
Method Article
Visualizing Actin and Microtubule Coupling Dynamics In Vitro by Total Internal Reflection Fluorescence (TIRF) Microscopy
In This Article
Summary
This protocol is a guide for visualizing dynamic actin and microtubules using an in vitro total internal fluorescence (TIRF) microscopy assay.
Abstract
Traditionally, the actin and microtubule cytoskeletons have been studied as separate entities, restricted to specific cellular regions or processes, and regulated by different suites of binding proteins unique for each polymer. Many studies now demonstrate that the dynamics of both cytoskeletal polymers are intertwined and that this crosstalk is required for most cellular behaviors. A number of proteins involved in actin-microtubule interactions have already been identified (i.e., Tau, MACF, GAS, formins, and more) and are well characterized with regard to either actin or microtubules alone. However, relatively few studies showed assays of actin-microtubule coordination with dynamic versions of both polymers. This may occlude emergent linking mechanisms between actin and microtubules. Here, a total internal reflection fluorescence (TIRF) microscopy-based in vitro reconstitution technique permits the visualization of both actin and microtubule dynamics from the one biochemical reaction. This technique preserves the polymerization dynamics of either actin filament or microtubules individually or in the presence of the other polymer. Commercially available Tau protein is used to demonstrate how actin-microtubule behaviors change in the presence of a classic cytoskeletal crosslinking protein. This method can provide reliable functional and mechanistic insights into how individual regulatory proteins coordinate actin-microtubule dynamics at a resolution of single filaments or higher-order complexes.
Introduction
Historically, actin and microtubules have been viewed as separate entities, each with their own set of regulatory proteins, dynamics behaviors, and distinct cellular locations. Abundant evidence now demonstrates that actin and microtubule polymers engage in functional crosstalk mechanisms that are essential to execute numerous cell processes including migration, mitotic spindle positioning, intracellular transport, and cell morphology1,2,3,4. The diverse coordinated behaviors that underlie these examples are dependent on an intricate balance of coupling factors, signals, and physical properties. However, the molecular details that underpin these mechanisms are still largely unknown because most studies focus on a single cytoskeletal polymer at a time1,2,5.
Actin and microtubules do not directly interact6,7,8. The coordinated dynamics of actin and microtubules seen in cells is mediated by additional factors. Many proteins thought to regulate actin-microtubule crosstalk have been identified and their activities are well characterized with regard to either cytoskeletal polymer alone1,2. Growing evidence suggests this single polymer approach has concealed the dual functions of some of the proteins/complexes that enable actin-microtubule coupling events7,8,9,10,11,12,13. Experiments where both polymers are present are rare and often define mechanisms with a single dynamic polymer and static stabilized version of the other6,8,9,10,11,14,15,16,17,18. Thus, methods are needed to investigate the emergent properties of actin-microtubule coordinating proteins that may only be fully understood in experimental systems that employ both dynamic polymers.
The combination of direct protein labeling approaches, genetically encoded affinity tags, and total internal reflection fluorescence (TIRF) microscopy has been applied with great success in biomimetic reconstitution systems19,20,21,22,23. Many bottom-up schemes do not contain all the factors that regulate proteins in cells. However, "biochemistry on a coverglass" technology has refined many mechanisms of actin and microtubule dynamics at high spatial and temporal scales, including the components required for polymer assembly or disassembly, and motor protein movement5,12,23,24,25,26,27. Here a minimal component single-filament approach to investigate actin-microtubule coupling in vitro is described. This protocol can be used with commercially available or highly pure purified proteins, fluorescently labeled proteins, perfusion chambers, and extended to more complicated schemes containing cell extracts or synthetic systems. Here, commercially available Tau protein is used to demonstrate how cytoskeletal dynamics change in the presence of an actin-microtubule coupling protein, but can be substituted for other putative actin-microtubule coordinating factors. The major advantage of this system over other approaches is the ability to simultaneously monitor the dynamics of multiple cytoskeletal polymers in one reaction. This protocol also provides users with examples and simple tools to quantify changes to cytoskeletal polymers. Thus, protocol users will produce reliable, quantitative, single-filament resolution data to describe mechanisms that underlie how diverse regulatory proteins coordinate actin-microtubule dynamics.
Protocol
1. Washing the coverslips
NOTE: Wash (24 mm x 60 mm, #1.5) coverslips according to Smith et al., 201328.
- Arrange coverslips in a plastic slide mailer container.
- Submerge coverslips sequentially in the following solutions and sonicate for 30-60 min, rinsing with ddH2O 10 times in between each solution: ddH2O with one drop of dish soap; 0.1 M KOH. Store coverslips in 100% ethanol for up to 6 months.
NOTE: Do not touch glass surfaces with ungloved fingers. Use forceps instead.
2. Coating cleaned (24 mm x 60 mm, #1.5) coverslips with mPEG- and biotin-PEG-silane
NOTE: This protocol specifically uses a biotin-streptavidin system to position actin and microtubules within the TIRF imaging plane. Other coatings and systems may be used (e.g., antibodies, poly-L-lysine, NEM myosin, etc.).
- Thaw aliquots of PEG-silane and biotin-PEG-silane powders.
- Dissolve PEG powders in 80% ethanol (pH 2.0) to generate coating stock solutions of 10 mg/mL mPEG-silane and 2-4 mg/mL biotin-PEG-silane, just before the use.
NOTE: PEG powders often appear dissolved but may not be at the microscopic level. Proper resuspension takes ~1-2 min with constant pipetting. Users are encouraged to pipette an additional 10 times following the appearance of powder dissolution.
CAUTION: Wear gloves to protect skin from concentrated HCl when making 80% ethanol (pH 2.0). - Remove the clean (24 mm x 60 mm, #1.5) coverslip from ethanol storage using forceps. Dry with nitrogen gas and store in a clean Petri dish.
- Coat coverslips with 100 µL of coating solution: a mixture of 2 mg/mL mPEG-silane (MW 2,000) and 0.04 mg/mL biotin-PEG-silane (MW 3,400) in 80% ethanol (pH 2.0).
NOTE: For sparse coating (recommended) use 2 mg/mL mPEG-silane and 0.04 mg/mL biotin-PEG-silane. For dense coating use 2 mg/mL mPEG-silane, 4 mg/mL biotin-PEG-silane. - Incubate coverslips at 70 °C for at least 18 h or until use.
NOTE: Coated coverslips degrade if stored at 70 °C for more than 2 weeks.
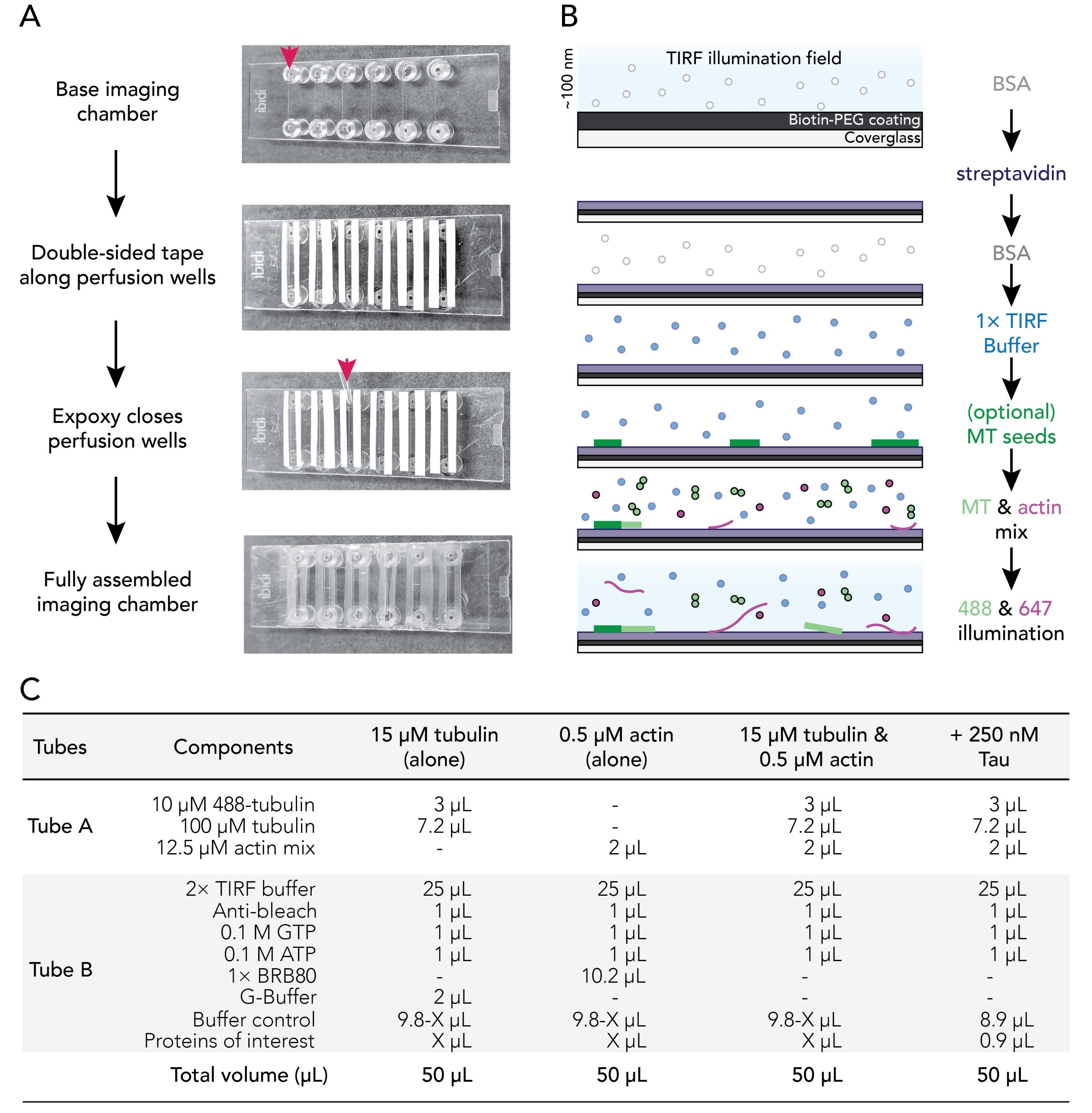
3. Assembling imaging flow chambers
- Cut 12 strips of double-backed double-sided tape to a length of 24 mm. Remove one side of the tape backing and fix pieces of tape adjacent to the six grooves present on a clean imaging chamber.
NOTE: Tape must be flat for proper assembly, otherwise imaging chambers will leak. Carefully remove the tape backing to avoid bumps. Sliding taped chambers on a clean surface to smooth tape-chamber contacts is recommended. - Remove the second piece of tape backing to expose the sticky side of the tape along each chamber groove. Place chamber tape side up on a clean surface.
- Mix epoxy resin and hardener solutions 1:1 (or according to manufacturer's instructions) in a small weigh boat.
- Use a P1000 tip to place a drop of mixed epoxy between the tape strips at the end of each imaging chamber groove (red arrow; Figure 1A). Place chamber tape/epoxy side up on a clean surface.
- Remove a coated coverslip from 70 °C incubator. Rinse coated and uncoated surfaces of coverslips with ddH2O six times, dry with filtered nitrogen gas, and then affix to the imaging chamber with the coverslip coating side toward the tape.
- Use a P200 or P1000 pipette tip to apply pressure on the tape-glass interface to ensure a good seal between the tape and the coverslip.
NOTE: With a proper seal, double-sided tape becomes translucent. Imaging chambers lacking sufficient tape-chamber contacts will leak. - Incubate assembled chambers at room temperature for at least 5-10 min to allow the epoxy to fully seal chamber wells before use. Perfusion chambers expire within 12-18 h of assembly.
NOTE: Depending on tape placement and the thickness of double-sided tape used, the assembled chamber will have a final volume of 20-50 µL.
4. Conditioning of perfusion chambers
- Use a perfusion pump (rate set to 500 µL/min) to sequentially exchange conditioning solutions in perfusion chamber as follows:
- Flow 50 µL of 1% BSA to prime the imaging chamber. Remove excess buffer from Luer lock fitting reservoir.
- Flow 50 µL of 0.005 mg/mL streptavidin. Incubate for 1-2 min at room temperature. Remove excess buffer from reservoir.
- Flow 50 µL of 1% BSA to block nonspecific binding. Incubate for 10-30 s. Remove excess buffer from reservoir.
- Flow 50 µL of warm (37 °C) 1x TIRF buffer (1x BRB80, 50 mM KCl, 10 mM DTT, 40 mM glucose, 0.25% (v/v) methylcellulose (4,000 cp)).
NOTE: Do not remove excess buffer from the reservoir. This prevents the chamber from drying out, which can introduce air bubbles into the system. - Optional: Flow 50 µL of stabilized29 and 50% biotinylated microtubule seeds diluted in 1x TIRF buffer.
NOTE: The proper dilution must be empirically determined and contain batch to batch variability. Protocols from27,29 are recommended as starting points. A dilution yielding 10-30 seeds per field of view works well with this setup.
5. Microscope preparation
NOTE: Biochemical reactions containing dynamic actin filaments and microtubules are visualized/performed using an inverted Total Internal Reflection Fluorescence (TIRF) microscope equipped with 120-150 mW solid-state lasers, a temperature corrected 63x oil immersion TIRF objective, and an EMCCD camera. Proteins in this example are visualized at the following wavelengths: 488 nm (microtubules) and 647 nm (actin).
- Set the stage/objective heater device to maintain 35-37 °C at least 30 min prior to imaging the first biochemical reaction.
- Set the image acquisition parameters as follows:
- Set acquisition interval to every 5 s for 15-20 min.
- Set 488 and 647 laser exposures to 50-100 ms at 5%-10% power. Set appropriate TIRF angle for microscope.
NOTE: Regardless of microscope setup, the simplest way to set the laser power, exposure, and TIRF angle is to make adjustments on images of either polymer alone (see 5.2.2.1 and 5.2.2.2, below). Users are strongly encouraged to use the lowest laser power and exposure settings that still permit detection.- Adjust the polymerization reaction (Figure 1C) to initiate actin filament assembly and acquire images at 647 nm. Make appropriate adjustments.
- Adjust the polymerization reaction in a second conditioned perfusion well to initiate microtubule assembly (Figure 1C) and visualize at 488 nm. Make appropriate adjustments.
6. Preparation of protein reaction mixes
- Prepare stock solution of fluorescently labelled tubulin.
- Determine the concentration of homemade unlabeled tubulin via spectrophotometry at Abs280, as follows:
- Blank spectrophotometer with 1xBRB80 lacking GTP.
- Calculate the concentration of tubulin using the determined extinction coefficient of 115,000 M-1 cm-1 and the following formula:

- Resuspend commercially-made lyophilized lysine-labeled 488-tubulin to 10 µM (1 mg/mL; 100% label) with 20 µL of 1x BRB80 lacking GTP.
- Thaw a 7.2 µL aliquot of 100 µM unlabeled recycled tubulin29 on ice.
NOTE: Recycled tubulin is critical for successful microtubule assembly in vitro because it removes polymerization-incompetent dimers formed in frozen protein stocks29,30. - Combine 3 µL of 10 µM 488-tubulin with the 7.2 µL aliquot of 100 µM unlabeled tubulin, no more than 15 min before use.
- Determine the concentration of homemade unlabeled tubulin via spectrophotometry at Abs280, as follows:
- Prepare the stock solution of fluorescently labeled actin.
- For homemade proteins, determine the concentration and percent label of actin via spectrophotometry Abs290 and Abs650, as follows:
- Blank spectrophotometer with G-buffer.
- Calculate the concentration of unlabeled actin using the determined extinction coefficient of 25,974 M-1 cm-1 and the following formula:

- Calculate the concentration of lysine labeled Alexa-647-actin using the extinction coefficient of unlabeled actin, the fluor correction factor of 0.03, and the following formula:
[Alexa-647 actin], µM = (Abs290 -
- Calculate the percent label of Alexa-647-actin using the determined ε for Alexa-647 of 239,000 M-1 cm-1, as follows:
% label of Alexa-647-actin = (Abs290 - (
- Thaw one 2 µL aliquot of 3 µM 100% labeled biotin-actin (labeled on lysine residues). Dilute 10-fold by adding 18 µL of G-buffer.
- Combine 3 µL of diluted biotinylated actin, appropriate volumes of unlabeled and labeled actin (above) such that the final mix will be 12.5 µM total actin with 10%-30% fluorescent label.
NOTE: Greater than 30% percent fluorescent actin monomers (final) can compromise imaging resolution as filaments become difficult to discern from the background.
- For homemade proteins, determine the concentration and percent label of actin via spectrophotometry Abs290 and Abs650, as follows:
- Prepare reaction mixes (Figure 1C).
- Prepare cytoskeleton mix (Tube A) by combining 2 µL of the 12.5 µM actin mix stock (6.2.3) with the tubulin stock mix (6.1.4), no more than 15 min prior to imaging. Store on ice until use.
- Prepare protein reaction mix (Tube B) by combining all other experimental components and proteins, including: 2x TIRF buffer, anti-bleach, nucleotides, buffers, and accessory proteins. An example is shown in Figure 1C.
NOTE: The final dilution results in a 1x TIRF buffer that contains ATP, GTP, and ionic strength within the estimated physiological range.
- Incubate Tube A and Tube B separately at 37 °C for 30-60 s. To initiate reaction, mix and add the contents of Tube B to Tube A (below).
7. Image actin and microtubule dynamics
- Condition perfusion well (Figure 1B; step 4, above).
- Initiate actin and microtubule assembly simultaneously by adding the contents of Tube B (reaction mix) to Tube A (cytoskeleton mix) (Figure 1C).
- Flow 50 µL of reaction containing 1x TIRF buffer supplemented with 15 µM free tubulin, 1 mM GTP, and 0.5 µM actin monomers and appropriate volumes of buffer controls.
- Record time-lapse movie using microscope software to acquire every 5 s for 15-20 min.
NOTE: Initiation of actin and microtubule dynamics occurs within 2-5 min (Figure 2). Longer delays indicate problems with temperature control or concentration-related problems of proteins in the reaction mix. - Condition a new perfusion well (step 4) and replace buffer volume with regulatory protein(s) of interest (i.e., Tau) and buffer controls (Figure 1C). Acquire as outlined in step 7 (above) to assess regulatory proteins for emergent actin-microtubule functions.
8. Process and analyze images using FIJI software31
- Open saved TIRF movies and view as a composite.
- Analyze microtubule dynamics (Figure 3A), as follows:
- Generate a time-based maximum Z-projection from the image stacks menu.
- Synchronize Z-projection window with the original TIRF movie from the Analyze> Tools> Synchronize Windows menu.
- Draw a line using the straight-line tool along a microtubule of interest on the time projected image.
- Open the region of interest (ROI) manager from the analyze menu (Analyze> Tools> ROI Manager).
- Save individual microtubule locations by pressing "t". Repeat for all microtubules of interest.
- Plot kymographs of selected lines using "/" or run the multi-kymo macro that generates a video and kymograph for every microtubule in the ROI manager31.
- Add both length (µm) and time (min) scale bars to kymographs from the Analyze> Tools> Scale bars menu.
- Measure microtubule growth speeds from kymograph slopes (Figure 3A, 1-2; slope of black lines).
- Count dynamic microtubule events (catastrophe or regrowth) from the generated kymograph or using available analysis macros5,8,18,25. Red dotted lines in Figure 3A, 1-2 represent catastrophe/rapid disassembly events.
- Analyze actin dynamics (Figure 3B), as follows:
- Measure actin nucleation, as follows:
- Count the number of actin filaments present in the field of view 100 s after the initiation of the reaction and express by the area (filaments per µm2).
- Record and save data in ROI manager, as in step 8.2.3.1, above.
- Measure actin filament elongation rates (Figure 3B), as follows:
- Draw a line along an actin filament of interest using the segmented-line tool.
- Add line to ROI manager as in step 8.2.3.1, above.
- Repeat following the line (adding each measurement to ROI manager) for at least four movie frames.
NOTE: Measuring seven to eight consecutive frames is recommended, however some conditions slow actin polymerization below the detectable limit that can be resolved by objective/microscope setups. In such a case, measurements can be made at regular intervals over non-consecutive frames, (e.g., every five frames). - Plot measured length values over elapsed time. The slope of the generated line is the actin elongation rate in microns/s.
- Convey final calculated rates as subunits s-1 µM-1 using a correction factor of 370 subunits to account for the number of actin monomers in a micron of filament32.
- Measure actin nucleation, as follows:
- Perform correlative analysis for regions of parallel actin-microtubule association (Figure 3C), as follows:
- Draw a line along a microtubule of interest at a specific time point (i.e., 300 s after reaction initiation), using the straight-line tool.
- Add line to ROI manager as in step 8.2.3.1, above.
- Plot the fluorescence intensity along the line in each channel.
- Select each channel with the image slider and plot the intensities along the line using "command k".
- Save or export values by clicking the "list" button in the output window.
- Express actin-microtubule coupling events as a ratio (actin overlapping with microtubules) from individual events or as a count of events in a given field of view at a consistent timepoint (Figure 3C).
- Alternative: Use software to determine the percent overlap of both channels5,12.
- Draw a line along a microtubule of interest at a specific time point (i.e., 300 s after reaction initiation), using the straight-line tool.
Results
With the conditions described above (Figure 1), actin and microtubule polymers should be visible (and dynamic) within 2 min of image acquisition (Figure 2). As with any biochemistry-based protocol, optimization may be required for different regulatory proteins or batches of protein. For these reasons, the TIRF angle and image exposures are set first with reactions containing each individual polymer. This confirms that stored proteins are functional and enough labeled protein is present for detection. While not always necessary (and not performed here), post-processing of movies (i.e., background subtraction, averaging, or Fourier transformations) can be used to enhance the image contrast (particularly of microtubules)5,25,33. The direct visualization of single actin filaments and microtubules afforded by this assay supports the quantitative determination of several dynamic measures for either cytoskeleton component alone or together, including polymerization parameters (i.e., nucleation or elongation rate), disassembly parameters (i.e., shrinkage rates or catastrophe events), and polymer coalignment/overlap (Figure 3). Further, these measures can be used as a starting point to decipher the binding or influence of regulatory ligands like Tau (Figure 3). Many measurements of single actin filaments or microtubules can be made from one TIRF movie. However, due to variations in coverslip coating, pipetting, and other factors, reliable measurements should also include multiple technical replicate reactions/movies.
Many facets of microtubule dynamics can be determined from example kymographs including the rate of microtubule elongation, as well as the frequency of catastrophe and rescue events (Figure 3A). Using kymographs to measure actin dynamics in this system is not as straightforward because actin filaments are more convoluted than microtubules. As a consequence, parameters of actin filament dynamics are measured by hand, which is time consuming and labor intensive. Nucleation counts are measured as the number of actin filaments present at a consistent timepoint for all conditions. These counts vary widely across TIRF imaging fields, but can be used with many replicates or to supplement observations from other polymerization assays. Nucleation counts may also be used for microtubules if trial conditions lack stabilized microtubule seeds. Actin filament elongation rates are measured as the length of filament over time from at least four movie frames. Rate values are conveyed per micromolar actin with a correction factor of 370 subunits to account for the number of actin monomers in a micron of filament (Figure 3B)32. Measurements to define the coordinated behaviors between actin and microtubules are less well defined. However, correlative analyses have been applied to measure the coincidence of both polymers including line scans (Figure 3C) or overlap software5,11,34.
Data Availability:
All datasets associated with this work have been deposited in Zenodo and are available with reasonable request at: 10.5281/zenodo.6368327.

Figure 1. Experimental schematics: flow chamber assembly to image acquisition. (A) Imaging chamber assembly. Top to bottom: IBIDI imaging chambers are taped along perfusion wells (denoted by arrow); the second (white) layer of tape backing (left on in the image shown to better orient users) is removed and Epoxy is applied at the edge of the perfusion chamber (arrow). Note: To more easily orient users where to place the epoxy, the white backing was left on in this image. The cleaned and coated coverslip is attached to the imaging chamber with the coating side facing the inside of the perfusion well. (B) Flow-chart illustrating the steps for conditioning imaging chambers for biotin-streptavidin linkages. (C) Examples of reactions used to acquire TIRF movies of dynamic microtubules and actin filaments. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Image sequences of growing actin filaments and microtubules in the absence or presence of Tau. Time-lapse image montage from TIRF assays containing 0.5 µM actin (10% Alexa-647-actin and 0.09% biotin-actin labeled) and 15 µM free tubulin (4% HiLyte-488 labeled) in the absence (A) or presence (B) of 250 nM Tau. Time elapsed from reaction initiation (mixing Tube A and Tube B) is shown. Scale bars, 25 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Example measurements of microtubules and actin filament dynamics. (A) Average time projection of the tubulin channel efficiently visualizes total microtubule lengths for the line scans used to generate kymograph plots. Black dotted lines correspond to the two example kymographs of dynamic microtubules shown on the right. The growth (solid black lines) and disassembly phases (doted pink lines; two denoted with pink arrows) of microtubules are shown on each kymograph. Time scale bar, 3 min. Length scale bar, 10 µm. Reaction contains 0.5 µM actin (10% 647-label) and 15 µM free tubulin (4% 488-HiLyte label). Only the tubulin channel is shown.(B) Two example time-lapse image montages depicting single-actin filaments actively polymerizing. Elongation rates are calculated as the slope of plots of the length of actin filaments over time per micromolar actin. Thus, a correction factor of two must be applied to 0.5 µM actin reactions for comparison for rates typically determined at the 1 µM actin concentration. Examples from five filaments are shown to the right. Scale bars, 10 µm. Reaction contains 0.5 µM actin (10% 647-label) and 15 µM free tubulin (4% 488-HiLyte label). Only the actin channel is shown. (C) TIRF images of dynamic microtubules (MT) (green) and actin filaments (purple) polymerizing in the absence (left) or presence of 250 nM Tau (middle). Blue dotted lines and arrows mark where a line was drawn for the line scan plots corresponding to each condition (below each image). Overlap between microtubules and actin regions (shown as black) can be scored at a set time point per area (right). Scale bars, 25 µm. Reactions contain 0.5 µM actin (10% 647-label) and 15 µM free tubulin (4% 488-HiLyte label) with or without 250 nM Tau. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The use of total internal reflection fluorescence (TIRF) microscopy to visualized purified proteins has been a fruitful and compelling approach to dissect unique mechanisms of cytoskeletal regulation5,23,24,25,26,27,35. Compared to traditional biochemical assays, TIRF reactions require very small volumes (50-100 µL), and quantitative measurements of cytoskeletal dynamics can be gleaned from an individual assay. Most studies of cytoskeletal dynamics focus on a single polymer system (i.e., actin filaments or microtubules), thus detailed measurements of the crosstalk or emergent behaviors between actin filaments and microtubules typically seen in cells have remained elusive and difficult to recapitulate in the test tube. To solve this problem, this protocol describes a single-filament TIRF microscopy system that enables the direct visualization of dynamic actin and microtubule polymers in the same biochemical reaction. Thus, this method goes beyond traditional assays that recapitulate the dynamic behavior of actin filaments or microtubules alone. This technique was also performed with Tau as an example of how several dynamic properties change in the presence of a cytoskeleton coupling factor. This protocol can be used with additional proteins known or suspected to coordinate actin or microtubule dynamics, including (but not limited to) MACF, GAS, formins, and more. Finally, provided example analyses can be used as a guide to quantify data acquired with this protocol.
"Seeing is believing" is a compelling reason to perform microscopy-based assays. However, caution is required in the execution and interpretation of TIRF microscopy experiments. One major challenge of cytoskeletal co-assembly assays is that many commonly used imaging conditions are not compatible each polymer. Microtubules and actin typically have different buffer, temperature, salt, nucleotide, and concentration requirements for polymerization. Actin, tubulin, regulatory proteins of interest, and the buffers utilized in this protocol are sensitive to freeze-thaw cycles. Therefore, careful handling of proteins and buffers is necessary to successfully execute this protocol. To alleviate many of these concerns, using freshly recycled tubulin (frozen for <6 weeks), and pre-clearing frozen/resuspended actins via ultracentrifugation is strongly recommended. These considerations also apply to the myriad of regulatory proteins to be assessed with this procedure, which may be sensitive to freeze-thaw cycles or the concentration of buffer salts5,11,36.
Unfortunately, no one-size-fits-all buffer without experimental tradeoffs exists. To appropriate more volume for proteins of lower concentration, ATP and GTP may be included in the 2x TIRF buffer solution (Figure 1C). However, because these nucleotides are extremely sensitive to freeze-thaw cycles, it is not recommended. The oxygen scavenging compounds used here (i.e., catalase and glucose-oxidase) are necessary to visualize proteins for long periods of time (minutes to hours), but are known to restrict microtubule polymerization at high concentrations5. Related to these buffer considerations, a limitation of this protocol is that some canonical microtubule-associated regulatory proteins may require more or less salt to recapitulate functions found in cells or assays using microtubules alone (without actin). Changing the nature or concentration of salt to address these concerns will likely influence rates of actin filament polymerization and/or parameters of microtubule dynamics. Measurements of multiple descriptive parameters (minimally, nucleation, elongation rates, and stability) (Figure 3) are required to confirm protocol success or to explicitly document the effects of specific buffers or regulatory proteins. For example, too much actin filament polymerization can obscure actin-microtubule coupling events within seconds. Consequently, fine-tuning experimental conditions by lowering the overall concentration of actin or including additional proteins to suppress actin nucleation (i.e., profilin) will extend the overall period that coordinated actin-microtubule activities can be viewed clearly. Controls addressing these prerequisites, and technical replicates (beyond multiple fields of view), are critical for users to generate reliable and reproducible results.
Cell-based studies provide limited opportunity to observe direct protein-protein relationships or the action of regulatory complexes. In contrast, some of the mechanisms gleaned from in vitro assays do not always reflect the exact behaviors of proteins seen in cells. This classic biochemist dilemma can be addressed in future applications of this technique with specific modifications. For example, adding functional fluorescently labeled coupling proteins expands this method from single-filament studies to single-molecule studies. Assays can be further modified to use cell extracts that may add the "missing" unknown key factors required to recapitulate cell-like phenomena. For example, TIRF-based assays employing yeast or Xenopus extracts have reconstituted contractile actomyosin rings37, mitotic spindles26,38, components of actin or microtubule assembly39,40, and even dynamics at the centrosome and kinetochores36,41,42,43. Moreover, such systems may pave the way toward artificial cell systems that have lipids or signaling factors present44,45,46.
Disclosures
There are no conflicts of interest to disclose.
Acknowledgements
I am grateful to Marc Ridilla (Repair Biotechnologies) and Brian Haarer (SUNY Upstate) for helpful comments on this protocol. This work was supported by the National Institutes of Health (GM133485).
Materials
| Name | Company | Catalog Number | Comments |
| 1% BSA (w/v) | Fisher Scientific | BP1600-100 | For this purpose (blocking TIRF chambers), BSA is resuspended in ddH20 and filtered through a 0.22 µm filter. |
| 1× BRB80 | Homemade | 80 mM PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8 with KOH | |
| 10 mg/mL (1000 U) glucose oxidase | Sigma Aldrich Inc, St. Louis, MO | G2133-50KU | Combined with catalase, aliquot and store at -80 oC until use |
| 100 µM tubulin | Cytoskeleton Inc, Denver, CO | T240 | Homemade tubulins should be recycled before use to remove polymerization-incompetent tubulin (Hyman et al. (1992)29; Li and Moore (2020)30). Commercially available tubulins are often too dilute to recycle, but function well if resuspended according to manufacturer’s instructions and pre-cleared via ultracentrifugation (278,000 × g) for 60 min, before use. |
| 100 mM ATP | Gold Biotechnology Inc, Olivette, MO | A-081 | Resuspended in ddH20 (pH 7.5) and filter sterilized. |
| 100 mM GTP | Fisher Scientific | AC226250010 | Resuspended in 1× BRB80 (pH 6.8) and filter sterilized. |
| 120-150 mW solid-state lasers | Leica Microsystems | 11889151; 11889148 | |
| 2 mg/mL catalase | Sigma Aldrich Inc, St. Louis, MO | C40-100 | Combined with glucose oxidase, aliquot and store at -80 oC until use |
| 2× TIRF buffer | Homemade | 2× BRB80, 100 mM KCl, 20 mM DTT, 80 mM glucose, 0.5% (v/v) methylcellulose (4,000 cp); Note: 1 µL of 0.1M GTP and 1 µL of 0.1M ATP added separately to TIRF reactions to avoid repeated freeze-thaw cycles. | |
| 24 × 60 mm, #1.5 coverglass | Fisher Scientific, Waltham, MA | 22-266882 | Coverglass must be extensively washed before use (Smith et al. (2014)22) |
| 37 oC heatblock | |||
| 37 oC water bath | |||
| 5 mg/mL Streptavidin (600x stock) | Avantor, Philadelphia, PA | RLS000-01 | Resuspended in Tris-HCl (pH 8.8); dilute the aliquot to 1× in HEK buffer on day of use |
| 5 min Epoxy resin and hardener | Loctite, Rocky Hill, CT | 1365736 | Combined resin and hardener may take up to 30 min to cure. |
| 50% biotinylated-GpCpp microtubule seeds | Cytoskeleton Inc; Homemade | T333P | (optional) GppCpp or Taxol stabilized microtubule seeds can more efficiently mediate microtubule polymerization. Taxol and GppCpp stabilize microtubules in different ways that can affect the microtubule lattice structure and ability of certain regulatory proteins to bind to the stabilized portion of the microtubule. A method to make diverse kinds of microtubule seeds is outlined in Hyman et al. (1992). |
| 70 oC incubator | |||
| Actin mix stock | Homemade; this protocol | A 12.5 µM actin mix comprised of labeled (fluorescent and biotinylated) and unlabeled actin for up to six reactions. 2 µL of stock is used in the final TIRF reaction. The final concentration of actin used in each reaction is 0.5 µM (10% Alexa-647; 0.09% biotin-labeled). | |
| Appropriate buffer controls | Homemade | Combination of buffers from all proteins being assessed | |
| Biotin-PEG-silane (MW 3,400) | Laysan Bio Inc | biotin-PEG-SIL-3400 | Dispensed into 2-5 mg aliquots, backfilled with nitrogen, parafilmed closed, and stored at -20 oC with desiccant until use |
| Biotinylated actin | Cytoskeleton Inc; Homemade | AB07 | Biotin-actin is made by labeling on lysine residues and thus assumed to be at least 100% labeled, but varies with different lots/preparations. Optimal biotinylated actin concentrations must be empirically determined for particular uses/experimental designs. Higher concentrations permit more efficient tracking, but may impede polymerization or interactions with regulatory proteins. Here a small percentage (0.09% or 900 pM) biotinylated actin is present in the final TIRF reaction. |
| Dishsoap | Dawn, Procter and Gamble, Cincinnati, OH | For unknown reasons, the blue version cleans coverslips more efficiently than other available colors. | |
| Dry ice | |||
| FIJI Software | www.https://imagej.net/software/fiji/downloads | Schneider et al. (2012)31. | |
| Fluorescently labeled actin | Cytoskeleton Inc; Homemade | AR05 | Homemade fluorescently labeled actin is stored in G-buffer supplemented with 50% glycerol at -20 oC (Spudich et al. (1971)47; Liu et al. (2022)48). Fluorescently labeled actin is dialyzed against G-buffer and precleared via ultracentrifugation for 60 min at 278,000 × g before use. |
| Fluorescently labeled tubulin | Cytoskeleton Inc | TL488M, TLA590M, TL670M | Resuspended in 20 µL 1× BRB80 (10 µM final concentration) and pre-cleared via ultracentrifugation (278,000 × g) for 60 min, before use. |
| G-buffer | Homemade | 3 mM Tris-HCl (pH 8.0), 0.2 mM CaCl2, 0.5 mM DTT, 0.2 mM ATP | |
| HEK Buffer | Homemade | 20 mM HEPES (pH 7.5), 1 mM EDTA (pH 8.0), 50 mM KCl | |
| Ice | |||
| Ice bucket | |||
| Imaging chambers | IBIDI, Fitchburg, WI | 80666 | Order chambers with no bottom to utilize different coverslip coatings |
| iXon Life 897 EMCCD camera | Andor, Belfast, Northern Ireland | 8114137 | |
| LASX Premium microscope software | Leica Microsystems | 11640611 | |
| Methylcellulose (4,000 cp) | Sigma Aldrich Inc | M0512 | |
| Microscope base equipped with TIRF module | Leica Microsystems, Wetzlar, Germany | 11889146 | |
| mPEG-silane (MW 2,000) | Laysan Bio Inc, Arab, AL | mPEG-SIL-2000 | Dispensed into 10-15 mg aliquots, backfilled with nitrogen, parafilmed closed, and stored at -20 oC with desiccant until use |
| Objective heater and heated stage insert | OKO labs, Pozzioli, Italy | 8113569 | Set temperature controls to 35-37 oC. Use manufacturer suggestions for accurate calibration. |
| Perfusion pump | Harvard Apparatus, Holliston, MA | 704504 | A syringe and tubing can be substituted. |
| Petri Dish, 100 x 15 mm | Genesee Scientific, San Diego, CA | 32-107 | |
| Plastic slide mailer container | Fisher Scientific | HS15986 | |
| SA-S-1L-SecureSeal 0.12 mm thick | Grace Biolabs, Bend, OR | 620001 | Double-sided tape of precise manufactured dimensions is strongly recommended. |
| Small styrofoam container | Abcam, Cambridge, UK | Reused from shipping | |
| Small weigh boat | Fisher Scientific | 02-202-100 | |
| Spectrophotometer | |||
| Tau | Cytoskeleton Inc | TA01 | Three isoforms of Tau are present in the commercially available preparation of Tau. The concentration in this protocol was determined from the highest molecular weight band (14.3 µM, when resuspended per manufacturer’s recommendations with 50 µL of ddH20). |
| Temperature corrected 63× Plan Apo 1.47 N.A. oil immersion TIRF objective | Leica Microsystems | 11506319 | |
| Tubulin stock | Homemade; this protocol | A tubulin stock consisting of 7.2 µL recycled 100 µM unlabeled tubulin and 3 µL of 10 µM resuspended commercially available fluorescently labeled tubulin. One tubulin stock is used per reaction and thawed/stored on ice. The final concentration of free tubulin in each reaction is 15 µM (4% labeled). More than 15 µM tubulin will result in hyperstabilized (not dynamic) microtubules, whereas concentrations below 7.5 µM free tubulin do not polymerize well. Careful determination of protein concentration and handling is required. | |
| Unlabeled actin (dark) | Cytoskeleton Inc; Homemade | AKL99 | Actin nucleates are almost always present in commercially available (lyophilized) or frozen actins and contribute to variability in quantitative measurements (Spudich et al. (1971)47; Liu et al. (2022)48). Rabbit muscle actin is stored in G-buffer at -80 oC and precleared via ultracentrifugation for 60 min at 278,000 × g before use. Several actin stock solutions are made throughout the day (making no more than enough for six reactions at a time is strongly recommended). |
References
- Pimm, M. L., Henty-Ridilla, J. L. New twists in actin-microtubule interactions. Molecular Biology of the Cell. 32 (3), 211-217 (2021).
- Dogterom, M., Koenderink, G. H. Actin-microtubule crosstalk in cell biology. Nature Reviews. Molecular Cell Biology. 20 (1), 38-54 (2019).
- Etienne-Manneville, S. Actin and microtubules in cell motility: which one is in control. Traffic. 5 (7), 470-477 (2004).
- Rodriguez, O. C., et al. Conserved microtubule-actin interactions in cell movement and morphogenesis. Nature Cell Biology. 5 (7), 599-609 (2003).
- Prezel, E., et al. TIRF assays for real-time observation of microtubules and actin coassembly: Deciphering tau effects on microtubule/actin interplay. Methods in Cell Biology. 141, 199-214 (2017).
- Griffith, L. M., Pollard, T. D. The interaction of actin filaments with microtubules and microtubule-associated proteins. The Journal of Biological Chemistry. 257 (15), 9143-9151 (1982).
- Henty-Ridilla, J. L., Rankova, A., Eskin, J. A., Kenny, K., Goode, B. L. Accelerated actin filament polymerization from microtubule plus ends. Science. 352 (6288), 1004 (2016).
- Elie, A., et al. Tau co-organizes dynamic microtubule and actin networks. Scientific Reports. 5 (1), 1-10 (2015).
- Preciado López, M., et al. Actin-microtubule coordination at growing microtubule ends. Nature Communications. 5 (1), 1-9 (2014).
- Oberhofer, A., et al. Molecular underpinnings of cytoskeletal cross-talk. Proceedings of the National Academy of Sciences of the United States of America. 117 (8), 3944-3952 (2020).
- Nakos, K., et al. Septins mediate a microtubule-actin crosstalk that enables actin growth on microtubules. bioRxiv. , (2022).
- Kučera, O., Gaillard, J., Guérin, C., Théry, M., Blanchoin, L. Actin-microtubule dynamic composite forms responsive active matter with memory. bioRxiv. , (2022).
- Kundu, T., Dutta, P., Nagar, D., Maiti, S., Ghose, A. Coupling of dynamic microtubules to F-actin by Fmn2 regulates chemotaxis of neuronal growth cones. Journal of Cell Science. 134 (13), 252916 (2021).
- Roth-Johnson, E. A., Vizcarra, C. L., Bois, J. S., Quinlan, M. E. Interaction between microtubules and the Drosophila formin Cappuccino and its effect on actin assembly. The Journal of Biological Chemistry. 289 (7), 4395-4404 (2014).
- Gaillard, J., et al. Differential interactions of the formins INF2, mDia1, and mDia2 with microtubules. Molecular Biology of the Cell. 22 (23), 4575-4587 (2011).
- Bartolini, F., et al. The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. The Journal of Cell Biology. 181 (3), 523-536 (2008).
- Sider, J. R., et al. Direct observation of microtubule-F-actin interaction in cell free lysates. Journal of Cell Science. 112 (12), 1947-1956 (1999).
- Alkemade, C., et al. Cross-linkers at growing microtubule ends generate forces that drive actin transport. Proceedings of the National Academy of Sciences of the United States of America. 119 (11), 2112799119 (2022).
- Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Methods in Enzymology. 361, 1-33 (2003).
- Amann, K. J., Pollard, T. D. Direct real-time observation of actin filament branching mediated by Arp2/3 complex using total internal reflection fluorescence microscopy. Proceedings of the National Academy of Sciences of the United States of America. 98 (26), 15009-15013 (2001).
- Al-Bassam, J. Reconstituting dynamic microtubule polymerization regulation by TOG domain proteins. Methods in Enzymology. 540, 131-148 (2014).
- Smith, B. A., Gelles, J., Goode, B. L. Single-molecule studies of actin assembly and disassembly factors. Methods in Enzymology. 540, 95-117 (2014).
- Wioland, H., Jégou, A., Romet-Lemonne, G. Celebrating 20 years of live single-actin-filament studies with five golden rules. Proceedings of the National Academy of Sciences of the United States of America. 119 (3), 2109506119 (2022).
- Ganzinger, K. A., Schwille, P. More from less - bottom-up reconstitution of cell biology. Journal of Cell Science. 132 (4), (2019).
- Mahamdeh, M., Howard, J. Implementation of interference reflection microscopy for label-free, high-speed imaging of microtubules. Journal of Visualized Experiments. (150), e59520 (2019).
- King, M. R., Petry, S. Phase separation of TPX2 enhances and spatially coordinates microtubule nucleation. Nature Communications. 11 (1), 270 (2020).
- Ramirez-Rios, S., et al. A TIRF microscopy assay to decode how tau regulates EB's tracking at microtubule ends. Methods in Cell Biology. 141, 179-197 (2017).
- Smith, B. A., et al. Three-color single molecule imaging shows WASP detachment from Arp2/3 complex triggers actin filament branch formation. eLife. 2, 01008 (2013).
- Hyman, A. A., Salser, S., Drechsel, D. N., Unwin, N., Mitchison, T. J. Role of GTP hydrolysis in microtubule dynamics: information from a slowly hydrolyzable analogue GMPCPP. Molecular Biology of the Cell. 3 (10), 1155-1167 (1992).
- Li, G., Moore, J. K. Microtubule dynamics at low temperature: evidence that tubulin recycling limits assembly. Molecular Biology of the Cell. 31 (11), 1154-1166 (2020).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Pollard, T. D., Blanchoin, L., Mullins, R. D. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annual Review of Biophysics and Biomolecular Structure. 29, 545-576 (2000).
- Kapoor, V., Hirst, W. G., Hentschel, C., Preibisch, S., Reber, S. MTrack: Automated detection, tracking, and analysis of dynamic microtubules. Scientific Reports. 9 (1), 3794 (2019).
- Willige, D., et al. Cytolinker Gas2L1 regulates axon morphology through microtubule-modulated actin stabilization. EMBO reports. 20 (11), (2019).
- Hirst, W. G., Kiefer, C., Abdosamadi, M. K., Schäffer, E., Reber, S. In vitro reconstitution and imaging of microtubule dynamics by fluorescence and label-free microscopy. STAR Protocols. 1 (3), 100177 (2020).
- Farina, F., et al. The centrosome is an actin-organizing centre. Nature Cell Biology. 18 (1), 65-75 (2016).
- Mishra, M., et al. In vitro contraction of cytokinetic ring depends on myosin II but not on actin dynamics. Nature Cell Biology. 15 (7), 853-859 (2013).
- Groen, A. C., Ngyuen, P. A., Field, C. M., Ishihara, K., Mitchison, T. J. Glycogen-supplemented mitotic cytosol for analyzing Xenopus egg microtubule organization. Methods in Enzymology. 540, 417-433 (2014).
- Pollard, L. W., Garabedian, M. V., Alioto, S. L., Shekhar, S., Goode, B. L. Genetically inspired in vitro reconstitution of Saccharomyces cerevisiae actin cables from seven purified proteins. Molecular Biology of the Cell. 31 (5), 335-347 (2020).
- Bergman, Z. J., Wong, J., Drubin, D. G., Barnes, G. Microtubule dynamics regulation reconstituted in budding yeast lysates. Journal of Cell Science. 132 (4), 219386 (2018).
- Inoue, D., et al. Actin filaments regulate microtubule growth at the centrosome. The EMBO Journal. 38 (11), (2019).
- Colin, A., Singaravelu, P., Théry, M., Blanchoin, L., Gueroui, Z. Actin-network architecture regulates microtubule dynamics. Current Biology. 28 (16), 2647-2656 (2018).
- Torvi, J. R., et al. Reconstitution of kinetochore and microtubule dynamics reveals a role for a kinesin-8 in establishing end-on attachments. bioRxiv. , (2022).
- Nguyen, P. A., et al. Spatial organization of cytokinesis signaling reconstituted in a cell-free system. Science. 346 (6206), 244-247 (2014).
- Abu Shah, E., Keren, K. Symmetry breaking in reconstituted actin cortices. eLife. 3, 01433 (2014).
- Vendel, K. J. A., Alkemade, C., Andrea, N., Koenderink, G. H., Dogterom, M. In vitro reconstitution of dynamic co-organization of microtubules and actin filaments in emulsion droplets. Cytoskeleton Dynamics. 2101, 53-75 (2020).
- Spudich, J. A., Watt, S. The regulation of rabbit skeletal muscle contraction. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. The Journal of Biological Chemistry. 246 (15), 4866-4871 (1971).
- Liu, X., Pimm, M. L., Haarer, B., Brawner, A. T., Henty-Ridilla, J. L. Biochemical characterization of actin assembly mechanisms with ALS-associated profilin variants. European Journal of Cell Biology. 101 (2), 151212 (2022).
- Pimm, M. L., Liu, X., Tuli, F., Lojko, A., Henty-Ridilla, J. L. Visualizing functional human profilin in cells and in vitro applications. bioRxiv. , (2021).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved