
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Mirata
In questo articolo
Riepilogo
A strategy for generating mutations in histone genes at their endogenous location in Saccharomyces cerevisiae is presented.
Abstract
We describe a PCR- and homologous recombination-based system for generating targeted mutations in histone genes in budding yeast cells. The resulting mutant alleles reside at their endogenous genomic sites and no exogenous DNA sequences are left in the genome following the procedure. Since in haploid yeast cells each of the four core histone proteins is encoded by two non-allelic genes with highly homologous open reading frames (ORFs), targeting mutagenesis specifically to one of two genes encoding a particular histone protein can be problematic. The strategy we describe here bypasses this problem by utilizing sequences outside, rather than within, the ORF of the target genes for the homologous recombination step. Another feature of this system is that the regions of DNA driving the homologous recombination steps can be made to be very extensive, thus increasing the likelihood of successful integration events. These features make this strategy particularly well-suited for histone gene mutagenesis, but can also be adapted for mutagenesis of other genes in the yeast genome.
Introduzione
I quattro proteine di base dell'istone H2A, H2B, H3, H4 e giocano un ruolo centrale nella compattazione, l'organizzazione e la funzione dei cromosomi eucariotiche. Due insiemi di ciascuna di tali istoni formano la ottamero istoni, un rocchetto molecolare che dirige l'avvolgimento ~ 147 paia di basi del DNA su se stessa, che si traduce nella formazione di un nucleosoma 1. I nucleosomi sono partecipanti attivi in una varietà di processi cromosomiche-based, come ad esempio la regolazione della trascrizione genica e la formazione di eucromatina e heterochromatin tra cromosomi, e come tali sono stati al centro di un'intensa ricerca nel corso degli ultimi decenni. Un certo numero di meccanismi sono stati descritti da cui nucleosomi può essere manipolato in modi che possono facilitare l'esecuzione di processi specifici - Questi meccanismi comprendono modificazione post-traslazionale di residui degli istoni, rimodellamento nucleosomi ATP-dipendente, e la riorganizzazione nucleosomi ATP-indipendentee montaggio / smontaggio 2, 3.
Il lievito Saccharomyces cerevisiae erba è particolarmente potente organismo modello per la comprensione della funzione istone negli eucarioti. Questo può essere in gran parte attribuito all'elevato grado di conservazione evolutiva delle proteine istone tutto il eucarioti dominio e la riconducibilità di lievito ad una varietà di sperimentale genetico e biochimico approcci 4. approcci Reverse-genetici nel lievito sono stati ampiamente utilizzati per studiare gli effetti di specifiche mutazioni istoni su vari aspetti della biologia della cromatina. Per questi tipi di esperimenti è spesso preferibile utilizzare cellule in cui gli istoni mutanti sono espressi dal loro loci genomici nativa, come espressione da plasmidi autonome può portare a anormali livelli intracellulari di proteine istoni (a causa di un numero variabile di plasmidi in cellule) e alterazione concomitante di cromatina environments, che alla fine possono confondere l'interpretazione dei risultati.
Qui, si descrive una tecnica PCR-based che consente di mutagenesi mirata di geni istone alle loro posizioni nativi genomiche che non richiede una fase di clonazione e risultati nella generazione della mutazione desiderata (s) senza rimanenti sequenze di DNA esogene nel genoma. Questa tecnica sfrutta l'efficiente sistema di ricombinazione omologa nel lievito e ha diverse caratteristiche in comune con altre tecniche simili sviluppati da altri gruppi - in particolare il Delitto Perfetto, genomica mutagenesi sito-specifica (SSG), e la clonazione senza PCR allele-based metodi sostitutivi 5, 6, 7. Tuttavia, la tecnica descriviamo ha un aspetto che lo rende particolarmente adatto per la mutagenesi dei geni istoni. In cellule di lievito aploidi, ciascuno dei quattro istoni nucleo è codificata da due non-ageni llelic e altamente omologhi: per esempio, l'istone H3 vengono codificati dai geni HHT1 e HHT2, e le fasi di lettura aperte (ORF) dei due geni sono oltre il 90% identica in sequenza. Questo elevato grado di omologia può complicare esperimenti progettati per indirizzare specificamente uno dei due geni istone-codifica per la mutagenesi. Considerando che i suddetti metodi richiedono spesso l'uso di almeno alcune sequenze all'interno della ORF del gene bersaglio di guidare ricombinazione omologa, la tecnica descriviamo qui fa uso di sequenze fiancheggianti le ORF dei geni istoni (che condividono molto meno omologia di sequenza) per la fase di ricombinazione, aumentando così la probabilità di successo di targeting mutagenesi al locus desiderata. Inoltre, le regioni omologhe che guidano la ricombinazione possono essere molto ampia, contribuendo ulteriormente alla efficiente ricombinazione omologa mirato.
Protocollo
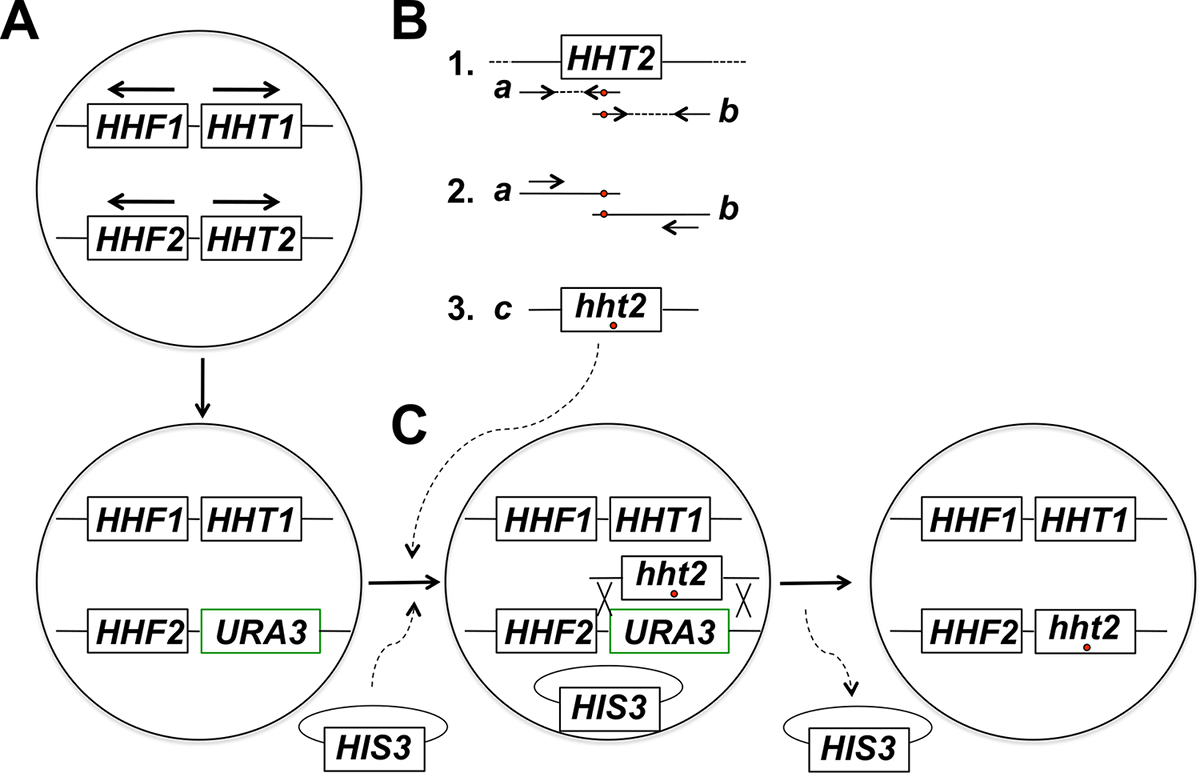
NOTA: La strategia sperimentale per mirata in situ mutagenesi gene istone comprende diversi passaggi (riassunti in Figura 1). Questi passaggi includono: (1) Sostituzione del gene bersaglio istone con il gene URA3, (2) Generazione e purificazione dei prodotti di PCR corrispondenti a due frammenti parzialmente sovrapposti del gene istone target usando primers della mutazione desiderata (s), (3 ) fusione PCR dei due frammenti parzialmente sovrapposti per ottenere pieni prodotti dimensione di PCR per l'integrazione, (4) Co-trasformazione dei prodotti di PCR a schermo intero e la spina dorsale plasmide, e la selezione per l'evidenziatore sulle plasmide, (5) dello schermo per 5-FOA-resistente trasformanti, (6) Purificazione di colonie 5-FOA-resistenti e perdita di backbone plasmide, e (7) Molecular analisi del dosaggio per la corretta integrazione del allele mutante.

Figura 1: Panoramica della strategia per la mirata in situ mutagenesi dei geni istoni in germogliamento lievito. In questo esempio il gene bersaglio è HHT2, ma qualsiasi altro gene nucleo istone può essere mutagenizzata usando questa strategia. (A) cellule di lievito aploidi porto due geni istone H3-encoding (HHT1 e HHT2) e due geni H4-codifica istone (HHF1 e HHF2) disposti come indicato in figura (i geni HHT1 e HHF1 si trovano sul cromosoma II e il HHT2 e geni HHF2 sono localizzati sul cromosoma XIV - in ogni caso, le frecce indicano la direzione di trascrizione). Nella prima fase della procedura, la ORF del gene HHT2 viene sostituito con il gene URA3, dando origine ad un ceppo hht2Δ :: URA3. (B) nella parte 1, una copia wild-type del gene HHT2 da un campione di DNA genomico viene utilizzato come modello per due reazioni PCR per generitE i due frammenti parzialmente sovrapposti del gene. Il primer inverso per la prima reazione comprende uno o più corrispondenti nucleotidi (indicati con un cerchio rosso) che corrispondono alla mutazione desiderata (s) da introdurre nel genoma. Il primer forward per la seconda reazione ha il disadattamento equivalente in una configurazione complementare inverso (anche indicato con un cerchio rosso). I due prodotti PCR generati nella parte 1 (prodotti A e B) vengono poi utilizzati come modelli per fusione PCR utilizzando due primer che ricottura per prodotti A e B nel modo mostrato nella parte 2. Ciò comporta la generazione di full-size PCR prodotti (prodotto C nella parte 3) che ospitano la mutazione desiderata (s). (C) La hht2Δ :: ceppo URA3 è poi co-trasformate con i prodotti di PCR in grandezza e un plasmide dorsale (a HIS3- contrassegnati plasmide in questo esempio), e le cellule vengono selezionate per la presenza del plasmide (su supporti privi histidina in questo esempio). I trasformanti sono poi sottoposti a screening per 5-FOA resistenza - cellule resistenti sono candidati che hanno subito un evento di ricombinazione omologa giungere all'integrazione del prodotto PCR e escissione del gene URA3, come mostrato. perdita successiva del plasmide spina dorsale per divisione cellulare mitotica conduce al istone desiderato ceppo mutante finale. Abbiamo trovato che la selezione del plasmide backbone seguita da screening per risultati di resistenza 5-FOA in una frequenza molto più elevata di identificazione di eventi di integrazione corretti rispetto alla selezione diretta su piastre 5-FOA, che identifica lo più cellule che hanno acquisito mutazioni URA3 spontanee. (Questa cifra è stata modificata dal riferimento 14). Clicca qui per vedere una versione più grande di questa figura.
{kind=link}
1. Sostituzione del target istone Gene con la URA3Gene
- Eseguire la PCR standard-mediata one-step gene distruzione di sostituire la ORF di gene istone bersaglio con il gene URA3 8, 9.
NOTA: L'uso di cellule di lievito che trasportano il ura3Δ0 è raccomandato come questa mutazione rimuove l'intero endogena URA3 ORF, evitando così l'integrazione del prodotto della PCR nella URA3 locus 8. In alternativa, il gene URA3 lactis K. può essere efficacemente utilizzato per la generazione di sostituzione istone in qualsiasi sfondo URA3 come è funzionale in S. cerevisiae ma ha solo omologia di sequenza parziale del gene URA3 di S. cerevisiae. Il ceppo deve essere auxotrophic per almeno un composto che permetterà la selezione del plasmide backbone nell'esperimento trasformazione (vedi punto 4 di questo protocollo). Questo passaggio non è necessario se un obiettivo istoni geneΔ :: URA3ceppo è già disponibile.
2. Generazione e purificazione dei prodotti di PCR corrispondente a due frammenti parzialmente sovrapposte del target istone Gene utilizzando primer della mutazione desiderata (s)
- Generare prodotti di PCR corrispondenti a due frammenti parzialmente sovrapposte del gene bersaglio istone.
- Preparare due reazioni di PCR come segue:
- Per generare prodotti di PCR corrispondenti alla prima metà del gene (prodotto a in Figura 1B), impostare la seguente reazione: 1 ml DNA stampo, 5 μl10 micron primer forward, 5 μl10 micron primer reverse, 0,5 ml (1,25 U) termostabile DNA polimerasi, 10 microlitri 5x DNA polimerasi di buffer, 5 ul miscela dNTP (2 mm ciascuno), e 23,5 ml dH 2 O.
NOTA: Il DNA modello può essere DNA genomico derivato da un ceppo wild-type per il gene bersaglio istone isolato utilizzando pro di serieprocedure 10. Per tenere conto di variazioni della concentrazione di DNA e il livello di impurità in diverse preparazioni genomiche, si raccomanda di ottimizzare le reazioni usando o DNA diluito o diverse diluizioni delle preparazioni genomici (ad esempio, 1:10 a 1: 100). Il primer forward dovrebbe ricottura ad una regione a monte del gene bersaglio. Il primer inverso dovrebbe ricottura all'interno della ORF, essere ~ 40 nucleotidi di lunghezza, e contiene la mutazione (s) desiderato da qualche parte nel mezzo di esso (vedi Figura 1B-1 e Rappresentante dei risultati sezione per gli esempi). L'uso di una DNA polimerasi alta fedeltà è raccomandato per ridurre i tassi di mutazioni indesiderate durante la sintesi dei prodotti di PCR. - Per generare prodotti di PCR corrispondenti alla seconda metà del gene (prodotto b nella Figura 1B), impostare una reazione come indicato in 2.1.1.1 ma con differenti primers.
NOTA: il PRI in avantimer dovrebbe ricottura all'interno della ORF, essere ~ 40 nucleotidi di lunghezza, e contiene la mutazione desiderata (s) da qualche parte nel mezzo di esso. Si noti che la mutazione (s) in questo primer è il complemento inverso della mutazione (s) in reverse primer nel passaggio 2.1.1.1. Il primer inverso deve ricottura ad una regione a valle del gene bersaglio (vedere Figura 1B-1 e Risultati rappresentativi sezione per gli esempi).
- Per generare prodotti di PCR corrispondenti alla prima metà del gene (prodotto a in Figura 1B), impostare la seguente reazione: 1 ml DNA stampo, 5 μl10 micron primer forward, 5 μl10 micron primer reverse, 0,5 ml (1,25 U) termostabile DNA polimerasi, 10 microlitri 5x DNA polimerasi di buffer, 5 ul miscela dNTP (2 mm ciascuno), e 23,5 ml dH 2 O.
- Mettere le reazioni in un termociclatore con le seguenti impostazioni: 94 C 30 sec; 30 cicli delle seguenti impostazioni: 98 C 10 sec, 60 ° C 5 sec, 72 c 1,5 min; e 72 c 10 min.
Nota: L'ottimizzazione dei parametri di PCR può essere richiesto per i set di primer specifici e bersaglio gene istone.
- Preparare due reazioni di PCR come segue:
- Eseguire 20 - 50 ml di materiale dalle reazioni di PCR su un gel di fusione basso punto di agarosio 0,9% a 89 mm di base Tris, 89 mM di acido borico, 2,5 mM EDTA (TBE) buffer.
- sezioni gel agarosio Tagliare contenenti il pr PCRodotti da gel utilizzando un bisturi o lametta pulito e trasferire ciascuna in una provetta da 1,5 ml microcentrifuga. Conservare sezioni agarosio contenenti prodotti di PCR a -20 ˚C fino al momento dell'uso.
3. Fusion PCR dei due frammenti parzialmente sovrapposte ottenere Full Size prodotti di PCR per l'integrazione
- Preparare modello per le reazioni PCR
- Sciogliere sezioni gel agarosio dal punto 2.3, ponendo le provette da microcentrifuga in un blocco set di calore a 65 ° C per 5 minuti (o fino a quando completamente sciolto). Tubi vortex ogni 1 - 2 min per facilitare il processo di fusione.
- Trasferire una certa quantità di fuso agarosio da ciascun campione (ad esempio, 50 ml ciascuno, per un totale di 100 ml) in un unico provetta e mescolare nel vortex. Utilizzare questo come il modello nelle reazioni PCR di fusione. Posizionare il tubo a -20 ˚C fino al momento dell'uso.
- Amplificare una grande quantità di piena prodotto di PCR (prodotto cin figura 1B)
- Impostare sei reazioni PCR, ognuno con i seguenti componenti: 2 ml modello di DNA, 10 microlitri 10 micron di primer in avanti, 10 microlitri 10 micron di primer inverso, 1 ml (2,5 U) termostabile DNA polimerasi, 20 l 5x tampone DNA polimerasi, 10 ml miscela dNTP (2 mm ciascuno) e 47 ml dH 2 O.
NOTA: Il numero di reazioni può essere modificato a seconda dell'efficienza PCR. Il DNA stampo (vedi 3.1.2), dovrebbe essere riscaldato a 65 ° C fino a fuso, mescolate con il vortex, e ha aggiunto infine alla miscela di reazione PCR. Una volta aggiunto, mescolare delicatamente ma accuratamente pipettando la soluzione su e giù parecchie volte. Per tenere conto di variazioni della concentrazione di DNA nei diversi campioni, si raccomanda di ottimizzare prima delle reazioni utilizzando template diluito o differenti diluizioni del modello (ad esempio, 1:10 a 1: 100). I due primer utilizzati dovrebbero temprare ai due frammenti parzialmente sovrapposte del gene bersaglio come malatiustrated in Figura 1B-2 ed essere progettato in modo tale che i prodotti di PCR finali avranno almeno 40 paia di basi su entrambi i lati omologhi alle regioni fiancheggianti il URA3 ORF che guideranno la fase di ricombinazione omologa (vedere la sezione Risultati rappresentativi per gli esempi). L'uso di una DNA polimerasi alta fedeltà è raccomandato per ridurre i tassi di mutazioni indesiderate durante la sintesi dei prodotti di PCR. - Mettere le provette in un termociclatore con le seguenti impostazioni: 94 C 30 sec; 30 cicli delle seguenti impostazioni: 98 C 10 sec, 50 C 15 sec, 72 c 1,5 min; e 72 c 10 min.
NOTA: può essere richiesta l'ottimizzazione dei parametri di PCR per i set di primer specifici e gene bersaglio istone.
- Impostare sei reazioni PCR, ognuno con i seguenti componenti: 2 ml modello di DNA, 10 microlitri 10 micron di primer in avanti, 10 microlitri 10 micron di primer inverso, 1 ml (2,5 U) termostabile DNA polimerasi, 20 l 5x tampone DNA polimerasi, 10 ml miscela dNTP (2 mm ciascuno) e 47 ml dH 2 O.
4. Co-trasformazione di Full Size prodotti di PCR e la spina dorsale plasmidi e selezione per Marker su plasmidi
- Concentrazione di prodotti di PCR
- Si riuniscono gli sReazioni ix PCR (600 ml totali) dal punto 3.2.2 in un unico provetta e mescolare nel vortex.
- Suddividere il campione in tre aliquote di 200 microlitri in tubi microcentrifuga. Precipitare il DNA in ciascun tubo aggiungendo 20 ml di sodio acetato 3M (pH 5,2) e 550 ml di etanolo al 100%. Mescolare la soluzione a fondo e mettere in ghiaccio per almeno 15 minuti. Raccogliere DNA per centrifugazione a circa 14000 xg per 10 minuti, lavare il pellet con 200 ml di etanolo al 70%, e aria secca.
- Risospendere ogni pellet di DNA in 25 ml di dH 2 O, e concentrare in un unico tubo (per un totale di 75 microlitri).
- Lievito co-trasformazione
- Preparare 10 ml di cultura durante la notte del ceppo generato nella sezione 1 di estratto di lievito Peptone destrosio (YPD) liquido di mezzo 11.
- La mattina seguente, inoculare 400 ml di YPD mezzo liquido con 8 ml di cultura durante la notte saturi e incubare agitandoa 30 ° C per 4 - 5 h per permettere alle cellule di entrare fase logaritmica di crescita.
- Raccogliere le cellule per centrifugazione a ~ 3.220 xg per 10 min, scartare il mezzo liquido, e risospendere le cellule in 1 volume di 10 mM Tris-HCl (pH 8,0), 1 mM EDTA, 0,1 soluzione M di litio acetato (TE / liac) .
- Raccogliere le cellule per centrifugazione a ~ 3.220 xg per 10 min, e scartare il TE / LIAC.
- Risospendere le cellule in 1 ml TE / LIAC.
- Impostare il seguente cocktail di reazione in una provetta: 800 ml di cellule dal punto 4.2.5, 40 ml di bollito / ml salmone DNA dello sperma di 10 mg, per un totale di 12,5 mg di spina dorsale DNA plasmide, e 75 ml di prodotto di PCR concentrato dal punto 4.1.3.
NOTA: Salmon DNA di sperma deve essere bollita per 5 min e posto in ghiaccio per almeno 5 minuti prima dell'uso nella reazione. Il volume totale di backbone plasmide DNA aggiunto deve essere ridotto al minimo (~ 80 microlitri o meno). Vedere la sezione Risultati Rappresentante per un esempio di un plasma spina dorsaleid. - Mescolare il tubo di cocktail a fondo e in modo uniforme aliquota in otto microprovette (Tubi 1-8).
- Impostare i seguenti due provette di reazione di trasformazione di controllo:
- Tubo 9 (senza controllo del prodotto PCR): 100 ml di cellule dal punto 4.2.5, 5 ml di bollito / ml sperma di salmone DNA 10 mg (bollite per 5 min; vedere il punto 4.2.6 Nota), per un totale di 1,56 mg di backbone plasmide DNA e nessun prodotto di PCR aggiunte.
- Tubo 10 (nessun controllo DNA): 100 ml di cellule dal punto 4.2.5, 5 ml di bollito 10 mg / mL di sperma di salmone DNA (vedi punto 4.2.6 Nota), senza spina dorsale plasmide aggiunto il DNA, e nessun prodotto di PCR aggiunto.
- Mescolare entrambi i tubi delicatamente ma accuratamente pipettando su e giù parecchie volte.
- Incubare le dieci provette a 30 C per 30 min.
- Per ogni tubo, aggiungere 1,2 ml di 40% polietilene glicole (PEG 3350) in TE / LIAC. Mescolare accuratamente con una pipetta P-1000 fino a che la soluzione risulti omogenea.
- Incubare le dieci provette a 30° C per 30 min. Mescolare delicatamente la soluzione pipettando su e giù e poi incubare le provette a 42 ° C per 15 min.
- Raccogliere le cellule facendo girare i tubi in microcentrifuga a ~ 14000 g per 30 sec. Eliminare il liquido e risospendere le cellule in 1 ml di dH sterile 2 O.
- Raccogliere le cellule facendo girare i tubi in microcentrifuga a ~ 14000 g per 30 sec. Eliminare il liquido e risospendere le cellule in 500 ml di dH sterili 2 O.
- tubi Pool 1-8 insieme (volume totale di 4 ml) e mescolare bene pipettando su e giù.
- Piatto 200 microlitri della miscela di cui sopra su ciascuno dei venti completi minima dropout piastre di terreno 11 (piastre 1-20) per la selezione del plasmide backbone.
- Piastra 200 ml di miscela da tubo 9 e 200 ml di miscela dalla fermata della 10 ciascuno sul proprio piatto di selezione (piatti, rispettivamente, 21 e 22,).
- Incubare le piastre a 22 30 ° C per 3 - 5 giorni aselezionare per trasformanti plasmidi.
- Ispezionare le piastre di trasformazione dopo 3 - 5 giorni di incubazione. Circa 5.000 colonie dovrebbero essere visibili su piastre 1-21 (vedi Rappresentante dei risultati per un esempio) e non colonie dovrebbero essere presenti sulla piastra 22.
5. schermo per trasformanti 5-FOA-resistenti
- Trasferire le cellule da lastre 1 - 20 (e la piastra di trasformazione 21 come controllo) a 5-fluoroorotic acido (5-FOA) piastre 11 dalla replica-plating 12 per lo screening per la perdita del gene URA3 come risultato dell'integrazione della prodotti di PCR nella posizione desiderata.
- Togliere il coperchio piatto e premere il piatto contenente colonie su un velluto sterile. Trasferire le cellule dal velluto per una piastra 5-FOA premendo la piastra sul velluto. Incubare le piastre a 30 C per 2 giorni.
- Dopo l'incubazione di 2 giorni, ispezionare accuratamente le lastre 5-FOA per growth.
NOTA: Un evento di integrazione candidato sarà rappresentato da un piccolo asimmetrica colonia "schiacciata" su una piastra 5-FOA - converso, piccole papille cresce su piastre 5-FOA probabilmente rappresentativi di mutazioni URA3 spontanee sollevata durante la crescita di colonie sulla piastre di trasformazione, e sono quindi difficilmente riescono a rappresentare l'evento di integrazione desiderato (vedere Figura 3 nella sezione Risultati rappresentativi per ulteriori elaborazioni su questo punto e per alcuni esempi).
6. Purificazione di colonie 5-FOA-resistenti e la perdita di Backbone plasmidi
- Utilizzando stuzzicadenti sterile, raccogliere le colonie candidati dalle piastre 5-FOA descritte al punto 5.2 e strisciare per singole colonie su piastre YPD. Incubare per 2 - 3 giorni a 30 ° C.
- Dopo l'incubazione, replica - Piastra ogni purificazione piatto YPD per un piatto fresco YPD, una piastra di drop-out manca uracile per verificare la presenza di perditedel gene URA3, ed una seconda piastra drop-out per monitorare la presenza o l'assenza del plasmide backbone. Incubare per 1 - 2 giorni a 30 ° C.
- Dopo l'incubazione, identificare una colonia di ciascun campione candidato che cresce sulla piastra YPD ma non cresce su entrambi piatto drop-out (questo tipo di colonie dovrebbe aver perso il gene URA3 attraverso l'evento di ricombinazione e perso il plasmide backbone durante mitotica divisione cellulare). Restreak tali colonie su piastre freschi YPD. Queste colonie sono i candidati di integrazione e saranno analizzati ulteriormente al punto 7.
7. Le analisi molecolari per test per l'integrazione corretta of the Mutant Allele
- Isolare il DNA genomico da campioni candidati utilizzando le procedure standard 10.
- Amplificare regione genomica che comprende il sito di destinazione.
- Impostare la seguente reazione PCR per ciascun campione: 0,5 ml modello di DNA, 5 ml10 micron di primer in avanti, 5 ml 10 micron di primer inverso, 0,5 ml (2,5 unità) Taq DNA polimerasi, 5 ml 10x Taq DNA polimerasi tampone, 5 ul miscela dNTP (2 mm ciascuno) e 29 ml dH 2 O.
NOTA: Modello del DNA è il DNA genomico derivato dai campioni candidati. Si raccomanda di includere anche due reazioni di controllo: uno utilizzando DNA genomico derivato dalle geneΔ istone originali :: URA3 ceppo come modello e un altro utilizzando DNA genomico da un ceppo selvatico istone come modello. Per tenere conto di variazioni della concentrazione di DNA e il livello di impurità in diverse preparazioni genomiche, si raccomanda di ottimizzare le reazioni usando o DNA diluito o diverse diluizioni delle preparazioni genomici (ad esempio, 1:10 a 1: 100). È importante assicurarsi che questi primer ricottura a sequenze di DNA di fuori della regione abbracciata dal prodotto putativamente integrato PCR - questo modo, la dimensione dei prodotti di PCR in questi reactions possono essere utilizzati come strumento diagnostico per l'integrazione dei prodotti nella posizione genomica corretta (vedi Risultati rappresentativi per esempio). - Mettere le reazioni in un termociclatore con le seguenti impostazioni: 94 ° C 3 min; 30 cicli delle seguenti impostazioni: 94 ° C 45 sec, 50 ° C 45 sec, 72 ° C 2 min; e 72 ° C 10 min.
NOTA: può essere richiesta l'ottimizzazione dei parametri di PCR per i set di primer specifici e gene bersaglio istone.
- Impostare la seguente reazione PCR per ciascun campione: 0,5 ml modello di DNA, 5 ml10 micron di primer in avanti, 5 ml 10 micron di primer inverso, 0,5 ml (2,5 unità) Taq DNA polimerasi, 5 ml 10x Taq DNA polimerasi tampone, 5 ul miscela dNTP (2 mm ciascuno) e 29 ml dH 2 O.
- Lavorazione dei prodotti di PCR
- Eseguire 20 microlitri da ogni reazione su un gel di agarosio 0,8% TBE.
- Valutare il formato dei prodotti PCR utilizzando standard DNA come riferimento per determinare se il gene URA3 è stato sostituito con successo dal gene mutato istone putativamente (vedi Risultati rappresentativi per esempio).
NOTA: In alcuni casi, la mutazione desiderata (s) introdotto nei geni istone creare o distruggere una restrizione sedersie. Se questo è il caso, la presenza della mutazione desiderata nei prodotti di PCR di dimensioni indicative della corretta integrazione può essere valutata sottoponendo i prodotti di digestione con l'enzima di restrizione corrispondente seguita da analisi elettroforesi su gel (vedi Risultati rappresentativi per esempio) . - Oggetto prodotti PCR di dimensioni indicative della corretta integrazione del sequenziamento del DNA per confermare la presenza della mutazione desiderata (s) e di assicurare che nessun ulteriori mutazioni sono state introdotte nel genoma.
Risultati
Descriviamo la generazione di un allele hht2 esprimere un istone H3 mutante proteina ospitare una sostituzione nella posizione 53 da un arginina ad acido glutammico (H3-R53E mutante) come un esempio rappresentativo della mirata strategia situ mutagenesi.
Abbiamo generato un ceppo in cui l'intera ORF di HHT2 è sostituito dal gene URA3 (vedi punto 1 del protocollo). Questo ceppo,...
Discussione
L'elevata omologia di sequenza tra due geni non alleliche che codificano ciascuno dei quattro proteine essenziali istoniche delle cellule S. cerevisiae aploidi possa rappresentare sfida per investigatori vogliano indirizzare specificamente uno dei due geni per mutagenesi. In precedenza descritto lievito metodologie di mutagenesi, compreso il Delitto Perfetto, genomica mutagenesi sito-specifica (SSG), e PCR-based metodi pezzi allele-free clonazione 5,
Divulgazioni
The authors declare that they have no competing financial interests.
Riconoscimenti
We thank Reine Protacio for helpful comments during the preparation of this manuscript. We express our gratitude to the National Science Foundation (grants nos. 1243680 and 1613754) and the Hendrix College Odyssey Program for funding support.
Materiali
| Name | Company | Catalog Number | Comments |
| 1 kb DNA Ladder (DNA standards) | New England BioLabs | N3232L | |
| Agarose | Sigma | A5093-100G | |
| Boric Acid | Sigma | B0394-500G | |
| dNTP mix (10 mM each) | ThermoFisher Scientific | R0192 | |
| EDTA solution (0.5 M, pH 8.0) | AmericanBio | AB00502-01000 | |
| Ethanol (200 Proof) | Fisher Scientific | 16-100-824 | |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma | E4884-500G | |
| Lithium acetate dihydrate | Sigma | L6883-250G | |
| MyCycler Thermal Cycler | BioRad | 170-9703 | |
| Poly(ethylene glycol) (PEG) | Sigma | P3640-1KG | |
| PrimeSTAR HS DNA Polymerase (high fidelity DNA polymerase) and 5x buffer | Fisher Scientific | 50-443-960 | |
| Salmon sperm DNA solution | ThermoFisher Scientific | 15632-011 | |
| Sigma 7-9 (Tris base, powder form) | Sigma | T1378-1KG | |
| Sodium acetate trihydrate | Sigma | 236500-500G | |
| Supra Sieve GPG Agarose (low metling temperature agarose) | AmericanBio | AB00985-00100 | |
| Taq Polymerase and 10x Buffer | New England BioLabs | M0273X | |
| Toothpicks | Fisher Scientific | S67859 | |
| Tris-HCl (1 M, pH 8.0) | AmericanBio | AB14043-01000 | |
| a-D(+)-Glucose | Fisher Scientific | AC170080025 | for yeast media |
| Agar | Fisher Scientific | DF0140-01-0 | for yeast media |
| Peptone | Fisher Scientific | DF0118-07-2 | for YPD medium |
| Yeast Extract | Fisher Scientific | DF0127-17-9 | for YPD medium |
| 4-aminobenzoic acid | Sigma | A9878-100G | for complete minimal dropout medium |
| Adenine | Sigma | A8626-100G | for complete minimal dropout medium |
| Glycine hydrochloride | Sigma | G2879-100G | for complete minimal dropout medium |
| L-Alanine | Sigma | A7627-100G | for complete minimal dropout medium |
| L-Arginine monohydrochloride | Sigma | A5131-100G | for complete minimal dropout medium |
| L-Asparagine monohydrate | Sigma | A8381-100G | for complete minimal dropout medium |
| L-Aspartic acid sodium salt monohydrate | Sigma | A6683-100G | for complete minimal dropout medium |
| L-Cysteine hydrochloride monohydrate | Sigma | C7880-100G | for complete minimal dropout medium |
| L-Glutamic acid hydrochloride | Sigma | G2128-100G | for complete minimal dropout medium |
| L-Glutamine | Sigma | G3126-100G | for complete minimal dropout medium |
| L-Histidine monohydrochloride monohydrate | Sigma | H8125-100G | for complete minimal dropout medium |
| L-Isoleucine | Sigma | I2752-100G | for complete minimal dropout medium |
| L-Leucine | Sigma | L8000-100G | for complete minimal dropout medium |
| L-Lysine monohydrochloride | Sigma | L5626-100G | for complete minimal dropout medium |
| L-Methionine | Sigma | M9625-100G | for complete minimal dropout medium |
| L-Phenylalanine | Sigma | P2126-100G | for complete minimal dropout medium |
| L-Proline | Sigma | P0380-100G | for complete minimal dropout medium |
| L-Serine | Sigma | S4500-100G | for complete minimal dropout medium |
| L-Threonine | Sigma | T8625-100G | for complete minimal dropout medium |
| L-Tryptophan | Sigma | T0254-100G | for complete minimal dropout medium |
| L-Tyrosine | Sigma | T3754-100G | for complete minimal dropout medium |
| L-Valine | Sigma | V0500-100G | for complete minimal dropout medium |
| myo-Inositol | Sigma | I5125-100G | for complete minimal dropout medium |
| Uracil | Sigma | U0750-100G | for complete minimal dropout medium |
| Ammonium Sulfate | Fisher Scientific | A702-500 | for complete minimal dropout medium |
| Yeast Nitrogen Base | Fisher Scientific | DF0919-07-3 | for complete minimal dropout medium |
| 5-Fluoroorotic acid (5-FOA) | AmericanBio | AB04067-00005 | for 5-FOA medium |
Riferimenti
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389 (6648), 251-260 (1997).
- Campos, E. I., Reinberg, D. Histones: annotating chromatin. Annu Rev Genet. 43, 559-599 (2009).
- Rando, O. J., Winston, F. Chromatin and transcription in yeast. Genetics. 190 (2), 351-387 (2012).
- Duina, A. A., Miller, M. E., Keeney, J. B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 197 (1), 33-48 (2014).
- Storici, F., Resnick, M. A. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y). 25, 189-207 (2003).
- Gray, M., Kupiec, M., Honigberg, S. M. Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7 (2004).
- Erdeniz, N., Mortensen, U. H., Rothstein, R. Cloning-free PCR-based allele replacement methods. Genome Res. 7 (12), 1174-1183 (1997).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
- Lundblad, V., Hartzog, G., Moqtaderi, Z. Manipulation of cloned yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Hoffman, C. S. Preparation of yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Treco, D. A., Lundblad, V. Preparation of yeast media. Curr Protoc Mol Biol. Chapter 13, (2001).
- Lederberg, J., Lederberg, E. M. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 63 (3), 399-406 (1952).
- Sikorski, R. S., Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122 (1), 19-27 (1989).
- Johnson, P., et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 5 (5), 741-749 (2015).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336-4343 (2013).
- Cross, S. L., Smith, M. M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol. 8 (2), 945-954 (1988).
- Libuda, D. E., Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 443 (7114), 1003-1007 (2006).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati