
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
المستهدفة
In This Article
Summary
A strategy for generating mutations in histone genes at their endogenous location in Saccharomyces cerevisiae is presented.
Abstract
We describe a PCR- and homologous recombination-based system for generating targeted mutations in histone genes in budding yeast cells. The resulting mutant alleles reside at their endogenous genomic sites and no exogenous DNA sequences are left in the genome following the procedure. Since in haploid yeast cells each of the four core histone proteins is encoded by two non-allelic genes with highly homologous open reading frames (ORFs), targeting mutagenesis specifically to one of two genes encoding a particular histone protein can be problematic. The strategy we describe here bypasses this problem by utilizing sequences outside, rather than within, the ORF of the target genes for the homologous recombination step. Another feature of this system is that the regions of DNA driving the homologous recombination steps can be made to be very extensive, thus increasing the likelihood of successful integration events. These features make this strategy particularly well-suited for histone gene mutagenesis, but can also be adapted for mutagenesis of other genes in the yeast genome.
Introduction
أربعة البروتينات هيستون الأساسية H2A، H2B، H3، H4 وتلعب دورا أساسيا في الضغط، والتنظيم، ووظيفة الصبغيات حقيقية النواة. مجموعتين من كل من هذه الهستونات تشكل octamer هيستون، بكرة الجزيئية التي توجه التفاف ~ 147 أزواج قاعدة الحمض النووي حول نفسها، مما أدى في نهاية المطاف إلى تشكيل جسيم نووي 1. جسيم نووي هي مشاركين نشطين في مجموعة متنوعة من العمليات القائمة على كروموسوم، مثل تنظيم النسخ الجيني وتشكيل كروماتين حقيقي والمغاير في الكروموسومات، وعلى هذا النحو تم التركيز من البحوث المكثفة على مدى العقود العديدة الماضية. وقد وصف عدد من الآليات التي يمكن التلاعب بها جسيم نووي في الطرق التي يمكن أن تسهل تنفيذ عمليات محددة - وتشمل هذه الآليات تعديل posttranslational من بقايا هيستون، ATP التي تعتمد على إعادة عرض جسيم نووي، وإعادة تنظيم جسيم نووي ATP مستقلةوالتجمع / التفكيك 2 و 3.
الخميرة في مهدها خميرة الخباز هي قوية ولا سيما نموذج حي لفهم وظيفة هيستون في حقيقيات النوى. ويمكن أن يعزى ذلك إلى حد كبير إلى درجة عالية من الحفظ التطوري من البروتينات هيستون في جميع أنحاء حقيقيات النوى المجال وقابليته للخميرة لمجموعة متنوعة من التجريبية الجينية والبيوكيميائية النهج 4. وقد استخدمت على نطاق واسع نهج عكس الوراثية في الخميرة لدراسة آثار الطفرات هيستون محددة بشأن مختلف جوانب علم الأحياء لونين. لهذه الأنواع من التجارب غالبا ما يكون من الأفضل استخدام الخلايا التي يتم التعبير عنها في الهستونات متحولة من مواضع الجيني الأصلي لها، والتعبير عن البلازميدات مستقلة يمكن أن يؤدي إلى مستويات غير طبيعية داخل الخلايا من البروتينات هيستون (بسبب وجود عدد من البلازميدات متفاوتة في الخلايا) و تغيير يصاحب ذلك من لونين أونvironments، والتي يمكن أن نخلط في نهاية المطاف تفسير النتائج.
هنا، نحن تصف تقنية PCR القائم الذي يسمح لالطفرات الموجهة للجينات هيستون في مواقع الجينومية الأصلية الخاصة بهم والتي لا تتطلب خطوة الاستنساخ والنتائج في جيل الطفرة المطلوبة (ق) من دون بقايا تسلسل الحمض النووي الخارجية في الجينوم. هذا الأسلوب يستفيد من نظام إعادة التركيب مثلي كفاءة في الخميرة، ولها عدة سمات مشتركة مع غيرها من التقنيات المشابهة التي وضعتها المجموعات الأخرى - وأبرزها Delitto بيرفيتو، الجينوم (SSG) الطفرات في مواقع محددة، وخالية استنساخ أليل القائم على PCR طرق استبدال 5، 6، 7. ومع ذلك، فإن تقنية وصفنا لها الجانب الذي يجعل من وبخاصة مناسبة تماما لالطفرات الجينات هيستون. في خلايا الخميرة فرداني، يتم ترميز كل من histones الأساسية الأربعة من قبل اثنين من غير على بعدالجينات llelic والمتجانسة للغاية: على سبيل المثال، يتم ترميز H3 هيستون بواسطة الجينات HHT1 وHHT2، وإطارات القراءة المفتوحة (ORFS) من الجينات هما أكثر من 90٪ مماثلة في التسلسل. هذه درجة عالية من التماثل يمكن تعقيد التجارب المصممة خصيصا لاستهداف واحد من اثنين من جينات ترميز هيستون عن الطفرات. في حين أن الطرق المذكورة أعلاه وغالبا ما تتطلب استخدام بعض ما لا يقل عن تسلسل داخل ORF من الجين المستهدف لحملة إعادة التركيب مثلي، والتقنية وصفنا هنا يجعل من استخدام تسلسل المرافقة ORFS من الجينات هيستون (التي تشترك أقل بكثير تسلسل تناظر) ل الخطوة إعادة التركيب، مما يزيد من احتمال استهداف ناجح من الطفرات إلى مكان المطلوب. وعلاوة على ذلك، يمكن للمناطق متماثلة التي تدفع إعادة التركيب تكون واسعة جدا، مما يسهم في كفاءة إعادة التركيب مثلي المستهدفة.
Protocol
ملاحظة: استراتيجية تجريبية لالمستهدفة في الموقع الطفرات هيستون الجين تتضمن عدة خطوات (ملخصة في الشكل 1). وتشمل الخطوات التالية: (1) استبدال الجين هيستون الهدف مع الجينات URA3، (2) جيل وتنقية المنتجات PCR المقابلة لاثنين من شظايا متداخلة جزئيا من الجين هيستون الهدف باستخدام بادئات إيواء الطفرة المطلوبة (ق) و (3 ) فيوجن PCR اثنين من شظايا متداخلة جزئيا للحصول على كامل حجم المنتجات PCR للتكامل، (4) المشاركة في التحول من المنتجات PCR بالحجم الكامل والبلازميد العمود الفقري، واختيار علامة على البلازميد، (5) شاشة لمقاومة-5-FOA transformants، (6) تنقية المستعمرات المقاومة لل5 FOA وفقدان البلازميد العمود الفقري، و (7) الجزيئية تحليلات لفحص للاندماج الصحيح للأليل متحولة.

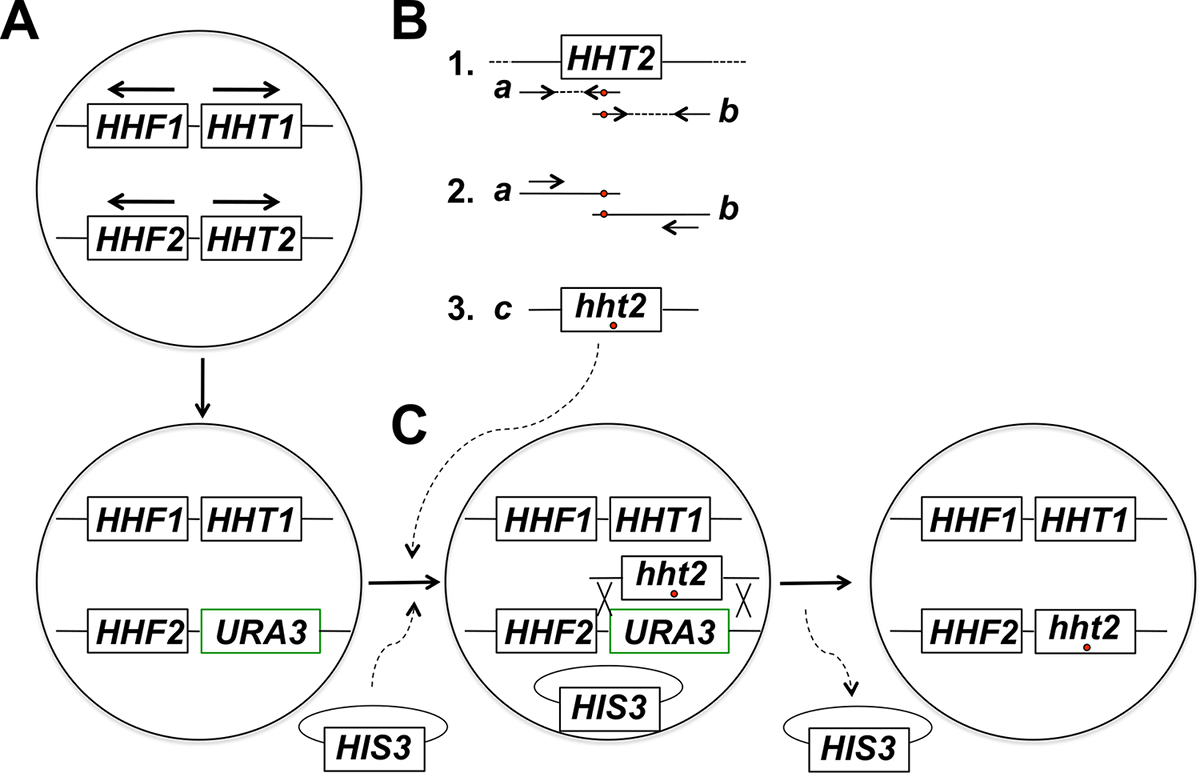
الشكل 1: نظرة عامة على استراتيجية المستهدفة في الموقع الطفرات من الجينات هيستون في مهدها الخميرة. في هذا المثال الجين المستهدف هو HHT2، ولكن يمكن أيضا أي الجينات هيستون الأساسية الأخرى أن mutagenized باستخدام هذه الاستراتيجية. (A) خلايا الخميرة فرداني تؤوي اثنين هيستون جينات ترميز H3 (HHT1 وHHT2) واثنين من الجينات ترميز H4 هيستون (HHF1 وHHF2) مرتبة كما هو مبين في الشكل (توجد جينات HHT1 وHHF1 على الكروموسوم الثاني وHHT2 وتقع الجينات HHF2 في الرابع عشر كروموسوم - في كل حالة على حدة، وتشير الأسهم في الاتجاه من النسخ). في الخطوة الأولى من هذا الإجراء، يتم استبدال ORF من الجين HHT2 مع الجين URA3، مما أدى إلى سلالة hht2Δ :: URA3. (ب) في الجزء 1، يتم استخدام نسخة من النوع البري من الجين HHT2 من عينة الحمض النووي الجيني كقالب لمدة تفاعلات PCR إلى أجناسالشركة المصرية للاتصالات اثنين شظايا متداخلة جزئيا من الجين. التمهيدي العكسي لأول رد فعل يتضمن واحد أو أكثر غير متطابقة النيوكليوتيدات (المشار إليها مع دائرة حمراء) التي تتوافق مع الطفرة المطلوبة (ق) ليتم إدخالها في الجينوم. التمهيدي إلى الأمام للتفاعل الثاني له فمنها ما يعادلها في تكوين تكميلي العكسي (المشار إليها أيضا مع دائرة حمراء). المنتجات PCR اثنين ولدت في الجزء 1 (منتجات أ و ب) يتم استخدامها بعد ذلك نماذج لانصهار PCR باستخدام بادئات أن يصلب لمنتجات أ و ب في الشكل المبين في الجزء 2. وهذا يؤدي إلى توليد كاملة الحجم PCR المنتجات (المنتج ج في جزء 3) بإيواء الطفرة المطلوبة (ق). (ج) hht2Δ :: سلالة URA3 ومن ثم شارك في تحويلها مع المنتجات PCR كاملة الحجم والبلازميد العمود الفقري (ملحوظ على HIS3- البلازميد في هذا المثال)، ويتم اختيار خلايا لوجود البلازميد (على وسائل الإعلام التي تفتقر إلى حistidine في هذا المثال). ثم يتم فحص Transformants لمدة 5-FOA المقاومة - الخلايا المقاومة هي المرشحين لأنه خضع لحدث إعادة التركيب مثلي مما يؤدي إلى تكامل المنتج PCR واستئصال الجين URA3، كما هو مبين. الخسارة اللاحقة من البلازميد العمود الفقري من قبل انقسام الخلية الإنقسامية يؤدي إلى هيستون المطلوب سلالة متحولة النهائي. لقد وجدنا أن اختيار البلازميد العمود الفقري تليها الكشف عن النتائج المقاومة 5 FOA في تردد أعلى بكثير من تحديد أحداث التكامل الصحيحة مقارنة الاختيار المباشر على لوحات 5 FOA، والذي يعرف في الغالب الخلايا التي اكتسبت طفرات URA3 عفوية. (تم تعديل هذا الرقم من مرجع 14). الرجاء انقر هنا لعرض نسخة أكبر من هذا الرقم.
{kind=link}
1. استبدال هدف هيستون جين مع URA3جينة
- أداء قياسي بوساطة PCR-خطوة واحدة تعطيل الجينات استبدال ORF الهدف الجينات هيستون مع الجين URA3 8 و 9.
ملاحظة: من المستحسن استخدام خلايا الخميرة تحمل ura3Δ0 لأن هذا التحور يزيل URA3 الذاتية كامل ORF، وبالتالي تجنب تكامل المنتج PCR في موضع URA3 8. بدلا من ذلك، K. اللبنية URA3 الجينات يمكن استخدامها بفعالية في توليد استبدال هيستون في أي خلفية ura3 كما هو وظيفي في البيرة س ولكن ليس لديها سوى جزئيا تسلسل تناظر مع الجين البيرة س URA3. وينبغي أيضا أن يكون من سلالة العوز الغذائي للمجمع واحد على الأقل من شأنها أن تسمح للاختيار من البلازميد العمود الفقري في تجربة التحول (راجع الخطوة 4 من هذا البروتوكول). هذه الخطوة غير ضرورية إذا كان geneΔ هيستون الهدف :: URA3سلالة متاح بالفعل.
2. الجيل وتنقية PCR المنتجات الموافق اثنان أجزاء متداخلة جزئيا من الهدف هيستون جين باستخدام الاشعال إيواء الطفرة المرجوة (ق)
- توليد منتجات PCR المقابلة لاثنين من شظايا متداخلة جزئيا من الجين هيستون الهدف.
- إعداد اثنين من ردود الفعل PCR على النحو التالي:
- لتوليد المنتجات PCR الموافق النصف الأول من هذا الجين (منتج في الشكل 1B)، إعداد التفاعل التالي: 1 ميكرولتر قالب الحمض النووي، 5 μl10 ميكرومتر التمهيدي إلى الأمام، 5 μl10 ميكرومتر التمهيدي العكسي 0.5 ميكرولتر (1.25 U) بالحرارة البلمرة DNA، 10 ميكرولتر 5X DNA العازلة البلمرة، 5 ميكرولتر dNTP خليط (2 مم لكل منهما)، و 23.5 ميكرولتر درهم 2 O.
ملاحظة: قالب الحمض النووي يمكن أن يكون الحمض النووي الجيني المستمدة من سلالة من النوع البري لجين هيستون الهدف معزولة باستخدام الموالية القياسيةcedures 10. لحساب التغيرات في تركيز الحمض النووي ومستوى الشوائب في الأعمال التحضيرية الجينومية مختلفة، فمن المستحسن لتحسين ردود الفعل باستخدام إما الحمض النووي مخفف أو التخفيفات مختلفة من الاستعدادات الجينوم (على سبيل المثال، 1:10 و 1: 100). التمهيدي إلى الأمام يجب أن يصلب إلى منطقة المنبع من الجين المستهدف. التمهيدي العكسي يجب أن يصلب داخل ORF، يكون ~ 40 النيوكليوتيدات في الطول، وتحتوي على الطفرة المرجوة (ق) في مكان ما في المنتصف منه (انظر الشكل 1B-1 وممثل النتائج القسم للحصول على أمثلة). من المستحسن استخدام عالية الدقة البلمرة DNA من أجل الحد من معدلات الطفرات غير مرغوب فيها أثناء تركيب المنتجات PCR. - لتوليد المنتجات PCR الموافق النصف الثاني من هذا الجين (المنتج ب في الشكل 1B)، إعداد رد الفعل كما هو مبين في 2.1.1.1 لكن مع الاشعال مختلفة.
ملاحظة: الحزب الثوري المؤسسي إلى الأماميجب مير يصلب داخل ORF، يكون ~ 40 النيوكليوتيدات في الطول، واحتواء الطفرة المطلوبة (ق) في مكان ما في منتصفها. لاحظ أن الطفرة (ق) في هذا الكتيب هو تكملة العكسي للطفرة (ق) في التمهيدي العكسي في الخطوة 2.1.1.1. التمهيدي العكس يجب أن يصلب إلى منطقة المصب من الجينات المستهدفة (انظر الشكل 1B-1 وممثل النتائج القسم للحصول على أمثلة).
- لتوليد المنتجات PCR الموافق النصف الأول من هذا الجين (منتج في الشكل 1B)، إعداد التفاعل التالي: 1 ميكرولتر قالب الحمض النووي، 5 μl10 ميكرومتر التمهيدي إلى الأمام، 5 μl10 ميكرومتر التمهيدي العكسي 0.5 ميكرولتر (1.25 U) بالحرارة البلمرة DNA، 10 ميكرولتر 5X DNA العازلة البلمرة، 5 ميكرولتر dNTP خليط (2 مم لكل منهما)، و 23.5 ميكرولتر درهم 2 O.
- وضع ردود الفعل في thermocycler مع الإعدادات التالية: 94 ج 30 ثانية؛ 30 دورات من الإعدادات التالية: 98 ج 10 ثانية و 60 ج 5 ثانية، 72 ج 1.5 دقيقة. و72 ج 10 دقيقة.
ملاحظة: قد تكون هناك حاجة الأمثل المعلمات PCR لمجموعات التمهيدي محددة واستهداف الجين هيستون.
- إعداد اثنين من ردود الفعل PCR على النحو التالي:
- تشغيل 20 - 50 ميكرولتر من المواد من ردود الفعل PCR على 0.9٪ انصهار منخفضة هلام نقطة الاغاروز في 89 ملي تريس قاعدة، 89 ملم حمض البوريك، 2.5 ملي EDTA (TBE) العازلة.
- قطع أقسام هلام الاغاروز التي تحتوي على العلاقات العامة PCRoducts العلاقات من هلام باستخدام نظيفة مشرط أو شفرة حلاقة ونقل كل لأنبوب microcentrifuge 1.5 مل. تخزين المقاطع الأغاروس المنتجات المحتوية على PCR عند درجة حرارة -20 مئوية لتصبح جاهزة للاستخدام.
3. فيوجن PCR من شظايا متداخلة جزئيا اثنين من الحصول على كامل الحجم PCR المنتجات للتكامل
- إعداد نموذج لردود الفعل PCR
- تذوب أقسام هلام الاغاروز من الخطوة 2.3 عن طريق وضع أنابيب microcentrifuge في مجموعة كتلة الحرارة عند 65 مئوية لمدة 5 دقائق (أو حتى ذاب تماما). أنابيب دوامة كل 1-2 دقيقة لتسهيل عملية الصهر.
- نقل مبلغ محدد من agarose ذاب من كل عينة (على سبيل المثال، 50 ميكرولتر لكل منهما، ليصبح المجموع 100 ميكرولتر) في أنبوب microcentrifuge واحد ومزيج من قبل vortexing. استخدام هذا القالب في الانصهار ردود الفعل PCR. وضع أنبوب في -20 مئوية لتصبح جاهزة للاستخدام.
- تضخيم كمية كبيرة من حجم كامل المنتج PCR (المنتج جفي الشكل 1B)
- إعداد ستة تفاعلات PCR، مع كل من العناصر التالية: 2 ميكرولتر الحمض النووي القالب، و 10 ميكرولتر 10 ميكرومتر التمهيدي إلى الأمام، 10 ميكرولتر 10 ميكرومتر التمهيدي العكسي، 1 ميكرولتر (2.5 U) بالحرارة البلمرة DNA، 20 5X ميكرولتر عازلة الحمض النووي بوليميريز، 10 ميكرولتر خليط dNTP (2 مم لكل منهما)، و 47 ميكرولتر درهم 2 O.
ملاحظة: يمكن تغيير عدد من ردود الفعل تبعا لكفاءة PCR. وينبغي تسخين الحمض النووي قالب (انظر 3.1.2) إلى 65 مئوية حتى ذاب، مختلطة من قبل vortexing، وأضاف آخر من مزيج تفاعل PCR. مرة واحدة وأضاف، مزيج بلطف ولكن بدقة من قبل pipetting الحل صعودا وهبوطا عدة مرات. لحساب التغيرات في تركيز الحمض النووي في العينات المختلفة، فمن المستحسن لأول مرة تحسين ردود الفعل باستخدام أي قالب غير مخفف أو التخفيفات مختلفة من القالب (على سبيل المثال، 1:10 و 1: 100). وهما الاشعال المستخدمة يجب أن يصلب لاثنين من شظايا متداخلة جزئيا من الجينات المستهدفة وسوءustrated في الشكل 1B-2 وتكون مصممة بحيث منتجات PCR النهائية سوف يكون لا يقل عن 40 أزواج قاعدة على أي مثلي الجانب للمناطق المحيطة وURA3 ORF من شأنها أن تدفع خطوة إعادة التركيب مثلي (انظر القسم ممثل النتائج على سبيل المثال). من المستحسن استخدام عالية الدقة البلمرة DNA من أجل الحد من معدلات الطفرات غير مرغوب فيها أثناء تركيب المنتجات PCR. - وضع أنابيب في thermocycler مع الإعدادات التالية: 94 ج 30 ثانية؛ 30 دورات من الإعدادات التالية: 98 ج 10 ثانية و 50 ج 15 ثانية، 72 ج 1.5 دقيقة. و72 ج 10 دقيقة.
ملاحظة: قد تكون هناك حاجة الأمثل المعلمات PCR لمجموعات التمهيدي محددة والجينات هيستون الهدف.
- إعداد ستة تفاعلات PCR، مع كل من العناصر التالية: 2 ميكرولتر الحمض النووي القالب، و 10 ميكرولتر 10 ميكرومتر التمهيدي إلى الأمام، 10 ميكرولتر 10 ميكرومتر التمهيدي العكسي، 1 ميكرولتر (2.5 U) بالحرارة البلمرة DNA، 20 5X ميكرولتر عازلة الحمض النووي بوليميريز، 10 ميكرولتر خليط dNTP (2 مم لكل منهما)، و 47 ميكرولتر درهم 2 O.
4. المشارك تحول كامل الحجم PCR المنتجات والعمود الفقري البلازميد، واختيار لعلامة على البلازميد
- تركيز المنتجات PCR
- تجميع الصورةردود الفعل التاسع PCR (600 مجموع ميكرولتر) من الخطوة 3.2.2 إلى أنبوب microcentrifuge واحد ومزيج من قبل vortexing.
- تقسيم العينة إلى ثلاث 200 مكل في أنابيب microcentrifuge. ترسيب الحمض النووي في كل أنبوب عن طريق إضافة 20 ميكرولتر من 3M خلات الصوديوم (الرقم الهيدروجيني 5.2) و 550 ميكرولتر من الإيثانول بنسبة 100٪. مزيج من حل شامل ووضع على الجليد لمدة 15 دقيقة على الأقل. جمع الحمض النووي عن طريق الطرد المركزي في ~ 14000 x ج لمدة 10 دقيقة، شطف بيليه مع 200 ميكرولتر من الايثانول 70٪، والهواء الجاف.
- resuspend كل بيليه الحمض النووي في 25 ميكرولتر من O 2 درهم، وتجمع في أنبوب واحد (أي ما مجموعه 75 ميكرولتر).
- الخميرة شارك في التحول
- إعداد 10 مل من الثقافة بين عشية وضحاها من سلالة ولدت في القسم 1 في استخراج الخميرة ببتون سكر العنب (YPD) السائل المتوسطة 11.
- في صباح اليوم التالي، تطعيم 400 مل من YPD المتوسطة السائل مع 8 مل من الثقافة بين عشية وضحاها المشبعة واحتضان التي تهزفي 30 درجة مئوية لمدة 4-5 ساعات للسماح للخلايا لدخول مرحلة لوغاريتمي من النمو.
- جمع الخلايا بواسطة الطرد المركزي في ~ 3220 x ج لمدة 10 دقيقة، تجاهل المتوسطة السائل، وresuspend الخلايا في 1 حجم 10 ملي تريس، حمض الهيدروكلوريك (الرقم الهيدروجيني 8.0)، 1 ملم EDTA، 0.1 م ليثيوم خلات الحل (TE / LiAc) .
- جمع الخلايا بواسطة الطرد المركزي في ~ 3220 x ج لمدة 10 دقيقة، وتجاهل TE / LiAc.
- Resuspend الخلايا في 1 مل TE / LiAc.
- إعداد كوكتيل التفاعل التالي في أنبوب microcentrifuge: 800 ميكرولتر من الخلايا من الخطوة 4.2.5، 40 ميكرولتر من مغلي 10 ملغ / مل السلمون الحمض النووي الحيوانات المنوية، أي ما مجموعه 12.5 ميكروغرام من العمود الفقري DNA البلازميد، و 75 ميكرولتر من الناتج PCR المركزة من الخطوة 4.1.3.
يجب أن تكون مسلوقة سمك السلمون الحمض النووي الحيوانات المنوية لمدة 5 دقائق ويوضع على الجليد لمدة 5 دقائق على الأقل قبل استخدامها في رد الفعل: ملاحظة. يجب أن تبقى إجمالي حجم الحمض النووي العمود الفقري البلازميد وأضاف إلى أدنى حد ممكن (~ 80 ميكرولتر أو أقل). انظر القسم ممثل النتائج للحصول على مثال من بلازما العمود الفقريهوية شخصية. - مزيج من أنبوب كوكتيل بدقة وقسامة بالتساوي إلى ثمانية أنابيب microcentrifuge (أنابيب 1-8).
- إعداد التحكم اثنين أنابيب رد فعل التحول التالية:
- أنبوب 9 (أي سيطرة المنتج PCR): 100 ميكرولتر من الخلايا من الخطوة 4.2.5، 5 ميكرولتر من مغلي 10 ملغ / مل السلمون الحمض النووي الحيوانات المنوية (المغلي لمدة 5 دقائق، وانظر الخطوة 4.2.6 ملاحظة)، أي ما مجموعه 1.56 ميكروغرام من وأضاف العمود الفقري DNA البلازميد وأي منتج PCR.
- أنبوب 10 (أي سيطرة DNA): 100 ميكرولتر من الخلايا من الخطوة 4.2.5، 5 ميكرولتر من مغلي 10 ملغ / مل السلمون الحمض النووي الحيوانات المنوية (راجع الخطوة 4.2.6 ملاحظة)، إضافة أي البلازميد العمود الفقري وأضاف الحمض النووي، وأي منتج PCR.
- خلط كل من أنابيب بلطف ولكن بشكل دقيق من قبل pipetting صعودا وهبوطا عدة مرات.
- احتضان الأنابيب عشرة في 30 مئوية لمدة 30 دقيقة.
- لكل أنبوب، إضافة 1.2 مل من 40٪ البولي ايثيلين جلايكول (PEG 3350) في TE / LiAc. تخلط جيدا باستخدام الماصة-P 1000 حتى الحل هو متجانس.
- احتضان الأنابيب عشرة في 30° مئوية لمدة 30 دقيقة. المزيج بلطف حل عن طريق pipetting صعودا وهبوطا ثم احتضان الأنابيب عند 42 درجة مئوية لمدة 15 دقيقة.
- جمع الخلايا من خلال الدوران الأنابيب في microcentrifuge في ~ 14000 x ج لمدة 30 ثانية. تجاهل السائل وresuspend الخلايا في 1 مل من درهم معقم 2 O.
- جمع الخلايا من خلال الدوران الأنابيب في microcentrifuge في ~ 14000 x ج لمدة 30 ثانية. تجاهل السائل وresuspend الخلايا في 500 ميكرولتر من درهم معقم 2 O.
- أنابيب بركة 1-8 معا (الحجم الكلي من 4 مل)، ومزيج دقيق من قبل pipetting صعودا وهبوطا.
- لوحة 200 ميكرولتر من الخليط المذكور أعلاه على كل عشرين لوحات كاملة المتوسطة الحد الأدنى من التسرب 11 (لوحات 1-20) لاختيار البلازميد العمود الفقري.
- لوحة 200 ميكرولتر من خليط من أنبوب 9 و 200 ميكرولتر من خليط من أنبوب 10 كل على لوحة اختيار الخاصة بها (لوحات 21 و 22، على التوالي).
- احتضان لوحات 22 حتى 30 درجة مئوية لمدة 3-5 أيام لحدد لtransformants البلازميد.
- تفقد لوحات التحول بعد 3-5 أيام من الحضانة. ما يقرب من يجب أن يكون 5000 المستعمرات مرئية على لوحات 1-21 (انظر ممثل النتائج للحصول على مثال) ويجب أن تكون هناك مستعمرات موجودة على لوحة 22.
5. الشاشة لTransformants مقاومة لل5 FOA
- نقل الخلايا من لوحات 1 - 20 (ولوحة تحول 21 كعنصر تحكم) إلى 5 fluoroorotic حمض (5 FOA) لوحات 11 من 12 من أجل كشف عن فقدان الجين URA3 نتيجة اندماج تصفيح نسخة منتجات PCR في الموقع المطلوب.
- إزالة غطاء لوحة واضغط على لوحة تحتوي على المستعمرات على المخملية العقيمة. نقل الخلايا من المخمل إلى لوحة 5-FOA عن طريق الضغط على لوحة على المخملية. احتضان لوحات عند 30 مئوية لمدة 2 أيام.
- بعد حضانة 2 يوما، وفحص بعناية لوحات 5 FOA للغرامowth.
ملاحظة: وسوف يمثل حدثا التكامل مرشح صغيرة غير المتماثلة مستعمرة "سحق" على طبق من 5 FOA - على العكس، حليمات صغيرة تنمو على لوحات 5 FOA وممثل المرجح الطفرات URA3 العفوية التي نشأت أثناء نمو المستعمرات على لوحات التحول، وبالتالي فهي غير المرجح أن يمثل هذا الحدث التكامل المنشود (انظر الشكل 3 في قسم ممثل النتائج لمزيد من التفصيل حول هذه النقطة وبالنسبة لبعض الأمثلة).
6. تنقية المستعمرات المقاومة لل5 FOA وفقدان البلازميد العمود الفقري
- استخدام المسواك العقيمة، واختيار المستعمرات مرشح من لوحات 5 FOA هو موضح في الخطوة 5.2 و خط لالمستعمرات واحد على لوحات YPD. احتضان لمدة 2-3 أيام عند 30 درجة مئوية.
- بعد الحضانة، نسخة طبق الاصل - لوحة كل YPD تنقية لوحة إلى لوحة YPD جديدة، لوحة التسرب تفتقر اليوراسيل للتحقق من الخسارةمن الجين URA3، وصفيحة التسرب الثانية لمراقبة وجود أو عدم وجود البلازميد العمود الفقري. احتضان لمدة 1-2 أيام عند 30 درجة مئوية.
- بعد الحضانة، وتحديد مستعمرة من كل عينة المرشح الذي ينمو على لوحة YPD ولكن لا ينمو على أي لوحة التسرب (من المتوقع مثل مستعمرة قد فقدت الجينات URA3 من خلال هذا الحدث إعادة التركيب وخسر البلازميد العمود الفقري خلال الإنقسامية انقسام الخلية). Restreak هذه المستعمرات على لوحات YPD جديدة. هذه المستعمرات هي المرشحين التكامل ووسيجري تحليل في الخطوة 7.
7. التحليلات الجزيئية لفحص للتكامل السليم للالمسخ أليل
- عزل الحمض النووي الجيني من العينات مرشح باستخدام إجراءات القياسية 10.
- تضخيم المنطقة الجينومية يشمل الموقع المستهدف.
- إعداد تفاعل PCR التالية لكل عينة: 0.5 ميكرولتر الحمض النووي القالب، 5 ميكرولتر10 ميكرومتر التمهيدي إلى الأمام، 5 ميكرولتر 10 ميكرومتر التمهيدي العكسي 0.5 ميكرولتر (2.5 وحدة) طق الحمض النووي بوليميريز، 5 ميكرولتر 10X طق DNA العازلة البلمرة، 5 ميكرولتر خليط dNTP (2 مم لكل منهما)، و 29 ميكرولتر درهم 2 O.
ملاحظة: قالب الحمض النووي هو الحمض النووي الجيني المستمدة من العينات مرشح. فمن المستحسن أن تشمل أيضا اثنين من ردود الفعل التحكم: واحد باستخدام الحمض النووي الجيني المستمدة من geneΔ هيستون الأصلي :: URA3 سلالة كقالب واستخدام الحمض النووي الجيني آخر من نوع السلالة البرية هيستون كقالب. لحساب التغيرات في تركيز الحمض النووي ومستوى الشوائب في الأعمال التحضيرية الجينومية مختلفة، فمن المستحسن لتحسين ردود الفعل باستخدام إما الحمض النووي مخفف أو التخفيفات مختلفة من الاستعدادات الجينوم (على سبيل المثال، 1:10 و 1: 100). من المهم للتأكد من أن هذه الاشعال يصلب لتسلسل الحمض النووي خارج المنطقة التي تشملها المنتج PCR متكاملة مزعومة - وبهذه الطريقة، وحجم المنتجات PCR في هذه صeactions يمكن أن تستخدم كأداة تشخيصية لتكامل المنتجات في الموقع الجيني الصحيح (انظر ممثل النتائج للحصول على مثال). - وضع ردود الفعل في thermocycler مع الإعدادات التالية: 94 ° C 3 دقائق. 30 دورات من الإعدادات التالية: 94 درجة مئوية و 45 ثانية و 50 درجة مئوية و 45 ثانية و 72 درجة مئوية 2 دقيقة. و 72 درجة مئوية مدة 10 دقيقة.
ملاحظة: قد تكون هناك حاجة الأمثل المعلمات PCR لمجموعات التمهيدي محددة والجينات هيستون الهدف.
- إعداد تفاعل PCR التالية لكل عينة: 0.5 ميكرولتر الحمض النووي القالب، 5 ميكرولتر10 ميكرومتر التمهيدي إلى الأمام، 5 ميكرولتر 10 ميكرومتر التمهيدي العكسي 0.5 ميكرولتر (2.5 وحدة) طق الحمض النووي بوليميريز، 5 ميكرولتر 10X طق DNA العازلة البلمرة، 5 ميكرولتر خليط dNTP (2 مم لكل منهما)، و 29 ميكرولتر درهم 2 O.
- تجهيز المنتجات PCR
- تشغيل 20 ميكرولتر من كل رد فعل على 0.8٪ TBE هلام الاغاروز.
- تقييم حجم المنتجات PCR باستخدام معايير الحمض النووي كمرجع لتحديد ما إذا تم استبدال الجينات URA3 بنجاح بواسطة الجينات هيستون تحور مزعومة (انظر ممثل النتائج للحصول على مثال).
ملاحظة: في بعض الحالات، الطفرة المطلوبة (الصورة) التي أدخلت الجينات هيستون إما إنشاء أو تدمير تقييد الجلوسه. إذا كان هذا هو الحال، فإن وجود الطفرة المرجوة في منتجات PCR من حجم يدل على التكامل الصحيح يمكن تقييمها من خلال إخضاع المنتجات للهضم مع انزيم التقييد المقابل تليها التحليل الكهربائي للهلام (انظر ممثل النتائج للحصول على مثال) . - منتجات PCR تخضع لحجم يدل على التكامل الصحيح لتسلسل الحمض النووي لتأكيد وجود طفرة المطلوب (ق) وضمان أن أدخلت أي طفرات إضافية في الجينوم.
النتائج
نحن تصف الجيل من أليل hht2 تعبر عن هيستون H3 البروتين متحولة إيواء إجراء تبديل في موقف 53 من أرجينين إلى حمض الجلوتاميك (H3-R53E متحولة) كمثال التمثيلي للالمستهدفة في استراتيجية الموقع الطفرات.
Discussion
على مستوى عال من تسلسل تناظر بين الجينين غير أليلية أن رمز لكل من البروتينات هيستون الأساسية الأربعة في الخلايا البيرة س فرداني يمكن أن تمثل تحديا للمحققين الذين يرغبون خصيصا لاستهداف واحد من اثنين من الجينات لالطفرات. سبق وصفها منهجيات الطفرات الخميرة، بما في ...
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
We thank Reine Protacio for helpful comments during the preparation of this manuscript. We express our gratitude to the National Science Foundation (grants nos. 1243680 and 1613754) and the Hendrix College Odyssey Program for funding support.
Materials
| Name | Company | Catalog Number | Comments |
| 1 kb DNA Ladder (DNA standards) | New England BioLabs | N3232L | |
| Agarose | Sigma | A5093-100G | |
| Boric Acid | Sigma | B0394-500G | |
| dNTP mix (10 mM each) | ThermoFisher Scientific | R0192 | |
| EDTA solution (0.5 M, pH 8.0) | AmericanBio | AB00502-01000 | |
| Ethanol (200 Proof) | Fisher Scientific | 16-100-824 | |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma | E4884-500G | |
| Lithium acetate dihydrate | Sigma | L6883-250G | |
| MyCycler Thermal Cycler | BioRad | 170-9703 | |
| Poly(ethylene glycol) (PEG) | Sigma | P3640-1KG | |
| PrimeSTAR HS DNA Polymerase (high fidelity DNA polymerase) and 5x buffer | Fisher Scientific | 50-443-960 | |
| Salmon sperm DNA solution | ThermoFisher Scientific | 15632-011 | |
| Sigma 7-9 (Tris base, powder form) | Sigma | T1378-1KG | |
| Sodium acetate trihydrate | Sigma | 236500-500G | |
| Supra Sieve GPG Agarose (low metling temperature agarose) | AmericanBio | AB00985-00100 | |
| Taq Polymerase and 10x Buffer | New England BioLabs | M0273X | |
| Toothpicks | Fisher Scientific | S67859 | |
| Tris-HCl (1 M, pH 8.0) | AmericanBio | AB14043-01000 | |
| a-D(+)-Glucose | Fisher Scientific | AC170080025 | for yeast media |
| Agar | Fisher Scientific | DF0140-01-0 | for yeast media |
| Peptone | Fisher Scientific | DF0118-07-2 | for YPD medium |
| Yeast Extract | Fisher Scientific | DF0127-17-9 | for YPD medium |
| 4-aminobenzoic acid | Sigma | A9878-100G | for complete minimal dropout medium |
| Adenine | Sigma | A8626-100G | for complete minimal dropout medium |
| Glycine hydrochloride | Sigma | G2879-100G | for complete minimal dropout medium |
| L-Alanine | Sigma | A7627-100G | for complete minimal dropout medium |
| L-Arginine monohydrochloride | Sigma | A5131-100G | for complete minimal dropout medium |
| L-Asparagine monohydrate | Sigma | A8381-100G | for complete minimal dropout medium |
| L-Aspartic acid sodium salt monohydrate | Sigma | A6683-100G | for complete minimal dropout medium |
| L-Cysteine hydrochloride monohydrate | Sigma | C7880-100G | for complete minimal dropout medium |
| L-Glutamic acid hydrochloride | Sigma | G2128-100G | for complete minimal dropout medium |
| L-Glutamine | Sigma | G3126-100G | for complete minimal dropout medium |
| L-Histidine monohydrochloride monohydrate | Sigma | H8125-100G | for complete minimal dropout medium |
| L-Isoleucine | Sigma | I2752-100G | for complete minimal dropout medium |
| L-Leucine | Sigma | L8000-100G | for complete minimal dropout medium |
| L-Lysine monohydrochloride | Sigma | L5626-100G | for complete minimal dropout medium |
| L-Methionine | Sigma | M9625-100G | for complete minimal dropout medium |
| L-Phenylalanine | Sigma | P2126-100G | for complete minimal dropout medium |
| L-Proline | Sigma | P0380-100G | for complete minimal dropout medium |
| L-Serine | Sigma | S4500-100G | for complete minimal dropout medium |
| L-Threonine | Sigma | T8625-100G | for complete minimal dropout medium |
| L-Tryptophan | Sigma | T0254-100G | for complete minimal dropout medium |
| L-Tyrosine | Sigma | T3754-100G | for complete minimal dropout medium |
| L-Valine | Sigma | V0500-100G | for complete minimal dropout medium |
| myo-Inositol | Sigma | I5125-100G | for complete minimal dropout medium |
| Uracil | Sigma | U0750-100G | for complete minimal dropout medium |
| Ammonium Sulfate | Fisher Scientific | A702-500 | for complete minimal dropout medium |
| Yeast Nitrogen Base | Fisher Scientific | DF0919-07-3 | for complete minimal dropout medium |
| 5-Fluoroorotic acid (5-FOA) | AmericanBio | AB04067-00005 | for 5-FOA medium |
References
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389 (6648), 251-260 (1997).
- Campos, E. I., Reinberg, D. Histones: annotating chromatin. Annu Rev Genet. 43, 559-599 (2009).
- Rando, O. J., Winston, F. Chromatin and transcription in yeast. Genetics. 190 (2), 351-387 (2012).
- Duina, A. A., Miller, M. E., Keeney, J. B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 197 (1), 33-48 (2014).
- Storici, F., Resnick, M. A. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y). 25, 189-207 (2003).
- Gray, M., Kupiec, M., Honigberg, S. M. Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7 (2004).
- Erdeniz, N., Mortensen, U. H., Rothstein, R. Cloning-free PCR-based allele replacement methods. Genome Res. 7 (12), 1174-1183 (1997).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
- Lundblad, V., Hartzog, G., Moqtaderi, Z. Manipulation of cloned yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Hoffman, C. S. Preparation of yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Treco, D. A., Lundblad, V. Preparation of yeast media. Curr Protoc Mol Biol. Chapter 13, (2001).
- Lederberg, J., Lederberg, E. M. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 63 (3), 399-406 (1952).
- Sikorski, R. S., Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122 (1), 19-27 (1989).
- Johnson, P., et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 5 (5), 741-749 (2015).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336-4343 (2013).
- Cross, S. L., Smith, M. M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol. 8 (2), 945-954 (1988).
- Libuda, D. E., Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 443 (7114), 1003-1007 (2006).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved