
Method Article
単一粒子極低温電子顕微鏡の最終スタックにおける粒子の大部分の除去による密度マップの強化
* これらの著者は同等に貢献しました
要約
クライオ電子顕微鏡の高度な粒子選択方法であるCryoSieveは、実世界のデータセットへの適用を通じて実証されているように、最終スタックの粒子の大部分を除去することにより、密度マップの解像度を向上させます。
要約
過去10年間で、極低温電子顕微鏡(クライオEM)の単粒子分析(SPA)の分野における技術と方法論の進歩により、生体高分子の高分解能構造検査の能力が大幅に向上しました。この進歩により、分子の洞察の新時代が到来し、X線結晶構造解析が主要な方法に取って代わり、生物学における長年の疑問に対する答えがもたらされました。クライオ電子顕微鏡は、X線結晶構造解析の大きな制限である結晶化に依存しないため、さまざまな品質の粒子を捕捉します。したがって、選択した粒子の品質は再構築された密度マップの解像度に直接影響するため、粒子の選択は非常に重要です。CryoSieveと呼ばれる粒子選択のための革新的な反復アプローチは、最終スタック内の粒子の数を効果的に減らすことにより、再構築された密度マップの品質を大幅に向上させます。実験的な証拠によると、この方法により、最終スタックの粒子の大部分を除去でき、密度マップの品質が大幅に向上します。この記事では、このアプローチの詳細なワークフローの概要を説明し、実際のデータセットでのその適用を紹介します。
概要
極低温電子顕微鏡(クライオEM)単粒子分析(SPA)は、生体高分子の高解像度3次元密度マップを決定するための主要な方法となっています。一連の技術革新1,2,3,4,5,6、すなわち分解能革命7により、クライオ電子顕微鏡は、前例のない速度で原子分解能まで生体高分子の構造を決定する能力を持っています。このブレークスルーは、分子洞察の新時代の幕開けであり、X線結晶構造解析を抜いて主要な技術となり、長年の生物学的な疑問に答えるものです。
クライオEM SPAは、生体高分子の結晶化を必要としないため、X線結晶構造解析とは異なります。代わりに、標的の生体高分子を含む溶液は、ガラス質の氷中で急速に凍結されます。次に、電子ビームでイメージングして一連の顕微鏡写真を作成し、結晶化の必要性を回避します8。続いて、粒子ピッキングアルゴリズムを利用して、これらの顕微鏡写真4、9、10、11、12から個々の生粒子を抽出する。クライオ電子顕微鏡は結晶化に依存しないため、抽出された粒子が主に損傷を受けたり、望ましくないコンフォメーション状態にあるのは当然であり、高解像度の密度マップを得るためには、粒子選択を複数回行う必要があります。したがって、クライオEM SPA画像処理では、高解像度の密度マップ13を得るために粒子の選択が重要です。
クライオ電子顕微鏡SPAでは、標準的な粒子選択方法には、2次元(2D)および3次元(3D)分類14が含まれます。2D 分類では、粒子が事前定義された数のグループに分類され、各クラスの平均画像と推定 2D 解像度が得られます。その後、研究者はこれらのクラスを目視で検査し、低解像度のグループから粒子を取り除き、残りの粒子を高解像度の再構築に使用できます。リファインメントアルゴリズムを使用して粒子のポーズが確立されると、研究者は粒子を複数のクラスにクラスタリングする3D分類を進めます。これにより、各クラスの再構築された密度マップを目視で検査でき、望ましくない粒子、たとえば望ましくない粒子を除外できます。複数回の分類ラウンドの後、比較的高品質の粒子を含む最終スタックが得られます。これらの最終スタックは、原子または原子に近い解像度の密度マップを作成するのに役立ちます。
Zhu氏と彼女の同僚は、これらの最終スタック15でさらなる粒子選択を行うことができることを実証しました。粒子選択のための革新的な反復方法であるCryoSieve15は、粒子の数を大幅に減らすことにより、最終的な密度マップの品質を向上させるために適用できます。正規化相互相関(NCC)法16、角度グラフ一貫性(AGC)アプローチ17、および非アライメント分類5などの他の粒子選別基準およびソフトウェアが現在、この分野で使用されていますが、この方法は、有効性の点でこれらのアルゴリズムよりも優れていることが示されています。
この研究では、プロセス全体の詳細なガイドを提供します。ケーススタディとして、この新しい方法をインフルエンザ赤血球凝集素三量体(EMPIARエントリ:10097)18のデータセットに適用しました。このデータセットには、最終スタックに130,000個の粒子が含まれています。私たちの手順では、このデータセットの最終スタックからパーティクルの約73.8%を無事に破棄し、再構築された密度マップの解像度を4.11 Åから3.62 Åに向上させました。インフルエンザ赤血球凝集素三量体に加えて、複数のデータセットの結果が以前の出版物15に示されており、生体分子のさまざまな解像度と分子量が示されています。
プロトコル
1. インストール
- GPUアクセラレーション環境の確認と設定
- ターミナルを開き、コマンド nvidia-smi を入力します。コマンドがGPUカードに関するすべての情報を正常に表示し、CUDAバージョンが10.2より後であることを確認してください。conda -V コマンドを実行して、Conda がインストールされているかどうかを確認します (補足図 1)。
- 仮想環境を構成する
- 次のコマンドを入力して仮想環境を設定し、CRYOSIEVE_ENV を目的の環境名に置き換えます: conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge。環境が正常に構成されるまで数分待ちます (補足図 2)。
注: ユーザーは、必要に応じて環境名を柔軟に変更できます。提供されるコマンドは CUDA 10.2 に固有です。別の CUDA バージョンが必要な場合は、cudatoolkit のバージョン番号を調整してください。
- 次のコマンドを入力して仮想環境を設定し、CRYOSIEVE_ENV を目的の環境名に置き換えます: conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge。環境が正常に構成されるまで数分待ちます (補足図 2)。
- CryoSieveをインストールする
- コマンド conda activate CRYOSIEVE_ENV を実行して、環境をアクティブ化します。pip install cryosieveまたはconda install -c mxhulab cryosieveを実行して、ソフトウェアをインストールします(補足図3)。cryosieve -h と入力し、ヘルプ情報が正しく表示されていることを確認します (補足図 4)。
2.粒子ふるい分け
- データを取得する
- EMPIAR-10097 最終スタックデータセットを EMPIAR からダウンロードします (資料の表を参照)。Github から Star ファイル、マスク ファイル (mask.mrc)、初期モデル (再推定ステップ用、initial.mrc) をダウンロードします (資料の表を参照)。これらすべてのファイルを一緒にフォルダに配置します(補足図5)。
注: https://github.com/mxhulab/cryosieve-demos のリポジトリは、Git Large File Storage (Git LFS) を利用しています。Git LFS のインストールは、リポジトリ全体をクローンするために不可欠です。または、GitHubリンクからファイルにアクセスし、[Download raw file ]ボタンをクリックして個々のファイルをダウンロードします。
- EMPIAR-10097 最終スタックデータセットを EMPIAR からダウンロードします (資料の表を参照)。Github から Star ファイル、マスク ファイル (mask.mrc)、初期モデル (再推定ステップ用、initial.mrc) をダウンロードします (資料の表を参照)。これらすべてのファイルを一緒にフォルダに配置します(補足図5)。
- プロセス粒子ふるい分け
- ターミナルを開き、コマンド cd FILEPATH を使用して、データセットが配置されているフォルダーに移動します。conda activate CRYOSIEVE_ENV を使用して Conda 環境をアクティブ化します。
- 次のコマンドを入力して、粒子ふるい分け実験を開始します: cryosieve --reconstruct_software relion_reconstruct --postprocess_software relion_postprocess --i T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star --o output/ --mask mask.mrc --angpix 1.3099979 --num_iters 10 --frequency_start 40 --frequency_end 3 --retention_ratio 0.8 --sym C3 --num_gpus 1 --balance (補足図5)。実行中、ターミナルには各イテレーションの出力ログが表示されます。
注: 各オプションの詳細な手順については、 補足ファイル 1 を参照してください。処理時間と実行の最小要件については、 補足ファイル 2 に詳しく説明されています。T40_HA_130K-Equalized_run-data_CryoSPARC_refined.starは、方位推定技術の進歩による影響を軽減するために、TCryoSPARCによってT40_HA_130K-Equalized_run-data.star(EMPIARからダウンロード)から改良されました。
3. 最適なイテレーションを見つける
- 解像度を確認する
- コマンド grep "+ FINAL RESOLUTION:" output/_postprocess*.txt を使用して、ふるい分けの 10 回の反復の解像度結果を印刷します (図 1)。7回目の 反復でフィルタリングされたパーティクルスタックは、最も少ないパーティクルで最高の解像度を持つため、最適な結果が得られる可能性があります。
注:廃棄されたパーティクル15から保持されたパーティクル15への意図しない情報転送を回避し、パーティクルスタックがポストする第7の反復が実際に最適であることを確保するために、ユーザは、近くの反復に対して再推定ステップを実行する必要があります。このプロトコルでは、イテレーション 4、5、6、7、および 8 が検証の対象となります。
- コマンド grep "+ FINAL RESOLUTION:" output/_postprocess*.txt を使用して、ふるい分けの 10 回の反復の解像度結果を印刷します (図 1)。7回目の 反復でフィルタリングされたパーティクルスタックは、最も少ないパーティクルで最高の解像度を持つため、最適な結果が得られる可能性があります。
- ふるいにかけた粒子のインポート
- CryoSPARCのWebインターフェースを開き、次の手順に従います:ワークスペースに入り、パネルの右上にある [ビルダー ]ボタンをクリックします。パネルで、[ Import Particle Stack ] オプションを選択してクリックします。[Particle Stack Import] パネルの [Parameters] セクションで、完成した結果の出力フォルダにある [Particle] メタパスを _iter{n}.star ファイルとして指定し、mrcs ファイルが保存されているフォルダへのパーティクル データ パスを指定します。「 Queue Job 」ボタンをクリックし、「 Queue 」ボタンをクリックしてプロセスを開始します。同じ方法を使用して、再推定が必要な残りの反復をインポートします (補足図 6A)。
- 初期モデルのインポート
- パネルの右上にある [ビルダー ]ボタンをクリックします。パネルで、[ Import 3D Volumes ]オプションを選択してクリックします。
- ボリューム・データ・パスをinitial.mrcファイルとして指定します。 [Queue Job ] ボタンをクリックし、[ Queue ] ボタンをクリックしてプロセスを開始します (補足図 6B)。
注: 初期モデルは、 ab initio 再構成 (補足ファイル 3) によって生成することもできます。
- 均質な改良 (ビルド ジョブ)
- パネルの右上にある [ビルダー ]ボタンをクリックします。パネルで、[ Homogeneous Refinement ]オプションを選択してクリックします。
注:不均一なリファインメントも適用されます。
- パネルの右上にある [ビルダー ]ボタンをクリックします。パネルで、[ Homogeneous Refinement ]オプションを選択してクリックします。
- 均質なリファインメント(インポート粒子)
- 左側のメインパネルで、5番目の 反復 (または目的の反復) のパーティクルスタックをインポートするジョブを開きます。インポートしたパーティクルモジュールをメインパネルの右側からドラッグし、右側のビルダーのパーティクルスタックセクションにドロップします。メインパネルの右上隅にある赤い X をクリックして、Import Particle Stackジョブを閉じます。
- 3Dボリュームをインポートするジョブを開きます。インポートしたボリュームモジュールをメインパネルの右側からドラッグし、右側のビルダーの初期ボリュームセクションにドロップします。
- 均質なリファインメント (パラメータの変更)
- [パラメータ]フォールドの下で、[シンメトリ]オプションを見つけて C3 に設定します。[GS分割を強制する]オプションを見つけて無効にします。「 Queue Job 」ボタンをクリックし、「 Queue 」ボタンをクリックして、同種リファインメントを開始します。同じ方法を使用して、残りの反復計算に対して均質な細分化を実行します(補足図6C-D)。
注: [GS 分割を強制再実行] オプションは重要です。このオプションを無効にすると、CryoSPARC はスターファイルによって与えられるゴールドスタンダードの分割を保持し、オーバーフィッティングを回避できます。Force Re-do GS Split を無効にする詳細な根拠は、 補足ファイル 4 にあります。
- [パラメータ]フォールドの下で、[シンメトリ]オプションを見つけて C3 に設定します。[GS分割を強制する]オプションを見つけて無効にします。「 Queue Job 」ボタンをクリックし、「 Queue 」ボタンをクリックして、同種リファインメントを開始します。同じ方法を使用して、残りの反復計算に対して均質な細分化を実行します(補足図6C-D)。
- すべてのジョブの実行が終了するまで待ってから、結果を取得します。この結果から、6回目の 反復でフィルタリングされたパーティクルスタックが実際の最適な結果であることが確認されます。
注:得られた結果が、このプロトコルで提供された結果からわずかにランダムな偏差を持つのは正常です。これらの逸脱は、全体的な結論に影響を与えません。
結果
このプロトコルでは、このプロセスの有効性のデモンストレーションとして、インフルエンザ赤血球凝集素三量体データセット(EMPIARエントリ:10097)を利用しました。サンプルの向きが好ましいため、データ取得には40°の傾きが必要でした。このタンパク質はC3対称性を示し、分子量は150 kDaです。
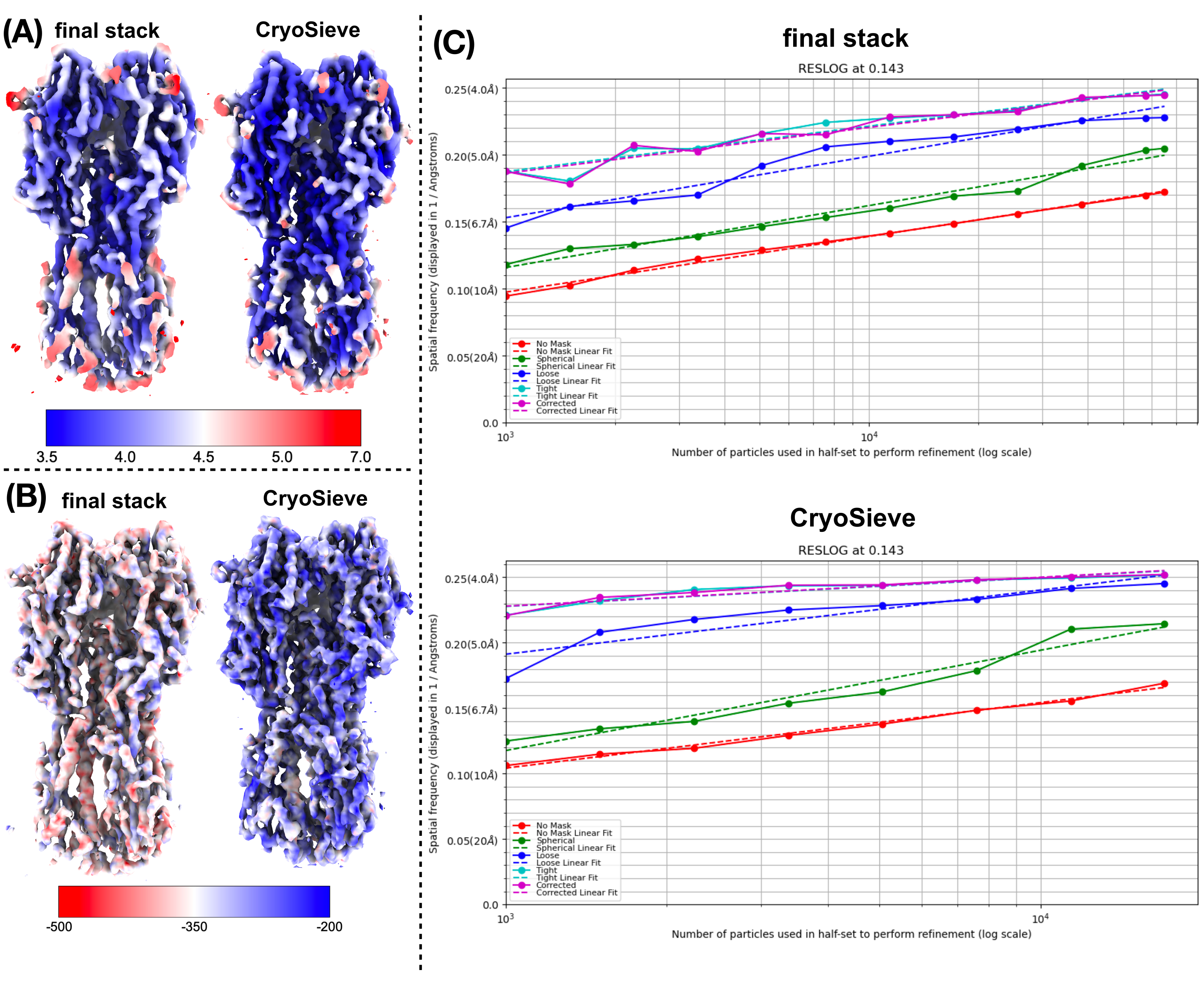
最終的なパーティクルスタックを処理するために、前述のプロトコルを実装しました。各反復で粒子の 20% を徐々に除去し、その結果、80.0%、64.0%、51.2% などの保持率が得られました。図1と図2に示すように、保持された粒子の分解能は最初は向上しましたが、最終的には減少しました。反復の中で、6番目の反復は、最も少ない粒子を含みながら最高の解像度を達成した最適のサブセットとして特定されました。私たちのアルゴリズムは、元のスタックのわずか26.2%を占める粒子のサブセットを特定することに成功し、図2に示すように、分解能が4.19 Åから3.62 Åに向上しました(CryoSPARCによる再推定)。さらに、図3では、CryoSieveを使用する前と使用した後の密度マップを比較しました。また、本法の前後で再構築した密度マップのモデル間フーリエシェル相関(FSC)曲線とハーフマップFSC曲線も示しています(図3A-B)。得られた生の密度マップとシャープな密度マップも比較され、同等の等高線レベルが適用されました(図3C)。シャープな密度マップの側鎖を比較し、再構築された密度マップの強化を示しました。推定されたローゼンタール・ヘンダーソンB因子も粒子品質の基準に採用されました19。最終スタックの粒子の大部分を除去した後、ローゼンタール・ヘンダーソンのB因子は226.9 Å2から146.2 Å2に上昇しました(図3D)。また、ローカル解像度、ローカルBファクター20、およびResLog21も比較に利用され、CryoSieveが密度マップと粒子の両方の品質を実際に向上させることが示されています(図4)。

図1:各反復の解像度。 報告された決議は、赤いボックスで強調表示されています。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:各反復の解像度。 均質な絞り込みジョブによって識別された解像度は、赤いボックスで強調表示されます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:密度マップ。(A) CryoSieveを使用する前と後で再構築された密度マップのモデル間FSC曲線の比較。y軸はFSC、x軸は解像度を表します。赤い破線は、FSCのしきい値0.5を示しています。縦の破線は、0.5 のしきい値で取得された密度マップの解像度を示しています。 (B) ハーフマップFSC曲線は、CryoSPARCを介してCryoSieveを使用する前後に再構築された密度マップから取得されました。y軸はFSC、x軸は解像度を表します。 (C) CryoSieveで保持された粒子と最終スタックの粒子の完全なセットの両方について、生の密度マップとシャープな密度マップが示されました。同等の等高線レベル 0.65 は、未加工の密度マップに適用されました。0.84 の等高線レベルは、シャープな密度マップに適用されました。シャープな密度マップは、CryoSPARCによって直接取得されました。シャープな密度マップは自動後処理され、最初に FSC で重み付けされました (CryoSPARC によって提供された FSC に基づく)。次に、自動決定されたBファクター(最終スタック内のすべての粒子で232.0 Å2 、CryoSieveで160.8 Å2 )を使用してBファクターをシャープにしました。シャープな密度マップの側鎖を比較し、参照用に原子モデルを組み込んでいました。赤い矢印は、改善された領域を強調しています。 (D) 推定されたローゼンタール・ヘンダーソンBファクターは、CryoSieveで保持された粒子と最終スタックの粒子の完全なセットの両方について示されました。y軸は使用される粒子の数を表し、x軸は解像度の2乗の逆数を表します。上から下に移動すると、各ポイントは前のポイントの半分の粒子を表します。決議は改良によって決定されました。B因子は、フィッティング曲線で示されるように、測定された点の最小二乗近似を使用して決定されました。推定されたローゼンタールとヘンダーソンのBファクターは凡例で示されています:オレンジ色はCryoSieveによって保持された粒子を表し、青色は最終スタック内のすべての粒子を示します。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:密度マップのさまざまなメトリックの比較(A)CryoSPARCによって得られたCryoSieveを使用する前と使用後のローカル解像度マップの比較。ローカル解像度の範囲は 7 Å (赤) から 3.5 Å (青) です。(B) LocBFactor が [20-3.5] Å の解像度範囲を使用して取得したローカル B ファクター マップで色付けした、CryoSieve 使用前と使用後の密度マップの比較 (C)、CryoSPARC で取得した CryoSieve 使用前と使用後の ResLog プロットの比較。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
補足図1:コマンドnvidia-smiおよびconda -Vを使用して前提条件を確認します。 前提条件が満たされている場合、コマンド nvidia-smi を入力すると、GPU ドライバーのバージョン、CUDA バージョン、および GPU カードのステータスが表示されます。同様に、コマンド conda -V を入力すると、インストールされているバージョンの Conda が正しく表示されます。 このファイルをダウンロードするには、ここをクリックしてください。
補足図 2: 新しい GPU アクセラレーション環境を作成するプロセス。 画面には、Conda 環境の作成に使用したコマンドによって生成された出力が表示されます。 このファイルをダウンロードするには、ここをクリックしてください。
補足図3:GPUアクセラレーション環境でのCryoSieveのインストール。 新しく作成したConda環境をアクティブ化すると、Pipを使用してCryoSieveをインストールするコマンドを実行した結果の出力が画面に表示されます。このファイルをダウンロードするには、ここをクリックしてください。
補足図4:ヘルプ情報。このファイルをダウンロードするには、ここをクリックしてください。
補足図 5: 実行中のプロセス。 コマンドラインからCryoSieveを実行すると、実行中のプロセスに関する情報が画面に表示されます。 このファイルをダウンロードするには、ここをクリックしてください。
補足図6:CryoSPARCのジョブの構成。(A) パーティクルスタックをインポートします。(B) 3Dボリュームをインポートします。 (C-D) 均質な洗練。 このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル1:CryoSieveのオプション。このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル2:Cryosieveを実行するための処理時間と最小要件。このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル3:CryoSPARCによる初期モデルの生成このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル4:GS分割の強制やり直しを無効にする根拠。このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル5:cryosieve-csrefineのオプション。このファイルをダウンロードするには、ここをクリックしてください。
補足ファイル6:cryosieve-csrhbfactorのオプション。このファイルをダウンロードするには、ここをクリックしてください。
ディスカッション
クライオ電子顕微鏡は、生体分子の構造を解明するための極めて重要な技術です。このプロセスでは、顕微鏡によるデータ収集後、顕微鏡写真から粒子を抽出し、その後、複数の段階で分類して最終的なスタックをまとめることが不可欠です。一般的な課題は、損傷した粒子や望ましくない粒子が優勢であることであり、高解像度の密度マップを得るためには、粒子選択を繰り返す必要があることが強調されています。そのため、クライオ電子顕微鏡スパでは、粒子選択が高品質の密度マップを実現するための重要なステップとなります。既存の粒子選択技術には、統計的非傾斜検証アルゴリズム22、zスコアベースのアプローチ23、および角度精度推定方法24が含まれる。
CryoSieveは、このコンテキストで貴重なツールとして浮上しており、最終的なスタックからかなりの数の無関係な粒子を排除することに長けています。この削減により、再構成の計算効率が向上するだけでなく、プロセスが合理化されます。これは、粒子選択のための包括的なスイートを提供し、粒子の破棄の程度とそれに伴う解像度の向上は、初期データ品質とデータ処理に採用される方法論に大きく依存します。
本稿では、インフルエンザ赤血球凝集素三量体(EMPIARエントリー:10097)の実例データセットを用いた粒子ふるい分けの完全なワークフローを紹介しました。ここで取り上げて説明する手順は、粒子ふるい分けと姿勢の再推定として要約できます。最終的な3D再構成ボリュームは3.62 Åの解像度を達成し、アルファヘリックスの側鎖は、公開された密度マップと比較して、後処理されたボリュームでより明確になりました。
CryoSieveは、GitHub(https://github.com/mxhulab/cryosieve)で公開されているオープンソースの方法です。詳細なチュートリアルは、ホームページでも見つけることができます。ユーザーは、チュートリアルに従ってインストールして使用できます。さらに、cryosieve-csrefineとcryosieve-csrhbfactorの2つのモジュールが提供されます。cryosieve-csrefineモジュールは、CryoSPARC(補足ファイル5)内のさまざまな操作の逐次実行を自動化するために特別に作成されています。これらの操作には、パーティクル スタックのインポートや、 ab initio、均質なリファインメント、または不均一なリファインメント ジョブの実行が含まれます。一方、cryosieve-csrhbfactor モジュールは、cryosieve-csrefine の機能を活用して Rosenthal-Henderson B-factor の決定を自動化するように設計されています (補足ファイル 6)。
現在、この手法の適用は単一のコンフォメーションシナリオに限定されています。したがって、粒子が複数のコンフォメーションを表す場合、その能力は制限されます。ユーザーは、最初に3D分類に従事して、異なるコンフォメーションの粒子を分離してから、それを精製された粒子の選択に使用することをお勧めします。さらに、この方法は、最終スタックから粒子の50%以上をろ過することに熟練していることを示していますが、これらの廃棄された粒子の起源と、それらが再構築品質にほとんど寄与しない根本的な理由は不明のままです。この理解のギャップは、この制限に包括的に対処し、潜在的に修正するために追加の研究を必要とします。
粒子選別または粒子ふるい分けには、3つの既存の方法があります。まず第一に、cisTEM4は3Dリファインメント後の各単一粒子画像のスコアを報告できます。ユーザーは、cisTEMスコアを使用して粒子を分類し、粒子を破棄できます。角度グラフ一貫性(AGC)アプローチ17は、位置がずれた粒子を破棄する方法でもある。さらに、非アライメント分類5は、3D分類を使用して粒子を破棄する従来の方法です。これらの方法で保持される粒子の品質をCryoSieveと比較したところ、CryoSieveの保持粒子はより高品質であることがわかりました15。ここで紹介する方法は、他の方法を大幅に上回り、同じ解像度で最小数の粒子を達成します。
この結果で示されたように、クライオ電子顕微鏡の最終スタック内の粒子の大部分は、密度マップの再構築に寄与しません。言い換えれば、画像取得中に収集されたすべての粒子のうち、選択された少数の粒子、つまり最も細かいサブセットのみが、最終的な再構成に実際に貢献します。したがって、収集された粒子の総数に対するこの最終サブセットの比率は、サンプルの品質を評価するための定量的な指標として役立つ可能性があります。この比率が高いほど、サンプルの品質は向上します。技術の進歩により、構造生物学者がクライオ電子顕微鏡をより利用しやすくなったにもかかわらず、サンプル調製は依然としてワークフローの大きなボトルネックとなっています。したがって、科学者とエンジニアは、この課題に力を注いでいます25。単粒子分析(SPA)では、サンプル調製は、サンプルの最適化とグリッド調製という2つの重要なステップで構成されています。前者は、試料を最適な生化学的状態を維持しながら精製することです。後者では、グリッドの化学的またはプラズマ処理、サンプルの堆積、ガラス化など、顕微鏡での分析のためにサンプルを準備します。高分子の不安定性に対処するために数多くの技術が提案されてきたが、あるアプローチが他のアプローチよりも有効かどうかは、サンプルの特性に依存する25,26。現在、グリッド調製の結果はユーザーの専門知識と経験に大きく影響されるため、プロセスが時間がかかり、困難になる可能性があります27,28。サンプルとグリッドの準備で遭遇する多数の変数は、研究者が顕微鏡を使用して分子レベルでしかサンプルを評価できないため、因果関係を確立する上で課題を提起します。その結果、異なるサンプルおよびグリッド調製プロトコルの比較による定量的統計はまだ不足しており、傾向を調査し、サンプル挙動の基本的なメカニズムを理解するための体系的なアプローチが必要である29。
開示事項
他のすべての著者は、競合する利益を宣言しません。
謝辞
本研究は、Shenzhen Academy of Research and Translation(M.H.氏)、Advanced Innovation Center for Structural Biology(M.H.氏)、Beijing Frontier Research Center for Biological Structure(M.H.氏)、National Key R&D Program of China(No.2021YFA1001300)(C.B.氏)、National Natural Science Foundation of China(No.12271291)(C.B.氏)、 中国国家自然科学基金会(No.12071244)(Z.S.へ)。
資料
| Name | Company | Catalog Number | Comments |
| CryoSPARC | Structura Biotechnology Inc. Toronto, Canada | CryoSPARC (Cryo-EM Single Particle Ab-Initio Reconstruction and Classification) is a state of the art HPC software solution for complete processing of single-particle cryo-electron microscopy (cryo-EM) data. CryoSPARC is useful for solving cryo-EM structures of membrane proteins, viruses, complexes, flexible molecules, small particles, phase plate data and negative stain data. | |
| EMPIAR-10097 Dataset | https://ftp.ebi.ac.uk/empiar/world_availability/10097/data/Particle-Stack/T40_HA_130K-Equalized-Particle-Stack.mrcs | This dataset comprises single-particle cryo-EM data of the Influenza Hemagglutinin trimer, characterized by its highly preferred orientation, collected using a 40-degree tilted collection strategy. | |
| initial.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| mask.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| RELION | 4.0-beta-2 | RELION (REgularised LIkelihood OptimisatioN) is an open-source software for cryo-electron microscopy (cryo-EM) data processing, particularly for refining macromolecular structures. Utilizing a Bayesian approach, it excels in separating signal from noise, enabling high-resolution structure determination. RELION supports single-particle analysis, tomography, and sub-tomogram averaging, and has become widely used in structural biology due to its effectiveness and user-friendly interface. | |
| T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | Metadata file for the final stack of particles from EMPIAR-10097 |
参考文献
- Bai, X. C., Fernandez, I. S., Mcmullan, G., Scheres, S. H. Ribosome structures to near-atomic resolution from thirty thousand cryo-em particles. elife. 2, 00461(2013).
- Campbell, M. G., et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 20 (11), 1823-1828 (2012).
- Li, X., et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-em. Nat Meth. 10 (6), 584-590 (2013).
- Grant, T., Rohou, A., Grigorieff, N. Cis tem, user-friendly software for single-particle image processing. eLife. 7, e35383(2018).
- Scheres, S. H. Relion: Implementation of a bayesian approach to cryo-em structure determination. J Str Biol. 180 (3), 519-530 (2012).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. Cryosparc: Algorithms for rapid unsupervised cryo-em structure determination. Nat Meth. 14 (3), 290-296 (2017).
- Kühlbrandt, W. The resolution revolution. Science. 343 (6178), 1443-1444 (2014).
- Dubochet, J., et al. Cryo-electron microscopy of vitrified specimens. Quart Rev Biophys. 21 (2), 129-228 (1988).
- Wagner, T., et al. Sphire-cryolo is a fast and accurate fully automated particle picker for cryo-EM. Comm Biol. 2 (1), 218(2019).
- Bepler, T., et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Meth. 16 (11), 1153-1160 (2019).
- Wang, F., et al. Deeppicker: A deep learning approach for fully automated particle picking in cryo-em. J Str Biol. 195 (3), 325-336 (2016).
- Heimowitz, A., Andén, J., Singer, A. Apple picker: Automatic particle picking, a low-effort cryo-em framework. J Str Biol. 204 (2), 215-227 (2018).
- Glaeser, R. M. How good can single-particle cryo-em become? What remains before it approaches its physical limits. Ann Rev Biophys. 48, 45-61 (2019).
- Diiorio, M. C., Kulczyk, A. W. A robust single-particle cryo-electron microscopy (cryo-em) processing workflow with cryosparc, relion, and scipion. J Vis Exp. (179), e63387(2022).
- Zhu, J., et al. A minority of final stacks yields superior amplitude in single-particle cryo-em. Nat Comm. 14 (1), 7822(2023).
- Zhou, Y., Moscovich, A., Bendory, T., Bartesaghi, A. Unsupervised particle sorting for high-resolution single-particle cryo-em. Inv Probl. 36 (4), 044002(2020).
- Méndez, J., Garduno, E., Carazo, J. M., Sorzano, C. O. S. Identification of incorrectly oriented particles in cryo-em single particle analysis. J Str Biol. 213 (3), 107771(2021).
- Tan, Y. Z., et al. Addressing preferred specimen orientation in single-particle cryo-em through tilting. Nat Meth. 14 (8), 793-796 (2017).
- Rosenthal, P. B., Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 333 (4), 721-745 (2003).
- Kaur, S., et al. Local computational methods to improve the interpretability and analysis of cryo-em maps. Nat Comm. 12 (1), 1240(2021).
- Stagg, S. M., Noble, A. J., Spilman, M., Chapman, M. S. Reslog plots as an empirical metric of the quality of cryo-em reconstructions. J Str Biol. 185 (3), 418-426 (2014).
- Vargas, J., Otón, J., Marabini, R., Carazo, J. M., Sorzano, C. Particle alignment reliability in single particle electron cryomicroscopy: A general approach. Sci Rep. 6 (1), 21626(2016).
- Vargas, J., et al. Particle quality assessment and sorting for automatic and semiautomatic particle-picking techniques. J Str Biol. 183 (3), 342-353 (2013).
- Vargas, J., Melero, R., Gomez-Blanco, J., Carazo, J. -M., Sorzano, C. O. S. Quantitative analysis of 3d alignment quality: Its impact on soft-validation, particle pruning and homogeneity analysis. Sci Rep. 7 (1), 6307(2017).
- Carragher, B., et al. Current outcomes when optimizing 'standard'sample preparation for single-particle cryo-em. J Microsc. 276 (1), 39-45 (2019).
- Drulyte, I., et al. Approaches to altering particle distributions in cryo-electron microscopy sample preparation. Acta Crystallographica Sec D: Str Biol. 74 (6), 560-571 (2018).
- Glaeser, R. M. How good can cryo-em become. Nat Meth. 13 (1), 28-32 (2016).
- Kim, L. Y., et al. Benchmarking cryo-em single particle analysis workflow. Front Mol Biosci. 5, 50(2018).
- Weissenberger, G., Henderikx, R. J., Peters, P. J. Understanding the invisible hands of sample preparation for cryo-em. Nat Meth. 18 (5), 463-471 (2021).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved