
JoVE 비디오를 활용하시려면 도서관을 통한 기관 구독이 필요합니다. 전체 비디오를 보시려면 로그인하거나 무료 트라이얼을 시작하세요.
Method Article
대상
요약
A strategy for generating mutations in histone genes at their endogenous location in Saccharomyces cerevisiae is presented.
초록
We describe a PCR- and homologous recombination-based system for generating targeted mutations in histone genes in budding yeast cells. The resulting mutant alleles reside at their endogenous genomic sites and no exogenous DNA sequences are left in the genome following the procedure. Since in haploid yeast cells each of the four core histone proteins is encoded by two non-allelic genes with highly homologous open reading frames (ORFs), targeting mutagenesis specifically to one of two genes encoding a particular histone protein can be problematic. The strategy we describe here bypasses this problem by utilizing sequences outside, rather than within, the ORF of the target genes for the homologous recombination step. Another feature of this system is that the regions of DNA driving the homologous recombination steps can be made to be very extensive, thus increasing the likelihood of successful integration events. These features make this strategy particularly well-suited for histone gene mutagenesis, but can also be adapted for mutagenesis of other genes in the yeast genome.
서문
네 개의 핵심 히스톤 단백질 H2A, H2B, H3 및 H4는 진핵 세포 염색체의 압축, 조직 및 기능에 중심 역할을한다. 이러한 히스톤 각각 두 세트의 궁극적 뉴 클레오 (1)의 형성의 결과로, 히스톤 옥타 자체 주변 DNA의 ~ 147 염기쌍의 감쌈 지시 분자의 스풀을 형성한다. 뉴 클레오 솜은 염색체 전체 유전자 전사의 조절과 euchromatin의 형성 및 이질 같은 염색체 기반 프로세스의 다양한 활성 참가자이며, 같은 과거 수십 년에 걸쳐 집중적 연구의 초점이되어왔다. 이러한 메커니즘은 히스톤 잔류의 번역 후 변형, ATP 의존적 인 뉴 클레오 리모델링 및 ATP 독립적 인 뉴 클레오 재구성을 포함 - 메커니즘의 수는있는 뉴 클레오는 특정 프로세스의 실행을 용이하게 할 수있는 방법으로 조작 할 수있는 기술되어있다조립 / 해체 (2, 3).
신진 효모 사카로 마이 세스 세레 비지는 진핵 생물에서 히스톤 함수의 이해에 특히 강력한 모델 생물이다. 이것은 주로 도메인 eukarya 걸쳐 히스톤 단백질의 진화 적 보전의 높은 유전 생화학 실험 다양 효모의 가공성에 기인 할 수있다 (4)에 접근한다. 효모 역방향 유전자 접근법 널리 염색질 생물학의 다양한 양상의 특정 히스톤 돌연변이의 효과를 연구하기 위해 사용되었다. 실험은 이러한 유형의 비정상적인 세포 (인해 세포에서 플라스미드의 수를 가변으로) 히스톤 단백질의 수준을 초래할 수 자율 플라스미드로부터 발현 같은 돌연변이 히스톤은 그 나라의 게놈 유전자좌로부터 발현 된 세포를 사용하는 것이 바람직하다 크로 엔의 수반 변경최종적으로 결과의 해석을 혼동 할 수 많은 환경.
여기서는 게놈 남은 외래 DNA 서열없이, 원하는 돌연변이 (들)의 발생에 클로닝 단계 및 결과를 필요로하지 않고 그 나라의 게놈 위치에서 히스톤 유전자 표적화 돌연변이 유발을 허용하는 PCR 계 기술을 설명한다. 이 기술은 효모에서 효율적인 상동 재조합 시스템을 활용하고 다른 그룹에 의해 개발 된 다른 유사한 기술과 공통점이 몇 가지 기능이있다 - 가장 특히 Delitto PERFETTO, 사이트 별 게놈 (SSG) 돌연변이 유발 및 복제없이 PCR 기반의 대립 유전자를 대체 방법 5, 6, 7. 그러나,이 설명 된 기술은 특히 히스톤 유전자의 돌연변이 유발에 적합하게 한 측면을 갖는다. 반수체 효모 세포에서, 네 개의 코어 히스톤의 각각은 두 개의 비 a로 인코딩llelic 높은 상 동성 유전자는 예를 들어, 히스톤 H3가 HHT1 HHT2 및 유전자에 의해 암호화되고,이 유전자의 오픈 리딩 프레임 (ORF를는) 시퀀스의 90 % 이상 동일하다. 상 동성이 높은 수준은 구체적으로 돌연변이 유발을위한 두 개의 히스톤를 코딩하는 유전자 중 하나를 목표로 설계된 실험을 복잡하게 할 수 있습니다. 상기 방법들은 상동 재조합을 유도하는 표적 유전자의 ORF 내의 적어도 몇몇 시퀀스의 사용을 필요로하는 반면, 우리는 여기서 설명하는 기술에 대한 (더 적은 서열 상 동성을 공유하는) 히스톤 유전자의 ORF를 플 랭킹 서열을 이용한다 재결합 단계, 따라서 원하는 궤적에 돌연변이를 성공적으로 표적의 가능성을 증가시킨다. 또한, 재조합 드라이브 상동 지역이 더 효율적 대상 상동 재조합에 기여하고, 매우 광범위 할 수 있습니다.
프로토콜
참고 : 현장 히스톤 유전자 돌연변이에 대상에 대한 실험 전략 (그림 1에 요약) 여러 단계를 포함한다. 이러한 단계는 다음을 포함한다 : URA3 유전자 표적 히스톤 유전자 (1)의 교환, (2) 생성 및 원하는 돌연변이 (들)을 보유하는 프라이머를 사용하여 대상 히스톤 유전자의 두 부분 오버랩하는 조각에 해당하는 PCR 산물의 정제 (3 ) 두 부분 오버랩 조각의 융합 PCR은 플라스미드에 마커 (4) 전체 크기의 PCR 제품 및 백본 플라스미드 공동 변환 및 선택 (5) 화면 5-FOA 방지를위한, 통합을위한 전체 크기의 PCR 제품을 얻었다 형질 전환, (6) 5-FOA 내성 콜로니 정제 골격 플라스미드의 손실, 및 (7) 분자는 돌연변이 대립 유전자의 적절한 통합 분석으로 분석한다.

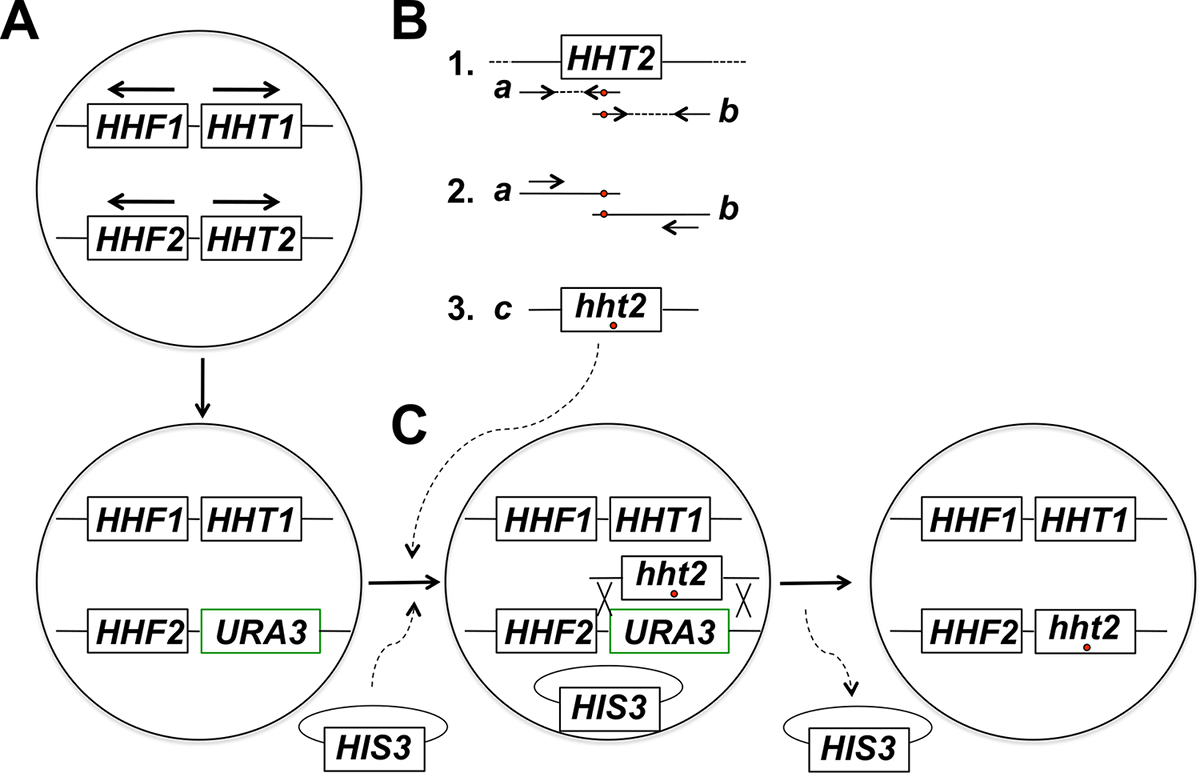
그림 1 : 신진 효모에서 히스톤 유전자의 현장 돌연변이 유발에 대상에 대한 전략의 개요. 이 예에서, 표적 유전자는 HHT2이지만, 다른 코어 히스톤 유전자의이 전략을 사용하여 돌연변이 될 수있다. 그림과 같이 (A) 반수체 효모 세포 배열이 히스톤 H3를 코딩하는 유전자 (HHT1 및 HHT2)와 두 개의 히스톤 H4를 코딩하는 유전자 (HHF1 및 HHF2를) 항구합니다 (HHT1 및 HHF1 유전자는 염색체 II와 HHT2에 있습니다 및 HHF2 유전자는 염색체 XIV에있다 - 각각의 경우에, 화살표)은 전사의 방향을 지적한다. 절차의 제 1 단계에서, HHT2 유전자의 ORF는 hht2Δ :: URA3 변형을 야기주는 URA3 유전자로 대체된다. 제 1 부 (B)는 게놈 DNA 시료로부터 HHT2 유전자의 야생형 카피 속 두 PCR 반응을위한 주형으로 사용유전자의 두 부분 오버랩 단편 테. 첫 번째 반응에 대한 역방향 프라이머는 게놈에 도입 할 원하는 돌연변이 (들)에 해당합니다 (빨간색 원으로 표시) 하나 이상의 일치하지 않는 뉴클레오티드를 포함한다. 제 2 반응의 정방향 프라이머 (또한 빨간색 원으로 표시) 역 보완적인 구성에서 해당 불일치가 있습니다. 두 PCR 1 부에서 생성 된 제품 (제품 A와 B)을 융합 PCR 템플릿이 방식으로 제품 A와 B에 어닐링이 최대 크기의 PCR 생성 결과 파트 2에 도시 된 두 개의 프라이머를 사용로서 사용 원하는 돌연변이 (들)을 숨겨 제품 (부품 3 제품 다). (C)가 hht2Δ는 :: URA3 균주는 인 공동 형질 부족 미디어 (전체 크기의 PCR 산물과 백본 플라스미드 (a HIS3-이 예에서 플라스미드로 표시)으로하고, 세포가 플라스미드의 존재를 선택 시간이 예에서 istidine). 형질 전환 후 5 FOA성에 대해 스크리닝 - 저항성 세포 같이 URA3 유전자의 PCR 생성물 및 절단 통합 선도 동성 재조합 이벤트를 실시한 후보이다. 유사 분열 세포 분열에 의해 골격 플라스미드이어서 손실은 최종 목적하는 히스톤 변이주로 이끈다. 우리는 주로 자연 URA3 돌연변이를 획득 한 세포를 식별 5 FOA 플레이트상에서 직접 선택에 비해 정확한 통합 이벤트 식별 훨씬 높은 주파수에서 5-FOA 저항 결과를 스크리닝 하였다 백본 플라스미드의 선택을 발견 하였다. (이 수치는 기준 (14)에서 수정되었습니다). 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
URA3와 대상 히스톤 유전자의 1. 교체유전자
- URA3 유전자 (8, 9)와 타겟 히스톤 유전자의 ORF를 대체 표준 PCR 매개 한 단계 유전자 파괴를 수행한다.
주 :이 돌연변이 따라서 URA3 궤적 8 내로 PCR 생성물을 통합 회피 전체 내인성 URA3 ORF 제거로 ura3Δ0 운반 효모 세포의 사용이 권장된다. 이 S. cerevisiae의 기능적이지만 S. cerevisiae의 URA3 유전자 부분적인 서열 상 동성을 갖는 한 대안으로, K. 락 티스 URA3 유전자는 URA3 배경 히스톤 여분의 발생을 효과적으로 사용할 수있다. 변형은 또한 변형 실험 (이러한 프로토콜의 단계 4)의 골격 플라스미드 선택할 수 있도록 적어도 하나의 화합물에 대해 영양 요해야한다. 이 단계는 타깃 히스톤 geneΔ 경우 :: URA3 필요하지균주는 이미 사용할 수 있습니다.
2. 생성하고 원하는 돌연변이를 품고 프라이머를 사용하여 대상 히스톤 유전자의 두 부분적으로 겹치는 조각에 해당하는 PCR 제품의 정제 (들)
- 대상 히스톤 유전자의 두 부분 오버랩하는 조각에 해당하는 PCR 산물을 생성한다.
- 다음과 같이 두 개의 PCR 반응을 준비합니다 :
- 내열성 1 μL 템플릿 DNA, 5 μl10 μM 정방향 프라이머, 5 μl10 μM 역방향 프라이머, 0.5 μL (1.25 U) : 다음과 같은 반응을 설정하는 유전자 (그림 1B에서 제품 A)의 첫 번째 절반에 해당하는 PCR 제품을 생성하려면 DNA 폴리머 라 아제 10 μL 배 DNA 중합 효소 완충액을 5 μL의 dNTP 혼합물 (2 mM의 각), 23.5 μL의 DH 2 O.
참고 : 템플릿 DNA가 대상 히스톤 유전자 변형 야생 형에서 파생 된 게놈 DNA가 될 수는 표준 프로를 사용하여 격리10 cedures. DNA 농도 및 다른 게놈 제제 중의 불순물 레벨의 변동을 고려하기 위해,이 희석 DNA 게놈 또는 다른 제제의 희석액을 사용하여 반응을 최적화하기 위해 추천 (예를 들어 1:10 내지 1 : 100). 정방향 프라이머는 표적 유전자의 업스트림 영역에 어닐링한다. 역방향 프라이머는 (예제 그림 1B-1 대표 결과 섹션 참조) 길이 ~ 수의 ORF에서 40 뉴클레오티드를 어닐링하고, 중간에 어딘가에 원하는 돌연변이 (들)을 포함해야합니다. 높은 충실도의 DNA 중합 효소의 사용은 PCR 산물의 합성 과정에서 바람직하지 않은 변이의 비율을 감소시키기 위하여 권장된다. - 2.1.1.1이 아니라 다른 프라이머로 나타낸 바와 같이 유전자 (도 1b에 제품 b)의 두 번째 반에 대응하는 PCR 산물을 생성하는 반응을 설정한다.
참고 : 앞으로 PRI메르의 길이는 ~ 할 수는 ORF에서 40 뉴클레오티드를 어닐링하고, 어딘가에 중간에 원하는 돌연변이 (들)을 포함해야합니다. 이 프라이머의 돌연변이 (들) 단계 2.1.1.1의 역방향 프라이머의 돌연변이 (들)의 역 보완합니다. 역방향 프라이머는 표적 유전자의 하류 영역에 어닐링한다 (예는도 1b-1 대표 결과 절 참조).
- 내열성 1 μL 템플릿 DNA, 5 μl10 μM 정방향 프라이머, 5 μl10 μM 역방향 프라이머, 0.5 μL (1.25 U) : 다음과 같은 반응을 설정하는 유전자 (그림 1B에서 제품 A)의 첫 번째 절반에 해당하는 PCR 제품을 생성하려면 DNA 폴리머 라 아제 10 μL 배 DNA 중합 효소 완충액을 5 μL의 dNTP 혼합물 (2 mM의 각), 23.5 μL의 DH 2 O.
- 다음 설정을 사용하여 열 순환기의 반응을 놓고 94 ℃에서 30 초; 다음 설정 중 30 회 : 98 ˚C 10 초, 60 ℃ 5 초, 72 ˚C 1.5 분; 72 ˚C 10 분.
PCR 매개 변수의 최적화 특정 프라이머 세트에 필요한 될 수 있으며, 히스톤 유전자를 대상으로 참고.
- 다음과 같이 두 개의 PCR 반응을 준비합니다 :
- 89 mM 트리스베이스, 89 mM의 붕산, 2.5 mM의 EDTA (TBE) 버퍼의 0.9 % 저 융점 아가로 오스 겔에 PCR 반응에서 물질의 50 μL - 20를 실행합니다.
- 상기 증폭 된 PCR (PR)를 포함하는 아가로 스겔 컷 부깨끗한 메스이나 면도기 블레이드를 사용하여 겔에서 oducts의는 1.5 ml의 microcentrifuge 관에 각각 전송합니다. 사용할 준비가 될 때까지 -20 ℃에서 PCR 제품을 포함하는 아가로 오스 부분을 저장합니다.
'두 부분적으로 겹치는 조각 3. 퓨전 PCR은 통합을위한 전체 크기 PCR 제품을 구하는
- PCR 반응에 대한 템플릿을 준비
- 5 분 동안 65 ℃에서 가열 블럭 세트의 마이크로 원심 튜브를 배치함으로써 단계 2.3에서 아가 로스 겔 부분 용융 (또는 완전히 용융 될 때까지). 보텍스 튜브마다 1-2 분의 용융 공정을 용이하게한다.
- 각 샘플에서 용융 아가 일정한 양의 전송 (예를 들면, 50 ㎕를 각 100 μL의 총)는 단일 마이크로 원심 튜브에 넣고 텍싱하여 혼합한다. 융합 PCR 반응의 템플릿으로 사용합니다. 사용할 준비가 될 때까지 -20 ℃에서 튜브를 놓습니다.
- 전체 크기의 PCR 산물의 대량 증폭 (제품 C) 그림 1B에
- 2 μL 템플릿 DNA, 10 μl를 10 μM 정방향 프라이머, 10 μl를 10 μM 역방향 프라이머, 1 μL (2.5 U) 내열성 DNA 중합 효소, 20 ㎕의 5 배 DNA 중합 효소 버퍼, 10 μl의 여섯 PCR 반응, 다음과 같은 구성 요소와 각을 설정합니다 의 dNTP 혼합물 (2 mM의 각), 47 μL의 DH 2 O.
비고 : 반응의 수는 PCR 효율에 따라 변경 될 수있다. 용융 텍싱 혼합하고, PCR 반응 혼합물에 첨가 마지막까지 주형 DNA는 (3.1.2 참조)를 65 ℃로 가열한다. 일단 추가 솔루션을 피펫 팅에 의해 여러 번 아래로 부드럽게 잘 섞는다. 다른 샘플 DNA 농도의 변동을 고려하기 위해, 먼저 희석 템플릿 또는 템플릿의 다양한 희석액을 사용하여 반응을 최적화하기 위해 추천 (예를 들어 1:10 내지 1 : 100). 병 같은 표적 유전자의 두 부분 오버랩 단편 어닐링해야 사용 두 프라이머도 1b-2 ustrated 최종 PCR 제품은 상동 재조합 단계 (예 대표적인 결과 절 참조)를 구동 할 URA3 ORF 측부 영역들 양쪽 상동에 적어도 40 개의 염기쌍을 가질 것이다되도록 설계 될 수있다. 높은 충실도의 DNA 중합 효소의 사용은 PCR 산물의 합성 과정에서 바람직하지 않은 변이의 비율을 감소시키기 위하여 권장된다. - 다음 설정을 사용하여 열 순환기의 튜브를 놓고 94 ℃에서 30 초; 다음 설정 중 30 회 : 98 ˚C 10 초, 50 ℃ 15 초, 72 ˚C 1.5 분; 72 ˚C 10 분.
주 : PCR 파라미터 최적화 특정 프라이머 세트 및 목표 히스톤 유전자 요구 될 수있다.
- 2 μL 템플릿 DNA, 10 μl를 10 μM 정방향 프라이머, 10 μl를 10 μM 역방향 프라이머, 1 μL (2.5 U) 내열성 DNA 중합 효소, 20 ㎕의 5 배 DNA 중합 효소 버퍼, 10 μl의 여섯 PCR 반응, 다음과 같은 구성 요소와 각을 설정합니다 의 dNTP 혼합물 (2 mM의 각), 47 μL의 DH 2 O.
플라스미드에 마커 4. 전체 크기 PCR 제품 및 백본 플라스미드 공동 변환 및 선택
- PCR 산물의 농도
- 의 S 수영장IX PCR 반응 하나의 microcentrifuge 튜브에 단계 3.2.2에서 (600 μL 총)와 텍싱하여 혼합한다.
- 마이크로 원심 튜브 세 200 μL 씩에 샘플을 분할합니다. 3M 아세트산 나트륨 20 μL (PH 5.2), 100 % 에탄올 550 μL를 첨가하여 각 튜브의 DNA를 침전. 철저 솔루션을 섞어 적어도 15 분 동안 얼음에 배치합니다. 10 분 ~ 14,000 × g으로 원심 분리하여 DNA를 수집 200 70 % 에탄올 μL, 공기 건조하여 펠렛을 헹군다.
- DH 2 O 25 μL로 각 DNA 펠렛을 재현 탁하고 (75 μl의 총) 단일의 튜브로 풀.
- 효모 공동 변환
- 효모 추출물 펩톤 덱 스트로스 (YPD) 11 매질 액 섹션 1에서 생성 된 균주의 하룻밤 문화 10 ㎖를 준비합니다.
- 다음 날 아침, 포화 하룻밤 문화의 8 ㎖로 YPD 액체 배지 400 mL를 접종하고 흔들어 배양4 30 ° C에서 - 5 시간 세포가 성장의 로그 단계를 입력 할 수 있습니다.
- 10 분 ~ 3220 XG에서 원심 분리하여 세포를 수집 한 액체 배지를 폐기하고, 10 mM 트리스 -HCl (pH 8.0), 1 mM의 EDTA 1 부피로 세포를 재현 탁 0.1 M 리튬 아세테이트 용액 (TE / LiAc들을) .
- 10 분 ~ 3220 XG에 원심 분리하여 세포를 수집하고, TE / LiAc들을 폐기합니다.
- 1 ㎖의 TE / LiAc들을 상기 세포를 재현 탁.
- 미세 원심 분리 튜브에 다음 반응 칵테일 설정 : 세포를 800 ㎕의 단계 4.2.5에서 비등 10 ㎎ / ㎖ 연어 정자 DNA, 40 μL, 골격 플라스미드 DNA 12.5 μg의 총, 농축 PCR 생성물의 75 μL 단계 4.1.3에서.
주 : 연어 정자 DNA을 5 분 동안 끓인 반응에 사용하기 전에 적어도 5 분 동안 얼음 상에 배치한다. 추가 백본 플라스미드 DNA의 총 부피는 최소 (~ 80 μL 이하)로 유지한다. 백본 원형질의 예를 들어 대표 결과 섹션을 참조하십시오신분증. - 균등 팔의 microcentrifuge 튜브 (- 8 튜브 1)에 칵테일 철저하게 튜브와 나누어지는을 섞는다.
- 다음과 같은 두 개의 제어 변환 반응 튜브를 설정합니다 :
- 튜브 9 (아무 PCR 제품 제어) : 단계 4.2.5에서 세포를 100 ㎕, 삶은 10 ㎎ / ㎖ 연어 정자 DNA의 5 μL;의 1.56 μg의 총 (5 분 끓인 단계 4.2.6 주 참조) 백본 플라스미드 DNA없이 PCR 제품 덧붙였다.
- 튜브 10 (NO DNA 제어) : 단계 4.2.5, 삶은 10 ㎎ / ㎖ 연어 정자 DNA (단계 4.2.6 참고 참조) 5 μL에서 세포를 100 ㎕, DNA가 추가 더 백본 플라스미드없이 PCR 제품은 추가되지 않습니다.
- 최대 피펫 팅에 의해 여러 번 아래로 부드럽게하지만 철저하게 모두 튜브를 혼합한다.
- 30 분 동안 30 ℃에서 열 튜브를 품어.
- 각 튜브에, TE / LiAc들을 40 %의 폴리에틸렌 글리콜 1.2 ㎖ (PEG 3350)를 추가합니다. 용액이 균질해질 때까지 철저히 P-1000 피펫을 이용하여 혼합한다.
- (30)에 열 튜브를 품어30 분 동안 C를 °. 부드럽게 아래로 피펫 팅에 의해 용액을 혼합 한 후 15 분 동안 42 ° C에서 튜브를 품어.
- 30 초 ~ 14,000 XG에서의 microcentrifuge에 튜브를 회전시켜 세포를 수집합니다. 액체를 버리고 멸균 DH 2 O. 1 ㎖로 세포를 재현 탁
- 30 초 ~ 14,000 XG에서의 microcentrifuge에 튜브를 회전시켜 세포를 수집합니다. 액체를 취소하고 멸균 DH 2 O. 500 μL의 세포를 재현 탁
- 풀 튜브 1-8 함께 (총 4 ml의 부피) 및로 pipetting 아래로 잘 섞는다.
- 접시 백본 플라스미드 선택 스물 완전한 최소 강하 중간 판 (11) (판 1-20) 각각에서, 상기 혼합물을 200 μL.
- 접시 튜브 (9)의 혼합물을 200 ㎕의 자체 선택 플레이트에 튜브 (10) 각각으로부터의 혼합물을 200 μL (플레이트를 각각 21, 22).
- 5 일 - 3 30 ° C에서 22 접시를 품어플라스미드 형질 전환을 위해 선택합니다.
- 배양 5 일 - 3 후 변환 판을 검사합니다. 약 5,000 식민지 판 1-21 (예를 들어에 대한 대표 결과를 참조)에 표시해야하며, 더 콜로니 판 (22)에 본 제품을 장착 할 수 없습니다.
5-FOA 내성 형질 전환 5. 화면
- 플레이트로부터의 세포를 전송 1-20 (및 변환 제어 등의 판 21) 5- 플루오로 오로 아세트산 (5-FOA) 플레이트 (11)에 의해 복제 도금의 통합의 결과로서 URA3 유전자의 손실을 선별하기 위하여 12 원하는 위치 PCR 제품.
- 플레이트 뚜껑을 제거하고 멸균 벨벳에 식민지를 포함하는 판을 누릅니다. 벨벳에있는 플레이트를 눌러 5 FOA 판에 벨벳에서 세포를 전송합니다. 2 일간 30 ℃에서 번호판을 품어.
- 2 일 배양 후, 조심스럽게 GR에 대한 5-FOA 판을 검사owth.
참고 : 후보 통합 이벤트가 5 FOA 판에 작은 비대칭 "숙청"식민지로 표시됩니다 - 반대로, 5-FOA 판에 성장하는 작은 용의자는 식민지의 성장 중에 발생한 자연 URA3 돌연변이의 가능성 대표는 변환 플레이트, 원하는 통합 이벤트를 (몇 가지 예이 시점에 더 정교의 대표 결과 섹션과 그림 3 참조) 표현하는 것이 어렵다.
5-FOA 방지 식민지 및 백본 플라스미드의 손실 6. 정화
- 멸균 이쑤시개를 사용하여 YPD 플레이트에 하나의 식민지에 대한 단계 5.2 및 행진에 설명 된 5-FOA 판에서 후보 식민지를 선택합니다. 30 ° C 3 일 - 2 품어.
- 배양, 복제 다음 - 접시에 신선한 YPD 판에 각 YPD 정화 판, 드롭 아웃 플레이트 손실을 확인 우라실 결여URA3 유전자, 및 두번째 드롭 아웃 플레이트의 골격 플라스미드의 존재 여부를 모니터링한다. 30 ° C에서 2 일 - 1 품어.
- 배양를 수행하면은 YPD 판에 성장하지만 중 하나 드롭 아웃 접시에 성장되지 않은 각 후보 샘플에서 식민지를 식별 (예 : 식민지는 재결합 이벤트를 통해 URA3 유전자를 잃은 것으로 기대하고 유사 분열하는 동안 백본 플라스미드를 분실 세포 분열). 신선한 YPD 접시에 이러한 식민지를 Restreak. 이 식민지는 통합 후보 및 7 단계에서 추가로 분석됩니다.
돌연변이 대립 유전자의 적절한 통합을위한 분석 7. 분자 분석
- 표준 절차 (10)를 사용하여 후보 샘플로부터 게놈 DNA를 분리.
- 대상 사이트를 포괄하는 유전자 영역을 증폭.
- 0.5 ㎕를 주형 DNA, 5 ㎕를 각 샘플에 대해 다음 PCR 반응을 설정10 μM 정방향 프라이머, 5 ㎕의 10 μM 역방향 프라이머, 0.5 μL (2.5 부)의 Taq DNA 중합 효소, 5 ㎕의 10 배의 Taq DNA 중합 효소 완충액 5 ㎕의 용의 dNTP 혼합물 (2 mM의 각), 29 μL의 DH 2 O.
주 : DNA 템플릿이 후보 샘플로부터 유래 된 게놈 DNA이다. 하나 원래 히스톤 geneΔ 유래의 게놈 DNA를 주형으로 :: URA3 균주를 사용하여 주형으로서 야생형 히스톤 균주로부터 게놈 DNA를 사용하여 다른 : 또한, 두 개의 제어 반응을 포함 할 것을 권장한다. DNA 농도 및 다른 게놈 제제 중의 불순물 레벨의 변동을 고려하기 위해,이 희석 DNA 게놈 또는 다른 제제의 희석액을 사용하여 반응을 최적화하기 위해 추천 (예를 들어 1:10 내지 1 : 100). 이 확인 것이 중요하다고 추정되는 집적 PCR 생성물에 의해 둘러싸인 영역 외부의 DNA 서열이 프라이머 어닐링 - 이러한 방식으로,이 연구에서 PCR 산물의 크기eactions 정확한 게놈 위치에있는 제품의 통합을위한 진단 도구로 사용할 수 있습니다 (예를 들어에 대한 대표 결과를 참조). - 다음 설정을 사용하여 열 순환기의 반응을 놓고 : 94 ° C 3 분; 다음 설정 중 30 회 : 94 ° C 45 초, 50 ℃ 45 초, 72 ℃에서 2 분; 72 ° C 10 분.
주 : PCR 파라미터 최적화 특정 프라이머 세트 및 목표 히스톤 유전자 요구 될 수있다.
- 0.5 ㎕를 주형 DNA, 5 ㎕를 각 샘플에 대해 다음 PCR 반응을 설정10 μM 정방향 프라이머, 5 ㎕의 10 μM 역방향 프라이머, 0.5 μL (2.5 부)의 Taq DNA 중합 효소, 5 ㎕의 10 배의 Taq DNA 중합 효소 완충액 5 ㎕의 용의 dNTP 혼합물 (2 mM의 각), 29 μL의 DH 2 O.
- PCR 제품 가공
- 0.8 % TBE 아가 로스 젤에 각 반응에서 20 μl를 실행합니다.
- URA3 유전자가 성공적으로 추정되는 히스톤 돌연변이 유전자로 대체 된 경우를 결정하는 기준으로서 DNA 수준을 사용하여 PCR 산물의 크기를 평가한다 (예를 들어 대표적인 결과를 참조).
주 : 특정 경우에서 원하는 돌연변이 (들)가 히스톤 유전자 도입하거나 생성하거나 앉아 제한 파괴이자형. 이 경우 정확한 적분을 나타내는 크기의 PCR 산물의 원하는 돌연변이의 존재는 겔 전기 영동 분석 하였다 해당 제한 효소로 소화 생성물을 실시함으로써 평가 될 수있다 (예를 들어 대표적인 결과를 참조) . - DNA 서열에 정확한 적분을 나타내는 크기의 PCR 대상 제품은 원하는 돌연변이 (들)의 존재를 확인하고 추가의 돌연변이가 게놈 내로 도입되지 않았 음을 보장하기 위해.
결과
우리는 시츄 돌연변이 전략 표적의 대표 예로서 글루탐산 (돌연변이 H3-R53E)에 아르기닌에서 위치 53에서의 치환을 보유하는 히스톤 H3 돌연변이 단백질을 표현한 hht2 대립 형질의 발생을 설명한다.
우리 HHT2 전체 ORF가 URA3 유전자 (프로토콜의 단계 1 참고)로 대체 된 균주를 생성. 이 균주 yAAD156 ...
토론
반수체 S. cerevisiae의 세포에서 4 개의 핵심 히스톤 단백질의 각각에 대한 코드가 구체적으로 돌연변이 유발을위한 두 개의 유전자 중 하나를 타겟팅하려는 연구자에 대한 도전을 나타낼 수있는 두 개의 비 대립 유전자 사이의 서열 상 동성의 높은 수준. 이전에 종종 의존 상기 Delitto PERFETTO, 부위 - 특이 적 유전자 (SSG) 돌연변이 유발, 클로닝없이 PCR 기반 대립 유전자 교환 방법
공개
The authors declare that they have no competing financial interests.
감사의 말
We thank Reine Protacio for helpful comments during the preparation of this manuscript. We express our gratitude to the National Science Foundation (grants nos. 1243680 and 1613754) and the Hendrix College Odyssey Program for funding support.
자료
| Name | Company | Catalog Number | Comments |
| 1 kb DNA Ladder (DNA standards) | New England BioLabs | N3232L | |
| Agarose | Sigma | A5093-100G | |
| Boric Acid | Sigma | B0394-500G | |
| dNTP mix (10 mM each) | ThermoFisher Scientific | R0192 | |
| EDTA solution (0.5 M, pH 8.0) | AmericanBio | AB00502-01000 | |
| Ethanol (200 Proof) | Fisher Scientific | 16-100-824 | |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma | E4884-500G | |
| Lithium acetate dihydrate | Sigma | L6883-250G | |
| MyCycler Thermal Cycler | BioRad | 170-9703 | |
| Poly(ethylene glycol) (PEG) | Sigma | P3640-1KG | |
| PrimeSTAR HS DNA Polymerase (high fidelity DNA polymerase) and 5x buffer | Fisher Scientific | 50-443-960 | |
| Salmon sperm DNA solution | ThermoFisher Scientific | 15632-011 | |
| Sigma 7-9 (Tris base, powder form) | Sigma | T1378-1KG | |
| Sodium acetate trihydrate | Sigma | 236500-500G | |
| Supra Sieve GPG Agarose (low metling temperature agarose) | AmericanBio | AB00985-00100 | |
| Taq Polymerase and 10x Buffer | New England BioLabs | M0273X | |
| Toothpicks | Fisher Scientific | S67859 | |
| Tris-HCl (1 M, pH 8.0) | AmericanBio | AB14043-01000 | |
| a-D(+)-Glucose | Fisher Scientific | AC170080025 | for yeast media |
| Agar | Fisher Scientific | DF0140-01-0 | for yeast media |
| Peptone | Fisher Scientific | DF0118-07-2 | for YPD medium |
| Yeast Extract | Fisher Scientific | DF0127-17-9 | for YPD medium |
| 4-aminobenzoic acid | Sigma | A9878-100G | for complete minimal dropout medium |
| Adenine | Sigma | A8626-100G | for complete minimal dropout medium |
| Glycine hydrochloride | Sigma | G2879-100G | for complete minimal dropout medium |
| L-Alanine | Sigma | A7627-100G | for complete minimal dropout medium |
| L-Arginine monohydrochloride | Sigma | A5131-100G | for complete minimal dropout medium |
| L-Asparagine monohydrate | Sigma | A8381-100G | for complete minimal dropout medium |
| L-Aspartic acid sodium salt monohydrate | Sigma | A6683-100G | for complete minimal dropout medium |
| L-Cysteine hydrochloride monohydrate | Sigma | C7880-100G | for complete minimal dropout medium |
| L-Glutamic acid hydrochloride | Sigma | G2128-100G | for complete minimal dropout medium |
| L-Glutamine | Sigma | G3126-100G | for complete minimal dropout medium |
| L-Histidine monohydrochloride monohydrate | Sigma | H8125-100G | for complete minimal dropout medium |
| L-Isoleucine | Sigma | I2752-100G | for complete minimal dropout medium |
| L-Leucine | Sigma | L8000-100G | for complete minimal dropout medium |
| L-Lysine monohydrochloride | Sigma | L5626-100G | for complete minimal dropout medium |
| L-Methionine | Sigma | M9625-100G | for complete minimal dropout medium |
| L-Phenylalanine | Sigma | P2126-100G | for complete minimal dropout medium |
| L-Proline | Sigma | P0380-100G | for complete minimal dropout medium |
| L-Serine | Sigma | S4500-100G | for complete minimal dropout medium |
| L-Threonine | Sigma | T8625-100G | for complete minimal dropout medium |
| L-Tryptophan | Sigma | T0254-100G | for complete minimal dropout medium |
| L-Tyrosine | Sigma | T3754-100G | for complete minimal dropout medium |
| L-Valine | Sigma | V0500-100G | for complete minimal dropout medium |
| myo-Inositol | Sigma | I5125-100G | for complete minimal dropout medium |
| Uracil | Sigma | U0750-100G | for complete minimal dropout medium |
| Ammonium Sulfate | Fisher Scientific | A702-500 | for complete minimal dropout medium |
| Yeast Nitrogen Base | Fisher Scientific | DF0919-07-3 | for complete minimal dropout medium |
| 5-Fluoroorotic acid (5-FOA) | AmericanBio | AB04067-00005 | for 5-FOA medium |
참고문헌
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389 (6648), 251-260 (1997).
- Campos, E. I., Reinberg, D. Histones: annotating chromatin. Annu Rev Genet. 43, 559-599 (2009).
- Rando, O. J., Winston, F. Chromatin and transcription in yeast. Genetics. 190 (2), 351-387 (2012).
- Duina, A. A., Miller, M. E., Keeney, J. B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 197 (1), 33-48 (2014).
- Storici, F., Resnick, M. A. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y). 25, 189-207 (2003).
- Gray, M., Kupiec, M., Honigberg, S. M. Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7 (2004).
- Erdeniz, N., Mortensen, U. H., Rothstein, R. Cloning-free PCR-based allele replacement methods. Genome Res. 7 (12), 1174-1183 (1997).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
- Lundblad, V., Hartzog, G., Moqtaderi, Z. Manipulation of cloned yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Hoffman, C. S. Preparation of yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Treco, D. A., Lundblad, V. Preparation of yeast media. Curr Protoc Mol Biol. Chapter 13, (2001).
- Lederberg, J., Lederberg, E. M. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 63 (3), 399-406 (1952).
- Sikorski, R. S., Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122 (1), 19-27 (1989).
- Johnson, P., et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 5 (5), 741-749 (2015).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336-4343 (2013).
- Cross, S. L., Smith, M. M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol. 8 (2), 945-954 (1988).
- Libuda, D. E., Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 443 (7114), 1003-1007 (2006).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기더 많은 기사 탐색
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유