
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
ממוקד
In This Article
Summary
A strategy for generating mutations in histone genes at their endogenous location in Saccharomyces cerevisiae is presented.
Abstract
We describe a PCR- and homologous recombination-based system for generating targeted mutations in histone genes in budding yeast cells. The resulting mutant alleles reside at their endogenous genomic sites and no exogenous DNA sequences are left in the genome following the procedure. Since in haploid yeast cells each of the four core histone proteins is encoded by two non-allelic genes with highly homologous open reading frames (ORFs), targeting mutagenesis specifically to one of two genes encoding a particular histone protein can be problematic. The strategy we describe here bypasses this problem by utilizing sequences outside, rather than within, the ORF of the target genes for the homologous recombination step. Another feature of this system is that the regions of DNA driving the homologous recombination steps can be made to be very extensive, thus increasing the likelihood of successful integration events. These features make this strategy particularly well-suited for histone gene mutagenesis, but can also be adapted for mutagenesis of other genes in the yeast genome.
Introduction
ארבעת החלבונים היסטון הליבה H2A, H2B, H3, ו- H4 ממלאים תפקידים מרכזיים הדחיסה, ארגון ותפקוד של כרומוזומים איקריוטיים. שני סטים של כל אחד היסטונים אלה יוצרים את octamer היסטון, סליל מולקולרי שמנתב את העטיפה של זוגות בסיסים ~ 147 של DNA סביב עצמו, בסופו של דבר וכתוצאה מכך ההיווצרות של הנוקלאוזום 1. נוקלאוזום הם משתתפים פעילים במגוון תהליכים מבוסס כרומוזום, כגון הסדרת שעתוק גנים לבין ההיווצרות של euchromatin ו heterochromatin פני הכרומוזומים, וככזה הוא העיקר של מחקר אינטנסיבי במהלך העשורים האחרונים. מספר המנגנונים תואר שבאמצעותו נוקלאוזום ניתן להשפיע בדרכים שיכולים להקל על ביצוע של תהליכים ספציפיים - מנגנונים אלה כוללים שינוי posttranslational של שאריות היסטון, שיפוץ הנוקלאוזום תלוי ATP, וארגון מחדש הנוקלאוזום ATP-עצמאיוהרכבה / פירוק 2, 3.
השמרים ניצני שמר האפייה הוא אורגניזם מודל חזק במיוחד להבנת תפקוד היסטון אאוקריוטים. זו ניתן לייחס במידה רבה את הרמה הגבוהה של שימור אבולוציוני של חלבונים היסטון ברחבי eukarya תחום רמת המוכנות של שמרים למגוון ניסיוני גנטיים ביוכימיים מתקרב 4. גישות Reverse-גנטיות בשמרים היו בשימוש נרחב כדי לחקור את ההשפעות של מוטציות ספציפיות היסטון על היבטים שונים של ביולוגיה הכרומטין. עבור אלו סוגים של ניסויים הוא לעתים קרובות עדיף להשתמש בתאים בם ההיסטונים המוטציה באים לידי ביטוי מן הלוקוסים הגנומי מולדתם, כמו ביטוי מן פלסמידים אוטונומיים יכול להוביל לרמות תאיים החריגות של חלבונים היסטון (עקב מספרים שונים של פלסמידים בתאים) ו שינוי מקביל של הכרומטין environments, אשר בסופו של דבר יכול לבלבל את הפרשנות של תוצאות.
כאן אנו מתארים טכניקה PCR מבוססת המאפשרת mutagenesis הממוקד של גני היסטון במקומות גנומי מולדתם שאינו דורשים צעד שיבוט ותוצאות בדור של המוטציה הרצויה (ים) ללא רצפי DNA אקסוגני שאריות בגנום. טכניקה זו מנצלת את המערכה ההומולוגית היעילה שמרים ויש לו כמה תכונות משותפות עם בטכניקות דומות אחרות שפותחו על ידי קבוצות אחרות - בעיקר Delitto Perfetto, אתר ספציפי גנומי (SSG) mutagenesis, שיבוט ללא אלל מבוסס PCR שיטות החלפה 5, 6, 7. עם זאת, הטכניקה נתאר יש היבט זה עושה את זה במיוחד מתאים היטב mutagenesis של גני היסטון. בתאי שמרים הפלואידים, כל אחד ההיסטונים הליבה ארבעה מקודד על ידי שתי שאינוגני llelic ו הומולוגיים מאוד: למשל, H3 היסטון מקודדים על ידי גני HHT1 ו HHT2, ואת מסגרות הקריאה הפתוחות (ORFs) משני הגנים הן מעל 90% זהות ברצף. רמה גבוהה זו של הומולוגיה יכולה לסבך ניסויים שנועדו למקד ספציפי אחד הגנים שני קידוד-היסטון עבור mutagenesis. בעוד השיטות הנ"ל לעתים קרובות דורשים שימוש לפחות כמה רצפים בתוך ORF של גן המטרה לנהוג הומולוגיים, הטכניקה שאנו מתארים כאן עושה שימוש רצפים איגוף ORFs של גנים היסטון (אשר חולקים הרבה פחות הומולוגיה ברצף) עבור צעד רקומבינציה, ובכך להגדיל את הסבירות של מיקוד המוצלח של mutagenesis אל המוקד הרצוי. יתר על כן, באזורים ההומולוגיים שמניעים רקומבינציה יכולים להיות מאוד נרחבים, ובכך תורמים עוד יותר הומולוגי ממוקדים ויעיל.
Protocol
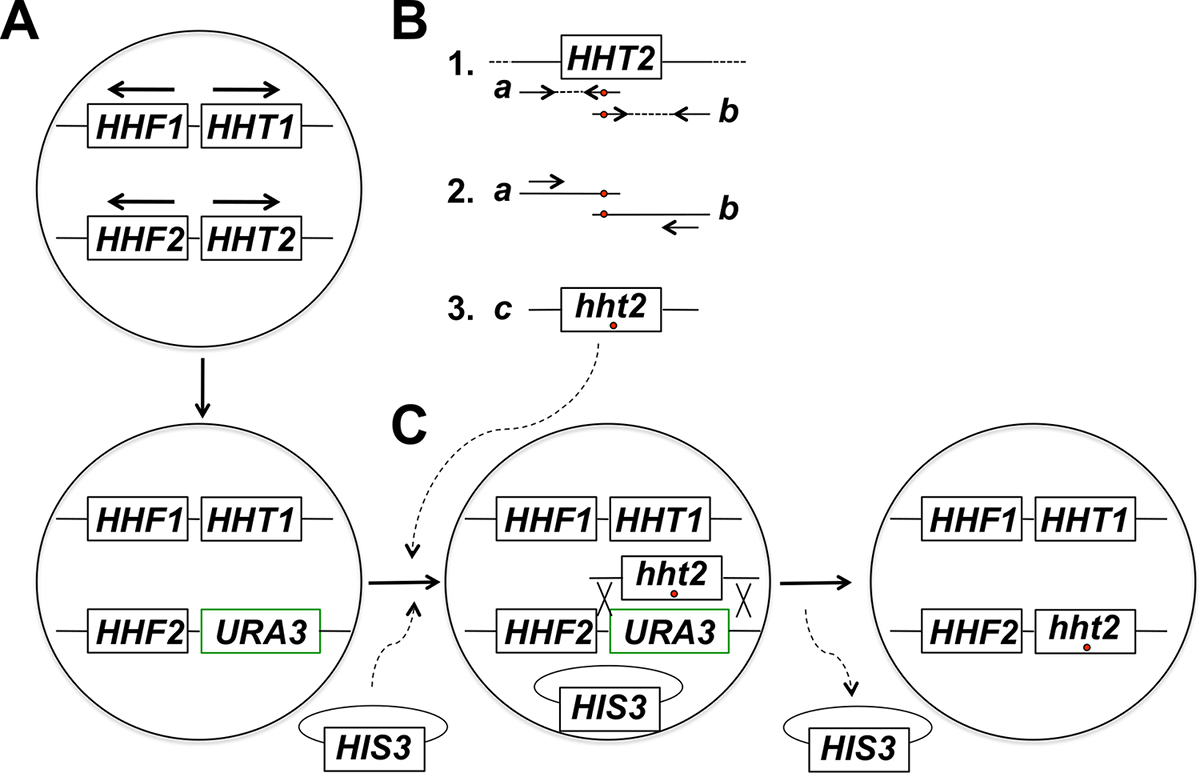
הערה: האסטרטגיה ניסיוני ממוקד mutagenesis גנים היסטון באתרו כוללת מספר שלבים (שהעיקריים בהם פורטו איור 1). צעדים אלה כוללים: (1) החלפת גן היסטון היעד עם גן URA3, (2) דור וטיהור של מוצרי ה- PCR המתאימים שני שברים חופף חלקי של גני היסטון היעד באמצעות פריימרים מחסה את המוטציה הרצויה (ים), (3 PCR Fusion) של שני שברים חופף חלקית להשיג מוצרי PCR בגודל מלאים עבור אינטגרציה, (4) Co-טרנספורמציה של מוצרי ה- PCR בגודל מלא פלסמיד עמוד שדרה, ובחירה עבור סמן על פלסמיד, (5) מסך עבור 5-פואה עמיד transformants, (6) טיהור של 5-פואה עמיד מושבות ואובדן פלסמיד עמוד השדרה, ו (7) מולקולרי מנתח assay לשילוב נכון של אלל מוטנטי.

איור 1: סקירה כללית של האסטרטגיה עבור ממוקד mutagenesis באתרו של גני היסטון שמרים ניצנים. בדוגמא זו הגן הממוקד הוא HHT2, אבל כל גן היסטון ליבה אחר יכול גם להיות mutagenized באמצעות אסטרטגיה זו. (א) בתאי שמרים הפלואידים הנמל שני גנים היסטון קידוד H3 (HHT1 ו HHT2) ושני גנים H4 קידוד היסטון (HHF1 ו HHF2) מסודרים כפי שמוצג באיור (הגנים HHT1 ו HHF1 ממוקמים על כרומוזום השנייה ואת HHT2 גני HHF2 ממוקמים על XIV כרומוזום - בכל מקרה, החצים מצביעים לכיוון של שעתוק). בשלב הראשון של ההליך, ORF של גן HHT2 מוחלף גן URA3, והוליד זן hht2Δ :: URA3. (ב) חלק 1, עותק wild-type של הגן HHT2 ממדגם הדנ"א הגנומי משמש כתבנית עבור שתי תגובות PCR לסוגיםte שני שברים חופפים חלקית של הגן. פריימר הפוכה התגובה הראשונה כוללת נוקלאוטידים אחד או יותר תואם (מסומנים בעיגול אדום) התואמים את המוטציה הרצויה (ים) יוכנס הגנום. פריימר קדימה עבור התגובה השנייה קיים אי ההתאמה המקבילה בתצורת משלימים הפוכה (מסומנת גם עם עיגול אדום). שני מוצרי PCR שנוצרו חלק 1 (א מוצרים ב) משמשים כתבניות היתוך PCR באמצעות שני פריימרים כי לחשל למוצרי A ו- B ב האופנה מוצגת חלק 2. התוצאה היא הדור בגודל מלא PCR מוצרים (ג מוצר חלקי 3) מחסה את המוטציה הרצויה (ים). (ג) hht2Δ :: זן URA3 אז שיתוף טרנספורמציה עם מוצרי ה- PCR בגודל מלא פלסמיד עמוד שדרה (א HIS3- מסומן פלסמיד בדוגמא זו), ותאים נבחרים עבור הנוכחות של פלסמיד (על התקשורת החסרה histidine בדוגמה זו). Transformants אז מוקרן על 5-פואה התנגדות - תאים עמידים הם מועמדים שעבר אירוע רקומבינציה הומולוגי המוביל אינטגרציה של מוצר ה- PCR כריתה של גן URA3, כפי שמוצג. הפסד בתקופות עוקבות של הפלסמיד עמוד השדרה על ידי חלוקת התא mitotic מוביל בזן העכברים המוטנטים היסטון הרצוי הסופי. מצאנו כי בחירה של פלסמיד עמוד השדרה ואחריו הקרנה עבור תוצאות התנגדות 5-פואה בתדירות גבוהה הרבה יותר של זיהוי של אירועי אינטגרציה נכונים לעומת בחירה ישירה על צלחות של 5 פואה, אשר רובם מזהה תאים רכשו מוטציות URA3 ספונטניות. (נתון זה כבר שונה מהתייחסות 14). אנא לחץ כאן כדי לצפות בגרסה גדולה יותר של דמות זו.
{kind=link}
1. החלפת ג'ין היסטון היעד עם URA3גֵן
- בצע סטנדרטי בתיווך PCR שיבוש גנטי צעד אחד החלפת ORF של גן היסטון יעד עם גן URA3 8, 9.
הערה: שימוש בתאי שמרים נושאים את ura3Δ0 מומלץ כמו מוטציה זו מסירה את URA3 אנדוגני כולו ORF, וכך למנוע שילוב של מוצר ה- PCR לתוך מוקד URA3 8. לחלופין, הגן ק lactis URA3 ניתן להשתמש ביעילות עבור הדור של החלפת היסטון בכל רקע URA3 כפי שהוא פונקציונלי cerevisiae ס אבל יש הומולוגיה ברצף חלקית בלבד עם הגן URA3 cerevisiae ס. הזן צריך להיות גם auxotrophic לפחות תרכובת כזו אשר תאפשר לבחירה של פלסמיד עמוד השדרה בניסוי טרנספורמציה (ראה שלב 4 של פרוטוקול זה). שלב זה אינו הכרחי אם geneΔ היסטון היעד :: URA3זן הוא כבר זמין.
הדור וטיהור 2. של PCR מוצרים המתאימים שני שברים חופפים חלקית של יעד היסטון ג'ין באמצעות פריימרים מחסה את המוטציה הרצויה (ים)
- צור מוצרי PCR המתאימים שני שברים חופף חלקי של גני היסטון היעד.
- הכן שתי תגובות PCR כדלקמן:
- כדי ליצור מוצרים PCR המתאים במחצית הראשונה של הגן (מוצר באיור 1B), להגדיר את התגובה הבאה: 1 תבנית ה- DNA μl, 5 μl10 מיקרומטר פריימר קדימה, 5 μl10 מיקרומטר פריימר הפוכה, 0.5 μl (1.25 U) thermostable DNA פולימרז, 10 μl חיץ פולימראז 5x DNA, 5 μl תערובת dNTP (2 מ"מ כל אחד), ו 23.5 μl dH 2 O.
הערה: תבנית ה- DNA יכול להיות הדנ"א הגנומי נגזר wild-type זן עבור גן היסטון היעד מבודד באמצעות פרו רגילפרוצדורות 10. כדי להסביר וריאציות בריכוז DNA ורמת זיהומים בהכנות גנומי שונים, מומלץ לייעל את התגובות או באמצעות DNA חי או דילולים שונים של ההכנות גנומי (למשל, 1:10 ו 1: 100). פריימר קדימה צריך לחשל לאזור במעלה הזרם של גן המטרה. פריימר ההפוכה צריכה לחשל בתוך ORF, להיות ~ 40 נוקלאוטידים אורך, ומכילה את המוטציה הרצויה (ים) אי שם באמצע של זה (ראה סעיף איור 1 ב- 1 ונציג תוצאות עבור דוגמאות). שימוש DNA פולימרז איכות גבוהה מומלץ על מנת להפחית את השיעורים של מוטציות לא רצויות במהלך הסינתזה של מוצרי ה- PCR. - כדי ליצור מוצרי PCR מתאימים במחצית השנייה של הגן (ב מוצר באיור 1B), להקים תגובה כמצוין 2.1.1.1 אבל עם פריימרים שונים.
הערה: פרי קדימהmer צריך לחשל בתוך ORF, להיות ~ 40 נוקלאוטידים אורך, ומכיל את המוטציה הרצויה (ים) אי שם באמצע של זה. ראוי לציין, כי מוטציה (ים) פריימר זה הוא המשלים השני של מוטציה (ים) של פריימר הפוכה בשלב 2.1.1.1. פריימר ההפוכה צריכה לחשל לאזור במורד זרם של גן המטרה (ראה איור 1 ב- 1 ו נציג תוצאות סעיף לדוגמות).
- כדי ליצור מוצרים PCR המתאים במחצית הראשונה של הגן (מוצר באיור 1B), להגדיר את התגובה הבאה: 1 תבנית ה- DNA μl, 5 μl10 מיקרומטר פריימר קדימה, 5 μl10 מיקרומטר פריימר הפוכה, 0.5 μl (1.25 U) thermostable DNA פולימרז, 10 μl חיץ פולימראז 5x DNA, 5 μl תערובת dNTP (2 מ"מ כל אחד), ו 23.5 μl dH 2 O.
- מניחים את התגובות ב thermocycler עם ההגדרות הבאות: 94 ג 30 שניות; 30 מחזורים של ההגדרות הבאות: 98 ג '10 שניות, 60 ג 5 שניות, 72 1.5 C דקות; ו -72 ג '10 דק'.
הערה: אופטימיזציה של פרמטרי PCR עשויה להידרש סטים פריימר ספציפיים ולכוון גני היסטון.
- הכן שתי תגובות PCR כדלקמן:
- הפעל 20 - 50 μl של החומר על פי התגובות PCR על ג'ל agarose נקודת התכה נמוכה 0.9% ב -89 בסיס מ"מ טריס, 89 מ"מ חומצת בור, 2.5 mM EDTA חיץ (TBE).
- חותכים סעיפים agarose ג'ל המכיל את pr PCRoducts מן ג'ל באמצעות סכין אזמל או גילוח נקי ולהעביר כל צינור 1.5 מ"ל microcentrifuge. אחסן סעיפי agarose המכילים מוצרי ה- PCR ב -20 C עד מוכן לשימוש.
3. Fusion PCR של שברי חופפים שני חלקית להשיג בגודל מלא מוצרים PCR עבור אינטגרציה
- הכן תבנית עבור PCR תגובות
- ממיסים סעיפים ג'ל agarose משלב 2.3 על ידי הנחת צינורות microcentrifuge ב סט גוש חום 65 צלזיוס למשך 5 דקות (או עד שהיא נמסה לחלוטין). צינורות וורטקס כל 1 - 2 דקות כדי להקל על תהליך ההיתוך.
- העבר סכום קבוע של נמס agarose מכל מדגם (למשל, 50 μl כל, עבור סכום כולל של 100 μl) לתוך צינור microcentrifuge יחיד ומערבבים ידי vortexing. השתמש באפשרות זו כתבנית בתגובות PCR ההיתוך. מניחים את הצינור ב -20 C עד מוכן לשימוש.
- להגביר כמות גדולה של מוצר ה- PCR בגודל מלא (ג המוצרבאיור 1B)
- הגדרת שישה התגובות PCR, כל עם הרכיבים הבאים: תבנית ה- DNA 2 μl, 10 μl 10 מיקרומטר פריימר קדימה, 10 μl 10 מיקרומטר פריימר הפוכה, 1 μl (2.5 U) DNA פולימרז thermostable, 20 μl חיץ פולימראז 5x DNA, 10 μl תערובת dNTP (2 מ"מ כל אחד), ו -47 μl dH 2 O.
הערה: מספר תגובות ניתן לשנות בהתאם את יעילות PCR. ה- DNA התבנית (ראה 3.1.2) צריך להיות מחומם ל -65 C עד שהיא נמסה, מעורבב ידי vortexing, והוסיף אחרון לתערובת תגובת PCR. לאחר ההוספה, ומערבבים בעדינות אך ביסודיות על ידי pipetting הפתרון מעלה ומטה מספר פעמים. כדי להסביר וריאציות בריכוז DNA בדגימות השונות, מומלץ ראשון לייעל את התגובות או באמצעות תבנית חייה או דילולים שונים של התבנית (למשל, 1:10 ו 1: 100). שני פריימרים המשמשים צריך לחשל את שני שברי חופפים חלקית של גן המטרה כחולהustrated באיור 1 ב- 2 ו להיות מתוכנן כך מוצרי PCR הסופיים יהיו לפחות 40 זוגות בסיסים משני הומולוגי צד לאזורי איגוף ORF URA3 אשר יניעו את הצעד ההומולוגי (ראה סעיף נציג תוצאות לדוגמות). שימוש DNA פולימרז איכות גבוהה מומלץ על מנת להפחית את השיעורים של מוטציות לא רצויות במהלך הסינתזה של מוצרי ה- PCR. - מניחים את צינורות thermocycler עם ההגדרות הבאות: 94 ג 30 שניות; 30 מחזורים של ההגדרות הבאות: 98 ג '10 שניות, 50 ג 15 שניות, 72 1.5 C דקות; ו -72 ג '10 דק'.
הערה: אופטימיזציה של פרמטרי PCR עשויה להידרש סטים פריימר ספציפיים היסטון יעד גן.
- הגדרת שישה התגובות PCR, כל עם הרכיבים הבאים: תבנית ה- DNA 2 μl, 10 μl 10 מיקרומטר פריימר קדימה, 10 μl 10 מיקרומטר פריימר הפוכה, 1 μl (2.5 U) DNA פולימרז thermostable, 20 μl חיץ פולימראז 5x DNA, 10 μl תערובת dNTP (2 מ"מ כל אחד), ו -47 μl dH 2 O.
4. Co-טרנספורמציה של גודל מלא מוצרים PCR ו עמוד השדרה פלסמיד, בחירה עבור מרקר על פלסמיד
- ריכוז של מוצרי ה- PCR
- פינת ה- Six PCR תגובות (600 סך μl) משלב 3.2.2 לתוך צינור יחיד microcentrifuge ומערבבים ידי vortexing.
- פיצול המדגם לשלוש 200 aliquots μl צינורות microcentrifuge. להאיץ את ה- DNA בצינור אחד על ידי הוספת 20 μl של נתרן אצטט 3M (pH 5.2) ו 550 μl של אתנול 100%. מערבבים את הפתרון ביסודיות ומניחים על קרח למשך לפחות 15 דקות. אסוף DNA על ידי צנטריפוגה ב ~ 14,000 XG במשך 10 דקות, לשטוף את הגלולה עם 200 μl של 70% אתנול, ו אוויר יבש.
- Resuspend כל גלולה DNA לתוך 25 μl של DH 2 O, ובריכת לתוך צינור אחד (עבור סכום כולל של 75 μl).
- שיתוף טרנספורמציה שמרים
- כן 10 מיליליטר של תרבות הלילה של המתח שנוצר בסעיף 1 ב תמצית שמרי Peptone דקסטרוז (YPD) נוזל 11 בינוני.
- למחרת בבוקר, לחסן 400 מ"ל של מדיום נוזלי YPD עם 8 מ"ל של תרבות לילה רווי דגירה ידי ניעורב 30 מעלות צלזיוס למשך 4 - 5 שעות על מנת לאפשר לתאים להיכנס בשלב לוגריתמים של צמיחה.
- אוספים את התאים על ידי צנטריפוגה ב ~ 3,220 XG במשך 10 דקות, לבטל את במדיום נוזלי, ו resuspend התאים 1 נפח של 10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.1 M פתרון Acetate ליתיום (TE / LiAc) .
- אוספים את התאים על ידי צנטריפוגה ב ~ 3,220 XG במשך 10 דקות, וזורקים את TE / LiAc.
- Resuspend התאים 1 מ"ל TE / LiAc.
- הגדר את קוקטייל התגובה הבאה בצינור microcentrifuge: 800 μl של תאים משלב 4.2.5, 40 μl של ה- DNA זרע 10 מ"ג מבושלים / מ"ל סלמון, סך של 12.5 מיקרוגרם של ה- DNA פלסמיד עמוד השדרה, ו -75 μl של מוצר ה- PCR מרוכז משלב 4.1.3.
הערה: DNA זרע סלמון צריך להיות מבושל במשך 5 דקות והניח על קרח דק לפחות 5 לפני השימוש התגובה. נפח כולל של פלסמיד דנ"א עמוד השדרה הוסיף צריכה להישמר עד למינימום (~ 80 μl או פחות). ראה סעיף נציג תוצאות עבור דוגמא plasm עמוד שדרהתְעוּדַת זֶהוּת. - מערבבים את קוקטייל צינור ביסודיות aliquot שווה לשמונה צינורות microcentrifuge (צינורות 1 - 8).
- הגדר את צינורות תגובת טרנספורמציה השנייה מלא הבאים:
- Tube 9 (אין שליטה המוצר PCR): 100 μl של תאים משלב 4.2.5, 5 μl של ה- DNA זרע מבושלים 10 מ"ג / מ"ל סלמון (מבושל במשך 5 דקות, ראה שלב 4.2.6 הערה), סך של 1.56 מיקרוגרם של פלסמיד דנ"א עמוד השדרה ולא מוצר PCR הוסיף.
- Tube 10 (אין שליטה DNA): 100 μl של תאים משלב 4.2.5, 5 μL של מבושלים 10 מ"ג / מ"ל סלמון זרע DNA (ראה הערה צעד 4.2.6), לא פלסמיד עמוד השדרה DNA הוסיף, ולא מוצר PCR הוסיף.
- מערבבים את שני צינורות בעדינות אך ביסודיות על ידי pipetting מעלה ומטה מספר פעמים.
- דגירת עשרת הצינורות ב 30 צלזיוס למשך 30 דקות.
- כדי צינור אחד, להוסיף 1.2 מ"ל של פוליאתילן גליקול 40% (PEG 3350) ב TE / LiAc. מערבבים היטב באמצעות pipet P-1000 עד הפתרון הוא הומוגני.
- דגירת עשרת הצינורות ב 30° C למשך 30 דקות. לערבב בעדינות את הפתרון על ידי pipetting למעלה ולמטה ואז דגירה צינורות ב 42 מעלות צלזיוס למשך 15 דקות.
- אוספים את התאים על ידי ספינינג צינורות microcentrifuge בבית ~ 14,000 XG במשך 30 שניות. מחק את נוזלי resuspend התאים 1 מ"ל של סטרילי DH 2 O.
- אוספים את התאים על ידי ספינינג צינורות microcentrifuge בבית ~ 14,000 XG במשך 30 שניות. מחק את נוזלי resuspend התאים 500 μl של א '2 סטרילי DH
- בריכת צינורות 1 - 8 יחד (הנפח הכולל של 4 מ"ל) ומערבבים היטב על ידי pipetting למעלה ולמטה.
- פלייט 200 μl של התערובת שלעיל בכל עשרים צלחות בינוניות נשירה מינימאלית מלאות 11 (צלחות 1-20) לבחירה של פלסמיד עמוד השדרה.
- פלייט 200 μl של התערובת מתחנת הרכבת התחתית 9 ו -200 μl של תערובת מתחנת הרכבת התחתית 10 כל בצלחת מבחר משלה (צלחות 21 ו -22, בהתאמה).
- דגירה 22 צלחות על 30 מעלות צלזיוס במשך 3 - 5 ימיםלבחור עבור transformants פלסמיד.
- בדוק צלחות שינוי לאחר 3 - 5 ימי דגירה. כ -5,000 מושבות צריכות להיות גלויות על צלחות 1-21 (ראה נציג תוצאות עבור דוגמא) ולא מושבות צריכות להיות נוכחים על הצלחה 22.
מסך 5. עבור 5-פואה עמיד transformants
- העברת תאים מצלחות 1 - 20 (צלחת טרנספורמציה 21 כביקורת) עד 5-fluoroorotic חומצה (5-פואה) 11 צלחות על ידי העתק-ציפוי 12 כדי להקרין על אובדן של הגן URA3 כתוצאה אינטגרציה של מוצרי ה- PCR במקום הרצוי.
- מסירים את המכסה צלחת ולחץ על צלחת המכילה מושבות על קטיפה סטרילית. מעבירים את התאים מן קטיפה לצלחת 5-פואה ידי לחיצה על הצלחת על קטיפה. דגירה צלחות על 30 צלזיוס למשך 2 ימים.
- לאחר דגירה 2 ימים, בזהירות לבדוק את הצלחות של 5 פואה עבור גרowth.
הערה: אירוע אינטגרצית המועמד יהיה מיוצג על ידי מושבה "פחוסה" אסימטרי קטנה על צלחת 5-פואה - ומנגד, גבשושיות קטנות גדלו על צלחות של 5 פואה המייצגים סבירים של מוטציות URA3 ספונטניות שהתעוררו במהלך הצמיחה של מושבות על צלחות שינוי, ועל כן הם לא סבירות כדי לייצג את אירוע אינטגרציה הרצוי (ראה איור 3 בקטע נציג התוצאות עבור פירוט נוסף על נקודה זו ובמשך כמה דוגמאות).
טיהור 6. 5-פואה עמיד המושבות והפסד של פלסמיד חוט שדרה
- באמצעות קיסמים סטרילי, לאסוף המושבות המועמדות מצלחות 5-פואה כמתוארות בשלב 5.2 פס עבור מושבות אחת על גבי צלחות YPD. דגירה של 2 - 3 ימים ב 30 מעלות צלזיוס.
- לאחר דגירה, העתק - צלחת כל צלחת טיהור YPD לצלחת YPD טרי, צלחת הנשירה חסר אורציל לבדוק הפסדשל גן URA3, וצלחת החוצה ושחרר שנייה כדי לפקח על קיומו או אי קיומו של פלסמיד עמוד השדרה. דגירה של 1 - 2 ימים ב 30 מעלות צלזיוס.
- לאחר דגירה, לזהות מושבה מכל מדגם מועמד אשר גדל על הצלחת YPD אבל לא גדל משני צלחת הנשירה (מושבה כזו צפויה איבדו את הגן URA3 דרך אירוע רקומבינציה ואיבד את הפלסמיד עמוד השדרה במהלך mitotic חלוקת תא). Restreak מושבות כאלה על צלחות YPD טריות. מושבות אלה הם המועמדים אינטגרציה ינותחו עוד בשלב 7.
7. ניתוח מולקולרי כדי Assay עבור שילוב הנכון של אלל מוטנטי
- לבודד הדנ"א הגנומי ממדגמים המועמד באמצעות נהלים סטנדרטיים 10.
- להגביר באזור גנומי המקיף לאתר היעד.
- הגדר את תגובת PCR הבא עבור כל דגימה: 0.5 תבנית ה- DNA μl, 5 μlפריימר 10 מיקרומטר קדימה, 5 μl 10 מיקרומטר פריימר הפוכה, 0.5 μl (2.5 יחידות) פולימראז תקי DNA, 5 μl 10x חיץ פולימראז תקי DNA, 5 μl תערובת dNTP (2 מ"מ כל אחד), ו -29 μl DH 2 O.
הערה: תבנית ה- DNA הוא הדנ"א הגנומי נגזר דגימות מועמד. מומלץ לכלול גם שתי תגובות שליטה: אחד באמצעות הדנ"א הגנומי נגזר geneΔ היסטון המקורי :: זן URA3 כתבנית ועוד הדנ"א הגנומי באמצעות מן זן היסטון wild-type כתבנית. כדי להסביר וריאציות בריכוז DNA ורמת זיהומים בהכנות גנומי שונים, מומלץ לייעל את התגובות או באמצעות DNA חי או דילולים שונים של ההכנות גנומי (למשל, 1:10 ו 1: 100). חשוב לוודא כי לחשל פריימרים אלה כדי רצפי DNA מחוץ לאזור מוקפת המוצר PCR משולב putatively - זו הדרך, בגודל של מוצרי ה- PCR ב אלה Reactions יכול לשמש ככלי אבחוני אינטגרציה של המוצרים במיקום הגנומי הנכון (ראה נציג תוצאות עבור דוגמא). - מניחים את התגובות ב thermocycler עם ההגדרות הבאות: 94 ° C 3 דקות; 30 מחזורים של ההגדרות הבאות: 94 ° C 45 שניות, 50 ° C 45 שניות, 72 ° C 2 דקות; ו -72 מעלות צלזיוס 10 דקות.
הערה: אופטימיזציה של פרמטרי PCR עשויה להידרש סטים פריימר ספציפיים היסטון יעד גן.
- הגדר את תגובת PCR הבא עבור כל דגימה: 0.5 תבנית ה- DNA μl, 5 μlפריימר 10 מיקרומטר קדימה, 5 μl 10 מיקרומטר פריימר הפוכה, 0.5 μl (2.5 יחידות) פולימראז תקי DNA, 5 μl 10x חיץ פולימראז תקי DNA, 5 μl תערובת dNTP (2 מ"מ כל אחד), ו -29 μl DH 2 O.
- עיבוד של מוצרי ה- PCR
- הפעל 20 μl מכל תגובה על ג'ל agarose 0.8% TBE.
- להעריך את גודל מוצרי PCR באמצעות סטנדרטי DNA כהתייחסות לקבוע אם גן URA3 הוחלף בהצלחה על ידי גן היסטון מוטצית putatively (ראה נציג תוצאות עבור דוגמא).
הערה: במקרים מסוימים, את המוטציה הרצויה (ים) מוחדרת גני היסטון ליצור או להרוס הגבלה לשבתדואר. אם זה מקרה, הנוכחות של המוטציה הרצויה מוצרי PCR של הגודל מעיד על אינטגרציה נכונה ניתן להעריך על ידי העמדת מוצרי עיכול עם אנזים ההגבלה המקביל ואחריו ניתוח ג'ל אלקטרופורזה (ראה נציג תוצאות עבור דוגמא) . - מוצרי ה- PCR נושא הגודל מעיד על שילוב נכון רצפי DNA כדי לאשר את קיומו של המוטציה הרצויה (ים) ו, כדי לוודא ששום מוטציות נוספות הוכנסו לתוך הגנום.
תוצאות
אנו מתארים את הדור של אלל hht2 לבטא חלבון מוטנטי H3 היסטון מחסה חילוף במיקום 53 מתוך ארגינין חומצת גלוטמית (H3-R53E מוטציה) בתור דוגמה מייצגת של ממוקדת באסטרטגיה mutagenesis באתרו.
יצרנו זן שב...
Discussion
הרמה הגבוהה של הומולוגיה ברצף בין שני גנים שאינם אללים שקוד לכל אחד החלבונים היסטון ארבע ליבות בתאים הפלואידים cerevisiae ס יכול לייצג אתגר עבור חוקרים המבקשים למקד ספציפית אחת משני הגנים עבור mutagenesis. שתואר לעיל מתודולוגיות mutagenesis שמרים, כולל Perfetto Delitto, אתר ספציפ...
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
We thank Reine Protacio for helpful comments during the preparation of this manuscript. We express our gratitude to the National Science Foundation (grants nos. 1243680 and 1613754) and the Hendrix College Odyssey Program for funding support.
Materials
| Name | Company | Catalog Number | Comments |
| 1 kb DNA Ladder (DNA standards) | New England BioLabs | N3232L | |
| Agarose | Sigma | A5093-100G | |
| Boric Acid | Sigma | B0394-500G | |
| dNTP mix (10 mM each) | ThermoFisher Scientific | R0192 | |
| EDTA solution (0.5 M, pH 8.0) | AmericanBio | AB00502-01000 | |
| Ethanol (200 Proof) | Fisher Scientific | 16-100-824 | |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma | E4884-500G | |
| Lithium acetate dihydrate | Sigma | L6883-250G | |
| MyCycler Thermal Cycler | BioRad | 170-9703 | |
| Poly(ethylene glycol) (PEG) | Sigma | P3640-1KG | |
| PrimeSTAR HS DNA Polymerase (high fidelity DNA polymerase) and 5x buffer | Fisher Scientific | 50-443-960 | |
| Salmon sperm DNA solution | ThermoFisher Scientific | 15632-011 | |
| Sigma 7-9 (Tris base, powder form) | Sigma | T1378-1KG | |
| Sodium acetate trihydrate | Sigma | 236500-500G | |
| Supra Sieve GPG Agarose (low metling temperature agarose) | AmericanBio | AB00985-00100 | |
| Taq Polymerase and 10x Buffer | New England BioLabs | M0273X | |
| Toothpicks | Fisher Scientific | S67859 | |
| Tris-HCl (1 M, pH 8.0) | AmericanBio | AB14043-01000 | |
| a-D(+)-Glucose | Fisher Scientific | AC170080025 | for yeast media |
| Agar | Fisher Scientific | DF0140-01-0 | for yeast media |
| Peptone | Fisher Scientific | DF0118-07-2 | for YPD medium |
| Yeast Extract | Fisher Scientific | DF0127-17-9 | for YPD medium |
| 4-aminobenzoic acid | Sigma | A9878-100G | for complete minimal dropout medium |
| Adenine | Sigma | A8626-100G | for complete minimal dropout medium |
| Glycine hydrochloride | Sigma | G2879-100G | for complete minimal dropout medium |
| L-Alanine | Sigma | A7627-100G | for complete minimal dropout medium |
| L-Arginine monohydrochloride | Sigma | A5131-100G | for complete minimal dropout medium |
| L-Asparagine monohydrate | Sigma | A8381-100G | for complete minimal dropout medium |
| L-Aspartic acid sodium salt monohydrate | Sigma | A6683-100G | for complete minimal dropout medium |
| L-Cysteine hydrochloride monohydrate | Sigma | C7880-100G | for complete minimal dropout medium |
| L-Glutamic acid hydrochloride | Sigma | G2128-100G | for complete minimal dropout medium |
| L-Glutamine | Sigma | G3126-100G | for complete minimal dropout medium |
| L-Histidine monohydrochloride monohydrate | Sigma | H8125-100G | for complete minimal dropout medium |
| L-Isoleucine | Sigma | I2752-100G | for complete minimal dropout medium |
| L-Leucine | Sigma | L8000-100G | for complete minimal dropout medium |
| L-Lysine monohydrochloride | Sigma | L5626-100G | for complete minimal dropout medium |
| L-Methionine | Sigma | M9625-100G | for complete minimal dropout medium |
| L-Phenylalanine | Sigma | P2126-100G | for complete minimal dropout medium |
| L-Proline | Sigma | P0380-100G | for complete minimal dropout medium |
| L-Serine | Sigma | S4500-100G | for complete minimal dropout medium |
| L-Threonine | Sigma | T8625-100G | for complete minimal dropout medium |
| L-Tryptophan | Sigma | T0254-100G | for complete minimal dropout medium |
| L-Tyrosine | Sigma | T3754-100G | for complete minimal dropout medium |
| L-Valine | Sigma | V0500-100G | for complete minimal dropout medium |
| myo-Inositol | Sigma | I5125-100G | for complete minimal dropout medium |
| Uracil | Sigma | U0750-100G | for complete minimal dropout medium |
| Ammonium Sulfate | Fisher Scientific | A702-500 | for complete minimal dropout medium |
| Yeast Nitrogen Base | Fisher Scientific | DF0919-07-3 | for complete minimal dropout medium |
| 5-Fluoroorotic acid (5-FOA) | AmericanBio | AB04067-00005 | for 5-FOA medium |
References
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389 (6648), 251-260 (1997).
- Campos, E. I., Reinberg, D. Histones: annotating chromatin. Annu Rev Genet. 43, 559-599 (2009).
- Rando, O. J., Winston, F. Chromatin and transcription in yeast. Genetics. 190 (2), 351-387 (2012).
- Duina, A. A., Miller, M. E., Keeney, J. B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 197 (1), 33-48 (2014).
- Storici, F., Resnick, M. A. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y). 25, 189-207 (2003).
- Gray, M., Kupiec, M., Honigberg, S. M. Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7 (2004).
- Erdeniz, N., Mortensen, U. H., Rothstein, R. Cloning-free PCR-based allele replacement methods. Genome Res. 7 (12), 1174-1183 (1997).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
- Lundblad, V., Hartzog, G., Moqtaderi, Z. Manipulation of cloned yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Hoffman, C. S. Preparation of yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Treco, D. A., Lundblad, V. Preparation of yeast media. Curr Protoc Mol Biol. Chapter 13, (2001).
- Lederberg, J., Lederberg, E. M. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 63 (3), 399-406 (1952).
- Sikorski, R. S., Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122 (1), 19-27 (1989).
- Johnson, P., et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 5 (5), 741-749 (2015).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336-4343 (2013).
- Cross, S. L., Smith, M. M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol. 8 (2), 945-954 (1988).
- Libuda, D. E., Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 443 (7114), 1003-1007 (2006).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved