber. (10) Transfer 1-2 million cells per well into a round-bottom 96-well plate. (11) Proceed with viability dye and antibodies staining. (12) Finally, transfer the samples into labeled FACS tubes. Keep the tubes on ice or in a fridge until processing with FACS analysis within 24 h. Images created with BioRender.com. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Vaginal plug (A) and absence of it (B) in C57BL/6 females at 0.5 day post mating. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Dissection to extract the uterus from a pregnant mouse. (A) The dam is pinned down with needles on a soft board to wipe the body with 70% ethanol. Two vertical incisions are made, as indicated by the blue dotted lines. (B) The skin is lifted to expose internal organs. The intestinal loops are gently moved up to visualize the uterus. (C) The uterus is sampled by cutting at three points: next to the ovaries and at the cervix, as indicated by the two blue dotted lines and the blue arrow, respectively. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Preparation of single-cell suspension. (A) Mechanical removal of embryos from their implantation site. (B) Percoll gradient overlay; the top layer contains the single-cell suspension in 40% of Percoll and the bottom layer 80% of Percoll. (C) Lymphocyte ring formation after centrifugation of the percoll gradient. Please click here to view a larger version of this figure.
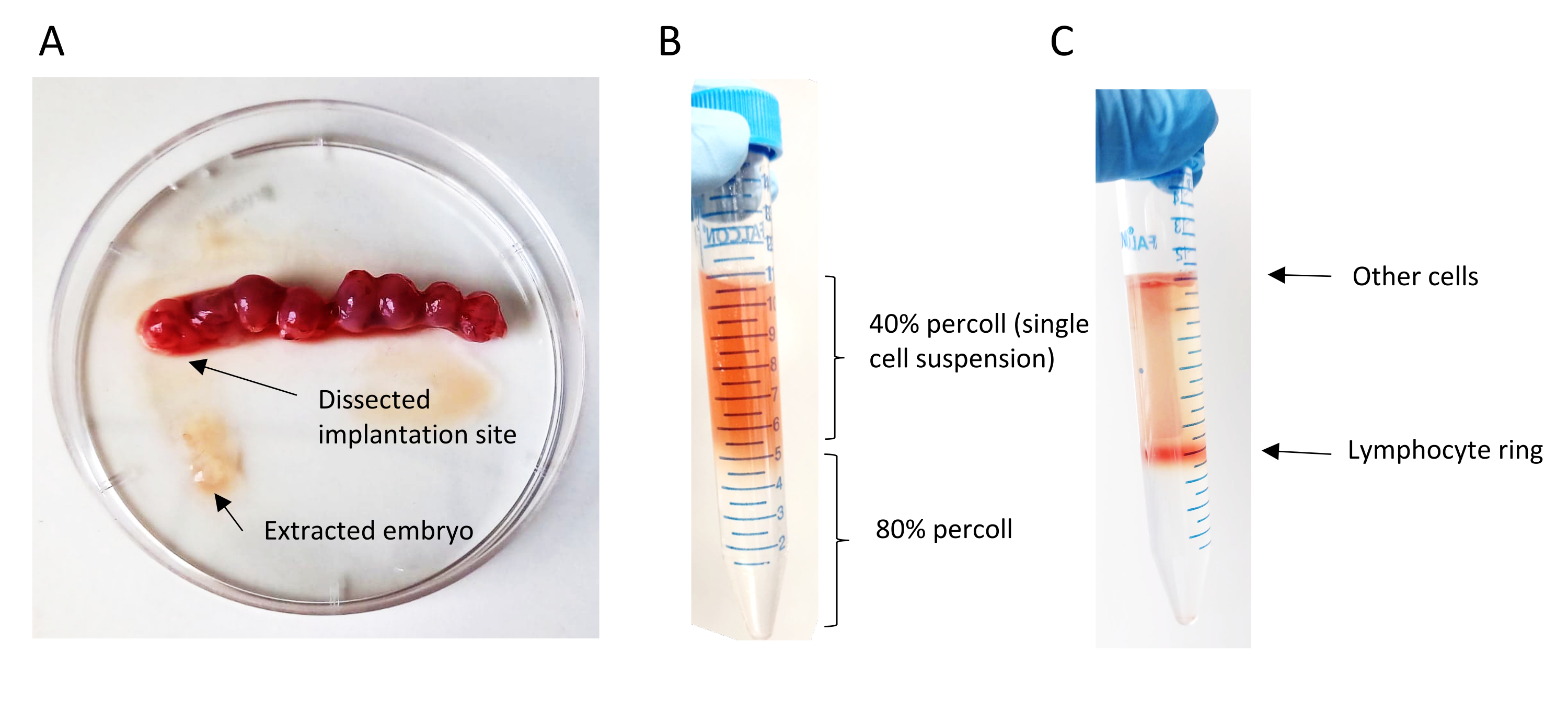{kind=link}

Figure 7: Representative FACS analysis of functional assay with group 1 ILCs. Intracellular IFN-ɣ and surface CD107a detection in group 1 ILCs expressing NK receptors for self-MHC (Ly49C, Ly49I, and NKG2A) compared to those that do not, after crosslinking NK1.1 with plate-bound antibodies. The cells were isolated from uterine tissues at gestation day 9.5. Tissue digestion was performed using a digestion medium containing Liberase DH. Shown are the raw values of all four quadrants (corners) as well as the relative percentage of responders among cells expressing receptors for self and responders that do not have self-receptors (bold numbers). Please click here to view a larger version of this figure.
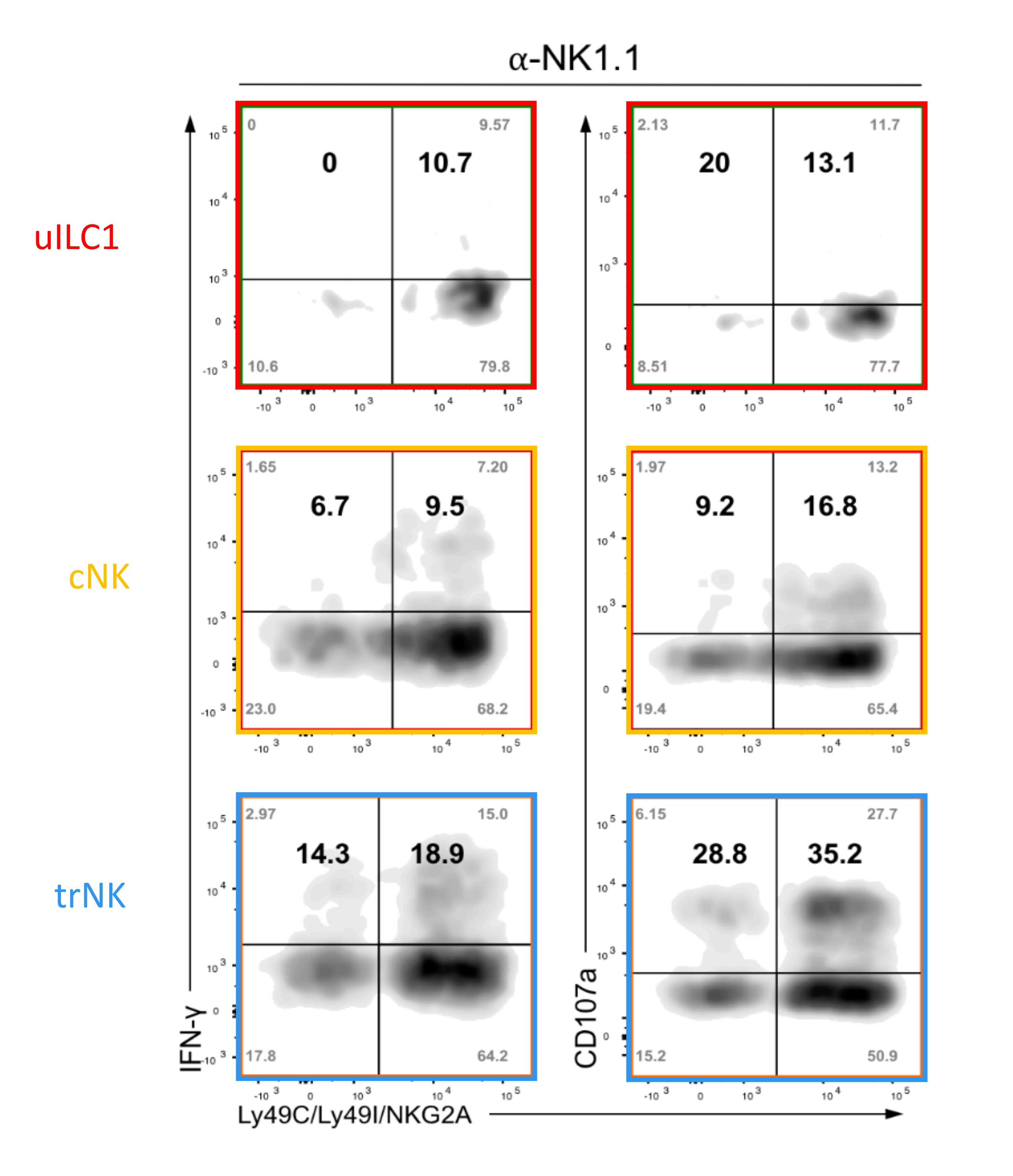{kind=link}

Figure 8: Staining splenic and uterine lymphocytes with anti-NKp46 and anti-NK1.1 antibodies. (A) Cell suspensions obtained from mouse spleen and (B) uterus at gestation day 10.5 were separated in two; one part was stained with NKp46-APC (red) and the other one with NK1.1-APC (blue). Note that the NKp46 staining of uterine lymphocytes does not separate NKp46+ and NKp46- cells as neatly as splenic lymphocytes. Please click here to view a larger version of this figure.
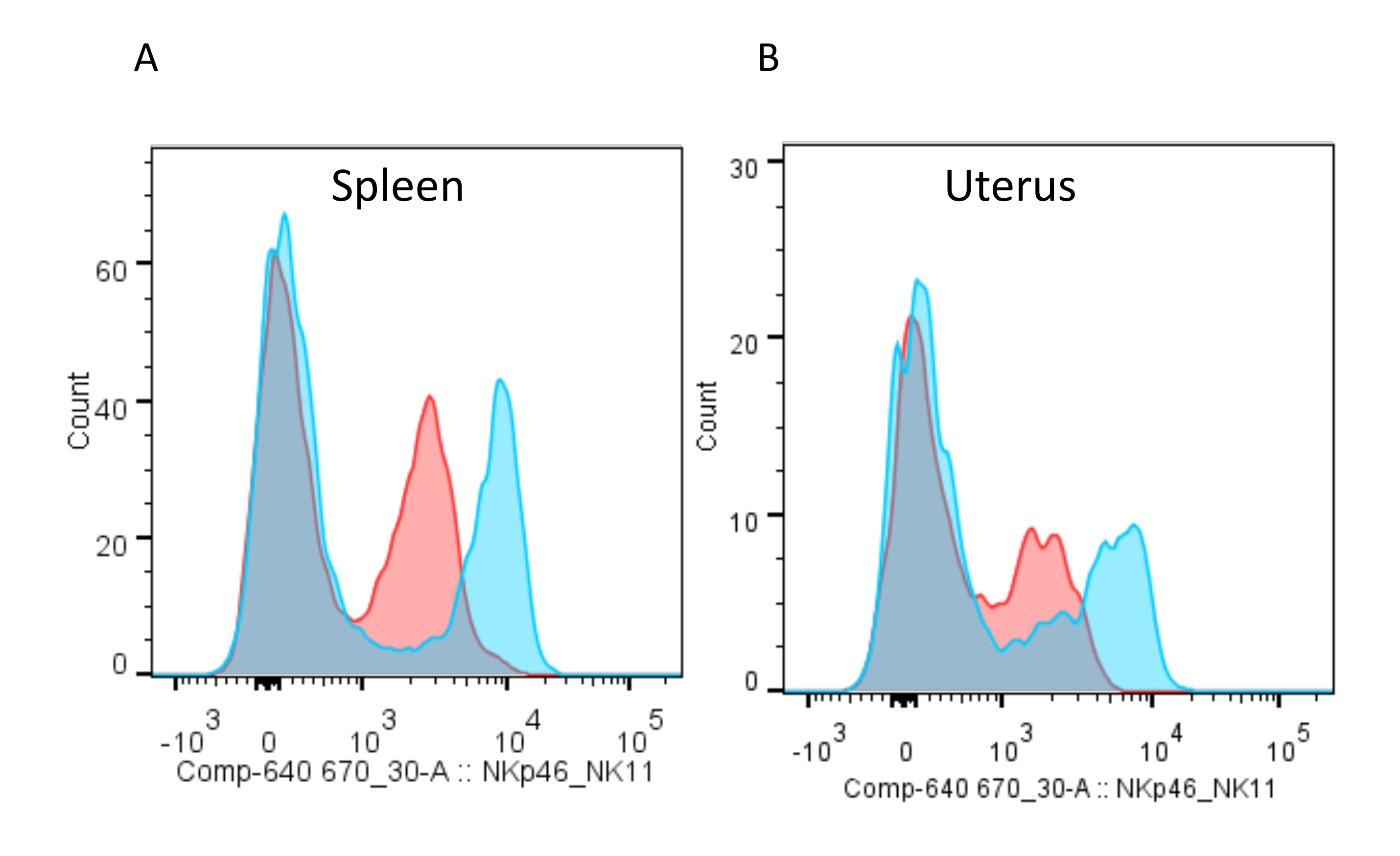{kind=link}

Figure 9: Decrease of NKG2A MFI (antibody clone: 16A11) by incubation with digestion medium. Cell suspension of C56BL/6 mouse splenocytes was divided into three parts. One part was incubated in Liberase DH digestion medium (HBSS containing 0.13 WU/mL Liberase DH and 30 µg/mL of DNAse), and another part was incubated with Liberase TM digestion medium (HBSS containing 0.52 WU/mL Liberase TM and 30 µg/mL of DNAse). The third part was treated with neat HBSS for 30 min at 37 °C. Expression of NKG2A marker on g1 ILCs was assessed by flow cytometry. Graph taken from Shreeve N. The role of uterine NK-cell inhibition in pregnancy (Thesis); Supervisor: Colucci F, 2020. Please click here to view a larger version of this figure.
{kind=link}

Figure 10: Intravital staining with anti-CD45 antibodies for exclusion of blood-derived g1 ILCs. A C57BL/6 dam mouse at gestation day 8.5 was culled 3 min after intravenous injection with 3 µg of CD45-AF647. The uterus, whole blood, and thymus were harvested and processed for FACS analysis. The X-axis shows the signal from intravenous staining with CD45-AF647, and Y-axis demonstrates signal from in vitro stained CD45-BUV395. The percentages of subpopulations are shown in quadrants. Please click here to view a larger version of this figure.
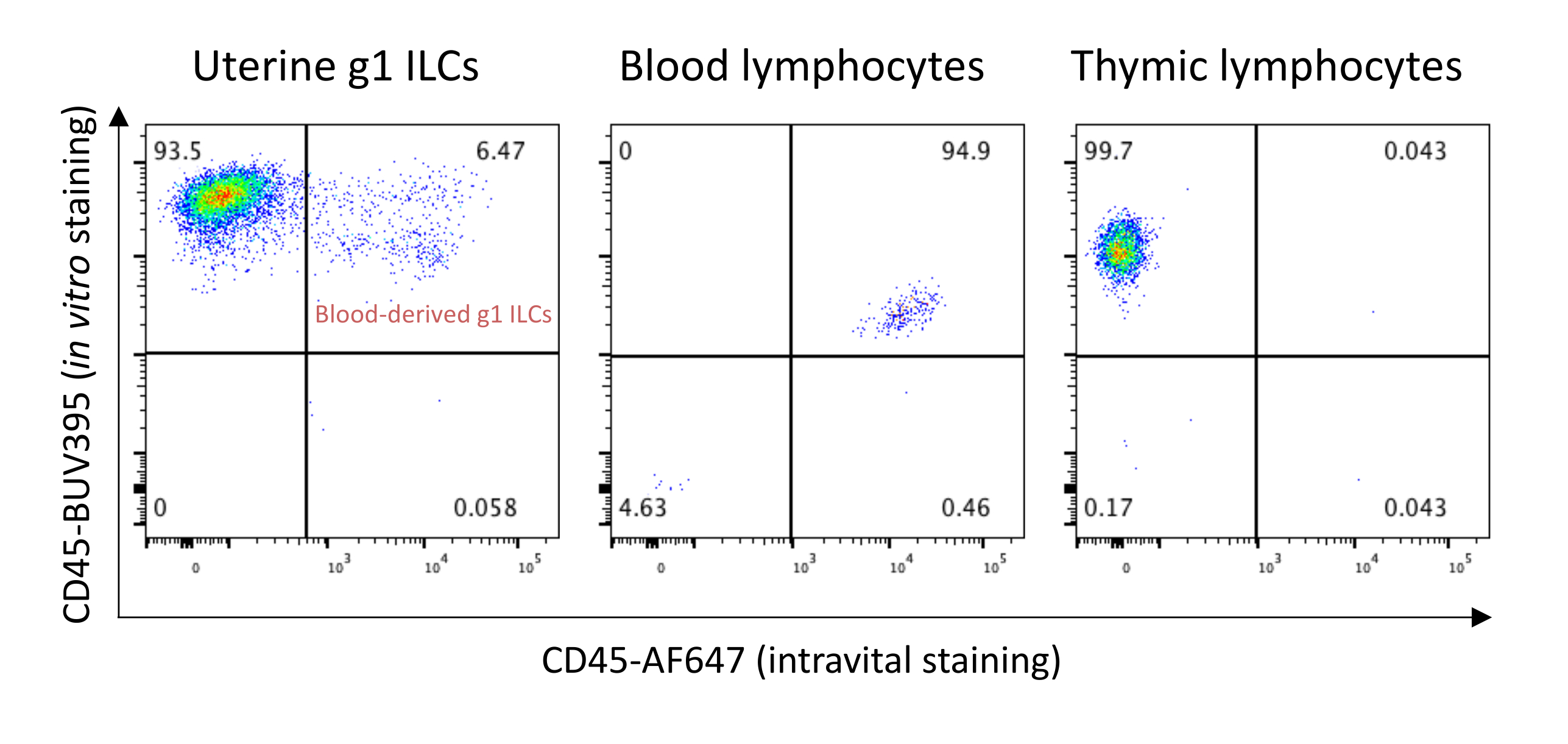{kind=link}
| Antibody / Dye | Clone | Fluorochrome | Laser | |
| 1 (Dump channel) | Zombie Violet Fixable Viability dye | Violet | ||
| CD19 | 1D3 | BV421 | ||
| CD3 | 145-2C11 | BV421 | ||
| 2 | CD45 | 30-F11 | FITC | Blue |
| 3 | NK1.1 | PK136 | BV605 | Violet |
| 4 | NKp46 | 29A1.4 | APC | Red |
| 5 | CD49a | Ha31/8 | BUV395 | Ultraviolet |
| 6 | EOMES | Dan11mag | PE | Green |
Table 1: An example of FACS panel for conventional 5-lasers cytometer.
| Problem | Possible cause | Suggestion |
| The cell ring is not visible at the interface of two percoll solutions | Poor layering of percoll solution or mixing two layers during sample handling | Take extra care to not break the 80% percoll cushion during overlaying. Pay attention during sample handling: do not disturb the percoll interface |
| Low numbers of leukocytes (for instance, when using a non-pregnant uterus) | The interface can be seen even when the cell number at the interface is very low. Even if the ring is not visible, collect liquid at between 40% and 80% percoll solutions, as there might still be enough cells for further processing | |
| Incomplete RBC lysis | Cells were not resuspended properly in the lysing buffer | Pipette cells up and down to break the clumps and fully resuspend cells in lysing buffer |
| Lysing solution is cold | Equilibrate the lysing solution to room temperature before use | |
| Prolong time of incubation with the lysing solution up to 15min | ||
| RBC lysis step can be repeated | ||
| Low cell yield | Poor enzymatic digestion | Check if enzymes are not out of date and have been stored according to their manuals |
| Cell loss during washing steps | Inspect the cell pellet after each washing step: the opaque pellet at the bottom of the well after spin. Using V-bottom instead U-bottom plates, swing rotor centrifuge, longer centrifugation time may reduce cell loss | |
| Tissue sample contains low numbers of lymphocytes (for instance, when using a non-pregnant uterus) | Pool several uteruses to obtain enough events for analysis. Consider using Option B of the protocol for cell isolation | |
| High variability of absolute leukocyte numbers obtained from mice of the same group | Inconsistent collection of cells at the interface of 40% and 80% percoll solutions | Make sure to collect whole cell fraction on the percoll interface. Consider using Option B of the protocol for cell isolation |
| Not able to detect expected lymphocyte subpopulations/markers or unusually low MFI for some cell surface markers | Enzymatic digestion affectes surface expression of some epitopes or their degradation | Optimize enzymatic digestion by changing: enzyme (e.g. to different type of liberase or collagenase) and/or length of incubation and/or enzyme concentration |
| High background noise in the flow cytometer | High proportion of cell debris or RBC contamination | Adjust FSC thresholding parameter. Consider using Option A of the protocol for cell isolation |
Table 2: Troubleshooting guide.
Discussion
The method contains several critical steps discussed hereafter. The first critical step is to obtain multiple synchronous pregnancies as the relative frequency of leukocyte populations changes through pregnancy. Having multiple dams at the same gestational day allows for either biological repeats in the same experiments or pooling lymphocytes from individual dams to obtain larger numbers required for downstream applications. Timed mating allows the researcher to pinpoint conception within a 24 h period. Although mice live for around 2.5 years, they will be of reproductive age from 4-7 weeks until 6-8 months old. As younger mice usually produce smaller pups, female mice are generally not mated until they are between 6-8 weeks, and male mice until they are between 8-10 weeks. Given that estrus lasts about 15 h in mice and occurs every 4-5 days, the typical mating rate (revealed by a vaginal plug, see Figure 4) is around 25%. It is therefore important to use mice in estrus and plan sufficient numbers to obtain the required number of dams for a given experiment. The estrus cycle phase can be determined by vaginal smear cytology22. The plug rate can be improved by resting males 48 h prior to mating and by taking advantage of the Whitten effect18. Alternatively, one can administer pregnant mare serum, which mimics the effect of the endogenous follicle-stimulating hormone, inducing oocyte maturation and, 42-50 h later, human chorionic gonadotropin, which mimics the effect of the endogenous luteinizing hormone, inducing ovulation. This hormonal treatment bypasses the requirement for estrus and makes virtually all treated females receptive.
A second critical step is to ensure the quality of the FACS staining. Antibodies used in flow cytometry must always be titrated and used at the optimal concentration, and it is necessary to check that the enzymatic digestion does not cleave crucial antigenic epitopes. To assess whether an enzyme will cleave an epitope, one could stain two fractions of the same sample in parallel, one undergoing enzymatic and the other mechanical digestion. Similarly, the use of appropriate controls and single stains is crucial to obtain reliable data. For rare events, beads can be used to generate single stain samples. It is recommended not to use beads for setting up voltages but rather a population of cells containing lymphocytes and other leukocytes, such as splenocytes. If beads are used, it is necessary to titrate antibodies for bead staining, so the fluorescence intensity of stained beads will be comparable to the fluorescence intensity of cells. In case of difficulty separating positive from negative cells for a particular marker, an FMO control can also be used to facilitate the gating for a specific marker. In the case of intracellular markers, an isotype control must be used as intracellular staining might result in residual unbound antibodies, which may still be present within cells after the washing steps and therefore increase the background signal. It is recommended to run the samples within 24 h of fixing the cells to obtain the best results in phenotyping by FACS analysis, as autofluorescence increases significantly over time and fluorescence intensity of some antibodies might decline over time.
Another crucial factor to consider is the downstream application of the single-cell suspension obtained with the protocol. For functional assays, it is essential to work in sterile conditions. Similarly, for subsequent omics studies, it is important to work in sterile and RNase, DNase and protease-free.
The protocol presented here focuses on phenotyping group 1 ILCs but can be adapted for phenotyping other cell types by modifying the antibody panel. It is recommended that all antibodies are tested against digested and non-digested cell suspension to detect the loss/alteration of surface epitopes by the enzymatic treatment. Similarly, different enzymes can be used to digest the tissue and increase cell yield, but its effect on crucial antigenic epitopes must be carefully studied. While NKp46 is a good marker for splenic NK cells and works in all strains of laboratory mice, the expression of NKp46 on uNK cells in C57BL/6 mice is considerably lower than on spleen NK cells. It is best to stain for both NK1.1 and NKp46 simultaneously. If multiple organs are to be compared directly, it is recommended to treat all samples equally, even if the enzymatic digestion is not required for tissues such as the spleen or the bone marrow. Although the method presented here is applicable to the non-pregnant uterus, the lymphocyte ring isolation by a two-phase Percoll gradient will be challenging, and the yield of isolated cells may be too low for reliable FACS analysis and therefore will require pooling together cells isolated from the uterus of individual, non-pregnant mice23.
There are limitations to the protocol to consider for the interpretation of the data. As it is the case for all tissues, circulating lymphocytes coming from the blood will be isolated alongside tissue-resident cells. If the exclusion of circulating lymphocytes is essential for data interpretation, intravital staining can be performed to label circulating cells. Furthermore, a second limitation to the protocol is that some cells will be lost as not all cells can be extracted from the tissue. The most common problems and their troubleshooting are presented in Table 2.
Historically, the study of cells in tissues has relied on histological examination of tissue sections. Sandra Peel's excellent review24 summarizes the work done over more than 100 years until the late '80s. Descriptions of cells later known as uNK cells indeed appear in manuscripts published more than half a century before lymphocytes were even discovered. So, before the discovery of NK cells in 1975, and the uNK cells have been indicated as maternal glycogen cells or granulated metrial gland cells. Anne Croy has made major contributions in the field25 and kindly taught the team the dissection she had optimized3, and that is used currently. Although it is instrumental in describing the morphology and tissue location of uNK cells, classical histological examination is limited to the detection of only a few markers on the cells of interest. In 2008, a flow cytometry-based method to simultaneously detect multiple markers on uterine lymphocytes was described26. This is essentially the method that has been described in this paper. More recent technologies such as spatial transcriptomics and imaging by mass cytometry combine the power of histology and flow-cytometry, allowing both the simultaneous detection of multiple genes or proteins, respectively, and the preservation of the normal tissue architecture.
The applications of the method described here are multiple and include FACS phenotyping, functional assay (such as ELISPOT, degranulation or cytotoxic assays), cell sorting, and subsequent transcriptomics or proteomics. Further applications that could be developed based on this method include the culture and expansion of decidual NK cells after cell sorting or enrichment by negative depletion. Currently, there is no protocol to culture and expand mouse uNK cells and preserve their viability and functionality for an extended period, in a similar fashion to human NK cells that can be cultivated and expanded for 7-14 days by addition of IL-2 or a combination IL-12 and IL-15. The optimization of such a method for mouse uNK cells would provide more flexibility when performing functional assays and allow for multiple conditions to be tested with a higher cell number. On the other hand, culture conditions are known to modify the unique phenotype of lymphocytes and potentially their function too.
Disclosures
The authors have nothing to disclose and no conflicts of interest.
Acknowledgements
We thank the previous and the current team members who have helped develop this method, including Jean-Marc Doisne, Norman Shreeve, Iva Filipovic, and Anita Qualls. This research was funded by the Wellcome trust [Grant number 200841/Z/16/Z ] and the Medical Research Council (MR/P001092/1). For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Materials
| Name | Company | Catalog Number | Comments |
| 70 µm cell strainers | Falcon | 352350 | |
| BSA | Sigma | A9647-100G | |
| CD19 antibody | BD | 562701 | |
| CD3 antibody | BD | 562600 | |
| CD45 antibody | BioLegend | 103108 | |
| CD49a antibody | BD | 740262 | |
| DNase I | Roche (Sigma) | 10104159001 | |
| EOMES antibody | eBioscience | 12-4875-82 | |
| Fc block Trustain fcx | BioLegend | 101320 | |
| Fetal Bovine Serum | Gibco | 10217-106 | |
| Fix/Perm buffer (part of BD Cytofix/Cytoperm Fixation/Permeabilization Kit) | BD | 554714 | |
| HBSS, calcium, magnesium, no phenol red | Gibco | 14025092 | |
| Liberase DH | Roche (Sigma) | 5401089001 | |
| Lysis buffer Pharmlyse | BD | 555899 | |
| NK1.1 antibody | BioLegend | 108739 | |
| NKp46 antibody | BioLegend | 137608 | |
| Paraformaldehyde 16% Solution (methanol-free) | Agar Scientific | AGR1026 | |
| PBS 10x | Gibco | 14030-048 | |
| PBS 1x (no Ca2+ or Mg2+) | Thermo Scientific | 14190144 | |
| Percoll | VWR international | 17-0891-01 | |
| Phosphate buffered saline | Sigma-Aldrich | P5368-10PAK | |
| Pre-Separation filters | Miltenyi | 130-095-823 | |
| RMPI-1640 medium + GlutaMAX | Gibco | 61870-010 | |
| UltraPure 0.5M EDTA, pH 8.0 | Thermo Scientific | 15575020 | |
| Zombie Violet Fixable Viability dye | BioLegend | 423113 |
References
- Colucci, F. The immunological code of pregnancy. Science. 365 (6456), New York, N.Y. 862-863 (2019).
- Mor, G., Aldo, P., Alvero, A. B. The unique immunological and microbial aspects of pregnancy. Nature reviews. Immunology. 17 (8), 469-482 (2017).
- Croy, A., Yamada, A., DeMayo, F., Adamson, S. L. The Guide to Investigation of Mouse Pregnancy. , Elsevier, Academic Press. (2014).
- Filipovic, I., et al. Molecular definition of group 1 innate lymphoid cells in the mouse uterus. Nature Communications. 9 (1), 4492(2018).
- Chen, Z., et al. DBA-lectin reactivity defines mouse uterine natural killer cell subsets with biased gene expression. Biology of Reproduction. 87 (4), 81(2012).
- Moffett, A., Colucci, F. Uterine NK cells: active regulators at the maternal-fetal interface. The Journal of Clinical Investigation. 124 (5), 1872-1879 (2014).
- Gaynor, L. M., Colucci, F. Uterine natural killer cells: Functional distinctions and influence on pregnancy in humans and mice. Frontiers in Immunology. 8, 467(2017).
- Sojka, D. K., Yang, L., Yokoyama, W. M. Uterine natural killer cells. Frontiers in immunology. 10, 960(2019).
- Wilkens, J., et al. Uterine NK cells regulate endometrial bleeding in women and are suppressed by the progesterone receptor modulator asoprisnil. The Journal of Immunology. 191 (5), 2226-2235 (2013).
- Ashkar, A. A., Di Santo, J. P., Croy, B. A. Interferon γ contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. Journal of Experimental Medicine. 192 (2), 259-270 (2000).
- Hanna, J., et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nature Medicine. 12 (9), 1065-1074 (2006).
- Chakraborty, D., Rumi, M. A. K., Konno, T., Soares, M. J. Natural killer cells direct hemochorial placentation by regulating hypoxia-inducible factor dependent trophoblast lineage decisions. Proceedings of the National Academy of Sciences of the United States of America. 108 (39), 16295-16300 (2011).
- Crespo, ÂC., et al. Decidual NK cells transfer granulysin to selectively kill bacteria in trophoblasts. Cell. 182 (5), 1125-1139 (2020).
- Shmeleva, E. V., Colucci, F. Maternal natural killer cells at the intersection between reproduction and mucosal immunity. Mucosal Immunology. , 1-15 (2020).
- Kather, A., et al. Neither lymphotoxin alpha nor lymphotoxin beta receptor expression is required for biogenesis of lymphoid aggregates or differentiation of natural killer cells in the pregnant mouse uterus. Immunology. 108 (3), 338-345 (2003).
- Croy, B. A., et al. Analysis of uterine natural killer cells in mice. Methods in Molecular Biology. 612, Clifton, N.J. 465-503 (2010).
- Vander lee, S., Boot, L. M. Spontaneous pseudopregnancy in mice. II. Acta Physiologica et Pharmacologica Neerlandica. 5 (2), 213-215 (1956).
- Whitten, W. K. Modification of the oestrous cycle of the mouse by external stimuli associated with the male. The Journal of Endocrinology. 13 (4), 399-404 (1956).
- Cossarizza, A., et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). European Journal of Immunology. 49 (10), 1457(2019).
- Kim, S., et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 436 (7051), 709-713 (2005).
- Anderson, K. G., et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nature Protocols. 9 (1), 209-222 (2014).
- Caligioni, C. S. Assessing reproductive status/stages in mice. Current Protocols in Neuroscience. , Appendix 4, Appendix 4I (2009).
- Doisne, J. -M., et al. Composition, development, and function of uterine innate lymphoid cells. Journal of Immunology. 195 (8), Baltimore, Md. 3937-3945 (2015).
- Peel, S. Fate of GMG Cells. Granulated Metrial Gland Cells Advances in Anatomy, Embryology and Cell Biology. 115, Springer. Berlin. (1989).
- Croy, B. A., vanden Heuvel, M. J., Borzychowski, A. M., Tayade, C. Uterine natural killer cells: a specialized differentiation regulated by ovarian hormones. Immunological Reviews. 214, 161-185 (2006).
- Yadi, H., Burke, S., Madeja, Z., Hemberger, M., Moffett, A., Colucci, F. Unique receptor repertoire in mouse uterine NK cells. Journal of Immunology. 181 (9), Baltimore, Md. 6140-6147 (2008).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved