
Method Article
Human Liver Microphysiological System for Assessing Drug-Induced Liver Toxicity In Vitro
In This Article
Summary
Drug-induced liver injury (DILI) is a major cause of drug failure. A protocol has been developed to accurately predict the DILI liability of a compound using a liver microphysiological system (MPS). The liver model uses the coculture of primary hepatic cells and translationally relevant endpoints to assess cellular responses to treatment.
Abstract
DILI is a major cause of attrition in drug development with over 1000 FDA-approved drugs known to potentially cause DILI in humans. Unfortunately, DILI is often not detected until drugs have reached clinical stages, risking patients' safety and leading to substantial losses for the pharma industry. Taking into account that standard 2D models have limitations in detecting DILI it is essential to develop in vitro models that are more predictive to improve data translatability. To understand causality and mechanistic aspects of DILI in detail, a human liver MPS consisting of human primary liver parenchymal and non-parenchymal cells (NPCs) and cultured in 3D microtissues on an engineered scaffold under perfusion has been developed. Cryopreserved primary human hepatocytes (PHHs) and Kupffer cells (HKCs) were cocultured as microtissues in the MPS platform for up to two weeks, and each compound of interest was repeatably dosed onto liver microtissues at seven test concentrations for up to four days. Functional liver-specific endpoints were analyzed (including clinical biomarkers such as alanine aminotransferase, ALT) to evaluate liver function. Acute and chronic exposure to compounds of various DILI severities can be assessed by comparing responses to single and multi-dosed microtissues. The methodology has been validated with a broad set of severe and mildly hepatotoxic compounds. Here we show the data for pioglitazone and troglitazone, well-known hepatotoxic compounds withdrawn from the market for causing liver failures. Overall, it has been shown that the liver MPS model can be a useful tool to assess DILI and its association with changes in hepatic function. The model can additionally be used to assess how novel compounds behave in distinct patient subsets and how toxicity profiles may be affected by liver disease states (e.g., viral hepatitis, fatty liver disease).
Introduction
DILI remains the most common cause for acute liver failure in the USA and Europe and is a leading cause of attrition of compounds in the drug development process1. Nearly all classes of medications can cause hepatotoxicity, with central nervous system agents and antibiotics being by far the most common treatments that cause DILI in patients2. Drug-induced hepatotoxicity is caused by a complex interaction of genetic, non-genetic, and environmental factors, leading to the death of hepatocytes and other liver cell types, including cholangiocytes and endothelial cells1,3.
DILI causing agents can be classified in two ways: those which cause predictable dose-dependent liver damage or those which cause idiosyncratic DILI that is rare and develops independently of drug dose, or route, or duration of administration, but is responsible for up to a sixth of all acute liver failures in the USA only4. Unfortunately, DILI is often not detected until drugs have reached the clinical stages of the drug development process. Drug-induced liver injury rank (or DILIrank) consists of more than a thousand FDA-approved medicines that are divided into four classes according to their potential for causing DILI, and their use in patients must be closely monitored5.
Studying mechanisms of drug hepatotoxicity remains very challenging, and therefore, many preclinical models have been developed to explore mechanisms of DILI. Current in vitro and in vivo models used to predict DILI in preclinical development have several limitations to providing insights into the complex, multifaceted interactions in a living human body. Cancerous hepatic cell lines (i.e., HepG2, HepaRG) cultured in 2D are still used in the early stages of drug development for evaluating the toxicity of candidate compounds6. Even so, these cell lines come from single donors and show abnormal levels of liver function, and do not always exhibit high sensitivity for detection of DILI7,8. As an alternative to cancerous hepatic cell lines, PHHs better represent human liver physiology if cultured appropriately in vitro, although several limitations exist with their culture, like short incubation time with drugs, relatively short life span, loss of hepatic gene expression, and changes of drug metabolic functions9,10,11. PHHs can be cultured on extracellular matrix proteins in standard 2D cell culture plates, but as a downside, the rapid decline in their function means they have low sensitivity (<50%) for DILI prediction12.
On the other hand, testing on animal models is slow, expensive, and needs a cross-species translation to extrapolate prediction to humans. Most newly developed drugs fail to gain approval making this process costly and risky5. Furthermore, for testing new human-specific modalities, animal models are less suitable due to gene sequence or immunological response differences versus humans13.
Consequently, interest in more advanced three-dimensional (3D) in vitro liver models has exponentially grown. Culturing PHHs as spheroidal structures generated by gravitational aggregation in hanging drops or on ultralow attachment surfaces represents a high-throughput method for assessing compound liabilities14. PHH spheroids have been used to assess DILI in a diseased background (e.g., steatosis and cholestasis)15. A wide variety of models have been developed to include plated micro-patterned cocultures of hepatocytes with stromal fibroblasts16, 3D bio-printed liver tissues17, 3D spheroid cultures with or without hepatic non-parenchymal cells15. However, all these methods still have drawbacks, and culturing PHHs in a more physiologically relevant microenvironment could provide them with higher levels of functionality for extended periods of time to enable the investigation of prolonged exposure to potential hepatotoxicants. Additionally, to improve the translational relevance of any advanced in vitro PHH culture, clinically relevant functional endpoints or toxicity output biomarkers must be utilized to allow data to be compared in vivo or clinical scenarios18.
In this study, we assessed whether a MPS, also known as an Organ-on-a-Chip (OOC), in vitro liver model could be used to understand the detailed mechanistic aspects of liver toxicity. The MPS has previously been shown to maintain highly functional 3D liver microtissues, under flow, for up to 4 weeks19. The system has been recently tested by FDA and shown to have high reproducibility when performing drug toxicity, metabolism, and intracellular accumulation20. Moreover, when compared with spheroids and sandwich cultures, the system had a more stable function and higher sensitivity in detecting the toxicity of several drugs20. To date, the MPS has been used in a wide range of applications that cover ADME21, disease modeling (HBV22, NAFLD23,24,25), and drug-drug interactions26, potentially making it highly suited to assessing acute and chronic DILI. The technology presented here offers an alternative to close the gap between more traditional cell cultures and animal models and human clinical trials, advancing towards the simulation of human biological conditions to support the assessment of candidate compounds' liver toxicity in preclinical stages of the drug development process.
Protocol
All the work was conducted in the laboratory following strict Health & Safety procedures and in accordance with its own laboratory risk assessments and SOPs. All equipment used is serviced according to the manufacture's guidelines. Microbiological safety cabinets (MBSCs) are serviced annually, and Ki-Discus (potassium iodide) tested to British standards. The protocol follows the United Kingdom Human Tissue Authority (HTA) Code of Practice and directives and uses ethically-sourced primary human cells supplied by vendors that fully comply with the general requirements for informed consent (45 CFR §46.116 and §46.117) and Good Clinical Practice (GLP), (ICH E6), and regulatory and ethics committees.
1. Preparing cell culture media
NOTE: Prepare Seeding Advanced DMEM medium on Day -1 and store at 4 °C. Prepare Maintenance Advanced DMEM medium on Day 1 and store at 4 °C for up to 1 week.
- Seeding Advanced DMEM medium for PHHs and HKCs coculture: Supplement one bottle of 500 mL Advanced DMEM medium (Table of Materials) with 18 mL of Cocktail A (final concentration of 3.6%) and with 25 mL of FBS (final concentration 5%).
- Maintenance Advanced DMEM medium for PHHs and HKCs coculture: Supplement one bottle of 500 mL Advanced DMEM medium (Table of Materials) with 20 mL of Cocktail B (final concentration of 4%) and 500 nM hydrocortisone.
NOTE: Hydrocortisone will be made fresh on the day of use, and the steps on how to prepare the stock solution and required dilutions are mentioned below. - Preparation of 500 nM hydrocortisone in Maintenance Advanced DMEM Medium
- Preparation of the starting stock solution (20 mM): Weigh 7.24 mg of hydrocortisone (Table of Materials) into a 1 mL glass vial. Record the exact amount of hydrocortisone weighed out and determine the volume of dimethyl sulfoxide (DMSO) using the following calculation:

- Preparation of the working 100 μM hydrocortisone stock solution: Add 5 µL of the starting 20 mM stock solution to 995 μL of Advanced DMEM.
NOTE: Diluting 25 µL of the DMSO solution from step 1 in water or media results in 0.5% DMSO concentration. In the final solution, the DMSO concentration will be 0.0025%. In this case, the additional volume of 5 µL results in an insignificant change in the total volume. - Preparation of the working 500 nM hydrocortisone solution in Advanced DMEM: To prepare 1 mL of 500 nM hydrocortisone solution in Advanced DMEM, add 5 μL of the stock solution of 100 μM hydrocortisone to 995 μL of the Maintenance Advanced DMEM medium.
- Preparation of the starting stock solution (20 mM): Weigh 7.24 mg of hydrocortisone (Table of Materials) into a 1 mL glass vial. Record the exact amount of hydrocortisone weighed out and determine the volume of dimethyl sulfoxide (DMSO) using the following calculation:
2. MPS set-up and priming (Day -1)
- Connect the controller to its docking station house in a cell culture incubator and ensure fresh desiccant (Table of Materials) is added into the desiccant jar located at the back of the controller.
NOTE: The controller unit draws moisture from the incubator over time and is kept dry using fresh desiccant. - Switch the controller ON by pressing the boat rocker switch located behind it and wait for 5 min for the system to stabilize and reach pressure. Then check the screen for the pneumatic report to ensure: (i) The Pressure Reservoir Output reached ~2000 mBar and (ii) The Vacuum Reservoir Output reached ~850 mBar.
- Remove each plate from packaging and visually inspect every well to check for possible defects (missing scaffolds, cracks etc.).
- Insert a driver (with a plate on) onto the docking station to check that the driver is recognized by the docking station and controller. Check the Pressure Reservoir Output has dropped by less than 100 mBar, and the Vacuum Reservoir Output has increased by less than 500 mBar.
- Prime each well by adding 500 µL of Seeding Advanced DMEM medium (pre-warmed to 37 °C) to the reservoir side.
- Select the Prime program on the controller screen (up flow for 3 min at 2.5 µL/s) until the fluid comes through the filter supports.NOTE: 'Up flow' is a setting on the controller that allows media to flow from the reservoir upwards through the scaffolds in the LC12 plate.
- Fill all wells with a further 1.1 mL of Seeding Advanced DMEM medium to cover the surface channel. All wells will then be at their full working volume of 1.6 mL.
- Place the drivers with plates in a 37 °C and 5% CO2 incubator, connect to the docking station and run the Incubate program.
NOTE: All programs used in the experiment (Prime, Incubate, Seed, Media Change) are pre-set in the MPS system. Prime the plates in the incubator until ready to seed.
3. Seeding liver cells into MPS (Day 0)
- Prevalidate all PHHS and HKCs. All PHHs and HKCs Lots are pre-validated in-house prior to performing the cell culture experiment (see Supplementary Material).
- Thaw vials of PHHs and HKCs cells (Table of Materials) by holding the vials steadily in a 37 °C water bath until only a small sliver of ice remains.
- Pipette PHHs directly into a tube of pre-warmed (37 °C) Cryopreserved Hepatocyte Recovery Medium CHRM media (two vials max per tube).
- Pipette the cells gently, then use 1 mL of CHRM to wash any remaining cells from the cryotube. Be very gentle with cells when thawing and transferring into a conical tube.
CAUTION Do not agitate the vials during thawing, and do not pipette their content up and down. - Pipette HKCs cells gently from the cryotube into 10 mL of ice-cold Seeding Advanced DMEM medium in a 15 mL centrifuge tube.
NOTE: Up to 2 vials of HKCs can be combined. - Centrifuge both cell types at room temperature (RT) at 100 x g for 10 min. Remove the supernatant.
- Resuspend the PHHs in warm Seeding Advanced DMEM medium and HKCs in ice-cold Seeding Advanced DMEM medium (to help reduce cell clumping), using 1 mL per vial of cells added to the tube and place the cells on ice. Use a gentle rocking action to resuspend the cells.
CAUTION: Do not resuspend PHHs by pipette action, as it can lead to cell death. - Combine the cell suspensions from multiple tubes (if applicable - i.e., if all PHHs are from the same donor), but do not mix cell types.
- Count cells. Record the viability (must be above 85% for both cell types, PHHs and HKCs) and the total number of cells. If cell viability falls below 85%, thaw a new vial of cells, and reassess cell viability.
- Calculate cell viability using the following formula:

- Calculate the desired volume of the cell suspension to seed into each well and additional volume of Seeding Advanced DMEM medium needed to take total seeding volume to 400 µL. Cell numbers per well: 0.4 x 106 PHHs and 0.04 x 106 HKCs per well, and cell density of 0.25 x 106 PHHs/mL, respectively 0.025 x 106 HKCs/mL.
- Disconnect the driver from the docking station and place it in the MBSC.
- Aspirate the media from the above scaffold to the stopping point (going down the deep notch on the retaining ring), channel and reservoir. Leave a "dead volume" of 0.2 mL in the culture well, reaching just above the scaffold. Care must be taken not to remove the total medium from above the scaffold to avoid air bubbles forming.
- Add 400 µL of Seeding Advanced DMEM medium into the well chamber, return the driver onto the docking station in the incubator and run the Media Change program for 3 min. The program will automatically pause after 3 min.
- Once complete, disconnect the driver from the docking station and place it back in the MBSC.
- Aspirate the media from the above scaffold down to the stopping point and at the reservoir end of each well.
- Carefully resuspend the PHHs by gently rocking the tube, then add the required volume of the cell suspension to each culture well. Carefully pipette the cell suspension, ensure tha the cells disperse evenly across the plate's scaffold.
NOTE: To ensure good coverage throughout scaffold, use a slow swirling motion to pipette cells down onto the scaffold. - Similarly, carefully resuspend HKCs and add the cell suspensions to each culture well.
NOTE: Use a slow swirling motion to seed HKCs to ensure good coverage throughout the scaffold. The two seeding sub-steps can be separated, or the two cell types can be pre-mixed at an appropriate density and seeded concomitantly. - Once all wells contain both cell types, place the MPS driver onto the docking station in the incubator without physically connecting it and leave it to stand for 1 h.
- After 1 h, fill up each well with the required volume of additional Seeding Advanced DMEM medium to reach 400 μL and run the Seed program.
- After 2 min, the program will automatically pause, remove the driver from the incubator and slowly add 1000 µL of the Seeding Advanced DMEM medium to the channel (closer to the reservoir end than the well chamber) to achieve a total volume of 1.4 mL (with a further 200 µL dead volume in the channels).
- Move the plates to the incubator and run the rest of the Seed program for 8 h.
NOTE: Flow will automatically switch to Incubate program after 8 h.
4. Media change (Day 1)
- Disconnect the driver from the docking station and place it in the MBSC.
- Perform Media Change by removing the Seeding Advanced DMEM medium in the well chamber down to the stopping point.
- Add 400 µL of the Maintenance Advanced DMEM medium into the well chamber, return the driver onto the docking station in the incubator and run the Media Change program for 3 min. The program will automatically pause after 3 min.
- Disconnect the driver from the docking station and, in the MBSC, aspirate away media from the reservoir chamber, channel, and stopping point above the scaffold. At this point, the culture well will be returned to the dead volume.
- Top up the reservoir chamber with 1.4 mL of fresh pre-warmed (37 °C) Maintenance Advanced DMEM medium.
- Return the driver onto the docking station in the incubator and run the Incubate program.
5. Liver microtissue quality control (QC), media collection, media change, and drug dosing (Day 4)
- On day 4 perform a Media Change using the Maintenance Advanced DMEM medium and QC checks to ensure seeding has been successful.
NOTE: QC is a process used to check the health of the formed microtissues by measuring Lactate dehydrogenase (LDH) and urea. - Prior to running the QC, prepare fresh stock solutions for each compound to test (either in Maintenance Advanced DMEM medium or Maintenance Advanced DMEM medium containing 0.1% DMSO, depending on the solubility of each compound). Prepare dilutions accordingly to yield test concentrations for each compound.
- Disconnect the driver and plate from the docking station and transfer to a MBSC.
- Transfer 50 µL of media from each well using a pipette to a 96 well plate to perform an LDH assay (Table of Materials) before sampling and 25 µL for a Urea assay (Table of Materials).
NOTE: LDH and urea assays will be performed following the manufacturer's instructions. - Continue the experiment after QC if LDH readings are lower than 2 AU/106 cells and urea is above 40 µg/day/106 cells.
NOTE: Albumin is not used as QC because it is a lengthy assay to run on the day and will be assayed later once the experiment is complete from the samples withdrawn on Day 4. - If any wells fail QC, remove them from the experimental design.
- Once the experimental layout is confirmed, sample the remaining media from each well, making sure not to disturb the cell culture by touching the scaffold. Store the collected media (labeled as pre-dose samples) at -80 °C for later assays.
- Perform Media Change in a MBSC following steps 4.3-4.5. Change the wells to Maintenance Advanced DMEM medium with the right drug concentration according to the experimental design.
- Once complete, return the driver onto the docking station in the incubator and run the Incubate program.
6. Media collection, media change and drug dosing (Day 6)
- Prepare fresh stock solutions for each compound to test (either in Maintenance Advanced DMEM medium or Maintenance Advanced DMEM medium containing 0.1% DMSO, depending on the solubility of each compound). Prepare dilutions accordingly to yield test concentrations for each compound according to the plate plan.
- Disconnect the driver and the plate from the docking station and transfer to a MBSC.
- Collect media from each well (~1 mL) manually with a pipette making sure not to disturb the cell culture by touching the scaffold, assay for LDH and Urea. Store the rest of the collected media at -80 °C for later assays, and label them 48 h post-dose samples.
- Re-dose each well with the same drug concentration as on Day 4 and according to the plate plan by performing Media Change following steps 4.3-4.5.
- Once complete, return the driver onto the docking station in the incubator and run the Incubate program.
7. Ending the experiment (Day 8)
- Disconnect the driver and the plate from the docking station and transfer to a MBSC.
- Sample media from each well manually using a pipette, making sure not to disturb the cell culture by touching the scaffold.
- Assay the withdrawn media for LDH and Urea and store the rest of the collected media at -80 °C for later assays.
- Running CYP3A4-glo assay.
- Measure the effects of the drugs tested on cytochrome P450 CYP3A4 activity in PHHs at the end of the experiment using this assay.
- Reconstitute the detection reagent (for CYP3A4 assay, see Table of Materials) following the manufacturer's instructions. If the detection reagent has previously been reconstituted and frozen, remove it from the -20 °C freezer and allow it to thaw at RT.
- Prepare 20 mM stock D-luciferin standard following the manufacturer's instructions.
- Prepare working luminogenic substrate medium with a 1:1000 dilution of Luciferin IPA in Maintenance Advanced DMEM medium (2 mL of luminogenic substrate medium per well).
- Perform a Media Change as described in steps 4.3-4.5 with luminogenic substrate medium. Save 500 µL of the luminogenic substrate medium in a 1.5 mL glass vial (Table of Materials) as input material.
- Return the driver onto the docking station in the incubator and run the Incubate program for 1.5 h.
- Prepare D-luciferin standard curve in culture medium in 1.5 mL tubes following the manufacturer's instructions and pipette 50 µL of each standard in duplicate on a white opaque 96-well plate (see Table of Materials), using culture medium as blank or 0 µM.
- Once the time has elapsed, remove the driver from the docking station and sample media for CYP3A4 assay following steps 7.4.9-7.4.13.
- After incubation, transfer 50 µL of the sample medium from each well and input material to the 96-well opaque white luminometer plate containing standards. Take care to leave at least two empty rows on the opaque plate between the standards and samples to avoid light carryover between the top standards and the samples readings.
- Add 50 µL of Luciferin Detection Reagent to each well to initiate a luminescent reaction.
- Incubate the plate at RT on a plate shaker for 20 min in the dark to stabilize the luminescent signal.
- Record the luminescence using a luminometer or CCD camera.
- Plot the standard curve by taking the average of each point and then subtracting the average of the blanks. Use the equation of the line to calculate the metabolic rate (pmol/min/106 cells) in the rest of the samples, remembering to include any dilutions done.
- Remove the scaffolds from the plates using a pair of tweezers and place them in a 24 well plate containing 500 µL of D-PBS (Without Ca++ and Mg++) in each well, taking care not to disturb the microtissue.
- Take snapshots of each scaffold using an inverted light microscope at magnification 10x.
- Running the ATP assay (see Table of Materials) following the manufacturer's instructions:
- Thaw the reagent at RT.
- Wash the scaffolds twice with 500 µL of D-PBS (Without Ca++ and Mg++) for each wash step.
- Add 120 μL of reagent and 120 μL of PBS to each scaffold and the same volumes to an empty well (this will serve as blank). Place the plate covered in aluminum foil on a shaker and shake vigorously (500 rpm) for 5 min followed by 30 min of incubation to stabilize the luminescence signal.
- Transfer 100 μL of the lysed samples in duplicate to a clear flat-bottom 96-well assay plate for measurement. Ensure that the blank wells are not placed next to the other measuring wells of high luminescence.
- Record the luminescence using a microplate reader.
- Compare the luminescence of the samples to the luminescence of the standards to determine ATP detected by the reagent in the samples.
Results
The manuscript describes a liver MPS model used for assessing DILI. The MPS facilitates the generation of 3D liver microtissues that are maintained highly functional under flow for up to 4 weeks. PHHs/HKCs are seeded onto collagen-coated scaffolds to form liver microtissues which are perfused with a growth medium and, after passing the QC check, are dosed with compounds. Here, we show data for troglitazone and pioglitazone, two structurally similar compounds but with different DILI severities.
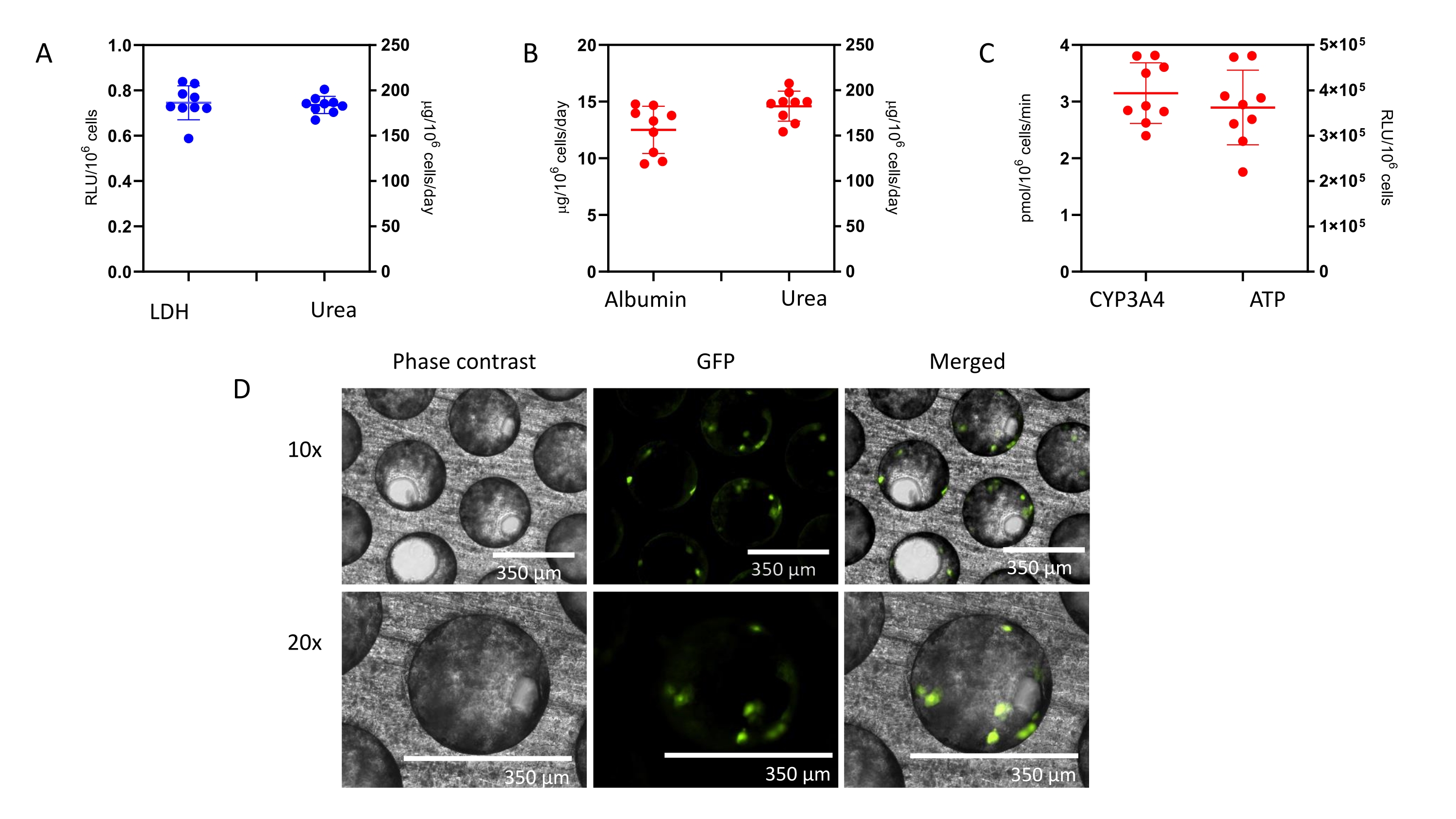
At Day 4, prior to drug dosing, a QC check of formed liver microtissues is assessed and consists of LDH release and urea synthesis (Figure 1A). The QC aims to confirm that the liver MPS produces highly consistent and functioning liver microtissues. The data presented here are generated from three experiments and shows good levels of reproducibility with low intra- and inter-study variability. After an 8-day culture, multiple health and hepatic metrics (albumin, urea, CYP3A4, ATP) are assessed and control microtissues show high levels of hepatic functionality and reproducibility (Figure 1B,C). Contrast phase microscopy and IF staining of the liver microtissues (See Supplementary Material) shows high seeding consistency throughout the scaffold's microchannels and reveal the distribution of HKCs in the PHH microtissues (Figure 1D)

Figure 1: Liver MPS produces highly reproducible data and consistent microtissues. (A) 3D Liver microtissue QC metrics at Day 4, and functionality assessment at the end of the study at Day 8 -(B) Albumin and Urea, (C) CY3A4 and ATP). Data is collected from 3 experiments; in each experiment, there were 3 vehicle control replicates. Data shown are Mean ± SD, N = 9. (D) Phase contrast microscopy (10x and 20x) and IF of 3D liver microtissues generated by coculturing PHHs and HKCs in liver MPS platform for assessing DILI. To visualize the HKCs, prior to seeding HKCs were transduced with an adenoviral vector expressing eGFP (see Supplementary Material). Representative photomicrographs are shown. The transduction and imaging were performed as a standalone experiment to demonstrate cell localization and not done with the DILI protocol described. HKCs cells are pre-validated in-house prior to use in experimental cell culture and must have low levels of post-thaw activation; this is assessed by measuring biomarkers IL-6 and TNF-alpha. Please click here to view a larger version of this figure.
{kind=link}
Troglitazone is known to cause severe DILI; following its license for the treatment of type 2 diabetes, it was withdrawn by the FDA after 3 years on the market because of the frequency of liver injury associated with its use. To date, published animal studies failed to predict troglitazone's potential to cause severe liver injury. The toxicity of this compound was also not detected in standard in vitro 2D hepatic assays14.
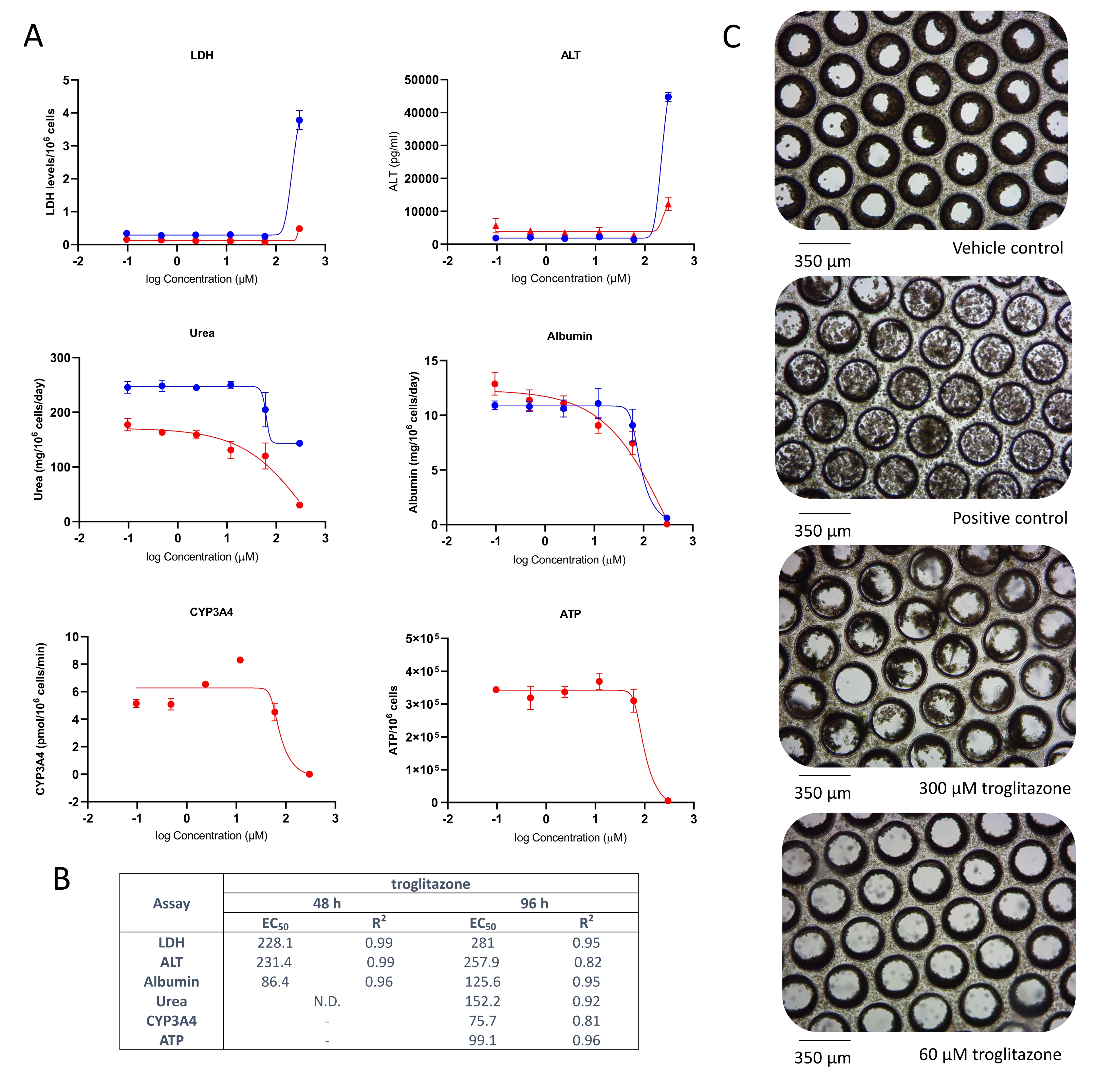
Liver microtissues in the MPS were dosed with troglitazone for 96 h, and it caused an acute toxic response, Cmax driven, which was detected by ALT and LDH release and a rapid reduction in albumin and urea production, at circa 15 x Cmax, following acute exposure to troglitazone (Figure 2A). Cellular endpoint (ATP content) and CYP3A4 activity (for assessing metabolic biotransformation), sampled after 96 h exposure, further confirmed toxicity caused by troglitazone and EC:50 values were highly comparable to other endpoints (Figure 2B). Brightfield microscopy images taken after 8-day culture in the MPS reveal a healthy liver microtissue, uniformly seeded throughout the scaffold (vehicle control) in contrast to generalized tissue death/degradation as seen in the replicates treated with positive control and troglitazone at the top two test concentrations (Figure 2C).

Figure 2: Determining DILI risk of troglitazone using multiple hepatotoxic endpoints. Liver microtissues were exposed to seven test concentrations of troglitazone for 96 h and compared for (A) LDH release, ALT release, Albumin production, Urea synthesis, CYP3A4 activity, and ATP content. Blue lines - 48 h exposure (media endpoints only), red lines - 96 h exposure. Positive control was 100 μM chlorpromazine. All endpoints are measured from the same liver MPS cultures. Data shown are mean ± SD, N = 3. (B) Summary of E:50 numbers generated from data. N.D. = data not plottable. Line = not assayed. (C) Representative brightfield microscopy of liver microtissues after 8-day culture (magnification 10x). Please click here to view a larger version of this figure.
{kind=link}
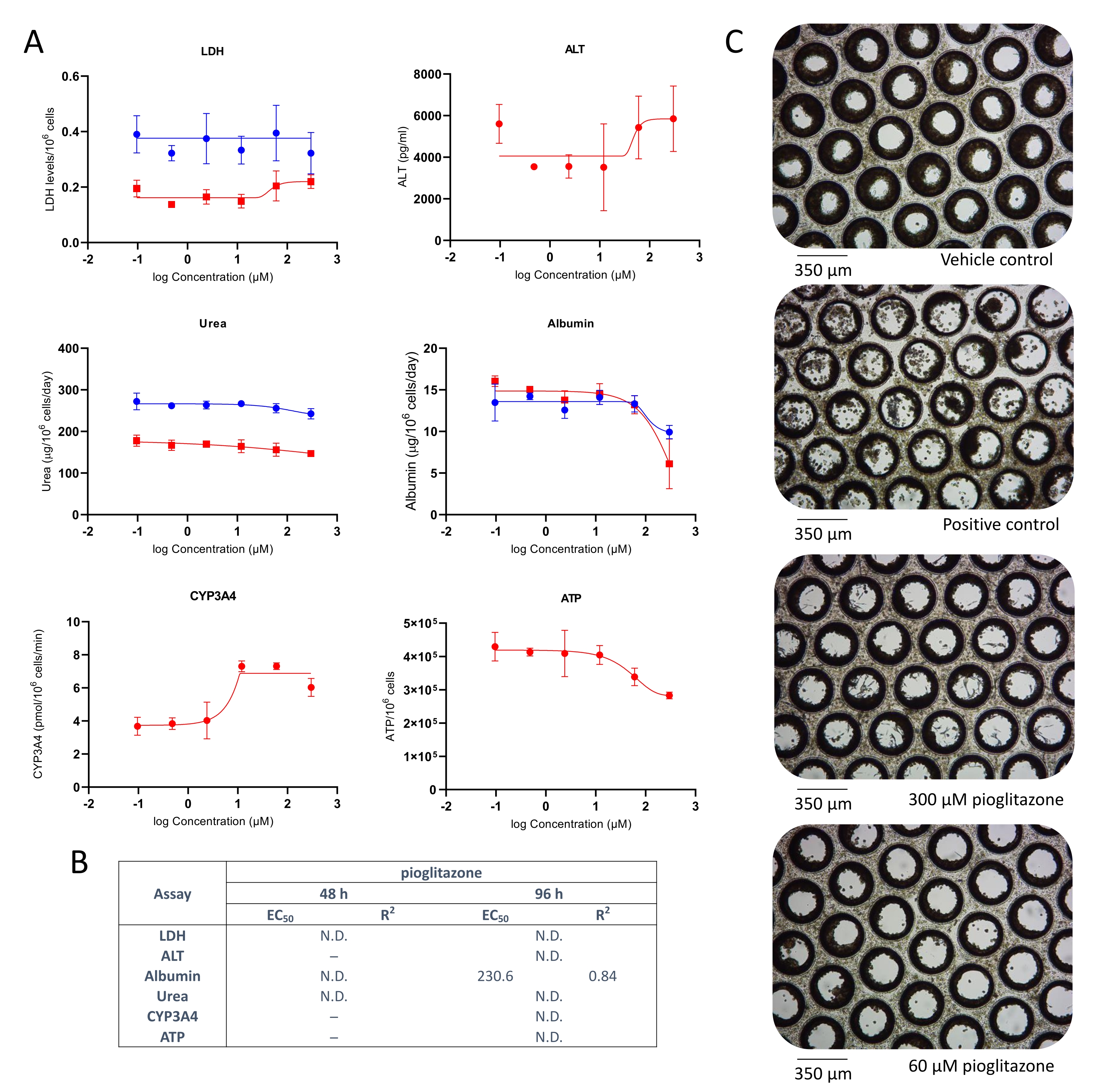
Liver toxicity following exposure to pioglitazone was also investigated. Pioglitazone is a compound known to be of low-DILI concern4 and did not exert hepatotoxicity in classic 2D primary hepatocytes cultures and even in some more advanced 3D models10, 11. Mild hepatotoxic effects were observed at both tested time points (Figure 3). No LDH or ALT release was detected; however, after 48 h, a mild reduction in albumin and urea production was observed, at approx. 25x Cmax (Figure 3A). Very minor reduction in ATP content was also observed at high pioglitazone concentrations, but this was not significant. EC:50 values generated from dose-response curves are presented in Figure 3B. Microscopy revealed slight microtissue alteration following 96 h exposure to pioglitazone at the two highest tested concentrations (Figure 3C). The results demonstrate the ability of the liver MPS to detect the toxicity of compounds with mild DILI concern.

Figure 3: Determining DILI risk of pioglitazone using multiple hepatotoxic endpoints. Liver microtissues were exposed to seven test concentrations of pioglitazone for 96 h and compared for (A) LDH release, ALT release, Albumin production, Urea synthesis, CYP3A4 activity, and ATP content. Blue lines - 48 h exposure (media endpoints only), red lines - 96 h exposure. Positive control was 100 μM chlorpromazine. All endpoints are measured from the same liver MPS cultures. Data shown are mean ± SD, N = 3. (B) Summary of EC:50 numbers generated from data. N.D. = data not plottable. Line = not assayed. (C) Representative brightfield microscopy of liver microtissues after 8-day culture (magnification 10x). Please click here to view a larger version of this figure.
{kind=link}
By assessing all functional endpoints and toxicity output biomarkers that might represent in vivo or clinical scenarios (LDH release, Urea synthesis, Albumin production, CYP3A4 activity, ATP content, ALT release) and corroborating the data generated for both tested compounds dosed at a seven-point dose range for 48 h and 96 h, a heatmap has been generated to yield a "signature of hepatotoxicity", helping to identify compounds with varying level of DILI concern (Figure 4).

Figure 4: Determining "signature of toxicity" with Liver MPS. Heatmap showing of troglitazone and pioglitazone from six functional liver-specific endpoints (LDH release, Urea synthesis, Albumin production, ALT release, CYP3A4 activity, and ATP content) following 48 h and 96 h exposure to seven-point dose range. Each value is generated as Mean, N = 3, and normalized to control samples. The values on the color bars represent a fold increase over baseline controls. Please click here to view a larger version of this figure.
{kind=link}
Supplementary Material: Fluorescent microscope imaging of microtissues and pre-qualification assessment of cells. Please click here to download this File.
Discussion
MPS are designed to recapitulate functional units of human organs in vitro and have been developed to address the limitations of conventional 3D cell culture models27. The liver is one of the most modeled organs using MPS, and a wide variety of systems have been developed. The human liver is responsible for drug metabolism and generation of toxic drug metabolites, and its function is a key element to model for drug development, including the assessment of DILI liability of compounds28. Here we have introduced a new method for assessing DILI using a liver MPS; the protocol enables mechanistic insights to be sought for each compound assayed to determine how it may cause DILI as well as being a highly sensitive and robust assay. Liver microtissues are formed in the MPS plates, which are a coculture of PHH and HKCs and are highly functional with high levels of albumin and urea production as well as high CYP3A4 activity compared to standard in vitro liver models20.
Although the DILI model described here can serve as a useful tool in later stages of preclinical testing in the drug development process, it also has several limitations. As the majority of MPS currently available on the market, it is a low-throughput platform and, therefore, more difficult to use for large-scale drug screening activities. Consisting of microtissues formed by coculturing PHHs and HKCs the DILI model also cannot entirely capture the complexity of the human liver, and further optimization by incorporating different types of cells (e.g., immune cells) would be beneficial to add value to the existing model. This single-organ MPS could also be combined with other organ platforms that can share a common medium and allow organ crosstalk at the cellular or endocrine level, and that can help to better understand the mechanistic insights of toxicity not limited only to the liver itself. Furthermore, as any relatively new technology, it might be considered costly and hence of limited accessibility.
MPS is a platform used to develop organotypic models of single or multi- human tissues. The system is composed of a controller, umbilical cable, and MPS driver into which the plate is inserted (Figure 5A). Each liver MPS plate has 12 independent open wells for culturing primary liver cells in 3D on engineered scaffolds. In summary, the system is QC checked, and the plates are primed at Day -1, the PHHs and HKCs are seeded on the plates at Day zero (Figure 5B, see 1). Embedded micropumps facilitate the circulation of cell culture media through the scaffolds to facilitate the formation of 3D microtissues (Figure 5B, see 2). Formed microtissues are QC'd at Day 4, dosed with different concentrations of each compound every 48 h for 4 days, and assayed for endpoint biomarkers at Day 8 (Figure 5C). The experimental timeline of the DILI assay in the MPS plate is depicted in Figure 5D.

Figure 5: The microphysiological systemand experimental timeline of a standard DILI assay. (A) The microphysiological system with its components: controller (1), umbilical cable (2), docking station (3), MPS driver (4) and LC12 plate (5). (B) Seeding of PHHs and HKCs on LC12 plate at Day 1 (1) and embedded micropumps facilitate circulation of cell culture media with tuneable flow rates through the 3D microtissues seeded on the scaffolds (2). (C) Taking down the scaffolds at the end of the study. (D) Experimental timeline. Please click here to view a larger version of this figure.
{kind=link}
When performing the protocol, it is important that a robust system QC check is performed prior to starting, checking the system is functioning pneumatically correctly and the consumable plates are visually inspected and primed efficiently to ensure even functionality across all wells. Having high-quality primary human cells is essential for this protocol, with hepatocytes known to adhere consistently in cell culture experiments and form 3D interactions. Thawing these cells is also a critical step, as primary hepatocytes should not be resuspended by pipetting action as this can rapidly lead to cell death. Having cell viability above 85% is critical for successful seeding, as large amounts of cellular debris will interfere with 3D microtissue formation. The QC check of formed liver microtissues at Day 4 is also important, and the user needs to ensure that acceptable levels of LDH and Urea are measured, as out-of-range parameters might be indicative of poor-quality tissue formation and allow straightforward troubleshooting. Finally, the hydrocortisone used in the cell culture media must be prepared fresh on the day of use to prevent any unwanted degradation that might impact cell culture functionality, as it is required to maintain the metabolic functionality of the hepatocytes.
Despite having significant complexity, the liver MPS does not contain all the cell types of the human liver. It is possible to add further cells types to the model24,29 to increase physiological relevance, but these should only be added with a clear justification for the context of use. For studying DILI PHH are the key cell type, and the incorporation of HKCs in this model allows some immunological responses to be determined. It should also be noted that PHHs isolated from human livers and commercially available cryopreserved PHHs tend to demonstrate some variations from lot to lot. We have demonstrated here that this protocol produces reproducible results when used with high-quality preparations of cells. However, some lot-lot variation would be expected, and this could be further overcome by using pooled lots of multiple donors. These limitations could be overcome by using hepatocyte-like cells differentiated from iPSC that recapitulate many functional properties of PHHs and that have been used in the drug development process30. HKCs also show a lot to lot variability and a high level of activation upon thaw; therefore, HKCs donors are pre-validated in-house prior to use in experimental cell culture (coculture with validated PHHs) and must have low levels of post-thaw activation; this is assessed by measuring biomarkers IL-6 and TNF-alpha (see Supplementary Material).
The data presented here confirm that the assay can detect DILI accurately, helping to identify hepatotoxicants that might not be detected by 2D10,11 and even some 3D models. Data generated from MPS are still not used as a standard by the pharmaceutical industry for regulatory submissions or drug screening purposes due to the lack of process standardization and harmonization, including inter-site reproducibility20. The data and experimental approaches demonstrated here address this, showing that the liver model can be used routinely and robustly in DILI screens to accurately predict the liability of novel compounds.
By measuring a range of endpoints to produce a "signature of hepatotoxicity", helping to identify compounds with different levels of DILI concern (including compounds not detectable by other in vitro methods) and their mechanisms of toxicity revealed. This technology can close the gap between traditional cell culture and animal models on one side and human clinical trials, advancing towards the simulation of human biological conditions for the preclinical assessment of liver toxicity as part of the drug development process.
Disclosures
All authors are employees of CN Bio Innovations Limited.
Acknowledgements
CN Bio Innovations Ltd. funded this study.
Materials
| Name | Company | Catalog Number | Comments |
| 24 well cell culture cluster plates flat bottom | Corning | 3524 | |
| 96 well clear assay plates, flat bottom clear plastic | Greiner | 655101 | |
| 96 well plates black flat bottom | Corning | 3915 | |
| 96 Well White/Clear Bottom Plate, TC Surface | ThermoScientific | 165306 | |
| Advanced DMEM (1x) | Gibco | 12491015 | Cell culture media. |
| AssayMax Albumin ELISA Kit | AssayPro | EA3201-1 | Dilution 1:250. Time point Day 4, 6, and 8. |
| Cell Maintenance Cocktail B, (Primary Hepatocyte Maintenance Supplements) | Gibco | CM4000 | |
| CellTiter-Glo 3D Cell Viability Assay | Promega | G9682 | Dilution 1:1. Time point Day 8. |
| Chlorpromazine HCl | Sigma Aldrich | C8138 | |
| Chromacol blue lids, 9 mm Autosampler Vial Screw Thread Caps | ThermoScientific | 9-SCK(B)-ST1 | glass vial |
| Chromacol vials, 9 mm Clear Glass Screw Thread Vials | ThermoScientific | 2-SVW | |
| Class 2 Microbiological Safety Cabinets - Trimat2 1500 exhaust | Contained Air Solutions | ||
| Conical tubes 50 mL | Greiner | 227261 | |
| Cryopreserved Hepatocyte Recovery Medium (CHRM) | ThermoFisher Scientific | Gibco CM7000 | |
| Cryopreserved primary human hepatocytes | BioIVT Europe | Lot. RAS | |
| CytoTox 96 Cytotoxicty (LDH) Assay Kit | Promega | G1781 | Dilution - none. Time point Day 4, 6 and 8 |
| >Data analysis model used to generate the graph and EC:50 curves was nonlinear regression (curve fit) asymmetric sigmoidal, 5PL, where X is log(concentration | GraphPad Prism 9 | ||
| Disposable PES Filter Units 500mL | Fisher Scientific | 15913307 | |
| Disposable Pipette Basins 50ml | Fisher Scientific | 12369175 | |
| DMSO (Dimethyl sulfoxide) | Sigma-Aldrich | Sigma D2650 | |
| Dulbeco’s Phosphate Buffered Saline without Ca2+ and Mg2+ (D-PBS) | ThermoFisher Scientific | 14190-144 | |
| Easy Reader Conical Polypropylene Centrifuge Tubes 15 mL | Fisher Scientific | 11889640 | |
| Foetal bovine serum | Gibco | 10500064 | |
| Human ALT ELISA Kit | Abcam | ab 234578 | Dilution 1:5. Time point Day 6 and 8. |
| Human Cryopreserved Kupffer Cells | Lonza Europe | Lot. 190088KC | |
| hydrocortisone | Merck | H0888-1G | |
| Incubators models: New Brunswick Galaxy 170 S, New Brunswick Galaxy 170 R and CellXpert® C170. | Eppendorf | All serviced yearly; paperwork available upon request. | |
| Inverted Microscope | Leica DMIL LED | ||
| MPS know as Organ-on-a-Chip (OOC) | CN Bio Innovations Ltd. | ||
| MPS LC-12 plate | CN Bio Innovations Ltd. | ||
| Neubauer Improved C-Chip Disposable Haemocytometer (2 channel) | Cambridge Bioscience | DHC-N01-50 | |
| P450-Glo CYP3A4 Assay and Screening System | Promega | V9002 | Dilution - none. Time point Day 8 |
| PhysioMimix MPS platform | CN Bio Innovations Ltd. | ||
| Pioglitazone | MedChemExpress Tocris | HY-13956/CS-1700 | |
| Quantichrom Urea Assay Kit – Bioassay systems | Bioassay Systems | DY970-05 | Dilution 1:2 if initial reading is too high. Time point Day 4, 6 and 8. |
| Silica gel | Sigma-Aldrich | S7625 | |
| Software used to analyse and generate all the graphs was | GraphPad Prism 9 | ||
| Stripettes 10 mL | Fisher Scientific | 11839660 | |
| Stripettes 25 mL | Fisher Scientific | 11839181 | |
| Thawing plate Cocktail A, (Primary Hepatocyte Thawing and Plating Supplements) | Gibco | CM3000 | |
| Troglitazone | MedChemExpress Tocris | 97322-87-7 | |
| Trypan Blue Solution, 0.4% | Gibco | 15250061 | |
| Tubes 1.5 mL | Greiner | 616201 | |
| Weighing balance - model PA214C and AV213C | Ohaus Corp |
References
- Lisi, D. M. Drug-induced liver injury: An overview. US Pharmacist. 41 (12), 30-34 (2016).
- Kuna, L., et al. Models of drug induced liver injury (DILI)-current issues and future perspectives. Current Drug Metabolism. 19 (10), 830-838 (2018).
- Katarey, D., Verma, S. Drug-induced liver injury. Clinical Medicine. 16 (6), 104-109 (2016).
- Kullak-Ublick, G. A., et al. Drug-induced liver injury: recent advances in diagnosis and risk assessment Recent advances in clinical practice. Gut. 66, 1154-1164 (2017).
- Dirven, H., et al. Performance of pre-clinical models in predicting drug-induced liver injury in humans: a systematic review. Scientific Reports. 11 (1), 6403 (2021).
- Donato, M. T., Lahoz, A., Castell, J. V., Gomez-Lechon, M. J. Cell lines: a tool for in vitro drug metabolism studies. Current Drug Metabolism. 9 (1), 1-11 (2008).
- Wilkening, S., Stahl, F., Bader, A. Comparison of primary human hepatocytes and hepatoma cell line HepG2 with regard to their biotransformation properties. Drug Metabolism and Disposition. 31 (8), 1035-1042 (2003).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28 (2), 69-87 (2012).
- Grainger, C. I., Greenwell, L. L., Lockley, D. J., Martin, G. P., Forbes, B. Culture of Calu-3 cells at the air interface provides a representative model of the airway epithelial barrier. Pharmaceutical Research. 23 (7), 1482-1490 (2006).
- Li, F., Cao, L., Parikh, S., Zuo, R. Three-dimensional spheroids with primary human liver cells and differential roles of kupffer cells in drug-induced liver injury. Journal of Pharmaceutical Sciences. 109 (6), 1912-1923 (2020).
- Proctor, W. R., et al. Utility of spherical human liver microtissues for prediction of clinical drug-induced liver injury. Archives of Toxicology. 91 (8), 2849-2863 (2017).
- Lin, C., Khetani, S. R. Advances in engineered liver models for investigating drug-induced liver injury. BioMed Research International. 2016, 1829148 (2016).
- Olson, H., et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regulatory Toxicology and Pharmacology. 32 (1), 56-67 (2000).
- Bell, C. C., et al. Comparison of hepatic 2D sandwich cultures and 3D spheroids for long-term toxicity applications: A multicenter study. Toxicological Sciences. 162 (2), 655-666 (2018).
- Bell, C. C., et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Scientific Reports. 6, 25187 (2016).
- Khetani, S. R., et al. Use of micropatterned co-cultures to detect compounds that cause drug-induced liver injury in humans. Toxicological Sciences. 132 (1), 107-117 (2013).
- Ma, X., et al. Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proceedings of the National Academy of Sciences of the united States of America. 113 (8), 2206-2211 (2016).
- Dieterle, P. Y. M., Dieterle, F. Tissue-specific, non-invasive toxicity biomarkers: translation from pre-clinical safety assessment to clinical safety monitoring. Expert Opinion on Drug Metabolism & Toxicology. 5 (9), 1023-1038 (2009).
- Rowe, C., et al. Perfused human hepatocyte microtissues identify reactive metabolite-forming and mitochondria-perturbing hepatotoxins. Toxicology in Vitro. 46, 29-38 (2018).
- Rubiano, A., et al. Characterizing the reproducibility in using a liver microphysiological system for assaying drug toxicity, metabolism, and accumulation. Clinical and Translational Science. 14 (3), 1049-1061 (2021).
- Tsamandouras, N., Kostrzewski, T., Stokes, C. L., Griffith, L. G., Hughes, D. J., Cirit, M. Quantitative assessment of population variability in hepatic drug metabolism using a perfused three-dimensional human liver microphysiological system. Journal of Pharmacology and Experimental Therapeutics. 360 (1), 95-105 (2017).
- Ortega-Prieto, A. M., et al. 3D microfluidic liver cultures as a physiological pre-clinical tool for hepatitis B virus infection. Nature Communications. 9 (1), 682 (2018).
- Kostrzewski, T., et al. Three-dimensional perfused human in vitro model of non-alcoholic fatty liver disease. World Journal of Gastroenterology. 23 (2), 204-215 (2017).
- Kostrzewski, T., et al. A microphysiological system for studying nonalcoholic steatohepatitis. Hepatology Communications. 4 (1), 77-91 (2020).
- Vacca, M., et al. Bone morphogenetic protein 8B promotes the progression of non-alcoholic steatohepatitis. Nature Metabolism. 2 (6), 514-531 (2020).
- Long, T. J., et al. Modeling therapeutic antibody-small molecule drug-drug interactions using a three-dimensional perfusable human liver co-culture platforms. Drug Metabolism and Disposition. 44, 1940-1948 (2016).
- Bai, J., Wang, C. Organoids and microphysiological systems: New tools for ophthalmic drug discovery. Frontiers in Pharmacology. 11, 407 (2020).
- Ribeiro, A. J. S., Yang, X., Patel, V., Madabushi, R., Strauss, D. G. Liver microphysiological systems for predicting and evaluating drug effects. Clinical Pharmacology & Therapeutics. 106 (1), 139-147 (2019).
- Clark, A. M., et al. A microphysiological system model of therapy for liver micrometastases hhs public access. Experimental Biology and Medicine (Maywood). 239 (9), 1170-1179 (2014).
- Qosa, H., Ribeiro, A. J. S., Hartman, N. R., Volpe, D. A. Characterization of a commercially available line of iPSC hepatocytes as models of hepatocyte function and toxicity for regulatory purposes. Journal of Pharmacological and Toxicological Methods. 110, 107083 (2021).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved