
Method Article
通过去除单颗粒低温电子显微镜最终堆栈中的大部分颗粒来增强密度图。
* 这些作者具有相同的贡献
摘要
一种先进的冷冻电镜颗粒选择方法,即 CryoSieve,通过去除最终堆栈中的大部分颗粒来提高密度图分辨率,正如其在真实世界数据集上的应用所证明的那样。
摘要
在过去的十年中,低温电子显微镜 (cryo-EM) 单颗粒分析 (SPA) 领域的技术和方法的进步大大提高了我们对生物大分子高分辨率结构检查的能力。这一进步开创了分子洞察的新时代,取代了 X 射线晶体学成为主导方法,并为生物学中长期存在的问题提供了答案。由于冷冻电镜不依赖于结晶,而结晶是 X 射线晶体学的一个重大限制,因此它可以捕获不同质量的颗粒。因此,颗粒的选择至关重要,因为所选颗粒的质量直接影响重建密度图的分辨率。一种创新的颗粒选择迭代方法,称为 CryoSieve,通过有效减少最终堆栈中的颗粒数量,显着提高了重建密度图的质量。实验证据表明,该方法可以消除最终堆栈中的大部分颗粒,从而显着提高密度图的质量。本文概述了这种方法的详细工作流程,并展示了其在真实数据集上的应用。
引言
低温电子显微镜(cryo-EM)单颗粒分析(SPA)已成为确定生物大分子高分辨率三维密度图的主要方法。由于一系列技术创新 1,2,3,4,5,6,称为分辨率革命 7,冷冻电子显微镜能够以前所未有的速度确定生物大分子的结构,最高可达原子分辨率。这一突破标志着分子洞察新时代的开始,取代了X射线晶体学成为主要技术,并回答了长期存在的生物学问题。
冷冻电子扫描与X射线晶体学不同,不需要生物大分子的结晶。取而代之的是,含有目标生物大分子的溶液在玻璃冰中迅速冷冻。然后用电子束对其进行成像,以产生一系列显微照片,从而绕过结晶的需要8。随后,利用颗粒拾取算法从这些显微照片中提取单个原始颗粒4,9,10,11,12。由于冷冻电镜不依赖于结晶,因此提取的颗粒主要受损或处于不希望的构象状态是很自然的,因此需要进行多轮颗粒选择才能获得高分辨率密度图。因此,在冷冻 EM SPA 图像处理中,颗粒选择对于获得高分辨率密度图至关重要13。
在冷冻 EM SPA 中,标准颗粒选择方法包括二维 (2D) 和三维 (3D) 分类14。二维分类将粒子分类为预定义数量的组,从而产生每个类别的平均图像和估计的 2D 分辨率。然后,研究人员可以目视检查这些类别,从较低分辨率的组中去除粒子,以使用剩余的粒子进行重建,以实现更高分辨率。一旦使用细化算法确定了粒子姿态,研究人员将进行 3D 分类,将粒子聚类到多个类别中。这样就可以对每一类重建的密度图进行目视检查,从而排除不需要的颗粒,例如来自不需要的构象的颗粒。经过多轮分类后,得到一个由相对高质量的颗粒组成的最终堆栈。这些最终堆栈有助于生成原子或近原子分辨率的密度图。
Zhu和她的同事们已经证明,可以在这些最终的堆栈上进行进一步的颗粒选择15。CryoSieve15 是一种创新的颗粒选择迭代方法,可用于通过显著减少颗粒数量来提高最终密度图的质量。虽然其他粒子分选标准和软件,如归一化互相关 (NCC) 方法16、角图一致性 (AGC) 方法17 和非对准分类5,目前已在该领域使用,但这种方法已被证明在有效性方面优于这些算法。
在这项研究中,我们提供了整个过程的详细指南。作为一个案例研究,我们将这种新方法应用于流感血凝素三聚体(EMPIAR 条目:10097)18 的数据集,该数据集在其最终堆栈中包括 130,000 个颗粒。我们的程序成功地从该数据集的最终堆栈中丢弃了约73.8%的颗粒,将重建的密度图的分辨率从4.11 Å提高到3.62 Å。除了流感血凝素三聚体外,早期出版物15 中还展示了多个数据集的结果,展示了生物分子的各种分辨率和分子量。
研究方案
1. 安装
- 检查并配置GPU加速环境
- 打开终端并输入命令:nvidia-smi。确保该命令成功显示有关 GPU 卡的所有信息,并且 CUDA 版本高于 10.2。执行命令:conda -V 检查是否已安装 conda(补充图 1)。
- 配置虚拟环境
- 输入以下命令以设置虚拟环境,并将 CRYOSIEVE_ENV替换为所需的环境名称:conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge。等待几分钟,直到成功配置环境(补充图 2)。
注意:用户可以根据需要灵活地修改环境名称。提供的命令特定于 CUDA 10.2。如果需要不同的 CUDA 版本,请调整 cudatoolkit 的版本号。
- 输入以下命令以设置虚拟环境,并将 CRYOSIEVE_ENV替换为所需的环境名称:conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge。等待几分钟,直到成功配置环境(补充图 2)。
- 安装 CryoSieve
- 通过执行以下命令激活环境:conda activate CRYOSIEVE_ENV。通过运行以下命令安装软件:pip install cryosieve 或 conda install -c mxhulab cryosieve(补充图 3)。输入 cryosieve -h 并确保正确显示帮助信息(补充图 4)。
2.颗粒筛分
- 检索数据
- 从 EMPIAR 下载 EMPIAR-10097 最终堆栈数据集(参见 材料表)。从 Github 下载 star 文件、mask 文件 (mask.mrc) 和初始模型 (用于重新估计步骤;initial.mrc)(参见 材料表)。将所有这些文件一起放在一个文件夹中(补充图 5)。
注意:https://github.com/mxhulab/cryosieve-demos 处的存储库使用 Git Large File Storage (Git LFS)。安装 Git LFS 对于克隆整个仓库至关重要。或者,通过 GitHub 链接访问文件,然后单击“下载原始文件 ”按钮以下载单个文件。
- 从 EMPIAR 下载 EMPIAR-10097 最终堆栈数据集(参见 材料表)。从 Github 下载 star 文件、mask 文件 (mask.mrc) 和初始模型 (用于重新估计步骤;initial.mrc)(参见 材料表)。将所有这些文件一起放在一个文件夹中(补充图 5)。
- 工艺颗粒筛分
- 打开终端并使用命令:cd FILEPATH 导航到数据集所在的文件夹。通过以下方式激活 Conda 环境:conda activate CRYOSIEVE_ENV。
- 输入以下命令开始我们的粒子筛分实验: cryosieve --reconstruct_software relion_reconstruct --postprocess_software relion_postprocess --i T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star --o output/ --mask mask.mrc --angpix 1.3099979 --num_iters 10 --frequency_start 40 --frequency_end 3 --retention_ratio 0.8 --sym C3 --num_gpus 1 --balance(补充图5)。在执行过程中,终端将显示每次迭代的输出日志。
注意:每个选项的详细说明可在 补充中找到 file 1.处理时间和执行的最低要求详见 补充文件2。T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star由CryoSPARC从T40_HA_130K-Equalized_run-data.star(从EMPIAR下载)中提炼出来,以减轻方向估计技术进步带来的影响。
3. 寻找最优迭代
- 检查分辨率
- 使用命令:grep “+ FINAL RESOLUTION:” output/_postprocess*.txt打印 10 次筛分迭代的分辨率结果(图 1)。由于在第 7次 迭代中过滤的粒子堆栈具有最高分辨率和最少的粒子,因此它可能会提供最佳结果。
注意:为避免无意中将信息从丢弃的粒子 15 传递到保留的粒子15 并确保粒子堆栈在第 7 次迭代中发布确实是最佳的,用户需要对附近的迭代执行重新估计步骤。在此协议中,迭代 4、5、6、7 和 8 需要进行验证。
- 使用命令:grep “+ FINAL RESOLUTION:” output/_postprocess*.txt打印 10 次筛分迭代的分辨率结果(图 1)。由于在第 7次 迭代中过滤的粒子堆栈具有最高分辨率和最少的粒子,因此它可能会提供最佳结果。
- 进口过筛的颗粒
- 打开 CryoSPARC Web 界面并按照以下步骤操作: 进入一个工作区,然后单击面板右上角的 Builder 按钮。在面板中,选择并单击“ 导入粒子堆栈 ”选项。在 Particle Stack Import 面板的 Parameters 部分中,将 Particle 元路径指定为位于已完成结果的输出文件夹中的 _iter{n}.star 文件,并将粒子数据路径指定为 mrcs 文件所在的文件夹。单击“ 队列作业 ”按钮,然后单击“ 队列 ”按钮以启动该过程。使用相同的方法导入需要重新估计的剩余迭代(补充图 6A)。
- 导入初始模型
- 单击面板右上角的 Builder 按钮。在面板中,选择并单击“ 导入 3D 体积” 选项。
- 将卷数据路径指定为 initial.mrc 文件。单击“ Queue Job ”按钮,然后单击“ Queue ”按钮以启动该过程(补充图6B)。
注:初始模型也可以通过 从头开始 重建(补充文件3)生成。
- 同质优化(构建作业)
- 单击面板右上角的 Builder 按钮。在面板中,选择并单击“ 同质细化 ”选项。
注意:非均匀细化也适用。
- 单击面板右上角的 Builder 按钮。在面板中,选择并单击“ 同质细化 ”选项。
- 均匀细化(进口颗粒)
- 在左侧的主面板中,打开用于导入第 5 次 迭代(或所需迭代)的粒子堆栈的作业。从主面板的右侧拖动导入的粒子模块,并将其拖放到右侧构建器的粒子堆栈部分。单击主面板右上角的红色 X 来关闭 Import Particle Stack 作业。
- 打开用于导入 3D 体积的作业。从主面板的右侧拖动导入的卷模块,然后将其拖放到右侧构建器的初始卷部分。
- 均匀细化(修改参数)
- 在“参数”折叠下,找到“对称”选项并将其设置为 C3。找到 强制重做 GS 拆分 选项并禁用它。单击“ 队列作业 ”按钮,然后单击“ 队列 ”按钮以启动同质优化。使用相同的方法对剩余的迭代执行均匀细化(补充图6C-D)。
注意:强制重做 GS 拆分选项至关重要。禁用此选项可确保 CryoSPARC 保留星形文件给出的黄金标准分割,从而避免过度拟合。禁用 Force Re-do GS Split 的详细理由可在 补充文件 4 中找到。
- 在“参数”折叠下,找到“对称”选项并将其设置为 C3。找到 强制重做 GS 拆分 选项并禁用它。单击“ 队列作业 ”按钮,然后单击“ 队列 ”按钮以启动同质优化。使用相同的方法对剩余的迭代执行均匀细化(补充图6C-D)。
- 等到所有作业都完成运行后才能获得结果。根据结果,确认了在第6次 迭代中滤除的粒子堆栈是实际的最优结果。
注意:获得的结果与本协议中提供的结果有微小的随机偏差是正常的。这些偏差不会影响总体结论。
结果
在该协议中,我们利用流感血凝素三聚体数据集(EMPIAR 条目:10097)作为该过程功效的证明。由于样品的优先取向,数据采集需要倾斜 40°。该蛋白质表现出 C3 对称性,分子量为 150 kDa。
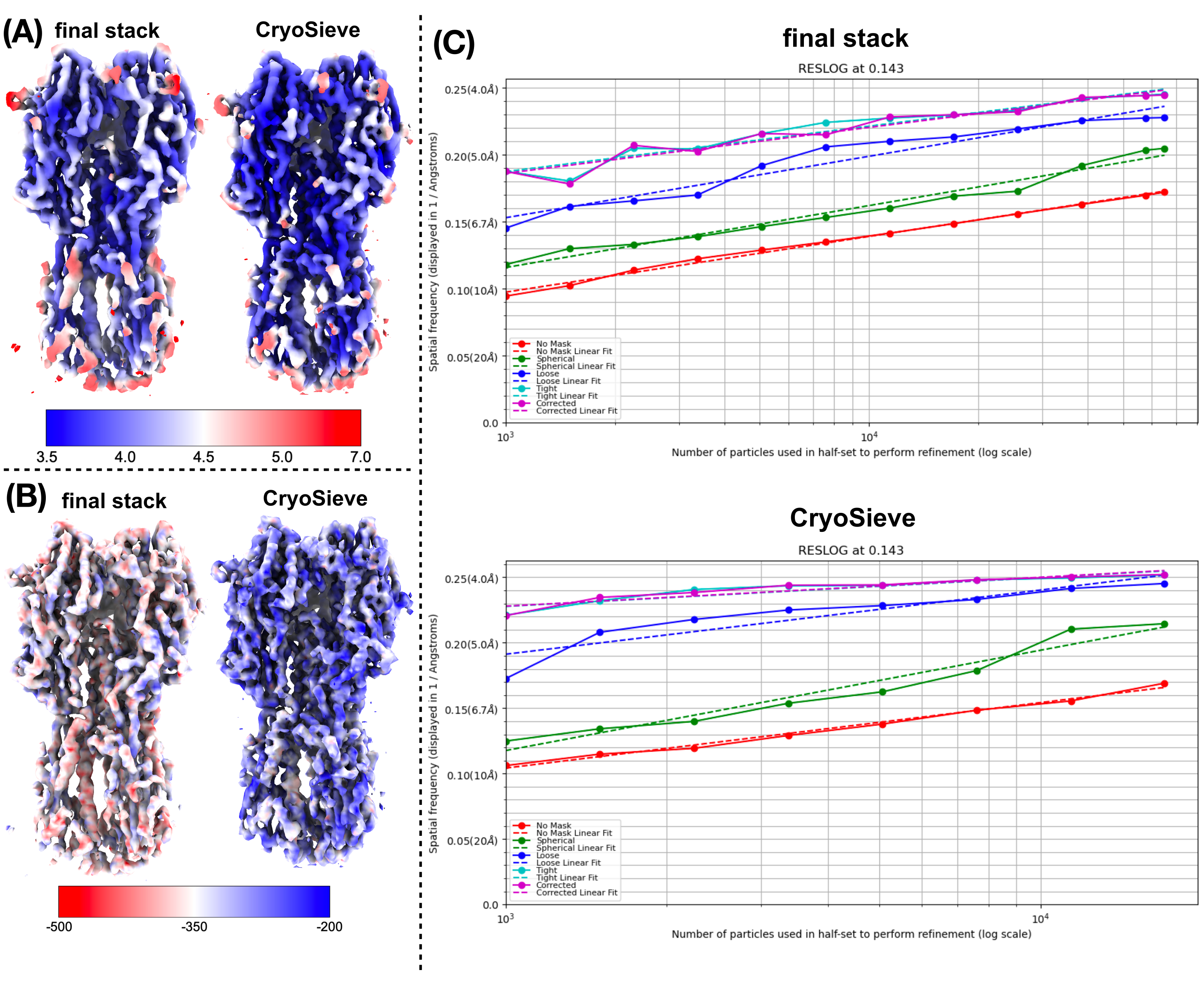
我们已经实施了前面描述的协议来处理最终的粒子堆栈。在每次迭代中逐步去除20%的颗粒,保留率为80.0%、64.0%、51.2%等。如图 1 和图 2 所示,截留颗粒的分辨率最初有所提高,但最终有所下降。在迭代中,第 6 次迭代被确定为最优的子集,包含的粒子最少,但分辨率最高。我们的算法成功识别了仅占原始堆栈 26.2% 的颗粒子集,从而将分辨率从 4.19 Å 提高到 3.62 Å(由 CryoSPARC 重新估计),如图 2 所示。此外,图 3 还比较了使用 CryoSieve 前后的密度图。还显示了该方法前后重建密度图的模型映射傅里叶壳相关(FSC)曲线和半图FSC曲线(图3A-B)。还比较了获得的原始密度图和锐利密度图,并应用了等效的等值线水平(图3C)。比较了锐利密度图的侧链,表明重建的密度图增强了。估计的Rosenthal-Henderson B因子也被采用为颗粒质量的标准19。在去除最终堆栈中的大部分颗粒后,Rosenthal-Henderson B因子从226.9 Å2提高到146.2 Å2(图3D)。还使用了局部分辨率、局部 B 因子20 和 ResLog21 进行比较,表明 CryoSieve 确实提高了密度图和颗粒的质量(图 4)。

图 1:每次迭代的分辨率。 已报告的决议在红色框中突出显示。 请点击这里查看此图的较大版本.
{kind=link}

图 2:每次迭代的分辨率。 由同构优化作业标识的分辨率在红色框中突出显示。 请点击这里查看此图的较大版本.
{kind=link}

图 3:密度图。(A) 使用CryoSieve前后重建密度图的模型到图谱FSC曲线的比较。y 轴代表 FSC,而 x 轴代表分辨率。红色虚线表示 FSC 的阈值 0.5。垂直虚线表示在阈值 0.5 下获得的密度图的分辨率。(B) 通过CryoSPARC使用CryoSieve重建前后重建的密度图获得FSC曲线的半图。y 轴代表 FSC,而 x 轴代表分辨率。(C) 显示了 CryoSieve 保留的颗粒和最终堆栈中的完整颗粒集的原始密度图和锐利密度图。等效等值线水平 0.65 应用于原始密度图。等效等值线水平 0.84 应用于锐密度图。锐利的密度图由CryoSPARC直接获得。锐利的密度图是自动后处理的,首先是FSC加权(基于CryoSPARC给出的FSC)。然后,使用自动确定的 B 因子(最终堆栈中的所有颗粒为 232.0 Å 2,CryoSieve 为 160.8 Å2)锐化 B 因子。比较了锐密度图中的侧链,并结合原子模型作为参考。红色箭头突出显示了改进的区域。(D) 显示了 CryoSieve 保留的颗粒和最终堆栈中的完整颗粒集的估计 Rosenthal-Henderson B 因子。y 轴表示使用的粒子数,x 轴表示分辨率平方的倒数。从上到下,每个点代表前一个点的一半粒子。这些决议是通过改进确定的。B 因子是使用测量点的最小二乘近似确定的,如拟合曲线所示。估计的 Rosenthal 和 Henderson 的 B 因子在图例中表示:橙色表示 CryoSieve 保留的颗粒,而蓝色表示最终堆栈中的所有颗粒。请点击这里查看此图的较大版本.
{kind=link}

图 4:密度图各种指标的比较。 (A) 使用CryoSPARC获得的CryoSieve前后的局部分辨率图的比较。本地分辨率范围在 7 Å(红色)和 3.5 Å(蓝色)之间。 (B) 使用CryoSieve前后的密度图比较,与LocBFactor获得的分辨率范围为[20-3.5] Å的局部B因子图着色。 ( C),使用CryoSPARC获得的CryoSieve前后的ResLog图的比较。 请点击这里查看此图的较大版本.
{kind=link}
补充图 1:使用命令 nvidia-smi 和 conda -V 验证先决条件。 如果满足先决条件,键入命令 nvidia-smi 将显示 GPU 驱动程序版本、CUDA 版本和 GPU 卡的状态。同样,输入命令 conda -V 应正确显示已安装的 Conda 版本。 请点击这里下载此文件。
补充图 2:创建新的 GPU 加速环境的过程。 屏幕显示用于创建 Conda 环境的命令生成的输出。 请点击这里下载此文件。
补充图 3:在 GPU 加速环境中安装 CryoSieve。 激活新创建的 Conda 环境后,屏幕会显示执行使用 Pip 安装 CryoSieve 的命令生成的输出。 请点击这里下载此文件。
补充图 4:帮助信息。请点击这里下载此文件。
补充图5:运行过程。 通过命令行执行 CryoSieve 后,屏幕会显示有关正在运行的过程的信息。 请点击这里下载此文件。
补充图 6:CryoSPARC 作业的配置。(A) 导入粒子堆栈。(B) 导入 3D 体积。 (C-D) 同质细化。 请点击这里下载此文件。
补充文件1:CryoSieve的选项。请点击这里下载此文件。
补充文件 2:运行 Cryosieve 的处理时间和最低要求。请点击这里下载此文件。
补充文件3:通过CryoSPARC生成初始模型。请点击这里下载此文件。
补充文件 4:禁用强制重做 GS 拆分的理由。请点击这里下载此文件。
补充文件 5:cryosieve-csrefine 的选项。请点击这里下载此文件。
补充文件 6:cryosieve-csrhbfactor 的选择。请点击这里下载此文件。
讨论
冷冻电子显微镜是阐明生物分子结构的关键技术。在这个过程中,在通过显微镜收集数据后,必须从显微照片中提取颗粒,然后分多个阶段对它们进行分类,以编译最终的堆栈。一个常见的挑战是损坏或不希望的顺形颗粒占主导地位,这凸显了需要重复选择颗粒以获得高分辨率的密度图。这使得颗粒选择成为冷冻电子显微镜SPA中实现高质量密度图的关键步骤。现有的粒子选择技术包括统计非倾斜验证算法22、基于z分数的方法23和角度精度估计方法24。
在这种情况下,CryoSieve成为了一个有价值的工具,擅长从最终堆栈中消除大量的外来颗粒。这种减少不仅提高了重建的计算效率,而且简化了过程。它为颗粒选择提供了一套全面的工具,其中颗粒丢弃的程度和随之而来的分辨率提高在很大程度上取决于初始数据质量和数据处理中采用的方法。
在这篇手稿中,我们使用流感血凝素三聚体的真实案例数据集(EMPIAR entry:10097)展示了一个完整的颗粒筛分工作流程。这里介绍和讨论的步骤可以概括为粒子筛分和姿态重新估计。最终的3D重建体积分辨率为3.62 Å,与已发布的密度图相比,后处理体积中的α-螺旋侧链更清晰。
CryoSieve 是一种开源方法,可在 GitHub (https://github.com/mxhulab/cryosieve) 上找到。详细的教程也可以在其主页上找到。用户可以按照教程进行安装和使用它。此外,还提供了两个模块,cryosieve-csrefine 和 cryosieve-csrhbfactor。cryosieve-csrefine 模块专门设计用于在 CryoSPARC(补充文件 5)中自动执行各种操作的顺序执行。这些操作包括导入粒子堆栈和执行 从头开始、均匀细化或非均匀细化作业。另一方面,cryosieve-csrhbfactor 模块旨在利用 cryosieve-csrefine(补充文件 6)的功能自动测定 Rosenthal-Henderson B 因子。
目前,该方法的应用仅限于单一构象场景。因此,在粒子代表多种构象的情况下,它们的能力是有限的。建议用户首先进行 3D 分类,以分离不同构象的颗粒,然后再将其用于精细的颗粒选择。此外,尽管该方法能够熟练地从最终堆栈中过滤掉超过50%的颗粒,但这些废弃颗粒的来源以及它们对重建质量的贡献可以忽略不计的根本原因仍不清楚。这种理解上的差距需要更多的研究来全面解决并可能纠正这一局限性。
现有的颗粒分选或颗粒筛分方法有三种可能。首先,cisTEM4 可以在 3D 细化后为每个单粒子图像报告一个分数。用户可以使用 cisTEM 评分对颗粒进行分类以丢弃颗粒。角度图一致性 (AGC) 方法17 也是一种丢弃错位粒子的方法。此外,非对准分类5 是一种使用 3D 分类丢弃粒子的传统方法。我们将这些方法保留的颗粒质量与 CryoSieve 进行了比较,发现 CryoSieve 保留的颗粒质量更高15。本文介绍的方法明显优于其他方法,并且在相同分辨率下实现了最小数量的粒子。
如结果所示,冷冻电子显微镜最终堆栈中的大多数颗粒对密度图重建没有贡献。换句话说,在图像采集过程中收集的所有粒子中,只有少数粒子,即最细的子集,实际上对最终的重建做出了贡献。因此,这个最终子集与收集颗粒总数的比率可以作为评估样品质量的定量指标。该比率越高,样品质量越好。尽管技术进步使冷冻电镜更容易为结构生物学家所接受,但样品制备仍然是工作流程中的主要瓶颈。因此,科学家和工程师们将精力集中在这一挑战上25.在单颗粒分析 (SPA) 中,样品制备包括两个关键步骤:样品优化和网格制备。前者涉及纯化标本,同时保持其最佳生化状态。后者需要准备样品以在显微镜下进行分析,包括网格的化学或等离子体处理、样品沉积和玻璃化。已经提出了许多技术来解决大分子不稳定性,但一种方法相对于另一种方法的有效性取决于样品的特性25,26。目前,电网准备结果在很大程度上受到用户的专业知识和经验的影响,这可能使该过程耗时且具有挑战性27,28。在样品和网格制备中遇到的众多变量给建立因果关系带来了挑战,因为研究人员只能使用显微镜在分子水平上评估样品。因此,仍然缺乏通过比较不同样本和网格制备方案进行定量统计,因此需要一种系统的方法来调查趋势和理解样本行为的基本机制29。
披露声明
所有其他作者均声明没有竞争利益。
致谢
这项工作得到了深圳研究转化研究院(to M.H.)、结构生物学高精尖创新中心(to M.H.)、北京生物结构前沿研究中心(to M.H.)、国家重点研发计划(No.2021YFA1001300)(to C.B.)、国家自然科学基金(No.12271291)(to C.B.)的支持。 国家自然科学基金面上项目(No.12071244)(授予Z.S.)。
材料
| Name | Company | Catalog Number | Comments |
| CryoSPARC | Structura Biotechnology Inc. Toronto, Canada | CryoSPARC (Cryo-EM Single Particle Ab-Initio Reconstruction and Classification) is a state of the art HPC software solution for complete processing of single-particle cryo-electron microscopy (cryo-EM) data. CryoSPARC is useful for solving cryo-EM structures of membrane proteins, viruses, complexes, flexible molecules, small particles, phase plate data and negative stain data. | |
| EMPIAR-10097 Dataset | https://ftp.ebi.ac.uk/empiar/world_availability/10097/data/Particle-Stack/T40_HA_130K-Equalized-Particle-Stack.mrcs | This dataset comprises single-particle cryo-EM data of the Influenza Hemagglutinin trimer, characterized by its highly preferred orientation, collected using a 40-degree tilted collection strategy. | |
| initial.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| mask.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| RELION | 4.0-beta-2 | RELION (REgularised LIkelihood OptimisatioN) is an open-source software for cryo-electron microscopy (cryo-EM) data processing, particularly for refining macromolecular structures. Utilizing a Bayesian approach, it excels in separating signal from noise, enabling high-resolution structure determination. RELION supports single-particle analysis, tomography, and sub-tomogram averaging, and has become widely used in structural biology due to its effectiveness and user-friendly interface. | |
| T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | Metadata file for the final stack of particles from EMPIAR-10097 |
参考文献
- Bai, X. C., Fernandez, I. S., Mcmullan, G., Scheres, S. H. Ribosome structures to near-atomic resolution from thirty thousand cryo-em particles. elife. 2, 00461(2013).
- Campbell, M. G., et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 20 (11), 1823-1828 (2012).
- Li, X., et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-em. Nat Meth. 10 (6), 584-590 (2013).
- Grant, T., Rohou, A., Grigorieff, N. Cis tem, user-friendly software for single-particle image processing. eLife. 7, e35383(2018).
- Scheres, S. H. Relion: Implementation of a bayesian approach to cryo-em structure determination. J Str Biol. 180 (3), 519-530 (2012).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. Cryosparc: Algorithms for rapid unsupervised cryo-em structure determination. Nat Meth. 14 (3), 290-296 (2017).
- Kühlbrandt, W. The resolution revolution. Science. 343 (6178), 1443-1444 (2014).
- Dubochet, J., et al. Cryo-electron microscopy of vitrified specimens. Quart Rev Biophys. 21 (2), 129-228 (1988).
- Wagner, T., et al. Sphire-cryolo is a fast and accurate fully automated particle picker for cryo-EM. Comm Biol. 2 (1), 218(2019).
- Bepler, T., et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Meth. 16 (11), 1153-1160 (2019).
- Wang, F., et al. Deeppicker: A deep learning approach for fully automated particle picking in cryo-em. J Str Biol. 195 (3), 325-336 (2016).
- Heimowitz, A., Andén, J., Singer, A. Apple picker: Automatic particle picking, a low-effort cryo-em framework. J Str Biol. 204 (2), 215-227 (2018).
- Glaeser, R. M. How good can single-particle cryo-em become? What remains before it approaches its physical limits. Ann Rev Biophys. 48, 45-61 (2019).
- Diiorio, M. C., Kulczyk, A. W. A robust single-particle cryo-electron microscopy (cryo-em) processing workflow with cryosparc, relion, and scipion. J Vis Exp. (179), e63387(2022).
- Zhu, J., et al. A minority of final stacks yields superior amplitude in single-particle cryo-em. Nat Comm. 14 (1), 7822(2023).
- Zhou, Y., Moscovich, A., Bendory, T., Bartesaghi, A. Unsupervised particle sorting for high-resolution single-particle cryo-em. Inv Probl. 36 (4), 044002(2020).
- Méndez, J., Garduno, E., Carazo, J. M., Sorzano, C. O. S. Identification of incorrectly oriented particles in cryo-em single particle analysis. J Str Biol. 213 (3), 107771(2021).
- Tan, Y. Z., et al. Addressing preferred specimen orientation in single-particle cryo-em through tilting. Nat Meth. 14 (8), 793-796 (2017).
- Rosenthal, P. B., Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 333 (4), 721-745 (2003).
- Kaur, S., et al. Local computational methods to improve the interpretability and analysis of cryo-em maps. Nat Comm. 12 (1), 1240(2021).
- Stagg, S. M., Noble, A. J., Spilman, M., Chapman, M. S. Reslog plots as an empirical metric of the quality of cryo-em reconstructions. J Str Biol. 185 (3), 418-426 (2014).
- Vargas, J., Otón, J., Marabini, R., Carazo, J. M., Sorzano, C. Particle alignment reliability in single particle electron cryomicroscopy: A general approach. Sci Rep. 6 (1), 21626(2016).
- Vargas, J., et al. Particle quality assessment and sorting for automatic and semiautomatic particle-picking techniques. J Str Biol. 183 (3), 342-353 (2013).
- Vargas, J., Melero, R., Gomez-Blanco, J., Carazo, J. -M., Sorzano, C. O. S. Quantitative analysis of 3d alignment quality: Its impact on soft-validation, particle pruning and homogeneity analysis. Sci Rep. 7 (1), 6307(2017).
- Carragher, B., et al. Current outcomes when optimizing 'standard'sample preparation for single-particle cryo-em. J Microsc. 276 (1), 39-45 (2019).
- Drulyte, I., et al. Approaches to altering particle distributions in cryo-electron microscopy sample preparation. Acta Crystallographica Sec D: Str Biol. 74 (6), 560-571 (2018).
- Glaeser, R. M. How good can cryo-em become. Nat Meth. 13 (1), 28-32 (2016).
- Kim, L. Y., et al. Benchmarking cryo-em single particle analysis workflow. Front Mol Biosci. 5, 50(2018).
- Weissenberger, G., Henderikx, R. J., Peters, P. J. Understanding the invisible hands of sample preparation for cryo-em. Nat Meth. 18 (5), 463-471 (2021).
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。