
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Gezielt
In diesem Artikel
Zusammenfassung
A strategy for generating mutations in histone genes at their endogenous location in Saccharomyces cerevisiae is presented.
Zusammenfassung
We describe a PCR- and homologous recombination-based system for generating targeted mutations in histone genes in budding yeast cells. The resulting mutant alleles reside at their endogenous genomic sites and no exogenous DNA sequences are left in the genome following the procedure. Since in haploid yeast cells each of the four core histone proteins is encoded by two non-allelic genes with highly homologous open reading frames (ORFs), targeting mutagenesis specifically to one of two genes encoding a particular histone protein can be problematic. The strategy we describe here bypasses this problem by utilizing sequences outside, rather than within, the ORF of the target genes for the homologous recombination step. Another feature of this system is that the regions of DNA driving the homologous recombination steps can be made to be very extensive, thus increasing the likelihood of successful integration events. These features make this strategy particularly well-suited for histone gene mutagenesis, but can also be adapted for mutagenesis of other genes in the yeast genome.
Einleitung
Die vier Kern Histonproteine H2A, H2B, H3 und H4 spielen eine zentrale Rolle in der Verdichtung, die Organisation und Funktion von eukaryotischen Chromosomen. Zwei Sätze von jeder dieser Histone bilden die Histonoktamer, ein Molekular Spule, die die Verpackung von ~ 147 Basenpaaren der DNA um sich ergeb leitet schließlich in der Bildung einer Nukleosomen 1. Nukleosomen sind aktive Teilnehmer in einer Vielzahl von Chromosom-basierte Prozesse, wie zum Beispiel die Regulation der Gen-Transkription und die Bildung von Euchromatin und Heterochromatin über Chromosomen, und als solche im Mittelpunkt intensiver Forschung im Laufe der vergangenen Jahrzehnte. Eine Anzahl von Mechanismen wurden durch die Nukleosomen beschrieben kann in einer Weise manipuliert werden, die Ausführung bestimmter Prozesse erleichtern können - diese Mechanismen posttranslationaler Modifikation von Histon-Reste, ATP-abhängige Nukleosom-Remodeling und ATP-unabhängige Nukleosom Reorganisation umfassenund Montage / Demontage 2, 3.
Die Bäckerhefe Saccharomyces cerevisiae ist ein besonders leistungsfähiges Modellorganismus für das Verständnis der Histon - Funktion in Eukaryoten. Dies kann weitgehend auf dem hohen Grad der evolutionären Konservierung der Histon - Proteine in der gesamten Domäne Eukarya und amenability von Hefe von genetischen und biochemischen experimentelle Ansätze 4 auf eine Vielzahl zurückzuführen. Reverse genetische Ansätze in Hefe wurden weitgehend verwendet, um die Auswirkungen der spezifischen Histon-Mutationen zu verschiedenen Aspekten der Chromatin Biologie zu studieren. Für diese Arten von Experimenten ist es oft bevorzugt, Zellen zu verwenden, bei dem die mutanten Histone aus ihren nativen genomischen Loci exprimiert werden, als Ausdruck von autonomen Plasmide zu abnormal intrazellulären Spiegel von Histon-Proteinen (durch eine unterschiedliche Anzahl von Plasmiden in Zellen) führen kann, und gleichzeitige Veränderung der Chromatinstruktur engebungen, die letztlich die Interpretation der Ergebnisse durcheinander bringen kann.
Hier beschreiben wir eine PCR-basierte Technik, die zur gezielten Mutagenese von Histon-Gene in ihrer nativen genomischen Stellen ermöglicht, die keine Klonierungsschritt und führt zur Erzeugung der gewünschten Mutation (en) ohne Überbleibsel exogenen DNA-Sequenzen in das Genom erfordert. Diese Technik nutzt die effiziente homologe Rekombinationssystem in Hefe und gemeinsam hat mehrere Funktionen mit anderen ähnlichen Techniken , die von anderen Gruppen entwickelt - insbesondere der Delitto Perfetto, ortsspezifischen genomischen (SSG) Mutagenese und Klonierung freien PCR-basierte Allel Ersatzmethoden 5, 6, 7. Jedoch hat die Technik, die wir beschreiben, einen Aspekt, dass es besonders gut geeignet für die Mutagenese von Histon-Gene macht. In haploiden Hefezellen, ist jeder der vier Core-Histone durch zwei nicht-a codiertllelic und hoch homologe Gene: zum Beispiel wird Histon H3 durch die HHT1 und HHT2 Gene kodiert, und die offenen Leserahmen (ORFs) der beiden Gene sind mehr als 90% in - Sequenz identisch. Dieser hohe Grad an Homologie kann Experimente komplizieren spezifisch eine der beiden für die Mutagenese Histon-kodierenden Gene entworfen abzuzielen. Während oft die oben genannten Verfahren die Verwendung von mindestens einigen Sequenzen innerhalb des ORF des Zielgens erfordern homologe Rekombination zu treiben, beschreiben die Technik, die wir hier macht die Verwendung von Sequenzen, die die ORFs der Histon-Gene flankieren (die viel weniger Sequenzhomologie teilen) für die Rekombinationsschritt, Steigerung somit die Wahrscheinlichkeit einer erfolgreichen Ausrichtung der Mutagenese an den gewünschten Ort. Darüber hinaus sind die homologen Regionen, die Rekombination fahren kann sehr umfangreich sein, weiter zu einer effizienten gezielte homologe Rekombination beiträgt.
Protokoll
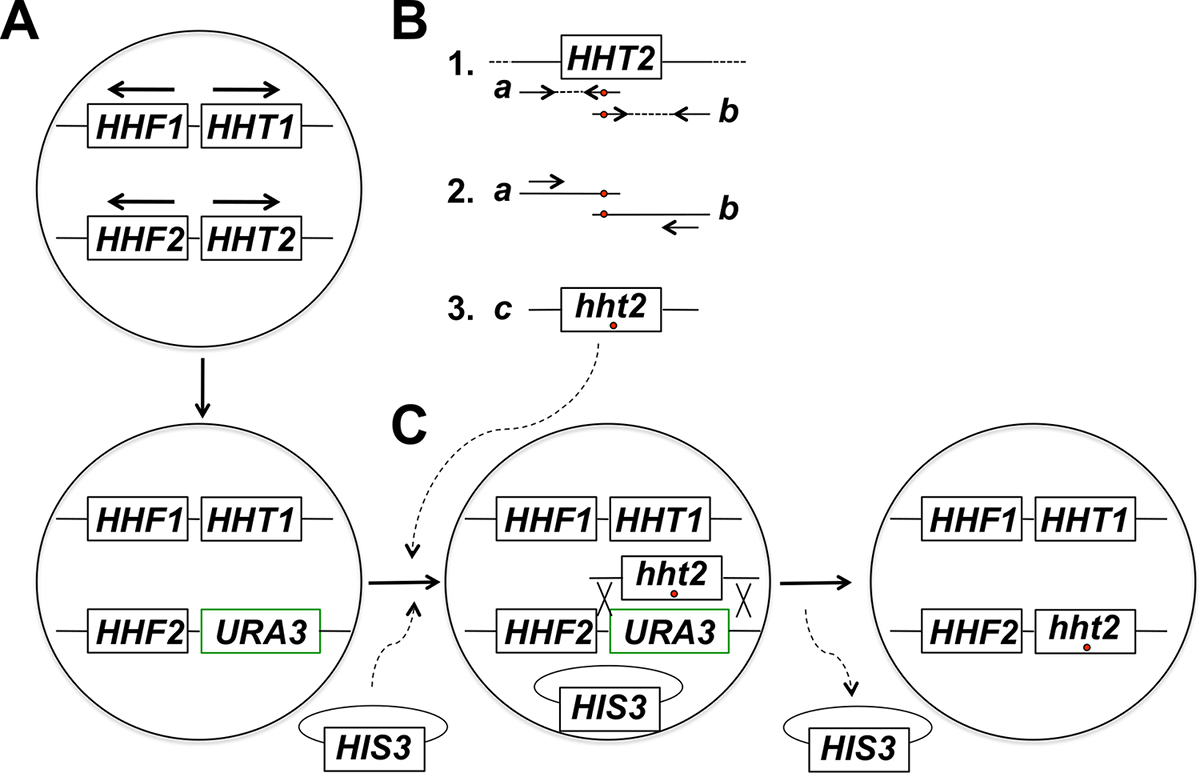
HINWEIS: Die experimentelle Strategie für die gezielte in - situ - Histon - Gen - Mutagenese umfasst mehrere Schritte (zusammengefasst in Figur 1). Diese Schritte umfassen: (1) Ersatz des Ziel Histon - Gens mit dem URA3 - Gen, (2) Erzeugung und Reinigung von PCR - Produkten auf zwei teilweise überlappenden Fragmenten des Ziel - Histon - Gens entspricht , unter Verwendung von Primern die gewünschte Mutation beherbergt (s), (3 ) Fusion PCR der beiden teilweise überlappende Fragmente PCR in voller Größe Produkte für die Integration zu erhalten, (4) Co-Transformation in voller Größe PCR-Produkte und Backbone-Plasmid und Selektion für Marker auf dem Plasmid, (5) Bildschirm für die 5-FOA-resistente Transformanten, (6) Reinigung von 5-FOA-resistenten Kolonien und Verlust des Rückgrats Plasmids, und (7) Molekularanalysen Assay für die richtige Integration des mutierten Allels.

Abbildung 1: Überblick über die Strategie für die in Bäckerhefe in situ Mutagenese von Histon - Gene Gezielte. In diesem Beispiel ist das Zielgen HHT2, aber jede andere Kern Histon - Gens können auch unter Verwendung dieser Strategie mutagenisiert werden. (A) Haploid Hefezellen zwei Histon H3-kodierenden Gene beherbergen (HHT1 und HHT2) und zwei Histon - H4-kodierenden Gene (HHF1 und HHF2) angeordnet sind, wie in der Figur gezeigt ist (die HHT1 und HHF1 Gene auf Chromosom II und dem HHT2 und HHF2 Gene auf dem Chromosom XIV befindet - in jedem Fall sind die Pfeile zeigen in Richtung der Transkription). Im ersten Schritt des Verfahrens wird das ORF des HHT2 Gens mit dem URA3 - Gen ersetzt, was zu einer hht2Δ :: URA3 - Stamm. (B) In Teil 1, ein Wildtyp - Kopie des HHT2 - Gens aus einer genomischen DNA - Probe als Matrize für zwei PCR - Reaktionen zu Gattungen verwendette, die zwei teilweise überlappenden Fragmente des Gens. Der Reverse-Primer für die erste Reaktion umfasst eine oder mehrere fehlgepaarte Nukleotide (mit einem roten Kreis angedeutet), die der gewünschten Mutation (en) entsprechen, in das Genom eingeführt werden. Der Vorwärtsprimer für die zweite Reaktion hat den äquivalenten Mismatch in einer umgekehrten komplementären Konfiguration (auch mit einem roten Kreis gekennzeichnet). Die beiden PCR - Produkte erzeugt , in Teil 1 (Produkte A und B) werden dann als Matrizen für die Fusions - PCR unter Verwendung von zwei Primern , die Ausheilung zu den Produkten A und B in der Art und Weise teil 2. Daraus ergibt sich die Erzeugung von Vollgröße PCR gezeigt ist, verwendet Produkte (Produkt C in Teil 3) beherbergt , das die gewünschte Mutation (en). (C) Die hht2Δ :: URA3 Stamm ist dann co-transformiert mit dem Full-Size - PCR - Produkte und ein Backbone - Plasmid (a HIS3- Plasmid in diesem Beispiel markiert), und die Zellen werden auf das Vorhandensein des Plasmids (auf Medien selektiert fehlt histidine in diesem Beispiel). Transformanten werden dann für 5-FOA - Resistenz gescreent - resistenten Zellen sind Kandidaten für ein homologes Rekombinationsereignis zur Integration des PCR - Produkts und Exzision des URA3 - Gens führt laufen hat, wie gezeigt. Die anschließende Verlust des Rückgrats Plasmid durch mitotische Zellteilung führt zur letzten Histon Mutantenstamm gewünscht. Wir haben gezeigt, dass die Auswahl des Rückgrats Plasmid gefolgt gefunden durch Screenen auf 5-FOA - Resistenz führt zu einer viel höheren Frequenz der Identifizierung der richtigen Integrationsereignisse im Vergleich zur direkten Selektion auf 5-FOA - Platten, die meist identifiziert Zellen , die spontan URA3 Mutationen erworben haben. (Diese Zahl wurde von Referenz 14 modifiziert). Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
{kind=link}
1. Ersatz des Ziel Histon - Gens mit dem URA3Gen
- Führen Sie Standard - PCR-vermittelte Ein-Schritt - Gen - Unterbrechung des ORF von Ziel Histon - Gens mit dem URA3 - Gen 8, 9 zu ersetzen.
HINWEIS: Die Verwendung von Hefezellen , die ura3Δ0 Durchführung wird empfohlen, da diese Mutation das gesamte endogene URA3 ORF entfernt, wodurch die Integration des PCR - Produkts in den URA3 - Locus 8 vermieden wird . Alternativ kann das K. lactis - URA3 - Gen effektiv zur Erzeugung des Histon - Ersatz in jedem ura3 Hintergrund verwendet werden , wie es in S. cerevisiae funktionell ist , sondern nur eine teilweise Sequenzhomologie mit der S. cerevisiae - URA3 - Gen. Der Stamm sollte auch auxotroph sein, mindestens eine Verbindung, die zur Auswahl des Rückgrats Plasmids in dem Transformationsexperiment erlaubt (Schritt 4 dieses Protokolls). Dieser Schritt ist nicht erforderlich , wenn ein Ziel Histon geneΔ :: URA3Stamm ist bereits verfügbar.
2. Erzeugung und Reinigung von PCR-Produkten entsprechend zwei teilweise überlappend Fragmente des Ziel Histone Gene Primers Beherbergen die gewünschte Mutation (en) aufnehmen
- Erzeugen PCR - Produkte zu zwei teilweise überlappenden Fragmenten des Ziel - Histon - Gens entspricht.
- Bereiten Sie zwei PCR-Reaktionen wie folgt:
- PCR - Produkte zu erzeugen , um die erste Hälfte des Gens (Produkt eine in 1B), eingerichtet , um die folgende Reaktion: 1 entspricht ul Templat - DNA, 5 μl10 uM Vorwärtsprimer, 5 μl10 uM Reverse - Primer, 0,5 ul (1,25 U) thermo DNA - Polymerase, 10 & mgr; l 5x - DNA - Polymerase - Puffer, 5 & mgr; l dNTP - Gemisch (jeweils 2 mM) und 23,5 & mgr; l dH 2 O.
HINWEIS: Die Template-DNA kann aus einem Wildtyp-Stamm für das Ziel-Histon-Gen abgeleitete genomische DNA isoliert Standard Pro mitverfahren 10. Zur Berücksichtigung Variationen in DNA - Konzentration und Menge an Verunreinigungen in unterschiedlichen genomischen Präparationen, wird empfohlen , um die Reaktionen zu optimieren , indem entweder unverdünnt DNA oder verschiedene Verdünnungen der genomischen Omen (beispielsweise 1:10 und 1: 100). Der Vorwärts-Primer anlagern sollte auf einen Bereich stromaufwärts des Zielgens. Der Reverse - Primer sollte innerhalb des ORF anlagern, werden ~ 40 Nukleotide lang und enthalten die gewünschte Mutation (n) irgendwo in der Mitte (siehe Abbildung 1B-1 und Repräsentative Ergebnisse Abschnitt für Beispiele). Die Verwendung einer High Fidelity DNA Polymerase wird empfohlen, um die Raten von unerwünschten Mutationen während der Synthese der PCR-Produkte zu reduzieren. - Zur Erzeugung einzurichten PCR - Produkte entsprechend der zweiten Hälfte des Gens (Produkt b in Abbildung 1B), eine Reaktion wie in 2.1.1.1 , jedoch mit unterschiedlichen Primer angegeben.
HINWEIS: Die vordere primer sollte innerhalb des ORF tempern, sein ~ 40 Nukleotide lang und enthalten die gewünschte Mutation (en) irgendwo in der Mitte. Man beachte, dass die Mutation (en) in diesem Primer das reverse Komplement der Mutation (en) in der Rückwärtsprimer in Schritt 2.1.1.1. Der reverse Primer des Zielgens stromabwärts (siehe 1B-1 und Repräsentative Ergebnisse Abschnitt Beispiele) zu einem Bereich anlagern soll.
- PCR - Produkte zu erzeugen , um die erste Hälfte des Gens (Produkt eine in 1B), eingerichtet , um die folgende Reaktion: 1 entspricht ul Templat - DNA, 5 μl10 uM Vorwärtsprimer, 5 μl10 uM Reverse - Primer, 0,5 ul (1,25 U) thermo DNA - Polymerase, 10 & mgr; l 5x - DNA - Polymerase - Puffer, 5 & mgr; l dNTP - Gemisch (jeweils 2 mM) und 23,5 & mgr; l dH 2 O.
- Legen Sie die Reaktionen in einem Thermocycler mit den folgenden Einstellungen: 94 ˚C 30 Sekunden; 30 Zyklen der folgenden Einstellungen: 98 C 10 sec, 60 ° C 5 s, 72 ° C 1,5 min; und 72 ° C 10 min.
Hinweis: Optimierung der PCR-Parameter können für spezifische Primersätze und Ziel Histon-Gens erforderlich.
- Bereiten Sie zwei PCR-Reaktionen wie folgt:
- Laufen 20-50 & mgr; l des Materials aus den PCR-Reaktionen auf einem 0,9% niedrigschmelzenden Agarosegel in 89 mM Tris-Base, 89 mM Borsäure, 2,5 mM EDTA (TBE) -Puffer.
- Schneiden Sie Agarosegel Abschnitte des PCR-pr enthältoducts aus Gel mit einem sauberen Skalpell oder einer Rasierklinge und übertragen die jeweils mit einem 1,5 ml Mikrozentrifugenröhrchen mit. Bewahren Sie Agarose Abschnitte mit PCR-Produkte bei -20 ° C bis zur Verwendung.
3. Fusion PCR der zwei teilweise überschneidende Fragmente zu erhalten Full Size PCR Produkte für die Integration
- Bereiten Matrize für die PCR - Reaktionen
- Melt Agarose-Gel-Abschnitte aus Schritt 2.3, indem die Reaktionsgefäße in einem Wärmeblocksatz bei 65 ° C für 5 Minuten (oder bis sie vollständig geschmolzen). Wirbelrohre alle 1 bis 2 min, den Schmelzvorgang zu erleichtern.
- Übertragen Sie eine festgelegte Menge des geschmolzenen Agarose aus jeder Probe (zB 50 & mgr; l jeweils für eine Gesamtmenge von 100 ul) in einem einzigen Reaktionsgefäß und mischen durch Vortexen. Verwenden Sie diese als Vorlage in den Fusions-PCR-Reaktionen. Das Röhrchen wird bei -20 ° C bis zur Verwendung.
- Amplify eine große Menge an in voller Größe PCR - Produkt (Produkt cin 1B)
- Set up sechs PCR-Reaktionen, die jeweils mit den folgenden Komponenten: 2 & mgr; l Matrizen-DNA, 10 ul 10 uM Vorwärtsprimer, 10 & mgr; l 10 & mgr; M Rückwärts-Primer, 1 & mgr; l (2,5 U) thermostabile DNA-Polymerase, 20 ul 5x DNA-Polymerasepuffer, 10 ul dNTP - Gemisch (jeweils 2 mM) und 47 & mgr; l dH 2 O.
HINWEIS: Die Anzahl der Reaktionen können in Abhängigkeit von der PCR-Effizienz verändert werden. Die Templat-DNA (siehe 3.1.2) sollte auf 65 C erhitzt werden, bis durch Vortexen geschmolzen, gemischt und schließlich zu dem PCR-Reaktionsgemisch zugesetzt. Nach dem Hinzufügen mischen vorsichtig, aber gründlich, indem die Lösung Auf- und Abpipettieren mehrmals. Zur Berücksichtigung Variationen in DNA - Konzentration in den verschiedenen Proben, wird empfohlen, zuerst die Reaktionen zu optimieren , indem entweder unverdünnt Vorlage oder unterschiedlichen Verdünnungen der Matrize (zB 1:10 und 1: 100). Die zwei Primer verwendet werden, sollten teilweise überlappenden Fragmente des Zielgens als schlecht an die beiden anlagernustrated in 1B-2 und so gestaltet sein , dass die endgültigen PCR - Produkte mindestens 40 Basenpaare auf beiden Seiten homolog zu den Regionen haben den URA3 ORF flankieren, die die homologe Rekombination Schritt zu fahren (siehe Abschnitt Repräsentative Ergebnisse für Beispiele). Die Verwendung einer High Fidelity DNA Polymerase wird empfohlen, um die Raten von unerwünschten Mutationen während der Synthese der PCR-Produkte zu reduzieren. - Die Röhrchen in einem Thermocycler mit den folgenden Einstellungen: 94 ˚C 30 Sekunden; 30 Zyklen der folgenden Einstellungen: 98 C 10 sec, 50 ° C 15 Sekunden, 72 ° C 1,5 min; und 72 ° C 10 min.
HINWEIS: Optimierung der PCR-Parameter können für spezifische Primer-Sets und Ziel Histon-Gens erforderlich.
- Set up sechs PCR-Reaktionen, die jeweils mit den folgenden Komponenten: 2 & mgr; l Matrizen-DNA, 10 ul 10 uM Vorwärtsprimer, 10 & mgr; l 10 & mgr; M Rückwärts-Primer, 1 & mgr; l (2,5 U) thermostabile DNA-Polymerase, 20 ul 5x DNA-Polymerasepuffer, 10 ul dNTP - Gemisch (jeweils 2 mM) und 47 & mgr; l dH 2 O.
4. Co-Transformation von Full Size PCR Produkte und Backbone Plasmid und Selektion für Marker auf dem Plasmid
- Konzentration von PCR - Produkten
- Pool, die six PCR-Reaktionen (600 ul insgesamt) aus Schritt 3.2.2 in einem einzigen Reaktionsgefäß und mischen durch Vortexen.
- Teilen Sie die Probe in drei 200 ul Aliquots in Mikrozentrifugenröhrchen. Auszufällen die DNA in jedem Röhrchen durch Zugabe von 20 ul 3 M Natriumacetat (pH 5,2) und 550 ul 100% Ethanol. Mischen Sie die Lösung gründlich und auf Eis für mindestens 15 min. Sammeln DNA durch Zentrifugation bei ~ 14.000 g für 10 min, spülen Sie das Pellet mit 200 ul 70% Ethanol und Luft trocknen lassen.
- Resuspendieren jedes DNA - Pellet in 25 & mgr; l dH 2 O, und Pool in einem einzigen Rohr (für insgesamt 75 & mgr; l).
- Hefe - Co-Transformation
- Bereiten Sie 10 ml über Nacht Kultur des in Abschnitt erzeugte Stamm 1 in Hefeextrakt Pepton Dextrose (YPD) flüssiges Medium 11.
- Am nächsten Morgen impfen 400 ml YPD-Flüssigmedium mit 8 ml der gesättigten über Nacht Kultur und Inkubation durch Schüttelnbei 30 ° C für 4 - 5 h Zellen zu ermöglichen logarithmischen Wachstumsphase einzutreten.
- Sammle die Zellen durch Zentrifugation bei ~ 3.220 xg für 10 min, Verwerfen des flüssigen Mediums und Resuspendieren der Zellen in 1 Volumen 10 mM Tris-HCl (pH 8,0), 1 mM EDTA, 0,1 M Lithium-Acetat-Lösung (TE / LiAc) .
- Sammeln Sie die Zellen durch Zentrifugation bei ~ 3.220 × g für 10 min, und entsorgen Sie die TE / LiAc.
- Resuspendieren der Zellen in 1 ml TE / LiAc.
- Legen Sie die folgende Reaktion Cocktail in ein Mikrozentrifugenröhrchen bis 800 & mgr; l von Zellen aus Schritt 4.2.5, 40 ul gekocht 10 mg / ml Lachssperma-DNA, insgesamt 12,5 ug Rückgrat Plasmid-DNA und 75 & mgr; l konzentrierter PCR-Produkt aus Schritt 4.1.3.
HINWEIS: Lachssperma-DNA sollte für 5 min und auf Eis für mindestens 5 min vor der Verwendung in der Reaktion abgekocht werden. Gesamtvolumen des DNA-Rückgrats Plasmid zugegeben wird, sollte auf ein Minimum (~ 80 & mgr; l oder weniger) gehalten werden. Siehe Repräsentative Ergebnisse Abschnitt für ein Beispiel eines Backbones plasmIch würde. - Mischen Sie die Cocktail-Rohr gründlich und aliquoten gleichmäßig in acht Mikrozentrifugenröhrchen (Röhrchen 1 bis 8).
- Legen Sie die folgenden zwei Steuer Transformation Reaktionsröhrchen nach oben:
- Rohr 9 (kein PCR-Produkt Kontrolle): 100 & mgr; l von Zellen aus Schritt 4.2.5, 5 ul gekocht 10 mg / ml Lachssperma-DNA (für 5 min gekocht, siehe Schritt 4.2.6 Hinweis), insgesamt 1,56 ug Backbone Plasmid-DNA und kein PCR-Produkt zugegeben.
- Rohr 10 (kein DNA-Kontrolle): 100 & mgr; l der Zellen aus Schritt 4.2.5, 5 & mgr; l gekochter 10 mg / ml Lachssperma-DNA (siehe Schritt 4.2.6 Note), keine Backbone-Plasmid-DNA gegeben, und kein PCR-Produkt zugegeben.
- Das Vermischen der beiden Röhren vorsichtig, aber gründlich durch Auf- und Abpipettieren mehrmals.
- Inkubieren Sie die zehn Röhrchen bei 30 ° C für 30 min.
- Zu jedem Röhrchen, mit 1,2 ml 40% Polyethylenglycol (PEG 3350) in TE / LiAc. Mischen Sie gründlich mit einem P-1000 pipettieren, bis die Lösung homogen ist.
- Inkubieren Sie die zehn Röhrchen bei 30° C für 30 min. durch Pipettieren von oben und unten und dann Röhrchen bei 42 ° C für 15 min vorsichtig die Lösung mischen.
- Sammle die Zellen durch die Röhrchen in einer Mikrozentrifuge bei ~ 14.000 xg für 30 sec dreht. Entsorgen Sie die Flüssigkeit und Resuspendieren der Zellen in 1 ml sterilem dH 2 O.
- Sammle die Zellen durch die Röhrchen in einer Mikrozentrifuge bei ~ 14.000 xg für 30 sec dreht. Entsorgen Sie die Flüssigkeit und Resuspendieren der Zellen in 500 ul sterilem dH 2 O.
- Pool Röhren 1 bis 8 zusammen (Gesamtvolumen von 4 ml) und gründlich durch Auf- und Abpipettieren mischen.
- Platte 200 ul des obigen Gemisches auf jeder der zwanzig vollständige minimal dropout Mediumplatten 11 (Platten 1-20) zur Auswahl des Rückgrats Plasmids.
- Platte 200 ul des Gemisches aus Rohr 9 und 200 & mgr; l der Mischung aus Rohr 10, die jeweils auf einer eigenen Selektionsplatte (Platten 21 bzw. 22).
- Inkubieren Sie die 22 Platten bei 30 ° C für 3 - 5 Tage zuWählen Sie für Plasmid-Trans.
- Inspizieren Transformationsplatten nach 3 bis 5 Tagen Inkubation. Rund 5.000 Kolonien sollten auf Platten 1-21 (siehe Repräsentative Ergebnisse für ein Beispiel) sichtbar sein und keine Kolonien sollten auf der Platte 22 vorhanden sein.
5. Bildschirm für die 5-FOA-resistente Trans
- Übertragungszellen von den Platten 1 bis 20 (und Transformationsplatte 21 als Kontrolle) zu 5-Fluororotsäure (5-FOA) Platten 11 durch Replika-Plattierung 12 um für den Verlust des URA3 - Gens als Ergebnis der Integration der zu screenen PCR-Produkte an der gewünschten Stelle.
- Entfernen Sie die Platte Deckel und drücken Sie die Platte Kolonien auf einem sterilen Samt enthält. Übertragen Sie die Zellen aus dem Samt zu einem 5-FOA Platte durch die Platte auf dem Samt drücken. Die Platten bei 30 ° C für 2 Tage.
- Im Anschluss an die 2-tägige Inkubation kontrollieren sorgfältig die 5-FOA-Platten für growth.
HINWEIS: Ein Kandidat Integrationsereignis wird von einem kleinen asymmetrischen "gestaucht" Kolonie auf einer 5-FOA Platte dargestellt werden - umgekehrt, kleine Papillen wachsen auf 5-FOA - Platten sind wahrscheinlich repräsentativ für spontane URA3 Mutationen , die auf die während des Wachstums der Kolonien entstanden Transformationsplatten und sind daher unwahrscheinlich , dass die gewünschte Integrationsereignis darzustellen (Abbildung 3 im Repräsentative Ergebnisse Abschnitt für weitere Ausführungen zu diesem Punkt und für einige Beispiele zu sehen).
6. Reinigung von 5-FOA-resistente Kolonien und Verlust der Backbone Plasmid
- Mit einer sterilen Zahnstochern, wählen Sie die Kandidaten Kolonien aus den 5-FOA-Platten in Schritt 5.2 beschrieben und Streifen für einzelne Kolonien auf YPD-Platten. Inkubieren für 2 - 3 Tage bei 30 ° C.
- Im Anschluss an die Inkubation Replik - Platte jede YPD Reinigung Platte auf eine frische YPD Platte, eine Drop-out - Platte ohne Uracil für den Verlust zu überprüfendes URA3 - Gens und eine zweite Drop-Out - Platte für die Anwesenheit oder Abwesenheit des Rückgrats Plasmids zu überwachen. Inkubieren für 1 bis 2 Tage bei 30 ° C.
- Nach der Inkubation eine Kolonie von jeder Kandidatenprobe zu identifizieren , die auf dem YPD Platte wächst , aber nicht wächst auf beiden Dropout-Platte ( eine solche Kolonie wird erwartet, haben das URA3 - Gen durch das Rekombinationsereignis verloren und verlor das Rückgrat Plasmids während der mitotischen Zellteilung). Restreak solche Kolonien auf frische YPD-Platten. Diese Kolonien sind die Integration Kandidaten und wird weiter in 7 Schritt analysiert werden.
7. Molekulare Analysen zur Assay für die ordnungsgemäße Integration des mutierten Allels
- Isolieren genomischer DNA aus den Kandidaten Proben unter Verwendung von Standardverfahren 10.
- Amplify genomischen Region der Zielstelle umfasst.
- Richten Sie die folgende PCR-Reaktion für jede Probe: 0,5 ul Templat-DNA, 5 ul10 & mgr; M Vorwärts - Primer, 5 & mgr; l 10 & mgr; M Rückwärts - Primer, 0,5 & mgr; l (2,5 Einheiten) Taq DNA - Polymerase, 5 ul 10x Taq - DNA - Polymerase - Puffer, 5 & mgr; l dNTP - Gemisch (jeweils 2 mM) und 29 & mgr; l dH 2 O.
HINWEIS: Template-DNA ist die genomische DNA aus den Kandidatenproben abgeleitet. Es wird empfohlen , auch Reaktionen umfassen zwei Kontrolle: eine genomische DNA aus den ursprünglichen Histon geneΔ abgeleitet unter Verwendung :: URA3 - Stamm als Matrize und eine andere unter Verwendung von genomischer DNA aus einem Wildtyp - Histon - Stamm als Vorlage. Zur Berücksichtigung Variationen in DNA - Konzentration und Menge an Verunreinigungen in unterschiedlichen genomischen Präparationen, wird empfohlen , um die Reaktionen zu optimieren , indem entweder unverdünnt DNA oder verschiedene Verdünnungen der genomischen Omen (beispielsweise 1:10 und 1: 100). Es ist darauf zu achten , dass diese Primer an DNA - Sequenzen außerhalb der Region durch die mutmaßlich integrierte PCR - Produkt umfasste - diese Weise die Größe der PCR - Produkte in diesen rTELLUNGNAHMEN kann als diagnostisches Werkzeug für die Integration der Produkte in der richtigen genomischen Stelle (siehe Repräsentative Ergebnisse für ein Beispiel) verwendet werden. - Legen Sie die Reaktionen in einem Thermocycler mit den folgenden Einstellungen: 94 ° C 3 min; 30 Zyklen mit den folgenden Einstellungen: 94 ° C 45 sec, 50 ° C 45 sec, 72 ° C 2 min; und 72 ° C 10 min.
HINWEIS: Optimierung der PCR-Parameter können für spezifische Primer-Sets und Ziel Histon-Gens erforderlich.
- Richten Sie die folgende PCR-Reaktion für jede Probe: 0,5 ul Templat-DNA, 5 ul10 & mgr; M Vorwärts - Primer, 5 & mgr; l 10 & mgr; M Rückwärts - Primer, 0,5 & mgr; l (2,5 Einheiten) Taq DNA - Polymerase, 5 ul 10x Taq - DNA - Polymerase - Puffer, 5 & mgr; l dNTP - Gemisch (jeweils 2 mM) und 29 & mgr; l dH 2 O.
- Verarbeitung von PCR - Produkten
- Laufen 20 ul von jeder Reaktion auf einem 0,8% TBE-Agarosegel.
- Beurteilung der Größe der PCR - Produkte unter Verwendung von DNA - Standards als Referenz zu bestimmen , ob das URA3 - Gen von der mutmaßlich mutierten Histon - Gen erfolgreich ersetzt wurde (siehe Repräsentative Ergebnisse für ein Beispiel).
HINWEIS: In bestimmten Fällen kann die gewünschte Mutation (en) eingeführt in die Histon-Gene entweder erstellen oder eine Beschränkung zerstören sitzene. Wenn dies der Fall ist, kann die Anwesenheit der gewünschten Mutation in den PCR-Produkten der Größe anzeigt korrekte Integration, indem die Produkte der Verdauung mit dem entsprechenden Restriktionsenzym durch Gelelektrophorese-Analyse (siehe Repräsentative Ergebnisse für ein Beispiel), gefolgt beurteilen . - Gegenstand PCR-Produkte der Größe anzeigt korrekte Integration der DNA-Sequenzierung die Gegenwart der gewünschten Mutation (en) zu bestätigen und sicherzustellen, dass keine zusätzlichen Mutationen in das Genom eingeführt worden ist.
Ergebnisse
Wir beschreiben die Erzeugung eines hht2 Allel ein Histon H3 mutiertes Protein beherbergt eine Substitution an Position 53 von Arginin zu einem einer Glutaminsäure (H3-R53E - Mutante) als repräsentatives Beispiel für die gezielte in - situ - Mutagenese - Strategie exprimieren.
Wir erzielten einen Stamm , in dem das gesamte ORF von HHT2 durch das URA3 - Gen ersetzt (Schritt 1 des ...
Diskussion
Die hohe Sequenzhomologie zwischen den zwei nicht allele Gene , die für jeden der vier Kern Histon - Proteine in haploiden S. cerevisiae - Zellen eine Herausforderung für die Forscher darstellen können , die wünschen speziell für die Mutagenese eines der beiden Gene abzuzielen. Zuvor Hefe Mutagenese Methodologien beschrieben, einschließlich der Delitto Perfetto, ortsspezifischen genomischen (SSG) Mutagenese und Klonierung freien PCR-basierten Allelaustausch Methoden 5,
Offenlegungen
The authors declare that they have no competing financial interests.
Danksagungen
We thank Reine Protacio for helpful comments during the preparation of this manuscript. We express our gratitude to the National Science Foundation (grants nos. 1243680 and 1613754) and the Hendrix College Odyssey Program for funding support.
Materialien
| Name | Company | Catalog Number | Comments |
| 1 kb DNA Ladder (DNA standards) | New England BioLabs | N3232L | |
| Agarose | Sigma | A5093-100G | |
| Boric Acid | Sigma | B0394-500G | |
| dNTP mix (10 mM each) | ThermoFisher Scientific | R0192 | |
| EDTA solution (0.5 M, pH 8.0) | AmericanBio | AB00502-01000 | |
| Ethanol (200 Proof) | Fisher Scientific | 16-100-824 | |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma | E4884-500G | |
| Lithium acetate dihydrate | Sigma | L6883-250G | |
| MyCycler Thermal Cycler | BioRad | 170-9703 | |
| Poly(ethylene glycol) (PEG) | Sigma | P3640-1KG | |
| PrimeSTAR HS DNA Polymerase (high fidelity DNA polymerase) and 5x buffer | Fisher Scientific | 50-443-960 | |
| Salmon sperm DNA solution | ThermoFisher Scientific | 15632-011 | |
| Sigma 7-9 (Tris base, powder form) | Sigma | T1378-1KG | |
| Sodium acetate trihydrate | Sigma | 236500-500G | |
| Supra Sieve GPG Agarose (low metling temperature agarose) | AmericanBio | AB00985-00100 | |
| Taq Polymerase and 10x Buffer | New England BioLabs | M0273X | |
| Toothpicks | Fisher Scientific | S67859 | |
| Tris-HCl (1 M, pH 8.0) | AmericanBio | AB14043-01000 | |
| a-D(+)-Glucose | Fisher Scientific | AC170080025 | for yeast media |
| Agar | Fisher Scientific | DF0140-01-0 | for yeast media |
| Peptone | Fisher Scientific | DF0118-07-2 | for YPD medium |
| Yeast Extract | Fisher Scientific | DF0127-17-9 | for YPD medium |
| 4-aminobenzoic acid | Sigma | A9878-100G | for complete minimal dropout medium |
| Adenine | Sigma | A8626-100G | for complete minimal dropout medium |
| Glycine hydrochloride | Sigma | G2879-100G | for complete minimal dropout medium |
| L-Alanine | Sigma | A7627-100G | for complete minimal dropout medium |
| L-Arginine monohydrochloride | Sigma | A5131-100G | for complete minimal dropout medium |
| L-Asparagine monohydrate | Sigma | A8381-100G | for complete minimal dropout medium |
| L-Aspartic acid sodium salt monohydrate | Sigma | A6683-100G | for complete minimal dropout medium |
| L-Cysteine hydrochloride monohydrate | Sigma | C7880-100G | for complete minimal dropout medium |
| L-Glutamic acid hydrochloride | Sigma | G2128-100G | for complete minimal dropout medium |
| L-Glutamine | Sigma | G3126-100G | for complete minimal dropout medium |
| L-Histidine monohydrochloride monohydrate | Sigma | H8125-100G | for complete minimal dropout medium |
| L-Isoleucine | Sigma | I2752-100G | for complete minimal dropout medium |
| L-Leucine | Sigma | L8000-100G | for complete minimal dropout medium |
| L-Lysine monohydrochloride | Sigma | L5626-100G | for complete minimal dropout medium |
| L-Methionine | Sigma | M9625-100G | for complete minimal dropout medium |
| L-Phenylalanine | Sigma | P2126-100G | for complete minimal dropout medium |
| L-Proline | Sigma | P0380-100G | for complete minimal dropout medium |
| L-Serine | Sigma | S4500-100G | for complete minimal dropout medium |
| L-Threonine | Sigma | T8625-100G | for complete minimal dropout medium |
| L-Tryptophan | Sigma | T0254-100G | for complete minimal dropout medium |
| L-Tyrosine | Sigma | T3754-100G | for complete minimal dropout medium |
| L-Valine | Sigma | V0500-100G | for complete minimal dropout medium |
| myo-Inositol | Sigma | I5125-100G | for complete minimal dropout medium |
| Uracil | Sigma | U0750-100G | for complete minimal dropout medium |
| Ammonium Sulfate | Fisher Scientific | A702-500 | for complete minimal dropout medium |
| Yeast Nitrogen Base | Fisher Scientific | DF0919-07-3 | for complete minimal dropout medium |
| 5-Fluoroorotic acid (5-FOA) | AmericanBio | AB04067-00005 | for 5-FOA medium |
Referenzen
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389 (6648), 251-260 (1997).
- Campos, E. I., Reinberg, D. Histones: annotating chromatin. Annu Rev Genet. 43, 559-599 (2009).
- Rando, O. J., Winston, F. Chromatin and transcription in yeast. Genetics. 190 (2), 351-387 (2012).
- Duina, A. A., Miller, M. E., Keeney, J. B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 197 (1), 33-48 (2014).
- Storici, F., Resnick, M. A. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y). 25, 189-207 (2003).
- Gray, M., Kupiec, M., Honigberg, S. M. Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7 (2004).
- Erdeniz, N., Mortensen, U. H., Rothstein, R. Cloning-free PCR-based allele replacement methods. Genome Res. 7 (12), 1174-1183 (1997).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
- Lundblad, V., Hartzog, G., Moqtaderi, Z. Manipulation of cloned yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Hoffman, C. S. Preparation of yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Treco, D. A., Lundblad, V. Preparation of yeast media. Curr Protoc Mol Biol. Chapter 13, (2001).
- Lederberg, J., Lederberg, E. M. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 63 (3), 399-406 (1952).
- Sikorski, R. S., Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122 (1), 19-27 (1989).
- Johnson, P., et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 5 (5), 741-749 (2015).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336-4343 (2013).
- Cross, S. L., Smith, M. M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol. 8 (2), 945-954 (1988).
- Libuda, D. E., Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 443 (7114), 1003-1007 (2006).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten