
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Targeted
Dans cet article
Résumé
A strategy for generating mutations in histone genes at their endogenous location in Saccharomyces cerevisiae is presented.
Résumé
We describe a PCR- and homologous recombination-based system for generating targeted mutations in histone genes in budding yeast cells. The resulting mutant alleles reside at their endogenous genomic sites and no exogenous DNA sequences are left in the genome following the procedure. Since in haploid yeast cells each of the four core histone proteins is encoded by two non-allelic genes with highly homologous open reading frames (ORFs), targeting mutagenesis specifically to one of two genes encoding a particular histone protein can be problematic. The strategy we describe here bypasses this problem by utilizing sequences outside, rather than within, the ORF of the target genes for the homologous recombination step. Another feature of this system is that the regions of DNA driving the homologous recombination steps can be made to be very extensive, thus increasing the likelihood of successful integration events. These features make this strategy particularly well-suited for histone gene mutagenesis, but can also be adapted for mutagenesis of other genes in the yeast genome.
Introduction
Les quatre protéines histones fondamentales H2A, H2B, H3, H4 et jouent un rôle central dans le compactage, l'organisation, et la fonction des chromosomes eucaryotes. Deux ensembles de chacun de ces histones forment l'octamère d'histone, une bobine moléculaire qui dirige l'emballage de ~ 147 paires de bases d'ADN autour de lui - même, entraînant finalement la formation d'un nucléosome 1. Les nucléosomes sont des participants actifs dans une variété de processus basés sur chromosomiques, telles que la régulation de la transcription du gène et la formation de l'euchromatine et l'hétérochromatine dans les chromosomes, et en tant que tels ont fait l'objet d'intenses recherches au cours des dernières décennies. Un certain nombre de mécanismes ont été décrits par lequel nucléosomes peuvent être manipulées d'une manière qui peut faciliter l'exécution des processus spécifiques - ces mécanismes comprennent la modification post-traductionnelle de résidus d'histones, dépendant de l'ATP nucléosome remodelage, et la réorganisation de nucléosome ATP-indépendanteet montage / démontage 2, 3.
La levure bourgeonnante Saccharomyces cerevisiae est un organisme modèle particulièrement puissant pour la compréhension de la fonction histone dans les eucaryotes. Cela peut être attribué en grande partie au degré élevé de conservation évolutive des protéines histones dans tout le eucaryotes de domaine et la susceptibilité de la levure à une variété de expérimentale génétiques et biochimiques approches 4. approches Reverse-génétiques dans la levure ont été largement utilisés pour étudier les effets des mutations histones spécifiques sur divers aspects de la biologie de la chromatine. Pour ces types d'expériences, il est souvent préférable d'utiliser des cellules dans lesquelles les histones mutantes sont exprimées à partir de leur locus génomique natif, comme expression de plasmides autonomes peuvent conduire à des niveaux anormaux intracellulaires de protéines histones (en raison du nombre de plasmides différents dans les cellules) et la modification concomitante de la chromatine environnements, qui peut finalement confondre l'interprétation des résultats.
Ici, nous décrivons une technique basée sur la PCR qui permet de mutagénèse ciblée des gènes d'histones à leurs emplacements génomiques natifs qui ne nécessite pas d'étape et les résultats du clonage dans la génération de la mutation (s) désirée sans restes de séquences d'ADN exogènes dans le génome. Cette technique profite du système de recombinaison homologue efficace dans la levure et a plusieurs caractéristiques en commun avec d' autres techniques similaires développées par d' autres groupes - notamment le Delitto Perfetto, génomique (SSG) mutagenèse spécifique du site, et allèle basée sur la PCR-clonage gratuit méthodes de substitution 5, 6, 7. Cependant, la technique nous décrivons a un aspect qui le rend particulièrement bien adapté pour la mutagenèse des gènes d'histones. Dans les cellules de levure haploïdes, chacun des quatre histones est codé par deux non unllelic gènes et hautement homologues , par exemple, l' histone H3 sont codées par les gènes et HHT1 HHT2, ainsi que les cadres de lecture ouverts (ORF) des deux gènes sont plus de 90% identique dans la séquence. Ce degré élevé d'homologie peut compliquer les expériences conçues pour cibler précisément l'un des deux gènes d'histones codant pour la mutagenèse. Alors que les procédés mentionnés ci-dessus nécessitent souvent l'utilisation d'au moins certaines séquences dans l'ORF du gène cible pour conduire une recombinaison homologue, la technique que nous décrivons ici fait appel à des séquences flanquant les ORFs des gènes d'histones (qui partagent beaucoup moins d'homologie de séquence) pour l'étape de recombinaison, augmentant ainsi la probabilité de ciblage réussi de mutagénèse sur le lieu souhaité. En outre, les régions homologues qui animent la recombinaison peut être très vaste, ce qui contribue à l'efficacité de recombinaison homologue ciblée.
Protocole
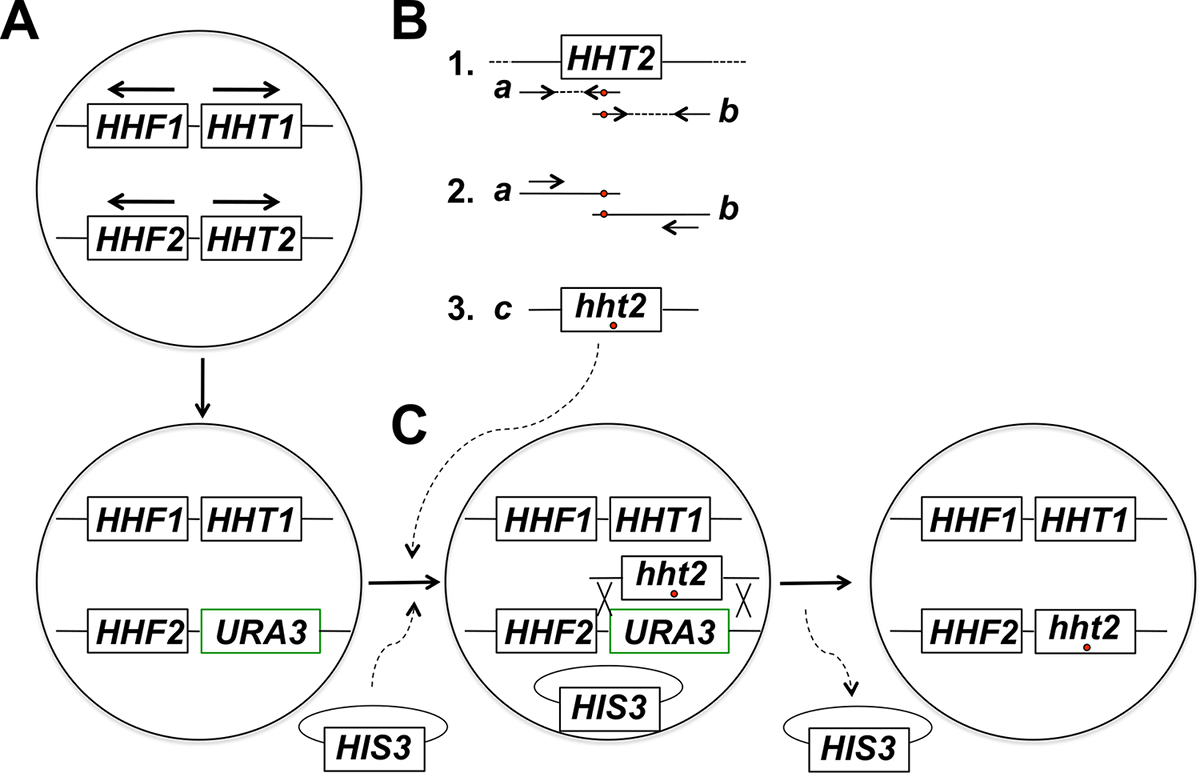
REMARQUE: La stratégie expérimentale pour ciblée in situ mutagenèse du gène d'histone comprend plusieurs étapes (résumées dans la figure 1). Ces étapes sont les suivantes: (1) Le remplacement du gène cible histone avec le gène URA3, (2) Production et purification des produits de PCR correspondant à deux fragments se chevauchant partiellement du gène d'histone cible en utilisant des amorces hébergeant la mutation désirée (s), (3 ) Fusion PCR des deux fragments se chevauchant partiellement pour obtenir des produits complets de PCR de taille pour l'intégration, (4) Co-transformation des produits de PCR pleine grandeur et la colonne vertébrale plasmide, et la sélection pour le marqueur sur le plasmide, (5) écran pour 5-FOA résistant les transformants (6) la purification de colonies 5 résistantes au-FOA et la perte de plasmide squelette, et (7) des analyses moléculaires de dosage pour une bonne intégration de l'allèle mutant.

Figure 1: Vue d' ensemble de la Stratégie pour ciblée in situ mutagenèse des gènes des histones dans la levure bourgeonnante. Dans cet exemple , le gène ciblé est HHT2, mais tout autre gène d'histone noyau peut également être soumis à une mutagenèse en utilisant cette stratégie. (A) Les cellules de levure haploïdes abritent deux gènes d'histone H3 codant (HHT1 et HHT2) et deux gènes H4 codant les histones (HHF1 et HHF2) disposés comme indiqué sur la figure (les gènes HHT1 et HHF1 sont situés sur le chromosome II et le HHT2 et les gènes HHF2 sont situés sur le chromosome XIV - dans chaque cas, les flèches pointent dans le sens de la transcription). Dans la première étape de la procédure, l'ORF du gène HHT2 est remplacé par le gène URA3 donnant lieu à une déformation hht2Δ :: URA3. (B) Dans la partie 1, une copie de type sauvage du gène HHT2 à partir d' un échantillon d'ADN génomique est utilisé comme matrice pour deux réactions de PCR à des genresTé les deux fragments se chevauchant partiellement du gène. L'amorce inverse pour la première réaction comprend un ou plusieurs nucléotides mésappariés (indiqués par un cercle rouge) qui correspondent à la mutation désirée (s) à introduire dans le génome. L'amorce sens pour la seconde réaction a le décalage équivalent dans une configuration complémentaire inverse (également indiqué par un cercle rouge). Les deux produits de PCR générés dans la partie 1 (produits a et b) sont ensuite utilisés comme modèles pour la fusion par PCR en utilisant deux amorces qui hybrident aux produits a et b de la manière indiquée dans la partie 2. Il en résulte la génération de pleine grandeur PCR produits (produits c dans la partie 3) abritant la mutation (s) souhaitée. (C) Le hht2Δ :: souche URA3 est alors co-transformées avec les produits de PCR pleine grandeur et un plasmide de squelette (un HIS3- marqué plasmide dans cet exemple), et les cellules sont sélectionnées pour la présence du plasmide (sur des milieux dépourvus histidine dans cet exemple). Les transformants sont ensuite criblés pour la résistance 5-FOA - cellules résistantes sont des candidats pour avoir subi un événement de recombinaison homologue conduisant à l' intégration du produit de PCR et l' excision du gène URA3, comme illustré. Après la perte du plasmide squelette par division cellulaire mitotique conduit à l'histone désiré souche mutante finale. Nous avons constaté que la sélection du plasmide de squelette suivi d' un tamisage pour obtenir des résultats de résistance à 5-FOA à une fréquence beaucoup plus élevée de l' identification des événements d'intégration correcte par rapport à la sélection directe sur des plaques de 5-FOA, qui identifie la plupart des cellules qui ont acquis des mutations URA3 spontanées. (Ce chiffre a été modifié de la référence 14). S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
1. Remplacement de la cible Histone Gene avec le URA3Gène
- Effectuer une PCR à médiation par une étape gène perturbation norme remplaçant l'ORF du gène d'histone cible avec le gène URA3 8, 9.
NOTE: L'utilisation de cellules de levure portant le ura3Δ0 est recommandé que cette mutation supprime la totalité de l' ORF URA3 endogène, ce qui évite ainsi l' intégration du produit de PCR dans le locus URA3 8. En variante, le gène URA3 K. lactis peut être utilisé efficacement pour la génération du remplacement des histones dans tous les milieux ura3 comme il est fonctionnel chez S.cerevisiae , mais a seulement une homologie de séquence partielle avec le gène URA3 de S. cerevisiae. La souche devrait également être auxotrophe pour au moins un composé qui permettra la sélection du plasmide de squelette dans l'expérience de la transformation (voir l'étape 4 de ce protocole). Cette étape est pas nécessaire si une cible histones geneΔ :: URA3souche est déjà disponible.
2. Production et purification des produits de PCR correspondant à deux fragments se chevauchant partiellement de la cible Histone Gene utilisant Amorces porteuse de la mutation souhaitée (s)
- Générer des produits de PCR correspondant à deux fragments se chevauchant partiellement du gène cible histone.
- Préparer deux réactions PCR comme suit:
- Pour générer des produits de PCR correspondant à la première moitié du gène (produit une figure 1B), mis en place la réaction suivante: 1 ul d' ADN matrice, 5 μl10 pM amorce avant, 5 μl10 pM amorce inverse, 0,5 pi (1,25 U) thermostable ADN polymérase, 10 ul de tampon de polymerase d' ADN 5x, 5 ul de mélange dNTP (2 mM chacun) et 23,5 ul dH 2 O.
NOTE: L'ADN matrice peut être de l'ADN génomique provenant d'une souche de type sauvage pour le gène cible de histone isolé en utilisant la norme pro10 procédures. Pour tenir compte des variations de la concentration d'ADN et le niveau d'impuretés présentes dans différentes préparations génomiques, il est recommandé d'optimiser les réactions en utilisant soit de l' ADN non dilué ou différentes dilutions des préparations génomiques (par exemple, 1:10 et 1: 100). L'amorce sens devrait recuire à une région en amont du gène cible. L'amorce inverse devrait recuire au sein de l'ORF, soit ~ 40 nucléotides de longueur, et contient la mutation (s) souhaitée quelque part au milieu de celui - ci (voir figure 1B-1 et résultats représentatifs section pour des exemples). L'utilisation d'un ADN polymérase haute fidélité est recommandée afin de réduire les taux de mutations indésirables lors de la synthèse des produits de PCR. - Pour générer des produits de PCR correspondant à la seconde moitié du gène (produit b dans la figure 1B), mis en place une réaction comme indiqué dans 2.1.1.1 mais avec des amorces.
NOTE: Le pri avantmer devrait recuire au sein de l'ORF, soit ~ 40 nucléotides de longueur, et contient la mutation (s) souhaitée quelque part au milieu de celui-ci. A noter que la mutation (s) dans cette amorce est le complément inverse de la mutation (s) dans l'amorce inverse de l'étape 2.1.1.1. L'amorce inverse devrait recuire à une région en aval du gène cible (voir la section 1B-1 et résultats représentatifs Figure pour des exemples).
- Pour générer des produits de PCR correspondant à la première moitié du gène (produit une figure 1B), mis en place la réaction suivante: 1 ul d' ADN matrice, 5 μl10 pM amorce avant, 5 μl10 pM amorce inverse, 0,5 pi (1,25 U) thermostable ADN polymérase, 10 ul de tampon de polymerase d' ADN 5x, 5 ul de mélange dNTP (2 mM chacun) et 23,5 ul dH 2 O.
- Placez les réactions dans un thermocycleur avec les paramètres suivants: 94 ° C 30 sec; 30 cycles des paramètres suivants: 98 ˚C 10 s, 60 ° C 5 sec, 72 ° C 1,5 min; et 72 ° C 10 min.
Remarque: L'optimisation des paramètres de la PCR peut être nécessaire pour des jeux d'amorces spécifiques et de cibler le gène d'histone.
- Préparer deux réactions PCR comme suit:
- Exécuter 20 - 50 ul de la matière provenant des réactions PCR sur un gel à 0,9% d'agarose à bas point de fusion dans 89 mM de base Tris, de l'acide borique 89 mM, EDTA (TBE) 2,5.
- sections de gel d'agarose Cut contenant le pr PCRoduits de gel à l'aide d'un scalpel ou une lame de rasoir propre et transfèrent chacun à un tube de 1,5 ml. Stockez sections d'agarose contenant des produits de PCR à -20 ° C jusqu'à utilisation.
3. Fusion PCR des deux fragments se chevauchant partiellement pour obtenir la pleine taille des produits de PCR pour l'intégration
- Préparer modèle pour les réactions PCR
- Faire fondre les sections de gel d'agarose de l'étape 2.3 en plaçant les tubes à centrifuger dans un ensemble de blocs de chaleur à 65 ° C pendant 5 minutes (ou jusqu'à ce que complètement fondu). Tubes vortex toutes les 1 - 2 minutes pour faciliter le processus de fusion.
- Transférer un montant fixe de fondu agarose de chaque échantillon (par exemple, 50 ul chacune, pour un total de 100 pi) dans un tube à centrifuger unique et mélanger par tourbillonnement. En utilisant ce comme matrice dans les réactions PCR de fusion. Placer le tube à -20 ° C jusqu'à utilisation.
- Amplifier une grande quantité de produit de PCR en taille réelle (produit cla figure 1B)
- Mise en place de six réactions de PCR, chacun avec les composants suivants: ADN matrice 2 pi, 10 pi de 10 pM de l'amorce sens, 10 ul 10 uM d'amorce inverse, 1 pi (2,5 U) thermostable d'ADN polymerase, 20 de tampon d'ADN polymerase ul 5x, 10 ul mélange de dNTP (2 mM chacun) et 47 ul dH 2 O.
NOTE: Le nombre de réactions peut être modifiée en fonction de l'efficacité de la PCR. L'ADN matrice (voir 3.1.2) doit être chauffé à 65 ° C jusqu'à ce que fondu, mélangé par tourbillonnement, et ajouté à la dernière mélange de PCR. Une fois ajouté, mélanger délicatement mais soigneusement par pipetage la solution de haut en bas plusieurs fois. Pour tenir compte des variations de la concentration d'ADN dans les différents échantillons, il est recommandé d'optimiser d' abord les réactions à l'aide de modèles non dilué ou différentes dilutions du modèle (par exemple, 1:10 et 1: 100). Les deux amorces utilisées doivent hybrider les deux fragments se chevauchant partiellement du gène cible maladeustrated à la figure 1B-2 et être conçu de telle sorte que les produits finaux de PCR auront au moins 40 paires de bases sur les deux homologues de côté pour les régions flanquant le URA3 ORF qui stimuleront l'étape de recombinaison homologue (voir la section Les résultats représentatifs pour des exemples). L'utilisation d'un ADN polymérase haute fidélité est recommandée afin de réduire les taux de mutations indésirables lors de la synthèse des produits de PCR. - Placer les tubes dans un thermocycleur avec les paramètres suivants: 94 ° C 30 sec; 30 cycles des paramètres suivants: 98 ˚C 10 s, 50 ° C 15 sec, 72 ° C 1,5 min; et 72 ° C 10 min.
REMARQUE: L'optimisation des paramètres de la PCR peut être nécessaire pour des jeux d'amorces spécifiques et le gène cible de l'histone.
- Mise en place de six réactions de PCR, chacun avec les composants suivants: ADN matrice 2 pi, 10 pi de 10 pM de l'amorce sens, 10 ul 10 uM d'amorce inverse, 1 pi (2,5 U) thermostable d'ADN polymerase, 20 de tampon d'ADN polymerase ul 5x, 10 ul mélange de dNTP (2 mM chacun) et 47 ul dH 2 O.
4. Co-transformation de produits Full Size PCR et Backbone plasmide, et la sélection pour le marqueur sur le plasmide
- La concentration des produits de PCR
- Réunissez le six réactions PCR (600 au total pi) de l'étape 3.2.2 dans un tube à centrifuger unique et mélanger par tourbillonnement.
- Diviser l'échantillon en trois 200 aliquotes dans des tubes de microcentrifugation. Précipiter l'ADN dans chaque tube par addition de 20 ul d'acétate de sodium 3M (pH 5,2) et 550 ul d'éthanol à 100%. Mélanger soigneusement la solution et placer sur la glace pendant au moins 15 min. Recueillir l'ADN par centrifugation à ~ 14.000 xg pendant 10 minutes, rincer le culot avec 200 pi de 70% d'éthanol, et l'air sec.
- Resuspendre chaque pastille d'ADN dans 25 pi de dH 2 O, et piscine dans un seul tube (pour un total de 75 pi).
- La levure co-transformation
- Préparer 10 ml de culture d'une nuit de la souche générée à l' article 1 dans l' extrait de levure peptone dextrose (YPD) milieu liquide 11.
- Le lendemain matin, inoculer 400 ml de milieu liquide YPD avec 8 ml de la culture d'une nuit saturée et incuber en agitantà 30 ° C pendant 4 - 5 heures pour permettre aux cellules d'entrer en phase de croissance logarithmique.
- Recueillir les cellules par centrifugation à ~ 3220 x g pendant 10 minutes, éliminer le milieu liquide, et remettre en suspension les cellules dans 1 volume de 10 mM de Tris-HCl (pH 8,0), 1 mM d'EDTA, 0,1 M de solution acétate de lithium (TE / LiAc) .
- Recueillir les cellules par centrifugation à ~ 3220 xg pendant 10 min, et jeter le TE / LiAc.
- Resuspendre les cellules dans 1 ml de TE / LiAc.
- Mettre en place le cocktail de réaction suivant dans un microtube de 800 pi de cellules provenant de l'étape 4.2.5, 40 ul d'ADN de sperme / saumon ml 10 mg bouillie, soit un total de 12,5 ug d'ADN de plasmide de squelette, et 75 pi de produit de PCR concentré de l'étape 4.1.3.
NOTE: L'ADN de sperme de saumon doit être porté à ébullition pendant 5 minutes et placée sur de la glace pendant au moins 5 minutes avant de l'utiliser dans la réaction. Le volume total de l'ADN ajouté squelette du plasmide doit être maintenu à un minimum (~ 80 pi ou moins). Voir la section Résultats représentant pour un exemple de plasme épine dorsaleid. - Mélanger le tube de cocktail à fond et aliquote uniformément dans huit tubes à centrifuger (Tubes 1-8).
- Mettre en place les deux tubes de réaction de transformation de contrôle suivants:
- Le tube 9 (pas de commande du produit de PCR): 100 ul de cellules de l'étape 4.2.5, 5 pi de bouillie d'ADN / ml de sperme de saumon 10 mg (bouillie pendant 5 min; voir l'étape 4.2.6 Note), un total de 1,56 pg squelette d'ADN plasmidique et aucun produit de PCR ajouté.
- Tube 10 (pas de contrôle de l'ADN): 100 pi de cellules de l'étape 4.2.5, 5 pi de bouillie 10 mg / ml d'ADN de sperme de saumon (voir étape 4.2.6 Note), pas de colonne vertébrale ADN plasmidique ajouté, et aucun produit PCR ajouté.
- Mélanger les deux tubes doucement mais complètement par pipetage de haut en bas plusieurs fois.
- Incuber les dix tubes à 30 ° C pendant 30 min.
- A chaque tube, ajouter 1,2 ml de 40% de polyéthylène glycol (PEG) 3350 dans TE / LiAc. Bien mélanger à l'aide d'une pipette P-1000 jusqu'à ce que la solution soit homogène.
- Incuber les dix tubes à 30° C pendant 30 min. Mélanger doucement la solution par pipetage de haut en bas, puis incuber les tubes à 42 ° C pendant 15 min.
- Recueillir les cellules en faisant tourner les tubes dans une microcentrifugeuse à ~ 14.000 xg pendant 30 sec. Jeter le liquide et de remettre les cellules dans 1 ml de dH 2 O stérile
- Recueillir les cellules en faisant tourner les tubes dans une microcentrifugeuse à ~ 14.000 xg pendant 30 sec. Jeter le liquide et remettre les cellules dans 500 pi de dH stérile 2 O.
- tubes Piscine 1-8 ensemble (volume total de 4 ml) et bien mélanger par pipetage de haut en bas.
- Plaque 200 ul du mélange ci - dessus sur chacun des vingt complets abandon milieu minimal plaques 11 (plaques 1-20) pour la sélection du plasmide de squelette.
- Plaque 200 pi du mélange de tube 9 et 200 pi de mélange de Tube 10 chacun sur sa propre plaque de sélection (plaques 21 et 22, respectivement).
- Incuber les 22 plaques à 30 ° C pendant 3 - 5 jours àsélectionner les transformants de plasmide.
- Inspecter les plaques de transformation après 3 - 5 jours d'incubation. Environ 5000 colonies doivent être visibles sur des plaques 1-21 (voir résultats représentatifs pour un exemple) et pas de colonies doivent être présents sur la plaque 22.
5. Ecran pour transformants 5-FOA-résistantes
- Les cellules de transfert à partir de plaques 1 - 20 (et la plaque de transformation 21 en tant que témoin) à 5-fluoroorotique d' acide (5-FOA) des plaques 11 en réplique de placage 12 pour cribler pour la perte du gène URA3 à la suite de l' intégration de la les produits de PCR à l'emplacement souhaité.
- Retirez le couvercle de la plaque et appuyez sur la plaque contenant des colonies sur un velours stérile. Transférer les cellules du velours sur une plaque de 5-FOA en appuyant sur la plaque sur le velours. Incuber les plaques à 30 ° C pendant 2 jours.
- Après l'incubation de 2 jours, inspecter soigneusement les plaques 5-FOA pour growth.
NOTE: Un événement d'intégration du candidat sera représentée par une petite asymétrique colonie "écrasé" sur une plaque 5-FOA - à l' inverse, de petites papilles de plus en plus sur des plaques 5-FOA sont susceptibles représentant des mutations URA3 spontanées qui se pose lors de la croissance des colonies sur la plaques de transformation, et sont donc peu susceptibles de représenter l'événement d'intégration souhaité (voir la figure 3 dans la section des résultats représentatifs pour plus de précisions sur ce point et pour quelques exemples).
6. Purification des colonies 5-FOA-résistantes et perte de Backbone plasmide
- En utilisant des cure-dents stériles, ramasser les colonies candidats à partir des plaques 5-FOA décrites à l'étape 5.2 et série pour des colonies isolées sur des plaques de YPD. Incuber pendant 2 - 3 jours à 30 ° C.
- Après l'incubation, réplique - plaque chaque purification de plaque YPD à une plaque YPD frais, une plaque d' abandon sans uracile pour vérifier la pertedu gène URA3, et une seconde plaque d' abandon pour surveiller la présence ou l' absence du plasmide squelette. Incuber pendant 1 - 2 jours à 30 ° C.
- Après l'incubation, identifier une colonie de chaque échantillon de candidat qui se développe sur la plaque YPD , mais pas de plus en plus sur chaque plaque d' abandon (une telle colonie devrait avoir perdu le gène URA3 par l'événement de recombinaison et a perdu le plasmide squelette pendant mitotique la division cellulaire). Restreak ces colonies sur des boîtes de YPD fraîches. Ces colonies sont les candidats d'intégration et seront analysées plus loin dans l'étape 7.
7. Analyses moléculaires à Assay pour une bonne intégration de l'allèle mutant
- Isoler l' ADN génomique à partir d' échantillons candidats en utilisant des modes opératoires classiques 10.
- Amplifier une région génomique englobant le site cible.
- Mettre en place la réaction PCR suivante pour chaque échantillon: 0,5 ADN matrice pi, 5 pi10 pM d'amorce sens, 5 ul 10 uM d' amorce inverse, 0,5 pi (2,5 unités) d' ADN Taq polymerase, 5 pi de 10 x tampon de Taq ADN polymerase, un mélange de dNTP 5 ul (2 mM chacun) et 29 ul dH 2 O.
REMARQUE: ADN matrice est dérivée de l'ADN génomique à partir des échantillons candidats. Il est recommandé d'inclure également deux réactions de contrôle: l' une en utilisant l' ADN génomique provenant des geneΔ histones originales :: souche URA3 comme matrice et un autre ADN génomique en utilisant d'une souche histone de type sauvage comme matrice. Pour tenir compte des variations de la concentration d'ADN et le niveau d'impuretés présentes dans différentes préparations génomiques, il est recommandé d'optimiser les réactions en utilisant soit de l' ADN non dilué ou différentes dilutions des préparations génomiques (par exemple, 1:10 et 1: 100). Il est important de veiller à ce que ces amorces hybrident aux séquences d'ADN en dehors de la région englobée par le produit de PCR putative intégré - de cette façon, la taille des produits de PCR dans ces reactions peuvent être utilisés comme un outil de diagnostic pour l' intégration des produits à l'emplacement génomique correcte (voir résultats représentatifs pour un exemple). - Placer les réactions dans un thermocycleur avec les paramètres suivants: 94 ° C 3 min; 30 cycles des paramètres suivants: 94 ° C 45 sec, 50 ° C 45 sec, 72 ° C 2 min; et 72 ° C 10 min.
REMARQUE: L'optimisation des paramètres de la PCR peut être nécessaire pour des jeux d'amorces spécifiques et le gène cible de l'histone.
- Mettre en place la réaction PCR suivante pour chaque échantillon: 0,5 ADN matrice pi, 5 pi10 pM d'amorce sens, 5 ul 10 uM d' amorce inverse, 0,5 pi (2,5 unités) d' ADN Taq polymerase, 5 pi de 10 x tampon de Taq ADN polymerase, un mélange de dNTP 5 ul (2 mM chacun) et 29 ul dH 2 O.
- Traitement des produits de PCR
- Exécuter 20 ul de chaque réaction sur un gel d'agarose 0,8% TBE.
- Évaluer la taille des produits de PCR en utilisant des étalons d'ADN de référence pour déterminer si le gène URA3 a été remplacé avec succès par le gène muté histone putative (voir les résultats représentatifs pour un exemple).
NOTE: Dans certains cas, la mutation (s) souhaitée introduite dans les gènes des histones créer ou détruire une restriction assise. Si tel est le cas, la présence de la mutation souhaitée dans les produits de PCR de la taille indicative d'intégration correcte peut être évaluée en soumettant les produits à une digestion par l'enzyme de restriction correspondante, suivie par une analyse par électrophorèse sur gel (voir les résultats représentatifs pour un exemple) . - Les produits de PCR de la taille de l'objet indicative d'une bonne intégration à un séquençage de l'ADN pour confirmer la présence de la mutation (s) désirée et pour veiller à ce qu'aucune des mutations supplémentaires ont été introduites dans le génome.
Résultats
Nous décrivons la production d'un allèle HHT2 exprimant une protéine mutante de l' histone H3 hébergeant une substitution à la position 53 d'une arginine en un acide glutamique (R53E mutant H3) comme un exemple représentatif de la stratégie de mutagenèse ciblée in situ.
Nous avons généré une souche dans laquelle l'ensemble de l' ORF de HHT2 est remplacé par le gène <...
Discussion
Le niveau élevé d'homologie de séquence entre les deux gènes non-alléliques codant pour chacune des quatre protéines histones fondamentales dans les cellules de S. cerevisiae haploïdes peut représenter un défi pour les chercheurs qui souhaitent cibler spécifiquement l' un des deux gènes pour la mutagenèse. Décrit précédemment levure méthodes de mutagenèse, y compris la Delitto Perfetto, génomique (SSG) mutagenèse spécifique du site, et les méthodes de remplacement de l'...
Déclarations de divulgation
The authors declare that they have no competing financial interests.
Remerciements
We thank Reine Protacio for helpful comments during the preparation of this manuscript. We express our gratitude to the National Science Foundation (grants nos. 1243680 and 1613754) and the Hendrix College Odyssey Program for funding support.
matériels
| Name | Company | Catalog Number | Comments |
| 1 kb DNA Ladder (DNA standards) | New England BioLabs | N3232L | |
| Agarose | Sigma | A5093-100G | |
| Boric Acid | Sigma | B0394-500G | |
| dNTP mix (10 mM each) | ThermoFisher Scientific | R0192 | |
| EDTA solution (0.5 M, pH 8.0) | AmericanBio | AB00502-01000 | |
| Ethanol (200 Proof) | Fisher Scientific | 16-100-824 | |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma | E4884-500G | |
| Lithium acetate dihydrate | Sigma | L6883-250G | |
| MyCycler Thermal Cycler | BioRad | 170-9703 | |
| Poly(ethylene glycol) (PEG) | Sigma | P3640-1KG | |
| PrimeSTAR HS DNA Polymerase (high fidelity DNA polymerase) and 5x buffer | Fisher Scientific | 50-443-960 | |
| Salmon sperm DNA solution | ThermoFisher Scientific | 15632-011 | |
| Sigma 7-9 (Tris base, powder form) | Sigma | T1378-1KG | |
| Sodium acetate trihydrate | Sigma | 236500-500G | |
| Supra Sieve GPG Agarose (low metling temperature agarose) | AmericanBio | AB00985-00100 | |
| Taq Polymerase and 10x Buffer | New England BioLabs | M0273X | |
| Toothpicks | Fisher Scientific | S67859 | |
| Tris-HCl (1 M, pH 8.0) | AmericanBio | AB14043-01000 | |
| a-D(+)-Glucose | Fisher Scientific | AC170080025 | for yeast media |
| Agar | Fisher Scientific | DF0140-01-0 | for yeast media |
| Peptone | Fisher Scientific | DF0118-07-2 | for YPD medium |
| Yeast Extract | Fisher Scientific | DF0127-17-9 | for YPD medium |
| 4-aminobenzoic acid | Sigma | A9878-100G | for complete minimal dropout medium |
| Adenine | Sigma | A8626-100G | for complete minimal dropout medium |
| Glycine hydrochloride | Sigma | G2879-100G | for complete minimal dropout medium |
| L-Alanine | Sigma | A7627-100G | for complete minimal dropout medium |
| L-Arginine monohydrochloride | Sigma | A5131-100G | for complete minimal dropout medium |
| L-Asparagine monohydrate | Sigma | A8381-100G | for complete minimal dropout medium |
| L-Aspartic acid sodium salt monohydrate | Sigma | A6683-100G | for complete minimal dropout medium |
| L-Cysteine hydrochloride monohydrate | Sigma | C7880-100G | for complete minimal dropout medium |
| L-Glutamic acid hydrochloride | Sigma | G2128-100G | for complete minimal dropout medium |
| L-Glutamine | Sigma | G3126-100G | for complete minimal dropout medium |
| L-Histidine monohydrochloride monohydrate | Sigma | H8125-100G | for complete minimal dropout medium |
| L-Isoleucine | Sigma | I2752-100G | for complete minimal dropout medium |
| L-Leucine | Sigma | L8000-100G | for complete minimal dropout medium |
| L-Lysine monohydrochloride | Sigma | L5626-100G | for complete minimal dropout medium |
| L-Methionine | Sigma | M9625-100G | for complete minimal dropout medium |
| L-Phenylalanine | Sigma | P2126-100G | for complete minimal dropout medium |
| L-Proline | Sigma | P0380-100G | for complete minimal dropout medium |
| L-Serine | Sigma | S4500-100G | for complete minimal dropout medium |
| L-Threonine | Sigma | T8625-100G | for complete minimal dropout medium |
| L-Tryptophan | Sigma | T0254-100G | for complete minimal dropout medium |
| L-Tyrosine | Sigma | T3754-100G | for complete minimal dropout medium |
| L-Valine | Sigma | V0500-100G | for complete minimal dropout medium |
| myo-Inositol | Sigma | I5125-100G | for complete minimal dropout medium |
| Uracil | Sigma | U0750-100G | for complete minimal dropout medium |
| Ammonium Sulfate | Fisher Scientific | A702-500 | for complete minimal dropout medium |
| Yeast Nitrogen Base | Fisher Scientific | DF0919-07-3 | for complete minimal dropout medium |
| 5-Fluoroorotic acid (5-FOA) | AmericanBio | AB04067-00005 | for 5-FOA medium |
Références
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389 (6648), 251-260 (1997).
- Campos, E. I., Reinberg, D. Histones: annotating chromatin. Annu Rev Genet. 43, 559-599 (2009).
- Rando, O. J., Winston, F. Chromatin and transcription in yeast. Genetics. 190 (2), 351-387 (2012).
- Duina, A. A., Miller, M. E., Keeney, J. B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 197 (1), 33-48 (2014).
- Storici, F., Resnick, M. A. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y). 25, 189-207 (2003).
- Gray, M., Kupiec, M., Honigberg, S. M. Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7 (2004).
- Erdeniz, N., Mortensen, U. H., Rothstein, R. Cloning-free PCR-based allele replacement methods. Genome Res. 7 (12), 1174-1183 (1997).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
- Lundblad, V., Hartzog, G., Moqtaderi, Z. Manipulation of cloned yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Hoffman, C. S. Preparation of yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Treco, D. A., Lundblad, V. Preparation of yeast media. Curr Protoc Mol Biol. Chapter 13, (2001).
- Lederberg, J., Lederberg, E. M. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 63 (3), 399-406 (1952).
- Sikorski, R. S., Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122 (1), 19-27 (1989).
- Johnson, P., et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 5 (5), 741-749 (2015).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336-4343 (2013).
- Cross, S. L., Smith, M. M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol. 8 (2), 945-954 (1988).
- Libuda, D. E., Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 443 (7114), 1003-1007 (2006).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.