
Method Article
Shotgun Lipidomics of Rodent Tissues
In This Article
Summary
Shotgun mass spectrometry-based lipidomics delivers a sensitive quantitative snapshot of a broad spectrum of lipid classes simultaneously in a single measurement from various rodent tissues.
Abstract
Lipids play a vital role as essential components of all prokaryotic and eukaryotic cells. Constant technological improvements in mass spectrometry have made lipidomics a powerful analytical tool for monitoring tissue lipidome compositions in homeostatic as well as disease states. This paper presents a step-by-step protocol for a shotgun lipid analysis method that supports the simultaneous detection and quantification of a few hundred molecular lipid species in different tissue and biofluid samples at high throughput. This method leverages automated nano-flow direct injection of a total lipid extract spiked with labeled internal standards without chromatographic separation into a high-resolution mass spectrometry instrument. Starting from sub-microgram amounts of rodent tissue, the MS analysis takes 10 min per sample and covers up to 400 lipids from 14 lipid classes in mouse lung tissue. The method presented here is well suited for studying disease mechanisms and identifying and quantifying biomarkers that indicate early toxicity or beneficial effects within rodent tissues.
Introduction
Cigarette smoke (CS) is recognized as a main risk factor associated with the development of chronic inflammatory diseases of the lung, including pulmonary carcinoma, bronchitis, and chronic obstructive pulmonary disease (COPD)1. Beyond the lung impact, CS exposure plays a significant role in the development of other diseases such as atherosclerotic coronary artery disease and peripheral vascular disease1. Cardiovascular disease, together with COPD, are the first and third leading causes of death worldwide, respectively. Toxicological risk assessment approaches have historically relied on the use of animal models such as rodents. In vivo nose-only or full-body rat and mouse models are commonly used for studying the long-term effects of exposure to CS.
For example, generally, 6 months of smoke exposure induces histological and functional abnormalities in murine lungs that mimic those of human disease, including emphysema, airway remodeling, and pulmonary hypertension, although the changes are relatively mild compared with those observed in long-term human smokers2. In both animal and human tissues, studies have observed a wide range of molecular changes in response to CS exposure, including oxidative stress responses, inflammation, and structural tissue changes3,4. Not surprisingly, CS exposure has also been reported to have a far-reaching impact on the lung lipidome, including effects on surfactant lipids, lipid signaling mediators, and structural lipids4,5,6.
To characterize the bulk lipid changes induced by the long-term CS exposure of mouse lungs, we performed a fast and quantitative shotgun direct infusion mass spectrometry analysis. After the introduction of the shotgun lipidomics method in 20057, this method has been effectively used to extend our knowledge on lipid cellular metabolism8,9,10 in model systems such as yeast11, Caenorhabditis elegans12, and Drosophila melanogaster13, as well as in a wide range of mammalian sample types such as cell lines14,15,16 various human17,18 and rodent tissues19,20 and body fluids21,22.
Over the last decades, studies have revealed the complexity of cellular responses to environmental changes, involving thousands of interconnected proteins, lipids, and metabolites. This has made it clear that using state-of-the-art analytical techniques is essential for gaining an in-depth view of the molecular machineries and for uncovering the full magnitude of exogenous physiological impacts. In this context, the comprehensive, quantitative lipid fingerprints produced by shotgun lipidomics approaches can effectively add to our knowledge on lipid cellular metabolism8,9,10.
Regarding CS exposure as a risk factor for several diseases, toxicological risk assessment approaches have historically relied on the use of animal models such as rodents. Shotgun lipidomics MS provides a fast, sensitive, and quantitative analytical tool for assessing lipidome perturbation in numerous sample types. The unique feature of shotgun lipidomics is the automated direct-injection analysis of a total lipid extract-spiked with labeled internal standards-without chromatographic separation into a high-resolution MS instrument by using a conductive nanochip generating an electrospray ionization (ESI) nanospray23.
The mass-to-charge ratio information that is simultaneously acquired in the MS1 mode provides the total lipid footprint of all intact endogenous lipids. Optionally, the MS2/MS3 mode, in which the parent lipid molecules are fragmented and analyzed, provides additional structural information. Data analysis requires specialized software and includes the deconvolution of spectra and peak assignments in pooled spectra that lead to lipid identification and putative chemical structure elucidation. Additionally, absolute quantification can be performed by spiking a labeled internal standards mixture containing at least one lipid standard per lipid class of interest. Overall, with the present technique, MS analysis taking a few minutes per sample can cover the identification and quantification of up to 800 lipids from 14 lipid classes24 in rodent tissues.
Protocol
All procedures involving animals were performed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International and licensed by the Agri-Food & Veterinary Authority of Singapore, with approval from an Institutional Animal Care and Use Committee and in compliance with the National Advisory Committee for Laboratory Animal Research Guidelines on the Care and Use of Animals for Scientific Purposes (NACLAR, 2004).
1. Sample collection
- Perform mouse lung dissections after 3 months and 6 months of exposure of ApoE−/− mice. Collect the tissues 16-24 h after exposure, snap-freeze them in liquid nitrogen, and store at −80 °C before initiating the lipidomics workflow. Ensure the collection of a total of nine samples from each "omics" dissection group.
2. Tissue - pulverization of the samples
- Prepare the magnetic hammer and consumables for tissue grinding. Precool tissue pouches, forceps, capped tissue transfer tubes with caps, a "V" plastic spatula, and 2 mL plastic collection tubes on dry ice. Install the corresponding tube holder on a magnetic hammer instrument. Switch on the magnetic hammer and set the impact power to High.
- Load the sample into a tissue pouch; insert the sample through the top opening of the tissue pouch using precooled forceps if needed. Place the sample at the center of the flexible pouch, away from the edges.
- Seal the tissue pouch by screwing a precooled collection tube onto the top of the tissue pouch.
- Snap-freeze the sample by immersing the flexible pouch into liquid nitrogen for 10 s.
- Vent the collection tube; loosen the tube for a 1/4 turn for venting to avoid rupturing the pouch when the magnetic hammer impacts.
- Load the frozen tissue pouch onto the magnetic hammer.
- Apply the magnetic hammer by pressing the activate button to pulverize the sample. If non-pulverized tissue pieces remain in the pouch, snap-freeze the sample in liquid nitrogen again and repeat for a second impact.
- Remove the tissue pouch from the magnetic hammer, keeping the pulverized sample at the bottom. To prevent the sample from melting, snap-freeze it again immediately.
- Transfer the sample to a collection tube. Quickly invert the tube so that the tissue pouch is at the top, and tap the pouch to transfer the tissue particles into the bottom of the collection tube.
NOTE: This step should be done quickly to avoid the melting of the tissue. - Transfer a 10 mg tissue aliquot into a 2 mL tube for further lipidomic analysis and store the sample at −80 °C until further steps are performed.
3. Tissue homogenization
- Switch on the tissue lyser instrument and set the shaking frequency at 30 Hz and the shaking duration at 2 min. Prepare the tissue homogenization buffer solution: 150 mM ammonium bicarbonate in water, with a pH of ~8.2.
- Take out the samples from the storage freezer, place on ice, and add four stainless steel beads into the tubes containing the tissue powder. Add 0.5 mL of 150 mM ammonium bicarbonate buffer to each sample.
- Place the sample tubes into the tissue lyser holders. Counterbalance both holders with the same number of tubes. Fix the holders into the tissue lyser holding arm. Disrupt the tissue.
- Place the tubes on ice and visually check that no leftover tissue aggregates are present in the tube. If tissue disruption is incomplete (aggregates are seen), repeat the disruption process by performing another treatment cycle. Place the samples on ice.
- Create an aliquot 1 of 100 µL of the sample in a 1.5 mL tube dedicated for protein determination. Centrifuge it at 18,200 × g for 5 min at 10 °C.
- Determine the protein content of aliquot 1 by the Bradford assay (see section 4). Store the samples on ice.
- Create an aliquot 2 of 20-100 µL of the stock homogenate in a new 2 mL microtube for lipid extraction (see section 5). If samples are not immediately analyzed, keep them on ice until the extraction is performed.
NOTE: The final extraction volume must be defined on the basis of the total protein amount. It must be in the range of 0.015-0.045 mg of protein per total aliquot.
4. Bradford protein determination assay
NOTE: Study samples should be randomized before the beginning of the analysis, and the randomization is respected for all steps of sample processing.
- Dilute centrifuged aliquot 1 supernatant 2x with ammonium bicarbonate buffer.
NOTE: The dilution factor depends on the concentration of the tissue homogenate. For more concentrated solutions, apply a higher factor so that the determined concentration falls in the dynamic range of the assay. - Prepare a bovine serum albumin (BSA) standard curve according to Table 1. Vortex the standard tubes. Centrifuge the tubes at 18,200 × g for 15 s at room temperature.
- From the standard tubes previously prepared, transfer 6 µL each of the blank and standards to a 96-well flat-bottom plate according to the plate layout shown in Figure 1.
- Transfer the previously prepared samples to the 96-well flat-bottom plate according to the plate layout described in Figure 1.
- Add 250 µL of the Bradford Reagent to each well by using a multichannel pipette and mix. Incubate for 5 min.
- Measure the absorbance at a 595 nm wavelength by using a plate reader.
- Calculate the protein concentrations in the aliquots based on the standard curve.
NOTE: The dilution factor used at the beginning of the procedure should be considered when calculating the protein concentrations.
5. BUME extraction
NOTE: Avoid the use of low-bind plastic tubes for lipidomics extraction, since the strong organic solvents release plasticizers and create a strong background during sample analysis.
- Prepare 1.5 mL tubes with 100 µL of ammonium bicarbonate solution in each tube of the total blank (TB), internal standard blank (ISB), and quality control samples (QC), considering 1 QC sample is placed between each set of 10 samples. Keep all samples on ice.
- Add 10 µL of pooled human plasma to all QC samples. Spike 10 µL of internal standard solution into all ISB, QC, and study samples (i.e., all samples except TB).
NOTE: For quality control material, use any pooled commercially available K2EDTA plasma. Quantify the lipid concentration in pooled plasma using the mentioned protocol upon receipt and keep these ranges as references for all analyses in which the QC material is used. Use NIST plasma only for more targeted applications or for global method verification, where the accuracy of the assay needs to be addressed. - Add 300 µL of −20 °C butanol/methanol mixture (3/1, v/v) to each sample. Shake at 450 × g for 10 min at room temperature on the thermomixer.
- Add 300 µL of heptane/ethyl acetate mixture (3/1, v/v). Shake at 450 × g for 10 min at room temperature on the thermomixer.
- Add 300 µL of 1% acetic acid mixture. Shake for 5 min at 450 × g at room temperature on the thermomixer. Centrifuge at 2,800 × g for 5 min at room temperature. Transfer 360 µL of the upper phase to a new 2 mL self-lock tube 2.
NOTE: Liquid-liquid extraction can result in shifting positions of the phase boundary in a sample-dependent manner, which can result in a slight volume discrepancy for the upper phase. Adjust the collection volume for the upper phase if necessary. - Add 320 µL of heptane/ethyl acetate (3/1, v/v) to the water phase tube 1. Shake at 450 × g for 5 min at room temperature on the thermomixer. Centrifuge at 2,800 × g for 5 min at room temperature. Transfer 320 µL of the upper phase and combine with the fraction from step 5.11.
NOTE: Adjust the collection volume for the upper phase if necessary. - Add 250 µL of heptane/ethyl acetate (3/1, v/v) to the water phase in tube 1. Shake at 450 × g for 5 min at room temperature on the thermomixer. Centrifuge at 2,800 × g for 5 min at room temperature. Transfer 200 µL of the upper phase and combine with the fractions from step 5.11 and step 5.15 in tube 2.
NOTE: Adjust the collection volume for the upper phase if necessary. - Evaporate to dryness at 35 °C in a vacuum concentrator.
NOTE: Dried samples can be stored at −20 °C until analysis, if required. - Redissolve in 300 µL of MS mix solution (7.5 mM ammonium acetate in isopropanol/methanol/chloroform, 4:2:1 solution). Vortex each tube for 5 s to ensure that everything is dissolved. Centrifuge at 18,200 × g for 5 min at 4 °C.
- Transfer an aliquot of 50 µL into the microliter plate according to the plate layout in Figure 2 for positive ionization mode analysis (columns 1-6) and dilute with 50 µL of MS mix solution.
- Transfer an aliquot of 20 µL into the MTP plate according to the plate layout in Figure 2 for negative ionization mode analysis (columns 7-12) and dilute with 80 µL of MS mix. Wrap the plate with foil and keep at 4 °C before the analysis.
6. MS analysis
- Calibrate a mass spectrometer (MS) both in positive and negative modes in accordance with the manufacturer's instructions 5 days or less before the analysis.
- Install the direct infusion nano-source upfront, ensuring that it is properly aligned against the transfer capillary of the MS. Install a 4.1 µm nozzle chip into the chip holder and configure it in the software.
- Use the following parameters for positive and negative mode method setup: gas pressure of 1.25 psi; source voltage of 1.1 kV; 5 µL of sample injection volume; output contact closure Rel1 for 2.5 s; 5 min of delivery time; plate cooling at 4 °C.
- Set up the analysis sequence queue for the direct infusion robot in positive mode.
- Set up the MS method for positive mode using the following MS settings:
- Set the capillary temperature at 250 °C and the S-lens RF level at 65.0. Perform full-scan MS acquisition from 0 to 1 min in the 550-1000 m/z range at 140,000 resolution with automated gain control of 1 ×106 and maximum injection time of 50 ms. Apply lock mass of 680.48022.
- Set data-independent MS/MS acquisition method between the 1 and 5 min time range at 17,500 resolution with a fixed first mass of 250 m/z. Use an automated gain control for MS2 at 1 × 105 and maximum injection time of 64 ms, a collision energy of 20 NCE, and an isolation window of 1 m/z. Use the inclusion mass list from 550 to 1,000 m/z, with a mass step of 1 Da.
- For the acquisition in negative mode, set the capillary temperature at 250 °C and the S-Lens RF level at 65.0 in the MS tune file.
- Use the following MS method settings: full-scan acquisition mode from 0 to 1 min at 140,000 resolution covering the 400-940 m/z range, automated gain control of 1 × 106; maximum injection time of 50 ms; use a lock mass of 529.46262.
- Set MS2 acquisition in DIA mode between 1 and 5 min of the run at a resolution of 17,500 with a fixed first mass of 150 m/z; automated gain control of 1 × 105; 64 ms of maximum injection time; and 35NCE of collision energy. Use the inclusion mass list from 400 to 940 with a mass step of 1 Da.
- Create the analysis sequence queue in MS software in positive mode. Wait for the sample acquisition to be triggered by a contact closure signal coming from the direct infusion robot for each sample.
- When positive mode analysis is complete, create the sequence queue for the direct infusion robot in negative mode.
- Set up the analysis sequence queue in MS software in negative mode.
7. Data processing
- To convert the data files from the positive and negative modes from .raw format to .mzML format, use the converter software.
NOTE: Data files from positive and negative modes must be converted separately (because of their different acquisition settings and different noise threshold).- Fill the following settings to perform file conversion: noise threshold factor is 3 and 5 for MS and MS2, respectively; activate the noise removal function for both MS and MS2, data compression, average MS2 scans, and peak picking. Set TIC thresholds for positive and negative modes (e.g., 10,000 and 5,000; to be defined based on the noise level of the particular sample produced by a specific instrument). Generate XLC and PDF reports at the end of the processing.
- Perform the lipid identification. Define the following (example) file import settings parameters: selection window ±0.5 Da (depends on the selection window in the MS method); time range 2-300 s (depends on scan time in the MS method); no calibration masses for MS and MS2; transfer and round up the mass range from the Peak by Peak software metadata files (min mass [MS]) and (min mass [MS2]); tolerance of 3 ppm and 10 ppm for MS and MS2, respectively; keep empty the MS and MS2 threshold field, frequency filter, MS1 offset and PMO; Isotopic correction for MS and MS2 selected; transfer resolution gradient from the converter metadata file as (Resolution linear fit [MS]) and as (Resolution linear fit [MS2]).
NOTE: Lipid identification in the positive and negative modes must be performed separately because of their different acquisition settings. - After the data are imported, go to the Run menu and upload the MFQL files for lipid identification.
NOTE: For detailed information on the structure of MFQLs, refer to the publication by Herzog et al.25. See some examples of MFQL files at https://wiki.mpi-cbg.de/lipidx/MFQL_library and see the discussion. All MFQLs used for the data processing are provided in the Supplementary Materials. - Quantify the identified species on the MS level by dividing the precursor mass intensity of the lipid feature by the respective intensity of the labeled internal standard and then multiplying the quotient by the concentration of the labeled internal standard. Normalize the final lipid concentration per amount of total protein measured by the Bradford assay.
8. QC check procedure
NOTE: A quality check procedure is an essential step to verify the technical reproducibility of the method. To this end, a commercial pooled plasma is analyzed to determine the endogenous levels of different lipids over 5 days in 15 identical aliquots per batch (total of five batches). Note that, in this case, a data evaluation pipeline was created as a Shiny web application using the R statistical software environment.
- Define the reference acceptance range as the average value calculated for all five batches ±3 standard deviations.
- Apply the "pass" acceptance rules for each analytical batch: 90% of the reference targets must pass, considering that one reference compound represents one lipid class. For example, if 15 reference targets are included in the QC verification, ensure that 13 targets pass for the batch to be accepted.
NOTE: In other words, 68% of QC samples per reference compound must pass for the data for this lipid class to be accepted. - If the study batch does not meet the acceptance criteria for the QC check, repeat the procedure.
9. Estimation of (relative) conjugated fatty acid amounts
- For each lipid class, estimate the amounts of conjugated fatty acids based on their MS2 intensities.
NOTE: Due to fragmentation and ionization differences, the derived values were only meant for relative abundance comparisons across sample groups rather than, for example, to estimate the relative contribution of a specific fatty acid to the overall conjugated fatty acid pool of a lipid class. - Normalize the MS2 intensities of the fatty acid fragments by the average intensity of the fatty acid fragments of the corresponding internal standard. Based on the concentration of the internal standard and the measured tissue weight, derive a "normalized concentration" for the conjugated fatty acids. Sum up these values per lipid class to estimate the total amount of each conjugated fatty acid per sample.
10. Statistical analysis
- Perform statistical analysis. Fit a linear model for each condition and the corresponding control group, and calculate p values from a t-statistic for log2-transformed data. Use the Benjamini-Hochberg false discovery rate method to correct for multiple testing effects.
NOTE: Lipids with adjusted p values < 0.05 are considered differentially abundant. Here, statistical analysis was performed in the R statistical environment26.
Results
Here, as representative results, we present data illustrating how shotgun lipidomics can be used to study the effects of CS on the lungs of mice. The shotgun lipid analysis protocol was successfully applied for an in vivo study for comparative analysis of two groups of samples: lung tissues dissected from nine female Apoe−/− mice exposed to CS (3R4F group; positive control) and an equal number of mice maintained under fresh air conditions (sham group) as negative control collected at 3 months, 4 months, and 6 months exposure time points. Animals were kept under filtered and conditioned fresh air at 22 °C ± 2 °C temperature and 55% ± 15% humidity and fed with a gamma-irradiated pellet diet. The light regime was maintained at 12 h per day. Up to eight mice were housed per each cage. 3R4F reference cigarettes for the exposure treatment were purchased from the University of Kentucky to generate the 3R4F CS aerosol (600 µg of total particulate matter TPM/L aerosol) for a target exposure concentration equivalent to 28 µg of nicotine/L. CS smoke from 3R4F cigarettes was produced using 30 port rotary smoking machines according to Phillips et al.27. Based on the Health Canada Intensive Smoking Protocol, 55 mL of puff volume, one puff per 30 s, and 100% blockage of ventilation holes were applied to 3R4F cigarettes to provide sufficient exposure. All additional details on the study design have been reported previously27,28.
Overall, ~400 molecular lipid species from the 14 most abundant lipid classes were identified and quantified (Figure 3). The total normalized lipid concentration per class was slightly elevated for the PE, PEO, PC, PCO, PI, and PG lipid classes, and some downregulation was observed for SM, LPE, and PA lipids (Figure 3A), while no difference could be seen for TAGs and PS. Interestingly, elevated levels of PCO and PEO plasmalogen lipids in the CS group have been reported in inflammatory cells29. One of the intracellular functions of plasmalogen lipids is antioxidant activity. Under exposure to reactive oxygen (ROS) and nitrogen species (RNS), selective oxidation of plasmalogens in comparison with diacyl phospholipids was reported30. The enyl-ether bond in the chemical structure of plasmalogens is sensitive to radical attack upon oxidative stress31. The major products of radical attack are hydroxylated eicosatetraenoic acids, 2-monoacylglycerol phospholipids, pentadecanol, formic acids, α-hydroxyaldehydes of various chain lengths, 1-formyl-2-arachidonoyl glycerophospholipid, and lysophospholipids.
These results show that, among the molecular features based on the total molecular formula, 100-120 compounds were significantly impacted by CS exposure (Figure 3D). The detailed deconvolution of the MS2 fragmentation information and normalization of the fragment signal intensities allowed the approximate definition of the total amount of each conjugated fatty acid (FA) represented in each lipid class (Figure 3E) and comparison of these data between exposed and control groups. A clear decrease in both fully saturated FAs and ones with only a few unsaturations, mostly in the context of lyso-lipids, was observed. In contrast, polyunsaturated fatty acids (PUFA) in the composition of PC, PE, and PG phospholipid classes were elevated in the CS group for all time points. The extreme cases of PC and PG conjugated with eicosapentanoic (EPA) and docosahexaenoic acid (DHA) were exclusively detected in the 3R4F CS exposure group (Figure 3E, red color).

Figure 1: Plate layout for sample distribution in the Bradford assay. Blank and standard samples are placed in triplicate in columns 1-3; unknown samples are placed in duplicate in columns 4-11. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Plate layout of the 96-well microtiter plate for shotgun mass spectrometry analysis. The distribution of blank, standard, unknown, and QC samples for acquisition in the positive ionization mode is mapped on the left side of the plate (columns 1-6); all samples for the negative ionization mode are placed on the right (columns 7-12). Abbreviations: pos = positive; QC = quality control; neg = negative; TB = total blank. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Shotgun lipidomics analysis of mouse lungs exposed to cigarette smoke. Apoe−/− mice were exposed to CS from the 3R4F reference cigarette or fresh air (sham). (A) Lipid class concentrations and the number of lipid species per class. For each class, the number of quantified lipid species is given in brackets. Mean and 95% confidence intervals for each lipid class, separately for each exposure type (aggregated across three time points). (B) Volcano plots showing the effect size (log2 fold change) and significance (-log10 FDR-adjusted p value) of the quantified lipid species for the 3R4F CS versus sham comparison at the three time points. Significantly elevated lipids are marked in yellow; significantly decreased lipids are marked in cyan (FDR-adjusted p value < 0.05). (C) Differential abundance of the 25 lipid species with the largest absolute mean fold changes. Log2 fold changes for the 3R4F CS versus sham comparison are color-coded, and statistical significance is indicated: *: FDR-adjusted p value < 0.01; X: FDR-adjusted p value < 0.05. (D) Comparison plots. Correlation coefficients for the fold-change comparisons are color-coded, and the number of shared differentially abundant lipids is indicated (total numbers in the margins). Pie charts show the percentage of shared differentially abundant lipids with the same direction of change (FC sign). Asterisk indicates a significant overlap of the differentially abundant lipids. (E) Differential abundance of conjugated FAs per lipid class. Heatmap as in panel C for conjugated fatty acids with significant abundance differences between 3R4F and sham exposure in all three time points (FDR-adjusted p value < 0.05). The amount of each fatty acid per lipid class was estimated based on the MS2 intensities of the fatty-acid fragments. Abbreviations: CS = cigarette smoke; FDR = false discovery rate; FC = fold change; FA = fatty acids; PC = phosphatidylcholine; PS = phosphatidylserine; TAG = triacylglycerol; SM = sphingomyelin; PEO = plasmalogen phosphatidylethanolamine; PE = phosphatidylethanolamine; PG = phosphatidyl glycerol; PI = phosphatidylinositol; DAG = diacylglycerol; SE = sterol/cholesteryl esters; PCO = plasmalogen phosphatidylcholine; LPC = lyso phosphatidylcholine; LPE = lyso phosphatidylethanolamine; PA = phosphatidic acid. Please click here to view a larger version of this figure.
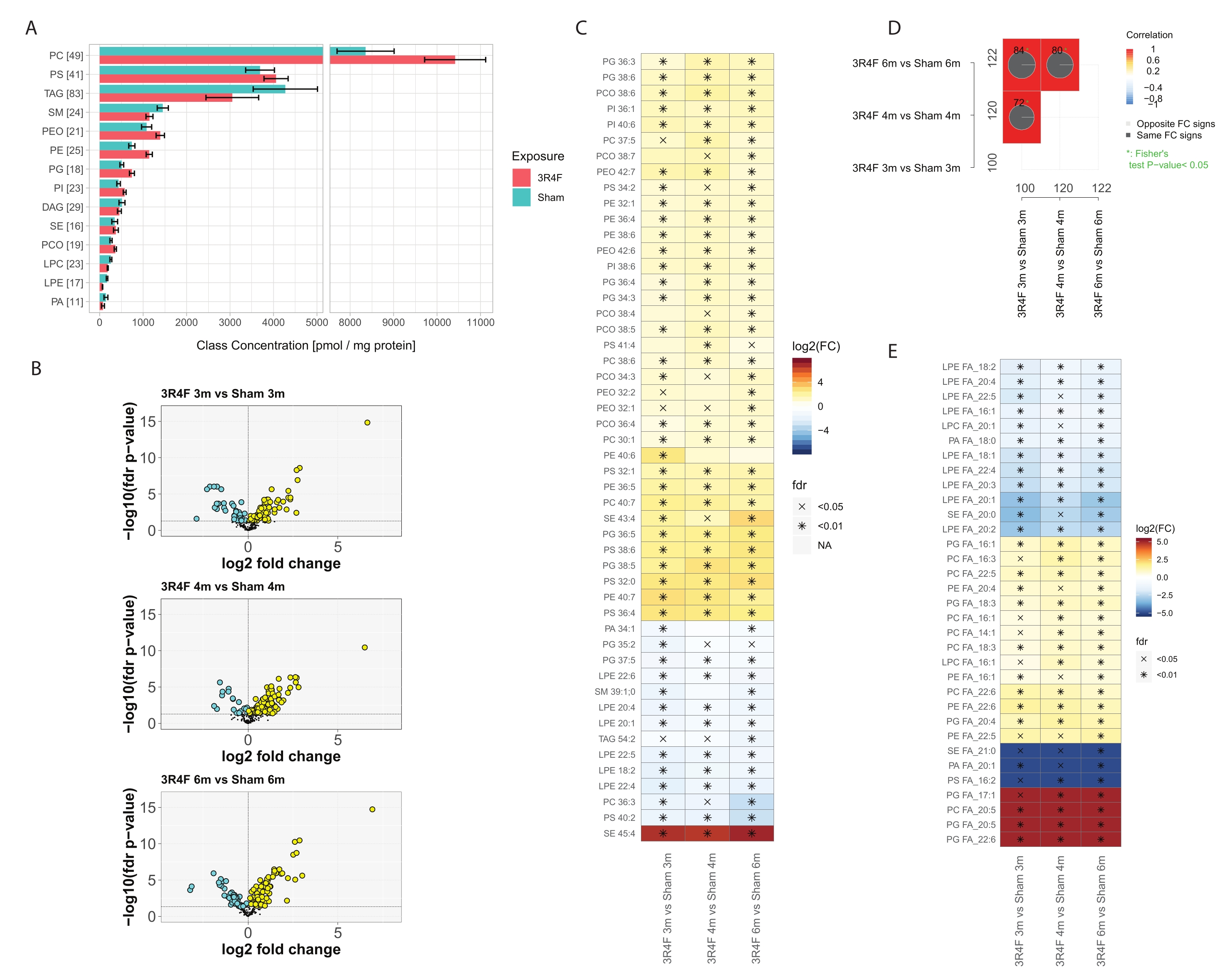{kind=link}
| BSA final concentration (mg/mL) | BSA solution at 2 mg/mL (µL) | Ammonium bicarbonate buffer (µL) |
| 1 | 50 | 50 |
| 0.75 | 37.5 | 62.5 |
| 0.5 | 25 | 75 |
| 0.25 | 12.5 | 87.5 |
| 0.125 | 12.5 | 187.5 |
| 0.0625 | 50 of 0.125 mg/mL solution | 50 |
| 0 | 0 | 100 |
Table 1: Dilution scheme of the BSA internal standard for the calibration curve for the Bradford assay. Abbreviation: BSA = bovine serum albumin.
Supplementary Information: MFQL files used for LipidXplorer lipid identification. The .zip package consists of the individual required MFQL files, each named following this pattern: <lipid class abbreviation>_<ionization mode>_MS2.mfql (e.g., PE_neg_MS2.mfql). The file names for labeled internal standard identification include the D7(D9) tag in the <lipid abbreviation> (e.g., PED7_neg_MS2.mfql). These MFQL files are used to verify the amount of deuterium in the internal standard. Please click here to download this File.
Discussion
Although advances in MS have yielded a variety of methods to accurately monitor many lipid species, the previous lipid profiling of various mammalian tissues did not show consistent results and, consequently, the particular tissue-specific functions of lipids remain unclear. In comparison with protein functional analysis, for which more robust approaches to knock out the expression of particular compounds are available, the majority of lipids cannot be selectively turned off or overexpressed in tissues, making the functional evaluation of lipids difficult. Advanced profiling of tissue lipidome concentrations may provide an alternative approach to identify associations between circulating lipids and human diseases.
In evaluating comprehensive lipidomics methods capable of qualitatively and quantitatively covering the endogenous lipid profile of any mouse tissue, we gave preference to the shotgun lipidomics method. In general, two opposite types of sample analysis are possible: either a completely untargeted screening of lipids using liquid chromatography (LC)-based separation with further mass spectrometric detection to reveal the total lipid complexity of the sample, or targeted approaches, which mostly allow for very precise quantification of specific lipids of interest. In contrast, the strong feature of the presented shotgun lipidomics workflow is the rapid comprehensive coverage of hundreds of endogenous lipids from predefined lipid classes, which can still be performed in a robust semi-quantitative manner.
The ionization efficiencies of different lipid classes are structure-dependent and can vary drastically depending on the different experimental ionization conditions. In contrast to LC-based separation methods, shotgun analysis minimizes these differences due to the direct simultaneous infusion of the whole lipid extract under the same ionization conditions into the MS instrument. The use of isotopically labeled lipid analogs or structurally similar non-endogenous standards allows semi-quantification of all lipid classes. Shotgun lipidomics provides low inter- and intra-run variability during MS analysis; as a result, this method produces lower coefficients of variation21 than untargeted liquid chromatography-based methods, which require multiple standards for adequate quantification. Importantly, while no external calibration curve is used in the current method, the method is still considered fully quantitative32.
One level of labeled internal standard (or unlabeled standard that is not endogenously expressed) per lipid class is sufficient for the quantification of most lipids. Only a few publications have reported a partial method validation for a shotgun lipidomics method. For example, in Gryzbek et al.17 and Surma et al.21, reversed calibration curves were prepared using internal standards and a fixed amount of sample matrix. Linearity was assessed by the linear regression of log-transformed lipid amounts and their intensities and reported as R2 and slope, respectively. The limit of detection (LOD) and limit of quantitation (LOQ) were determined by weighted linear regression based on a signal-to-noise ratio of 3 for LOD and 10 for LOQ. For most lipid classes, LOQ was defined between 2-9.8 pmol for adipose tissue and 0.05-5µM in plasma. In both cases, non-endogenous single internal standards per class were used to derive estimates for all lipids in the class. However, in this work, we do not provide LOD/LOQ due to several concerns: the endogenous matrix is not compound-free, and the surrogate matrix for tissues is unavailable-with this, the spike of small known amounts of standards is not possible. We do not perform a classical targeted quantification approach with the use of a calibration curve series of a particular compound normalized by its identical isotopically labeled standard due to the nonexistence of pure standards and the very limited availability of isotopically labeled lipids. Orbitrap detectors automatically perform the conversion of the transient signal by applying a Fourier transformation, and some signal is already substituted-as a result, the lower concentration range will be only linear down to some minimum signal, below which the molecule will not be detectable anymore. Xcalibur software signal-to-noise values depend on the m/z ratio of the molecule; as a result, the compounds of each lipid class, containing different combinations of fatty acids, will have different noise values. Moreover, LOQ/LOQ values are strictly linked to the type of matrix, and when quantification of the lipidome is done in different rodent tissues, it should be reflected by the assessment of LOQs for each tissue type individually.
In fact, the method offers a high linear dynamic quantification range of up to four orders of magnitude33 and very good sensitivity to cover the most important endogenous structural lipids, which can be further increased by technical improvements in MS acquisition32. The coefficients of variation of the mean lipid concentrations were mostly below 15%, meaning that shotgun lipidomics complies with Food and Drug Administration (FDA) requirements as a method to be considered for good laboratory practice (GLP) and good clinical practice (GCLP) studies34.
However, it must be noted that, due to their different polarity, some lipid classes become much more impacted by the contribution of specific conjugated FAs. This leads to the distortion in the intensity response in equimolar mixtures containing a broad range of conjugated FAs, resulting in quantification bias, as highlighted by Koivusalo et al.35 for phospholipid classes. Of note, these authors presented data for a broad range of FAs, from 24-48 chain length, which likely does not reflect the situation in a real biological sample. The response for polyunsaturated species was 40% higher than for fully saturated species, but this effect was only observed for higher concentrations. When the mixture was progressively diluted, the effect of unsaturation gradually diminished and virtually disappeared at 0.1 pmol/µL per species. Additionally, all measurements were done on ion trap and triples quadrupoles instruments and not on Q-Exactive equipment.
Another advantage of the presented workflow is its technical flexibility, which allows adaptation to specific project requirements. For example, any lipid extraction protocol - such as the modified Bligh and Dyer36, methyl tert-butyl ether37, or butanol-methanol38 methods - can be used for preparing a total lipid extract before MS analysis. The main limitation of chloroform-methanol extraction is that the lower phase contains the lipid fraction, which complicates routine work and especially automation; in addition, the toxicity of chloroform must be considered. Tert-butyl ether extraction is widely used for lipid analysis of plasma samples37, and an automated version has been proposed21. In this case, we chose the BUME method because it provides even better recoveries for the spiked PG, PI, PA, and PS lipid classes38, lower solvent consumption, and the possibility of automation39, while the overall concentrations quantified for tissue samples extracted by all three methods were comparable. Additionally, while the sample extraction was performed manually in the current work, it is also possible to obtain reproducible and precise results from large sample sets by automated sample preparation and lipid extraction in a 96-well format40,41. This allows one to implement lipidomics analysis in large-scale clinical and toxicological studies.
In the current work, we performed the MS acquisition of positive and negative modes separately without polarity switching as described by Schuhmann et al.42. The stability of the nanomate signal is better in negative mode for a slightly less concentrated solution than in positive mode. Additionally, we developed a fully traceable procedure with converter software from .raw files to mzML, which provides the values to be specified in the LipidXplorer software - with this, manual resolution slope calculations are not required. We also applied different noise setting substitutions because, in positive mode, the noise levels of the spectra are higher than in negative mode. All the steps were optimized for traceable, high-throughput routine analyses.
For identification, shotgun lipidomics analysis can exploit the unique behavior of different lipid classes, which form unique adducts in different polarity modes. In this method, PC and PE species with the same molecular mass that overlap in the positive ionization mode can be fully separated in the negative ionization mode, as PC forms acetate or formate adducts, and PE is ionized in a deprotonated form. Moreover, (partial) structural deconvolution is possible for the method utilizing not only the molecular formula but also the bulk fatty acid structure. For example, FA identification on the level of total carbon and total unsaturation count works for all phospholipids, DAGs, TAGs, and lyso-phospholipids. The bottom-up quantification of each isomeric form can be partially performed for some of the phospholipid classes43 but is much more complex for DAGs and TAGs due to the unequal signal response of different FAs in the MS2 spectra.
It is also important to emphasize the need to implement appropriate quality control procedures, fully aligned with recent initiatives in this field44. As we wish to ensure proper data traceability and reproducibility between and within the laboratory, a number of steps have been taken such as proper randomization of the samples for all steps of the analysis, working with supplier-certified standard mixtures, the inclusion of quality control samples, procedures to verify batch acceptance or rejection, and the creation of an internal database to track QC performance over the long term. Also consistent with these initiatives is the need for a standardized method to address sample stability. In general, the majority of structural lipids are not impacted by lipid oxidation, which is more relevant for oxilipins, oxidized lipids, and polyunsaturated fatty acids that are, therefore, much more sensitive to storage and handling conditions. However, the correct assessment of sample stability is still a technically challenging task.
However, this protocol has a limitation when it comes to determining submolecular levels of compounds. Considering that there is no separation of the total extract, all isoforms of lipids with the same molecular mass but different fatty acid compositions are merged in the MS analysis. For most classes, it is possible to achieve partial deconvolution of the structure by using the fragmentation ratios of residual fatty acid fragments in MS2. However, the independent quantification of each isoform remains a particularly challenging task owing to the big differences in the fragmentation behaviors of different isoforms and the fact that pure chemical standards are not available in large enough variety to define the compensation values. Another limitation is that the ESI process inevitably generates artifacts, resulting in the artificial generation of peaks for some lipids such as DAGs, Pas, and FAs, which can lead to erroneous quantification.
Next, we summarize the most critical parts of the protocol based on our experience. The first one is related to the fact that each mouse tissue type has a unique lipid profile in terms of both the lipid amount and class ratios. With this, the starting amounts of the tissue based on the total protein content before extraction must be carefully determined in order not to saturate the MS signal and not to leave the dynamic quantification range due to lipid aggregation at high concentrations33 or - at the opposite extreme - to provide enough MS signal to cover the major lipid compounds for each lipid class.
The second critical aspect is to ensure a proper alignment of the position of the direct infusion nano-source chip outlet with the transfer capillary of the mass spectrometer. Considering that full calibration of the mass spectrometer in both modes is performed weekly, swapping between the calibration source and the nano-source chip setup can be the reason for dramatic variability in signal intensity due to misalignment during installation.
Another critical part of the protocol is the careful handling of the internal standard mix. As this mixture contains a significant amount of dichloromethane, once opened, it should be consumed quickly to avoid long storage and multiple uses leading to evaporation and artificial concentration change. Moreover, consistent handling of the standard mixture after removal from −20 °C storage is important, since temperature differences can lead to volume inconsistencies during the pipetting with air-cushion pipettes. An option would be to replace dichloromethane in the standard resuspension buffer with pure methanol, which could improve the handling convenience but might negatively impact the solubility of some lipid classes and, thus, decrease quantification accuracy for these lipid classes.
The final critical part is data processing. The data processing workflow is combining the Peak by Peak software conversion from .raw to .mzML format, applying MS2 scan averaging and MS1 and MS2 noise filtration, as well as peak picking and data compression. As an alternative, the Proteowizard software can be also used for data conversion, but, in this case, several settings in LipidXplorer have to be defined manually. All the complexity of shotgun lipidomics is particularly concentrated on the step of deconvolution of direct injection MS1 and MS2 spectra. The open-source LipidXplorer software imports the converted spectra from mzML file format based on mass accuracy, mass resolution, and the slope of its change with increasing m/z. The software merges multiple individual MS and MS/MS spectra acquired within an analysis run. Afterward, it aligns individual peaks within different sample runs, and, in each cluster of aligned peaks, it replaces their masses with their single intensity-weighted average mass, while their abundances in each data file remain untouched. Representative masses of aligned peak clusters and individual peak intensities are stored in a master scan database.The master scan database contains all MS1 and MS2 spectra generated for all samples in the batch and can be deconvoluted to lipid identifications by queries written in the molecular fragmentation query language (MFQL).
Overall, the method covers the identification of DAG, TAG, and SE lipids based on positive mode and PC, PE, PS, PI, PA, PG, SM, LPC, and LPE lipids based on negative mode acquisition. During lipid identification, isotopic correction is performed for MS1 and MS2, and adjusted intensities are reported in the .xlsx output file. Alternatively, several other software are available to process shotgun data, such as ALEX45 and LipidHunter46.
Fatty acids - major nuclear and cellular membrane components - are stored in the form of phospholipids for further conversion into bioactive molecules. They can be converted by lysophospholipid acyltransferase enzymes into lysophospholipids through the Lands pathway47. The LPCAT3 enzyme, for example, is known to show high specificity to incorporate AA into lysophosphatidylcholine and lysophosphatidylserine intermediates. The relatively high expression of these enzymes was reported within inflammatory cells, such as alveolar macrophages and bronchial epithelial cells47, leading to the release of larger relative proportions of AA-containing phospholipids in those cells. However, following their liberation from membrane phospholipids by phospholipase A2, the conversion of PUFAs into various types of lipid mediators is catalyzed by many enzymes48. Further, these PUFAs can become the substrate for the production of pro-inflammatory and anti-inflammatory prostaglandins via the action of cyclooxygenase (COX)-1 and COX-2 enzymes47. Phospholipids are one of the sources of lipid mediators released at particular locations where these mediators are required to demonstrate their biological effects.
The lung is a complex organ comprising multiple cell types, each playing overlapping and niche roles in facilitating normal lung development and function. Only a few studies have been done to isolate and profile different cell types in the mouse or human lung (e.g., alveolar type 2 cells)49,50; other major lung cell types have not been characterized. Another interesting study51 was carried out to isolate and perform lipid analysis of endothelial, epithelial, mesenchymal, and mixed immune cells in the mouse lung. It was observed that the concentration of PUFAs incorporated in PCO and PG was enriched in the immune cells. Considering the fact that CS induces an increase in immune cells within the lung (4-5 fold), as assessed by bronchoalveolar lavage (BAL)52, the total lipid changes observed in this study could be explained by a cumulative increase in immune cell recruitment in the mouse lung.
In conclusion, the observed distortion of monounsaturated fatty acids and PUFAs incorporated in phospholipids can reflect the excess of certain FAs within the cell and/or constitute an intracellular resource of oxilipin precursors that are overproduced under oxidative stress and inflammatory conditions. However, additional data on free EPA, DHA, and other oxilipins are essential to clarify this point and are outside the scope of the current method application.
Disclosures
All authors are employees of Philip Morris International. Philip Morris International is the sole source of funding and sponsor of this project.
Acknowledgements
The authors would like to thank the study team and especially acknowledge the technical assistance and support of the bioresearch and aerosol teams at PMI R&D, Philip Morris International Research Laboratories Pte. Ltd., Singapore, and PMI R&D, Philip Morris Products S.A., Neuchâtel, Switzerland. The authors thank Sam Ansari for managing the biobanking and acknowledge the support of Sindhoora Bhargavi Gopala Reddy for editing a draft of the manuscript.
Materials
| Name | Company | Catalog Number | Comments |
| 1, 5, and 2 mL self-lock tubes | Eppendorf | 30120086, 30120094 | |
| 3 mm stainless still beads | Qiagen | 69997 | |
| 4.1 µm nozzle chip | Advion | HD-D-384 | |
| Acetic acid | Sigma Aldrich | 45754-100ML-F | |
| Ammonium acetate | Honeywell | 14267-25G | |
| Ammonium bicarbonate | Sigma Aldrich | 09830-500G | |
| Bovine serum albumin standard, 2 mg/mL | Thermo Scientific | 23209 | |
| Butanol | Honeywell | 33065-2.5L | |
| Chloroform | Sigma Aldrich | 650498-1L | |
| Dichloromethane | Honeywell | 34856-1L | |
| Ethyl acetate | Honeywell | 33211 | |
| Greiner CELLSTAR 96 well plates | Sigma | M9686 | |
| Heptane | Sigma Aldrich | 34873-2.5L | |
| Isopropanol | Fisher Scientific | A461 | |
| Methanol | Fisher Scientific | A456 | |
| Mouse pooled plasma | BioIvt | ||
| Mouse SPLASH standard | Avanti Polar Lipids | 330710X | Internal standard |
| Nunc 96-flat bottom well transparent plates | VWR | 62409-068 | |
| Plastic spatula | Sigma | Z560049-300EA | |
| Quick Start Bradford 1x Dye reagent | BioRad | 5000205 | |
| Serum diluent | Sigma Aldrich | D5197 | |
| Equipment/software | |||
| CryoPrep CP02 impactor instrument | Covaris | Magnetic hammer | |
| Centrifuge 5427R | Eppendorf | . | Centrifuge |
| ChipSoft 8.3 | Advion Biosciences | . | Software to set up method and acquisition on the Triversa nanomate robot |
| LipidXplorer 1.2.8.1 | N/A | . | Software to identify lipids |
| Peak By Peak | SpectroSwiss | . | Software to convert .raw data from MS to .mzml format |
| ProteoWizard | ProteoWizard | . | Alternative (open source) software to convert .raw data from MS to .mzml format |
| Q-Exactive MS | Thermo Fisher | . | High resolution orbitrap mass spectrometer |
| Qiagen Tissue Lyser II | Qiagen | . | Tissue lyser |
| SpeedVac SPD140DDA | Thermo Fisher | . | Vacuum concentrator |
| Tecan Infinite M nano plus | Tecan | . | Plate reader |
| ThermoMixer C | Eppendorf | . | Thermomixer |
| TriVersa Nanomate | Advion Biosciences | . | Direct infusion nano-source |
| Xcalibur 4.3 | Thermo Scientific | . | Software to set up method and acquisition on the Q-Exactive MS |
References
- Salehi, N., Janjani, P., Tadbiri, H., Rozbahani, M., Jalilian, M. Effect of cigarette smoking on coronary arteries and pattern and severity of coronary artery disease: A review. Journal of International Medical Research. 49 (12), 3000605211059893 (2021).
- Churg, A., Sin, D. D., Wright, J. L. Everything prevents emphysema: Are animal models of cigarette smoke-induced chronic obstructive pulmonary disease any use. American Journal of Respiratory Cell and Molecular Biology. 45 (6), 1111-1115 (2011).
- Lee, G., Walser, T. C., Dubinett, S. M. Chronic inflammation, chronic obstructive pulmonary disease, and lung cancer. Current Opinion in Pulmonary Medicine. 15 (4), 303-307 (2009).
- Titz, B., et al. Effects of cigarette smoke, cessation, and switching to two heat-not-burn tobacco products on lung lipid metabolism in C57BL/6 and Apoe-/- mice-An integrative systems toxicology analysis. Toxicological Sciences. 149 (2), 441-457 (2016).
- Morissette, M. C., Shen, P., Thayaparan, D., Stampfli, M. R. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. European Respiratory Journal. 46 (5), 1451-1460 (2015).
- Jubinville, E., et al. Interplay between cigarette smoking and pulmonary reverse lipid transport. European Respiratory Journal. 50 (3), 1700681 (2017).
- Han, X., Gross, R. W. Shotgun lipidomics: Electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrometry Reviews. 24 (3), 367-412 (2005).
- Titz, B., et al. Multi-omics systems toxicology study of mouse lung assessing the effects of aerosols from two heat-not-burn tobacco products and cigarette smoke. Computational and Structural Biotechnology Journal. 18, 1056-1073 (2020).
- Hartung, T., et al. Systems toxicology: Real world applications and opportunities. Chemical Research in Toxicology. 30 (4), 870-882 (2017).
- Talikka, M., et al., Lyubimov, A., et al. Systems Toxicology. Encyclopedia of Drug Metabolism and Interactions. , (2016).
- Ejsing, C. S., et al. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 106 (7), 2136-2141 (2009).
- Papan, C., et al. Systematic screening for novel lipids by shotgun lipidomics. Analytical Chemistry. 86 (5), 2703-2710 (2014).
- Carvalho, M., et al. Effects of diet and development on the Drosophila lipidome. Molecular Systems Biology. 8, 600 (2012).
- Strittmatter, N., et al. Shotgun lipidomic profiling of the NCI60 cell line panel using rapid evaporative ionization mass spectrometry. Analytical Chemistry. 88 (15), 7507-7514 (2016).
- Lydic, T. A., et al. Rapid and comprehensive 'shotgun' lipidome profiling of colorectal cancer cell derived exosomes. Methods. 87, 83-95 (2015).
- Sampaio, J. L., et al. Membrane lipidome of an epithelial cell line. Proceedings of the National Academy of Sciences of the United States of America. 108 (5), 1903-1907 (2011).
- Grzybek, M., et al. Comprehensive and quantitative analysis of white and brown adipose tissue by shotgun lipidomics. Molecular Metabolism. 22, 12-20 (2019).
- Stegemann, C., et al. Comparative lipidomics profiling of human atherosclerotic plaques. Circulation: Cardiovascular Genetics. 4 (3), 232-242 (2011).
- Tajima, Y., et al. Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer's disease. Lipids in Health and Disease. 12, 68 (2013).
- Gross, R. W., Han, X. Shotgun lipidomics of neutral lipids as an enabling technology for elucidation of lipid-related diseases. American Journal of Physiology: Endocrinology and Metabolism. 297 (2), 297-303 (2009).
- Surma, M. A., et al. An automated shotgun lipidomics platform for high throughput, comprehensive, and quantitative analysis of blood plasma intact lipids. European Journal of Lipid Science and Technology. 117 (10), 1540-1549 (2015).
- Heiskanen, L. A., Suoniemi, M., Ta, H. X., Tarasov, K., Ekroos, K. Long-term performance and stability of molecular shotgun lipidomic analysis of human plasma samples. Analytical Chemistry. 85 (18), 8757-8763 (2013).
- Zhang, S., Van Pelt, C. K. Chip-based nanoelectrospray mass spectrometry for protein characterization. Expert Review of Proteomics. 1 (4), 449-468 (2004).
- Surma, M. A., et al. Mouse lipidomics reveals inherent flexibility of a mammalian lipidome. Scientific Reports. 11 (1), 19364 (2021).
- Herzog, R., Schwudke, D., Shevchenko, A. LipidXplorer: Software for quantitative shotgun lipidomics compatible with multiple mass spectrometry platforms. Current Protocols in Bioinformatics. 43, 11-30 (2013).
- R: A language and environment for statistical computing. R: Foundation for Statistical Computing Available from: https://www.R-project.org/ (2018)
- Phillips, B., et al. A 7-month cigarette smoke inhalation study in C57BL/6 mice demonstrates reduced lung inflammation and emphysema following smoking cessation or aerosol exposure from a prototypic modified risk tobacco product. Food and Chemical Toxicology. 80, 328-345 (2015).
- Phillips, B., et al. A six-month systems toxicology inhalation/cessation study in ApoE(-/-) mice to investigate cardiovascular and respiratory exposure effects of modified risk tobacco products, CHTP 1.2 and THS 2.2, compared with conventional cigarettes. Food and Chemical Toxicology. 126, 113-141 (2019).
- Zemski Berry, K. A., Murphy, R. C. Electrospray ionization tandem mass spectrometry of glycerophosphoethanolamine plasmalogen phospholipids. Journal of the American Society for Mass Spectrometry. 15 (10), 1499-1508 (2004).
- Engelmann, B. Plasmalogens: Targets for oxidants and major lipophilic antioxidants. Biochemical Society Transactions. 32, 147-150 (2004).
- Nagan, N., Zoeller, R. A. Plasmalogens: Biosynthesis and functions. Progress in Lipid Research. 40 (3), 199-229 (2001).
- Southam, A. D., Weber, R. J., Engel, J., Jones, M. R., Viant, M. R. A complete workflow for high-resolution spectral-stitching nanoelectrospray direct-infusion mass-spectrometry-based metabolomics and lipidomics. Nature Protocols. 12 (2), 310-328 (2016).
- Yang, K., Han, X. Accurate quantification of lipid species by electrospray ionization mass spectrometry - Meet a key challenge in lipidomics. Metabolites. 1 (1), 21-40 (2011).
- Zullig, T., Trotzmuller, M., Kofeler, H. C. Lipidomics from sample preparation to data analysis: a primer. Analytical and Bioanalytical Chemistry. 412 (10), 2191-2209 (2020).
- Koivusalo, M., Haimi, P., Heikinheimo, L., Kostiainen, R., Somerharju, P. Quantitative determination of phospholipid compositions by ESI-MS: Effects of acyl chain length, unsaturation, and lipid concentration on instrument response. Journal of Lipid Research. 42 (4), 663-672 (2001).
- Bligh, E. G., Dyer, W. J. A rapid method of total lipid extraction and purification. Canadian Journal of Biochemistry and Physiology. 37 (8), 911-917 (1959).
- Matyash, V., Liebisch, G., Kurzchalia, T. V., Shevchenko, A., Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. Journal of Lipid Research. 49 (5), 1137-1146 (2008).
- Lofgren, L., Forsberg, G. B., Stahlman, M. The BUME method: A new rapid and simple chloroform-free method for total lipid extraction of animal tissue. Scientific Reports. 6, 27688 (2016).
- Lofgren, L., et al. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. Journal of Lipid Research. 53 (8), 1690-1700 (2012).
- Jung, H. R., et al. High throughput quantitative molecular lipidomics. Biochimica et Biophysica Acta. 1811 (11), 925-934 (2011).
- Lavrynenko, O., et al. Ceramide ratios are affected by cigarette smoke but not heat-not-burn or e-vapor aerosols across four independent mouse studies. Life Sciences. 263, 118753 (2020).
- Schuhmann, K., et al. Shotgun lipidomics on a LTQ Orbitrap mass spectrometer by successive switching between acquisition polarity modes. Journal of Mass Spectrometry. 47 (1), 96-104 (2012).
- Schuhmann, K., et al. Quantitative fragmentation model for bottom-up shotgun lipidomics. Analytical Chemistry. 91 (18), 12085-12093 (2019).
- Lipidomics Standards Initiative Consortium. Lipidomics needs more standardization. Nature Metabolism. 1 (8), 745-747 (2019).
- Husen, P., et al. Analysis of lipid experiments (ALEX): A software framework for analysis of high-resolution shotgun lipidomics data. PloS One. 8 (11), 79736 (2013).
- Ni, Z., Angelidou, G., Lange, M., Hoffmann, R., Fedorova, M. LipidHunter identifies phospholipids by high-throughput processing of LC-MS and shotgun lipidomics datasets. Analytical Chemistry. 89 (17), 8800-8807 (2017).
- Zemski Berry, K. A., Murphy, R. C., Kosmider, B., Mason, R. J. Lipidomic characterization and localization of phospholipids in the human lung. Journal of Lipid Research. 58 (5), 926-933 (2017).
- Ghosh, M., Tucker, D. E., Burchett, S. A., Leslie, C. C. Properties of the Group IV phospholipase A2 family. Progress in Lipid Research. 45 (6), 487-510 (2006).
- Besnard, V., et al. Deletion of Scap in alveolar type II cells influences lung lipid homeostasis and identifies a compensatory role for pulmonary lipofibroblasts. Journal of Biological Chemistry. 284 (6), 4018-4030 (2009).
- Plantier, L., et al. Activation of sterol-response element-binding proteins (SREBP) in alveolar type II cells enhances lipogenesis causing pulmonary lipotoxicity. Journal of Biological Chemistry. 287 (13), 10099-10114 (2012).
- Kyle, J. E., et al. Cell type-resolved human lung lipidome reveals cellular cooperation in lung function. Scientific Reports. 8 (1), 13455 (2018).
- Lugg, S. T., Scott, A., Parekh, D., Naidu, B., Thickett, D. R. Cigarette smoke exposure and alveolar macrophages: mechanisms for lung disease. Thorax. 77 (1), 94-101 (2022).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved