
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Visadas
Neste Artigo
Resumo
A strategy for generating mutations in histone genes at their endogenous location in Saccharomyces cerevisiae is presented.
Resumo
We describe a PCR- and homologous recombination-based system for generating targeted mutations in histone genes in budding yeast cells. The resulting mutant alleles reside at their endogenous genomic sites and no exogenous DNA sequences are left in the genome following the procedure. Since in haploid yeast cells each of the four core histone proteins is encoded by two non-allelic genes with highly homologous open reading frames (ORFs), targeting mutagenesis specifically to one of two genes encoding a particular histone protein can be problematic. The strategy we describe here bypasses this problem by utilizing sequences outside, rather than within, the ORF of the target genes for the homologous recombination step. Another feature of this system is that the regions of DNA driving the homologous recombination steps can be made to be very extensive, thus increasing the likelihood of successful integration events. These features make this strategy particularly well-suited for histone gene mutagenesis, but can also be adapted for mutagenesis of other genes in the yeast genome.
Introdução
Os quatro proteínas histonas centrais H2A, H2B, H3, H4 e desempenham papéis centrais na compactação, organização e função dos cromossomas eucarióticos. Dois conjuntos de cada uma destas histonas formar o octâmero da histona, um carretel molecular que dirige o invólucro de ~ 147 pares de bases de ADN em torno de si, resultando na formação de um nucleossoma 1. Nucleossomas são agentes activos numa variedade de processos à base de cromossomas, tais como a regulação da transcrição de genes e a formação de eucromatina e heterocromatina entre cromossomas, e, como tal, têm sido o foco de intensa investigação ao longo de várias décadas passadas. Um número de mecanismos têm sido descritos por nucleossomas que podem ser manipulados em formas que podem facilitar a execução dos processos específicos - estes mecanismos de modificação pós-tradução incluem os resíduos de histonas, remodelar nucleossoma dependente de ATP, e reorganização nucleossoma ATP-independentee montagem / desmontagem 2, 3.
A levedura de germinação Saccharomyces cerevisiae é um organismo particularmente poderoso modelo para a compreensão da função da histona em eucariotas. Isto pode ser em grande parte atribuído ao elevado grau de conservação evolutiva das proteínas histona eukarya todo o domínio e a receptividade de levedura para uma variedade de genética e bioquímica experimental aproxima 4. abordagens Reverse-genéticos nas leveduras têm sido amplamente utilizada para estudar os efeitos de mutações específicas de histona sobre vários aspectos da biologia da cromatina. Para estes tipos de experiências é frequentemente preferível a utilização de células em que as histonas mutantes são expressos a partir de loci genómico nativo, como a expressão a partir de plasmídeos autónomos podem levar a níveis anormais intracelular de proteínas histona (devido a diferentes números de plasmídeos em células) e alteração concomitante da cromatina enam-, que em última instância pode confundir a interpretação dos resultados.
Aqui, nós descrevemos uma técnica baseada em PCR que permite a mutagénese direccionada de genes de histonas nas suas localizações genómicas nativas, que não requerem um passo de clonagem e resulta na produção da mutação (ões) desejada, sem sequências de ADN exógeno que sobram no genoma. Esta técnica tira proveito do sistema de recombinação homóloga eficiente em levedura e tem várias características em comum com outras técnicas semelhantes desenvolvidas por outros grupos - mais notavelmente o Delitto Perfetto, genômica mutagénese específica (SSG), e alelo à base de PCR clonagem-livre métodos de reposição de 5, 6, 7. No entanto, a técnica que descrevem tem um aspecto que o torna particularmente adequado para a mutagénese de genes de histonas. Em células de levedura haplóides, cada um dos quatro histonas nucleares é codificada por dois não-Agenes llelic e altamente homólogas: por exemplo, histona H3 é codificado pelos genes HHT1 e HHT2, e as grelhas de leitura abertas (ORFs) dos dois genes são mais de 90% idênticos na sequência. Este elevado grau de homologia pode complicar experiências concebidas para visar especificamente um dos dois genes codificadores de histonas para mutagénese. Considerando que os métodos acima mencionados exigem frequentemente a utilização de, pelo menos, algumas sequências dentro da ORF do gene alvo para dirigir recombinação homóloga, a técnica aqui descrita faz uso das sequências que flanqueiam as ORFs dos genes de histonas (que partilhem menos homologia de sequência) para o passo de recombinação, aumentando assim a probabilidade de sucesso de mutagénese direccionamento ao local desejado. Além disso, as regiões homólogas que impulsionam a recombinação pode ser muito extensa, contribuindo para a eficiente recombinação homóloga alvo.
Protocolo
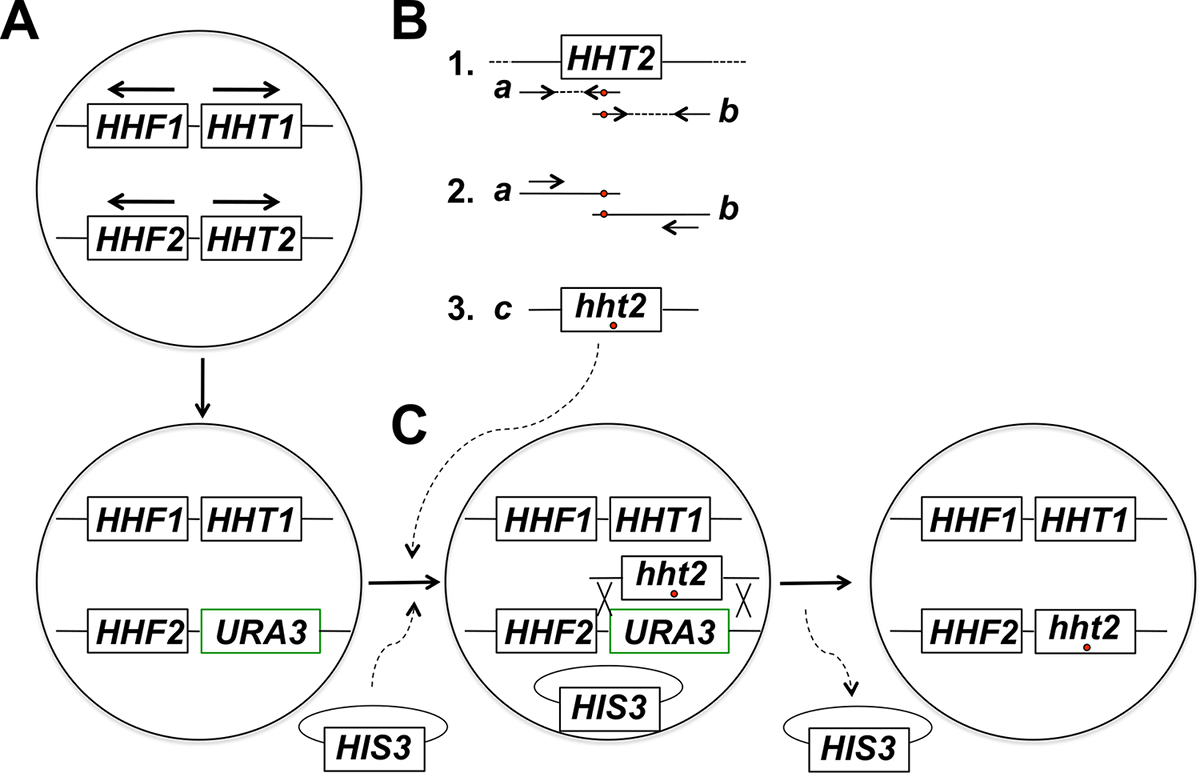
NOTA: A estratégia experimental para o alvo de mutagénese in situ do gene histona inclui várias etapas (resumidos na Figura 1). Estes passos incluem: (1) A substituição do gene alvo de histona com o gene URA3, (2) Geração e purificação de produtos de PCR correspondentes a dois fragmentos que se sobrepõem parcialmente do gene histona alvo utilizando iniciadores que albergam a mutação desejada (s), (3 ) fusão por PCR de dois fragmentos que se sobrepõem parcialmente de obter produtos de PCR completa tamanho para a integração, (4) co-transformação de produtos de PCR de tamanho completo e plasmídeo de esqueleto, e de selecção para o marcador no plasmídeo, (5) tela para-resistente a 5-FOA transformantes, (6) purificação de colónias resistentes a 5-FOA e perda de plasmídeo de esqueleto, e (7) as análises moleculares para ensaio de integração correcta do alelo mutante.

Figura 1: Visão geral da Estratégia para alvejado in situ Mutagênese de Genes histona em brotamento levedura. Neste exemplo, o gene alvo é HHT2, mas qualquer outro gene de histona de núcleo também pode ser mutagenizados usando esta estratégia. Células de levedura haplóides (A) abrigar dois genes da histona-codifica H3 (HHT1 e HHT2) e dois genes que codificam para histona H4 (HHF1 e HHF2) dispostos como mostrado na figura (os genes HHT1 e HHF1 estão localizados no cromossoma II e o HHT2 e genes HHF2 estão localizados no cromossoma XIV - em cada caso, as setas apontam na direcção da transcrição). No primeiro passo do processo, a ORF do gene HHT2 é substituído com o gene URA3, dando origem a uma estirpe hht2Δ :: URA3. (B) Na parte 1, uma cópia de tipo selvagem do gene HHT2 a partir de uma amostra de ADN genómico é utilizado como molde para duas reacções de PCR para géneroste os dois fragmentos que se sobrepõem parcialmente do gene. O iniciador inverso para a primeira reacção inclui um ou mais nucleótidos desemparelhados (indicado por um círculo vermelho) que correspondem à mutação (ões) desejada a ser introduzida no genoma. O iniciador directo para a segunda reação tem a incompatibilidade equivalente em uma configuração complementar reversa (também indicado com um círculo vermelho). Os dois produtos de PCR gerados na parte 1 (produtos a e b) são então utilizados como moldes para PCR de fusão utilizando dois iniciadores que se ligam aos produtos a e b na forma mostrados na parte 2. Isto resulta na geração de PCR de tamanho completo produtos (produto C na parte 3) que abrigam a mutação desejada (s). (C) A estirpe hht2Δ :: URA3 é, em seguida, co-transformada com os produtos de PCR de tamanho completo e um plasmídeo de esqueleto (um plasmídeo HIS3- marcado neste exemplo), e as células são seleccionadas quanto à presença do plasmídeo (em meios carentes histidine neste exemplo). Os transformantes são então pesquisadas para 5-FOA resistência - células resistentes são candidatos a ter sido submetido a um acontecimento de recombinação homóloga conduz à integração do produto de PCR e a excisão do gene URA3, como mostrado. subsequente perda do plasmídeo de esqueleto por divisão mitótica da célula leva à histona desejada estirpe mutante final. Encontraram-se que a selecção do plasmídeo de esqueleto seguido por rastreio para resultados de resistência 5-FOA numa frequência muito mais elevada de identificação de eventos de integração correctos em comparação com a selecção directa em placas de 5-FOA, que principalmente identifica células que adquiriram mutações URA3 espontâneas. (Esta figura foi modificado a partir da referência 14). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
1. A substituição da histona do gene alvo com o gene URA3Gene
- Realizar padrão mediada por PCR de uma etapa de disrupção do gene substituindo a ORF do gene da histona-alvo com o gene URA3 de 8, 9.
NOTA: O uso de células de levedura que transportam o ura3Δ0 é recomendada como esta mutação remove toda a ORF URA3 endógena, evitando assim a integração do produto de PCR para o gene URA3 locus de 8. Alternativamente, o gene de K. lactis URA3 pode ser utilizado eficazmente para a geração da substituição da histona em todo o fundo da URA3 como é funcional em S. cerevisiae, mas tem apenas homologia de sequência parcial com o gene URA3 de S. cerevisiae. A estirpe também deve ser auxotrófica para, pelo menos, um composto que irá permitir a selecção do plasmídeo de esqueleto na experiência de transformação (ver passo 4 deste protocolo). Este passo não é necessário se um alvo de histonas geneΔ :: URA3estirpe já está disponível.
2. Geração e purificação de produtos de PCR correspondentes a dois fragmentos que se sobrepõem parcialmente da histona do gene alvo utilizando iniciadores que albergam a mutação desejada (s)
- Gerar produtos de PCR correspondentes a dois fragmentos que se sobrepõem parcialmente de histona do gene alvo.
- Prepare a duas reacções de PCR como se segue:
- Para gerar produtos de PCR que corresponde à primeira metade do gene (produto A na Figura 1B), definir-se a reacção seguinte: 1 uL de ADN molde, 5 μl10 uM iniciador directo, 5 μl10 uM iniciador inverso, 0,5 ul (1,25 U) termoestável a polimerase de ADN, 10 uL de tampão de polimerase de ADN de 5x, 5 ul de mistura de dNTP (2 mM de cada), e 23,5 jil dH2O
NOTA: O molde de ADN pode ser ADN genómico derivado de uma estirpe de tipo selvagem para o gene alvo de histona isolado utilizando pró padrãoprocedimentos 10. Para ter em conta as variações na concentração de ADN e nível de impurezas em preparações diferentes genómicas, recomenda-se a optimizar as reacções utilizando um ADN não diluído ou diferentes diluições das preparações genómicas (por exemplo, 1:10 e 1: 100). O iniciador directo deve hibridar com uma região a montante do gene alvo. O iniciador inverso deve emparelhar dentro do ORF, ser ~ 40 nucleotídeos de comprimento, e conter a mutação desejada (s) em algum lugar no meio disso (veja a Figura secção 1B-1 e resultados representativos para exemplos). A utilização de uma polimerase DNA de alta fidelidade é recomendado, a fim de reduzir as taxas de mutações indesejáveis durante a síntese dos produtos de PCR. - Para gerar produtos de PCR que corresponde à segunda metade do gene (b produto na Figura 1B), definir-se uma reacção, como indicado em 2.1.1.1, mas com diferentes iniciadores.
NOTA: A pri frentemer devem emparelhar dentro do ORF, ser ~ 40 nucleótidos de comprimento, e contêm a mutação desejada (s), no meio do mesmo. Note-se que a mutação (s) neste iniciador é o complemento reverso da mutação (ões) na sequência iniciadora reversa no passo 2.1.1.1. O iniciador inverso deve hibridar com uma região a jusante do gene alvo (ver Figura secção 1B-1 e resultados representativos para exemplos).
- Para gerar produtos de PCR que corresponde à primeira metade do gene (produto A na Figura 1B), definir-se a reacção seguinte: 1 uL de ADN molde, 5 μl10 uM iniciador directo, 5 μl10 uM iniciador inverso, 0,5 ul (1,25 U) termoestável a polimerase de ADN, 10 uL de tampão de polimerase de ADN de 5x, 5 ul de mistura de dNTP (2 mM de cada), e 23,5 jil dH2O
- Coloque as reacções num termociclador com as seguintes definições: 94 ° C 30 s; 30 ciclos de as seguintes definições: 98 C 10 s, 60 ° C 5 segundos, 72 ° C 1,5 min; e 72 ° C 10 min.
Nota: Optimização dos parâmetros de PCR pode não ser necessária para conjuntos de iniciadores específicos e alvo do gene da histona.
- Prepare a duas reacções de PCR como se segue:
- Executar 20 - 50 ul do material a partir das reacções de PCR sobre um gel de agarose de baixo ponto de fusão a 0,9% em 89 mM de base Tris, ácido bórico 89 mM, 2,5 mM de EDTA (TBE) de tampão.
- secções de gel de agarose contendo a cortar PCR PRodutos de gel utilizando um bisturi ou lâmina de barbear limpo e transferir cada um para um tubo de microcentrífuga de 1,5 ml. Armazenar seções de agarose contendo produtos de PCR a -20 ° C até estar pronto para usar.
3. Fusão PCR dos dois fragmentos que se sobrepõem parcialmente obter Full Size produtos de PCR para a Integração
- Prepare modelo para reacções de PCR
- Derreter secções de gel de agarose do passo 2.3, colocando os tubos de microcentrífuga num conjunto de bloco de calor a 65 ° C durante 5 minutos (ou até totalmente fundido). tubos de vórtice cada 1 - 2 minutos para facilitar o processo de fusão.
- Transferir uma quantidade de agarose fundida a partir de cada amostra (por exemplo, 50 ul de cada, para um total de 100 ul) para um único tubo de microcentrífuga e misturar por vortex. Utilizar esta como molde nas reacções de PCR de fusão. Colocar o tubo a -20 ° C até estar pronto para usar.
- Amplificar uma grande quantidade de tamanho grande produto de PCR (produto cna Figura 1B)
- Defina-se seis reacções de PCR, cada um com os seguintes componentes: ADN molde 2 ul, 10 ul de 10 mM de iniciadores para a frente, 10 ul de 10 mM iniciador de sentido reverso, um ul (2,5 U) de polimerase de ADN termoestável, 20 de tampão de polimerase de ADN ul 5x, 10 ul mistura de dNTP (2 mM de cada), e 47 uL de dH 2 O.
NOTA: O número de reacções pode ser alterada dependendo da eficiência da PCR. O ADN molde (ver 3.1.2) deve ser aquecido a 65 ° C até derreter, misturado por vórtex, e adicionado por último à mistura de reacção de PCR. Uma vez adicionado, misture delicadamente mas completamente por pipetagem a solução cima e para baixo várias vezes. Para ter em conta as variações na concentração de DNA nas diferentes amostras, recomenda-se a primeira optimizar as reacções usando um molde não diluído ou diferentes diluições do modelo (por exemplo, 1:10 e 1: 100). Os dois iniciadores utilizados devem emparelhar com os dois fragmentos que se sobrepõem parcialmente de o gene alvo como malustrated na Figura 1B-2 e ser concebido de tal modo que os produtos de PCR finais terão pelo menos 40 pares de bases de cada lado homóloga às regiões que flanqueiam o gene URA3 ORF que irá conduzir o passo de recombinação homóloga (ver a secção de resultados representativos para exemplos). A utilização de uma polimerase DNA de alta fidelidade é recomendado, a fim de reduzir as taxas de mutações indesejáveis durante a síntese dos produtos de PCR. - Colocar os tubos em um termociclador com as seguintes definições: 94 ° C 30 s; 30 ciclos de as seguintes definições: 98 C 10 seg, 50 C 15 seg, 72 ° C 1,5 min; e 72 ° C 10 min.
NOTA: Optimização dos parâmetros de PCR pode não ser necessária para conjuntos de iniciadores específicos do gene alvo e histona.
- Defina-se seis reacções de PCR, cada um com os seguintes componentes: ADN molde 2 ul, 10 ul de 10 mM de iniciadores para a frente, 10 ul de 10 mM iniciador de sentido reverso, um ul (2,5 U) de polimerase de ADN termoestável, 20 de tampão de polimerase de ADN ul 5x, 10 ul mistura de dNTP (2 mM de cada), e 47 uL de dH 2 O.
4. Co-transformação da Full Size produtos de PCR e Backbone plasmídeo e selecção para Marcador no plasmídeo
- A concentração dos produtos de PCR
- Reúnem-se os sreações ix PCR (600 no total ul) a partir do passo 3.2.2 em um único tubo de microcentrífuga e misturar em vortex.
- Dividir a amostra em três 200 mL alíquotas em tubos de microcentrífuga. Precipitar o ADN em cada tubo por adição de 20 ul de acetato de sódio 3M (pH 5,2) e 550 ul de etanol a 100%. Misture a solução cuidadosamente e colocar em gelo durante pelo menos 15 min. Recolha de ADN por centrifugação a 14000 xg durante ~ 10 min, lavar o sedimento com 200 ul de etanol a 70%, e secar ao ar.
- Ressuspender cada pelete de ADN em 25 uL de dH2O, e reunir num único tubo (para um total de 75 uL).
- Levedura de co-transformação
- Prepare 10 ml de cultura durante a noite da estirpe gerado na seção 1 em extrato de levedura peptona dextrose (YPD) líquido do meio 11.
- Na manhã seguinte, inocular 400 ml de meio YPD líquido com 8 ml da cultura durante a noite saturado e incubar por agitaçãoa 30 ° C durante 4 - 5 h, para permitir que as células entram na fase logarítmica de crescimento.
- Recolher as células por centrifugação a ~ 3220 xg durante 10 min, descartar o meio líquido, e voltar a suspender as células em um volume de Tris-HCl a 10 (pH 8,0), EDTA 1 mM, solução 0,1 M de acetato de lítio (TE / LiAc) .
- Recolher as células por centrifugação a 3220 xg durante ~ 10 min, e o descarte de TE / LiAc.
- Ressuspender as células em 1 ml de TE / LiAc.
- Defina-se o seguinte cocktail de reacção num tubo de microcentrífuga: 800 ul de células a partir do passo 4.2.5, 40 ul de 10 mg de ADN de esperma de fervida / ml de salmão, um total de 12,5 ug de ADN plasmídeo de esqueleto, e 75 ul de produto de PCR concentrada a partir do passo 4.1.3.
NOTA: ADN de esperma de salmão deve ser fervidas durante 5 minutos e colocada em gelo durante pelo menos 5 minutos antes de utilização na reacção. O volume total de ADN de plasmídeo de esqueleto adicionada deve ser mantido a um mínimo (~ 80 ul ou menos). Consulte a seção de resultados representativos para um exemplo de um plasma de backboneidentidade. - Misture o tubo de cocktail completamente e alíquota de maneira uniforme em oito microtubos (tubos 1-8).
- Configurar as duas seguintes tubos de reacção de transformação de controle:
- Tubo 9 (sem controle de produto de PCR): 100 ul de células a partir do passo 4.2.5, 5 ul de 10 mg de ADN / ml de esperma de salmão fervido (fervida durante 5 min; ver passo 4.2.6 Nota), um total de 1,56 g de DNA plasmídeo backbone e nenhum produto PCR acrescentou.
- Tubo de 10 (sem controle de DNA): 100 ml de células a partir do passo 4.2.5, 5 mL de 10 mg mL DNA fervida / esperma de salmão (veja o passo 4.2.6 Nota), não plasmídeo backbone DNA adicionado, e nenhum produto PCR acrescentou.
- Misture os dois tubos suave mas completamente por pipetagem cima e para baixo várias vezes.
- Incubar os tubos de dez a 30 ° C durante 30 min.
- Para cada tubo, adicionar 1,2 ml de 40% de polietileno glicol (PEG 3350) em TE / LiAc. Misturar bem utilizando uma pipeta P-1000 até a solução ficar homogénea.
- Incubar os tubos a 30 dez° C durante 30 min. Misturar suavemente a solução pipetando para cima e para baixo e, em seguida, incubar os tubos a 42 ° C durante 15 min.
- Recolher as células por centrifugação dos tubos numa microcentrífuga a 14000 xg durante ~ 30 seg. Descartar o líquido e ressuspender as células em 1 ml de dH 2 O. estéril
- Recolher as células por centrifugação dos tubos numa microcentrífuga a 14000 xg durante ~ 30 seg. Descartar o líquido e ressuspender as células em 500 uL de dH 2 O. estéril
- tubos Piscina 1-8 juntos (volume total de 4 ml) e misture bem por pipetagem cima e para baixo.
- Placa de 200 ul da mistura anterior em cada um de vinte placas de meio completo mínima de evasão 11 (placas 1-20) para a selecção do plasmídeo de esqueleto.
- Placa 200 ul da mistura de tubo 9 e 200 mL de mistura de tubo 10 cada em seu próprio prato selecção (placas 21 e 22, respectivamente).
- Incubar as placas a 22 30 ° C durante 3 - 5 dias paraseleccionar os transformantes plasmídeo.
- Inspeccionar placas de transformação ao fim de 3 - 5 dias de incubação. Aproximadamente 5.000 colónias devem ser visíveis em placas de 1-21 (ver resultados representativos para um exemplo) e não colónias deve estar presente na placa 22.
5. Tela de transformantes resistentes à 5-FOA
- Transferência de células a partir de placas de 1 - 20 (e da placa de transformação 21 como um controlo) a 5-fluoroor�ico ácido (5-FOA) placas de 11 por réplica-plaqueamento 12, a fim de avaliar a perda do gene URA3 como um resultado da integração do os produtos de PCR no local desejado.
- Remover a tampa de placa e pressionar a placa contendo as colónias em um veludo esterilizado. Transferir as células a partir da placa de veludo com um 5-FOA, pressionando a placa sobre a veludo. Incubar as placas a 30 C durante 2 dias.
- Após a 2 dias de incubação, examine cuidadosamente as placas 5-FOA para growth.
NOTA: Um evento de integração candidato será representada por um pequeno assimétrica de colónias "achatado" numa placa de 5-FOA - Por outro lado, pequenas papilas crescer em placas de 5-FOA são susceptíveis representante de mutações URA3 espontâneas que surgiram durante o crescimento de colónias na placas de transformação, e são, portanto, pouco provável para representar o evento de integração desejado (veja a Figura 3 na seção resultados representativos para posterior elaboração sobre este ponto e para alguns exemplos).
6. Purificação de colónias resistentes a 5-FOA e perda de plasmídeo Backbone
- Usando palitos estéreis, escolher as colônias candidatos das placas 5-FOA descritos no passo 5.2 e raia para as colónias individuais em placas de YPD. Incubar durante 2 - 3 dias a 30 ° C.
- Após a incubação, réplica - placa cada purificação de placa YPD a uma placa YPD fresco, uma placa de drop-out sem uracilo para verificar se há perdado gene URA3, e uma segunda placa de desistência para monitorizar a presença ou ausência do plasmídeo de esqueleto. Incubar durante 1 - 2 dias a 30 ° C.
- Após a incubação, identificar uma colónia de cada amostra candidato que está crescendo sobre a placa de YPD, mas não a crescer em cada placa de abandono (tal colónia é esperado que perderam o gene URA3 através do acontecimento de recombinação e perdido o plasmídeo de esqueleto durante mitótico divisão celular). Restreak tais colônias em placas de YPD frescos. Estas colónias são os candidatos de integração e será analisado ainda na etapa 7.
7. As análises moleculares para testar a integração adequada do Mutant Alelo
- Isolar o ADN genómico a partir de amostras candidatos utilizando procedimentos padrão 10.
- Amplificar a região genómica que engloba o local alvo.
- Configure a seguinte reação de PCR para cada amostra: 0,5 DNA template l, 5 ul10 uM de iniciadores para a frente, 5 ul de 10 mM iniciador inverso, 0,5 ul (2,5 unidades) de Taq ADN polimerase, 5 ul de ADN de Taq 10x tampão de polimerase, mistura de dNTP 5 ul (2 mM de cada), e 29 uL de dH 2 O.
NOTA: ADN matriz é o DNA genómico derivadas das amostras candidatos. Recomenda-se a incluir também duas reacções de controlo: um utilizando ADN genómico derivados dos geneΔ histona originais :: URA3 estirpe como molde e outro utilizando ADN genómico de uma estirpe de histona de tipo selvagem como molde. Para ter em conta as variações na concentração de ADN e nível de impurezas em preparações diferentes genómicas, recomenda-se a optimizar as reacções utilizando um ADN não diluído ou diferentes diluições das preparações genómicas (por exemplo, 1:10 e 1: 100). É importante ter a certeza de que estes iniciadores emparelham com as sequências de DNA fora da região englobados pelo produto de PCR putativamente integrado - Deste modo, o tamanho dos produtos de PCR nos estes reactions pode ser utilizado como uma ferramenta de diagnóstico para a integração dos produtos no local genómico correcto (ver resultados representativos para um exemplo). - Coloque as reacções num termociclador com as seguintes definições: 94 ° C 3 min; 30 ciclos de as seguintes definições: 94 ° C 45 s, 50 ° C 45 s, 72 ° C 2 min; e 72 ° C 10 min.
NOTA: Optimização dos parâmetros de PCR pode não ser necessária para conjuntos de iniciadores específicos do gene alvo e histona.
- Configure a seguinte reação de PCR para cada amostra: 0,5 DNA template l, 5 ul10 uM de iniciadores para a frente, 5 ul de 10 mM iniciador inverso, 0,5 ul (2,5 unidades) de Taq ADN polimerase, 5 ul de ADN de Taq 10x tampão de polimerase, mistura de dNTP 5 ul (2 mM de cada), e 29 uL de dH 2 O.
- Processamento de produtos de PCR
- Executar 20 uL de cada reacção sobre um gel de agarose a 0,8% TBE.
- Avaliar o tamanho dos produtos de PCR utilizando padrões de ADN como uma referência para determinar se o gene URA3 foi substituída com sucesso pelo gene histona putativamente mutado (ver resultados representativos para um exemplo).
NOTA: Em certos casos, a mutação desejada (s) introduzido nos genes da histona criar ou destruir uma restrição sentare. Se este for o caso, a presença da mutação desejada nos produtos de PCR do tamanho indicativo da correcta integração pode ser avaliada sujeitando os produtos de digestão com a enzima de restrição correspondente, seguido por análise de electroforese em gel (ver resultados representativos para um exemplo) . - Os produtos de PCR do tamanho Assunto indicativo de integração correcta para a sequenciação de ADN para confirmar a presença da mutação desejada (s) e para garantir que não há mutações adicionais foram introduzidas no genoma.
Resultados
Descrevemos a geração de um alelo hht2 expressando uma proteína mutante da histona H3 abrigando uma substituição na posição 53 a partir de uma arginina para um ácido glutâmico (H3-R53E mutante) como um exemplo representativo do alvo na estratégia de mutagénese in situ.
Geramos uma estirpe em que toda a ORF de HHT2 é substituído pelo gene URA3 (veja o passo 1 do...
Discussão
O nível elevado de homologia de sequências entre os dois genes não alélicos que codificam para cada uma das quatro proteínas histona de núcleo em células haplóides de S. cerevisiae pode representar um desafio para os investigadores que desejam atingir especificamente um dos dois genes para a mutagénese. Anteriormente descreveu metodologias de mutagénese de levedura, incluindo a Delitto Perfetto, genómico de mutagénese específica do local (SSG), e métodos de substituição alelo baseada em...
Divulgações
The authors declare that they have no competing financial interests.
Agradecimentos
We thank Reine Protacio for helpful comments during the preparation of this manuscript. We express our gratitude to the National Science Foundation (grants nos. 1243680 and 1613754) and the Hendrix College Odyssey Program for funding support.
Materiais
| Name | Company | Catalog Number | Comments |
| 1 kb DNA Ladder (DNA standards) | New England BioLabs | N3232L | |
| Agarose | Sigma | A5093-100G | |
| Boric Acid | Sigma | B0394-500G | |
| dNTP mix (10 mM each) | ThermoFisher Scientific | R0192 | |
| EDTA solution (0.5 M, pH 8.0) | AmericanBio | AB00502-01000 | |
| Ethanol (200 Proof) | Fisher Scientific | 16-100-824 | |
| Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) | Sigma | E4884-500G | |
| Lithium acetate dihydrate | Sigma | L6883-250G | |
| MyCycler Thermal Cycler | BioRad | 170-9703 | |
| Poly(ethylene glycol) (PEG) | Sigma | P3640-1KG | |
| PrimeSTAR HS DNA Polymerase (high fidelity DNA polymerase) and 5x buffer | Fisher Scientific | 50-443-960 | |
| Salmon sperm DNA solution | ThermoFisher Scientific | 15632-011 | |
| Sigma 7-9 (Tris base, powder form) | Sigma | T1378-1KG | |
| Sodium acetate trihydrate | Sigma | 236500-500G | |
| Supra Sieve GPG Agarose (low metling temperature agarose) | AmericanBio | AB00985-00100 | |
| Taq Polymerase and 10x Buffer | New England BioLabs | M0273X | |
| Toothpicks | Fisher Scientific | S67859 | |
| Tris-HCl (1 M, pH 8.0) | AmericanBio | AB14043-01000 | |
| a-D(+)-Glucose | Fisher Scientific | AC170080025 | for yeast media |
| Agar | Fisher Scientific | DF0140-01-0 | for yeast media |
| Peptone | Fisher Scientific | DF0118-07-2 | for YPD medium |
| Yeast Extract | Fisher Scientific | DF0127-17-9 | for YPD medium |
| 4-aminobenzoic acid | Sigma | A9878-100G | for complete minimal dropout medium |
| Adenine | Sigma | A8626-100G | for complete minimal dropout medium |
| Glycine hydrochloride | Sigma | G2879-100G | for complete minimal dropout medium |
| L-Alanine | Sigma | A7627-100G | for complete minimal dropout medium |
| L-Arginine monohydrochloride | Sigma | A5131-100G | for complete minimal dropout medium |
| L-Asparagine monohydrate | Sigma | A8381-100G | for complete minimal dropout medium |
| L-Aspartic acid sodium salt monohydrate | Sigma | A6683-100G | for complete minimal dropout medium |
| L-Cysteine hydrochloride monohydrate | Sigma | C7880-100G | for complete minimal dropout medium |
| L-Glutamic acid hydrochloride | Sigma | G2128-100G | for complete minimal dropout medium |
| L-Glutamine | Sigma | G3126-100G | for complete minimal dropout medium |
| L-Histidine monohydrochloride monohydrate | Sigma | H8125-100G | for complete minimal dropout medium |
| L-Isoleucine | Sigma | I2752-100G | for complete minimal dropout medium |
| L-Leucine | Sigma | L8000-100G | for complete minimal dropout medium |
| L-Lysine monohydrochloride | Sigma | L5626-100G | for complete minimal dropout medium |
| L-Methionine | Sigma | M9625-100G | for complete minimal dropout medium |
| L-Phenylalanine | Sigma | P2126-100G | for complete minimal dropout medium |
| L-Proline | Sigma | P0380-100G | for complete minimal dropout medium |
| L-Serine | Sigma | S4500-100G | for complete minimal dropout medium |
| L-Threonine | Sigma | T8625-100G | for complete minimal dropout medium |
| L-Tryptophan | Sigma | T0254-100G | for complete minimal dropout medium |
| L-Tyrosine | Sigma | T3754-100G | for complete minimal dropout medium |
| L-Valine | Sigma | V0500-100G | for complete minimal dropout medium |
| myo-Inositol | Sigma | I5125-100G | for complete minimal dropout medium |
| Uracil | Sigma | U0750-100G | for complete minimal dropout medium |
| Ammonium Sulfate | Fisher Scientific | A702-500 | for complete minimal dropout medium |
| Yeast Nitrogen Base | Fisher Scientific | DF0919-07-3 | for complete minimal dropout medium |
| 5-Fluoroorotic acid (5-FOA) | AmericanBio | AB04067-00005 | for 5-FOA medium |
Referências
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 389 (6648), 251-260 (1997).
- Campos, E. I., Reinberg, D. Histones: annotating chromatin. Annu Rev Genet. 43, 559-599 (2009).
- Rando, O. J., Winston, F. Chromatin and transcription in yeast. Genetics. 190 (2), 351-387 (2012).
- Duina, A. A., Miller, M. E., Keeney, J. B. Budding yeast for budding geneticists: a primer on the Saccharomyces cerevisiae model system. Genetics. 197 (1), 33-48 (2014).
- Storici, F., Resnick, M. A. Delitto perfetto targeted mutagenesis in yeast with oligonucleotides. Genet Eng (N Y). 25, 189-207 (2003).
- Gray, M., Kupiec, M., Honigberg, S. M. Site-specific genomic (SSG) and random domain-localized (RDL) mutagenesis in yeast. BMC Biotechnol. 4, 7 (2004).
- Erdeniz, N., Mortensen, U. H., Rothstein, R. Cloning-free PCR-based allele replacement methods. Genome Res. 7 (12), 1174-1183 (1997).
- Brachmann, C. B., et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 14 (2), 115-132 (1998).
- Lundblad, V., Hartzog, G., Moqtaderi, Z. Manipulation of cloned yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Hoffman, C. S. Preparation of yeast DNA. Curr Protoc Mol Biol. Chapter 13, (2001).
- Treco, D. A., Lundblad, V. Preparation of yeast media. Curr Protoc Mol Biol. Chapter 13, (2001).
- Lederberg, J., Lederberg, E. M. Replica plating and indirect selection of bacterial mutants. J Bacteriol. 63 (3), 399-406 (1952).
- Sikorski, R. S., Hieter, P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 122 (1), 19-27 (1989).
- Johnson, P., et al. A systematic mutational analysis of a histone H3 residue in budding yeast provides insights into chromatin dynamics. G3 (Bethesda). 5 (5), 741-749 (2015).
- DiCarlo, J. E., et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 41 (7), 4336-4343 (2013).
- Cross, S. L., Smith, M. M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol Cell Biol. 8 (2), 945-954 (1988).
- Libuda, D. E., Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature. 443 (7114), 1003-1007 (2006).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados