
Method Article
Mejora de los mapas de densidad mediante la eliminación de la mayoría de las partículas en las pilas finales de microscopía electrónica criogénica de una sola partícula
* Estos autores han contribuido por igual
En este artículo
Resumen
Un método avanzado de selección de partículas para crio-EM, a saber, CryoSieve, mejora la resolución del mapa de densidad al eliminar la mayoría de las partículas en las pilas finales, como se demostró a través de su aplicación en un conjunto de datos del mundo real.
Resumen
Durante la última década, los avances en tecnología y metodología dentro del campo del análisis de partículas individuales (SPA) de microscopía electrónica criogénica (crio-EM) han mejorado sustancialmente nuestra capacidad para el examen estructural de alta resolución de macromoléculas biológicas. Este avance ha marcado el comienzo de una nueva era de conocimientos moleculares, reemplazando a la cristalografía de rayos X como método dominante y proporcionando respuestas a preguntas de larga data en biología. Dado que la crio-EM no depende de la cristalización, que es una limitación significativa de la cristalografía de rayos X, captura partículas de calidad variable. En consecuencia, la selección de las partículas es crucial, ya que la calidad de las partículas seleccionadas influye directamente en la resolución del mapa de densidad reconstruido. Un innovador enfoque iterativo para la selección de partículas, denominado CryoSieve, mejora significativamente la calidad de los mapas de densidad reconstruidos al reducir eficazmente el número de partículas en la pila final. La evidencia experimental muestra que este método puede eliminar la mayoría de las partículas en las pilas finales, lo que resulta en una mejora notable en la calidad de los mapas de densidad. En este artículo se describe el flujo de trabajo detallado de este enfoque y se muestra su aplicación en un conjunto de datos del mundo real.
Introducción
El análisis de una sola partícula (SPA) de microscopía electrónica criogénica (crio-EM) se ha convertido en un método dominante para determinar mapas de densidad tridimensional de alta resolución de macromoléculas biológicas. Debido a una serie de innovaciones tecnológicas 1,2,3,4,5,6, denominadas revolución de resolución 7, la crio-EM tiene la capacidad de determinar las estructuras de macromoléculas biológicas con una resolución atómica de hasta un ritmo sin precedentes. Este avance marca el comienzo de una nueva era en los conocimientos moleculares, superando a la cristalografía de rayos X como técnica predominante y respondiendo a preguntas biológicas de larga data.
Cryo-EM SPA difiere de la cristalografía de rayos X al no requerir la cristalización de macromoléculas biológicas. En su lugar, una solución que contiene las macromoléculas biológicas objetivo se congela rápidamente en hielo vítreo. A continuación, se obtienen imágenes con un haz de electrones para producir una serie de micrografías, evitando la necesidad de cristalización8. Posteriormente, se utilizan algoritmos de selección de partículas para extraer partículas crudas individuales de estas micrografías 4,9,10,11,12. Como la crio-EM no depende de la cristalización, es natural que las partículas extraídas estén predominantemente dañadas o en estados conformacionales no deseados, lo que requiere múltiples rondas de selección de partículas para lograr un mapa de densidad de alta resolución. Por lo tanto, en el procesamiento de imágenes crio-EM SPA, la selección de partículas es crucial para obtener mapas de densidad de alta resolución13.
En la crio-EM SPA, los métodos estándar de selección de partículas incluyen la clasificación bidimensional (2D) y tridimensional (3D)14. La clasificación 2D clasifica las partículas en un número predefinido de grupos, lo que produce una imagen promedio y una resolución 2D estimada para cada clase. Luego, los investigadores pueden inspeccionar visualmente estas clases, eliminando partículas de grupos de menor resolución para usar las restantes en reconstrucciones destinadas a lograr una resolución más alta. Una vez que se establezcan las poses de las partículas mediante algoritmos de refinamiento, los investigadores procederán con la clasificación en 3D, agrupando las partículas en múltiples clases. Esto permite la inspección visual del mapa de densidad reconstruido para cada clase, lo que permite la exclusión de partículas indeseables, como las de conformaciones no deseadas. Después de múltiples rondas de clasificación, se obtiene una pila final que comprende partículas de calidad relativamente alta. Estas pilas finales son fundamentales para producir mapas de densidad de resolución atómica o casi atómica.
Zhu y sus colegas han demostrado que se puede llevar a cabo una mayor selección de partículas en estas pilas finales15. CryoSieve15, un innovador método iterativo para la selección de partículas, se puede aplicar para mejorar la calidad del mapa de densidad final al reducir significativamente el número de partículas. Si bien otros criterios y software de clasificación de partículas, como el método de correlación cruzada normalizada (NCC)16, el enfoque de consistencia de grafo angular (AGC)17 y la clasificación de no alineación5, se utilizan actualmente en el campo, se ha demostrado que este método supera a estos algoritmos en términos de efectividad.
En este estudio, presentamos una guía detallada de todo el proceso. Como caso de estudio, aplicamos este nuevo método al conjunto de datos del trímero de hemaglutinina de la gripe (entrada EMPIAR: 10097)18, que incluye 130.000 partículas en su pila final. Nuestro procedimiento descartó con éxito alrededor del 73,8% de las partículas de la pila final de este conjunto de datos, mejorando la resolución del mapa de densidad reconstruido de 4,11 Å a 3,62 Å. Además del trímero de hemaglutinina de la gripe, los resultados de múltiples conjuntos de datos se presentan en la publicación anterior15, que muestra una variedad de resoluciones y pesos moleculares de las biomoléculas.
Protocolo
1. Instalación
- Comprobación y configuración del entorno de aceleración de GPU
- Abra el terminal e ingrese el comando: nvidia-smi. Asegúrese de que el comando muestre correctamente toda la información sobre las tarjetas GPU y que la versión de CUDA sea superior a la 10.2. Ejecute el comando: conda -V para comprobar si Conda está instalado (Figura complementaria 1).
- Configurar el entorno virtual
- Ingrese el siguiente comando para configurar el entorno virtual, reemplazando CRYOSIEVE_ENV con el nombre de entorno deseado: conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge. Espere unos minutos hasta que el entorno se haya configurado correctamente (Figura complementaria 2).
NOTA: Los usuarios tienen la flexibilidad de modificar el nombre del entorno según sea necesario. El comando proporcionado es específico de CUDA 10.2. Si desea una versión diferente de CUDA, ajuste el número de versión de cudatoolkit.
- Ingrese el siguiente comando para configurar el entorno virtual, reemplazando CRYOSIEVE_ENV con el nombre de entorno deseado: conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge. Espere unos minutos hasta que el entorno se haya configurado correctamente (Figura complementaria 2).
- Instalar CryoSieve
- Active el entorno ejecutando el comando: conda activate CRYOSIEVE_ENV. Instale el software ejecutando: pip install cryosieve o conda install -c mxhulab cryosieve (Figura complementaria 3). Ingrese cryosieve -h y asegúrese de que la información de ayuda se muestre correctamente (Figura complementaria 4).
2. Tamizado de partículas
- Recuperar los datos
- Descargue el conjunto de datos de pila final EMPIAR-10097 de EMPIAR (consulte la Tabla de materiales). Descargue el archivo estrella, el archivo de máscara (mask.mrc) y el modelo inicial (para el paso de reestimación; initial.mrc) de Github (ver Tabla de Materiales). Coloque todos estos archivos juntos en una carpeta (Figura complementaria 5).
NOTA: El repositorio de https://github.com/mxhulab/cryosieve-demos utiliza Git Large File Storage (Git LFS). La instalación de Git LFS es esencial para clonar todo el repositorio. Alternativamente, acceda al archivo a través del enlace de GitHub y haga clic en el botón Descargar archivo sin procesar para descargar un archivo individual.
- Descargue el conjunto de datos de pila final EMPIAR-10097 de EMPIAR (consulte la Tabla de materiales). Descargue el archivo estrella, el archivo de máscara (mask.mrc) y el modelo inicial (para el paso de reestimación; initial.mrc) de Github (ver Tabla de Materiales). Coloque todos estos archivos juntos en una carpeta (Figura complementaria 5).
- Tamizado de partículas de proceso
- Abra el terminal y use el comando: cd FILEPATH para navegar a la carpeta donde se encuentra el conjunto de datos. Active el entorno de Conda de la siguiente manera: conda activate CRYOSIEVE_ENV.
- Ingrese el siguiente comando para comenzar nuestro experimento de tamizado de partículas: cryosieve --reconstruct_software relion_reconstruct --postprocess_software relion_postprocess --i T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star --o output/ --mask mask.mrc --angpix 1.3099979 --num_iters 10 --frequency_start 40 --frequency_end 3 --retention_ratio 0.8 --sym C3 --num_gpus 1 --balance (Figura suplementaria 5). Durante la ejecución, el terminal mostrará los registros de salida para cada iteración.
NOTA: Las instrucciones detalladas para cada opción se pueden encontrar en el Archivo Complementario 1. El tiempo de tramitación y los requisitos mínimos para su ejecución se detallan en el Expediente Complementario 2. T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star fue refinado por CryoSPARC a partir de T40_HA_130K-Equalized_run-data.star (descargado de EMPIAR) para mitigar los efectos provocados por los avances en las técnicas de estimación de la orientación.
3. Encontrar la iteración óptima
- Comprobar resoluciones
- Utilice el comando: grep "+ FINAL RESOLUTION:" output/_postprocess*.txt para imprimir los resultados de resolución para las 10 iteraciones de tamizado (Figura 1). Dado que la pila de partículas filtrada en laséptima iteración tiene la resolución más alta con la menor cantidad de partículas, es probable que proporcione el resultado óptimo.
NOTA: Para evitar la transferencia involuntaria de información de las partículas15 descartadas a las retenidas y para garantizar que la pila de partículas publique la 7ª iteración sea realmente óptima, los usuarios deben ejecutar un paso de reestimación para las iteraciones cercanas. En este protocolo, las iteraciones 4, 5, 6, 7 y 8 están sujetas a verificación.
- Utilice el comando: grep "+ FINAL RESOLUTION:" output/_postprocess*.txt para imprimir los resultados de resolución para las 10 iteraciones de tamizado (Figura 1). Dado que la pila de partículas filtrada en laséptima iteración tiene la resolución más alta con la menor cantidad de partículas, es probable que proporcione el resultado óptimo.
- Importación de partículas tamizadas
- Abra la interfaz web de CryoSPARC y siga estos pasos: Ingrese a un espacio de trabajo y haga clic en el botón Builder en la parte superior derecha del panel. En el panel, seleccione y haga clic en la opción Importar pila de partículas . En la sección Parámetros del panel Importación de pila de partículas, especifique la ruta de metadatos de partículas como el archivo _iter{n}.star ubicado en la carpeta de salida de los resultados completados y la ruta de datos de partículas a la carpeta donde se almacena el archivo mrcs. Haga clic en el botón Trabajo en cola y, a continuación, haga clic en el botón Cola para iniciar el proceso. Utilice de la misma manera para importar las iteraciones restantes que necesitan reestimación (Figura complementaria 6A).
- Importar modelo inicial
- Haga clic en el botón Builder en la parte superior derecha del panel. En el panel, seleccione y haga clic en la opción Importar volúmenes 3D .
- Especifique la ruta de datos del volumen como el archivo initial.mrc. Haga clic en el botón Trabajo en cola y, a continuación, haga clic en el botón En cola para iniciar el proceso (Figura complementaria 6B).
NOTA: El modelo inicial también puede generarse a través de la reconstrucción ab initio (Legajo Suplementario 3).
- Refinamiento homogéneo (trabajo de compilación)
- Haga clic en el botón Builder en la parte superior derecha del panel. En el panel, seleccione y haga clic en la opción Refinamiento homogéneo .
NOTA: También se aplica el refinamiento no uniforme.
- Haga clic en el botón Builder en la parte superior derecha del panel. En el panel, seleccione y haga clic en la opción Refinamiento homogéneo .
- Refinamiento homogéneo (Importar partículas)
- En el panel principal de la izquierda, abra el trabajo para importar la pila de partículas de la5ª iteración (o la iteración deseada). Arrastre el módulo de partículas importadas desde el lado derecho del panel principal y suéltelo en la sección Pilas de partículas del Builder a la derecha. Cierre el trabajo Importar pila de partículas haciendo clic en la X roja en la esquina superior derecha del panel principal.
- Abra el trabajo para importar volúmenes 3D. Arrastre el módulo de volúmenes importados desde el lado derecho del panel principal y suéltelo en la sección Volumen inicial del Builder a la derecha.
- Refinamiento homogéneo (Modificar los parámetros)
- En el pliegue Parámetros, localice la opción Simetría y establézcala en C3. Busque la opción Forzar rehacer la división GS y desactívela. Haga clic en el botón Trabajo en cola y, a continuación, haga clic en el botón Cola para iniciar el refinamiento homogéneo. Realice el refinamiento homogéneo para las iteraciones restantes utilizando el mismo método (Figura complementaria 6C-D).
NOTA: La opción Forzar rehacer la división GS es fundamental. La desactivación de esta opción garantiza que CryoSPARC conserve la división estándar de oro dada por el archivo en estrella, evitando el sobreajuste. Una justificación detallada para deshabilitar Force Re-do GS Split se puede encontrar en el Archivo Complementario 4.
- En el pliegue Parámetros, localice la opción Simetría y establézcala en C3. Busque la opción Forzar rehacer la división GS y desactívela. Haga clic en el botón Trabajo en cola y, a continuación, haga clic en el botón Cola para iniciar el refinamiento homogéneo. Realice el refinamiento homogéneo para las iteraciones restantes utilizando el mismo método (Figura complementaria 6C-D).
- Espere hasta que todos los trabajos terminen de ejecutarse para obtener los resultados. Sobre la base de los resultados, se confirma que la pila de partículas filtrada en la6ª iteración es el resultado óptimo real.
NOTA: Es normal que los resultados obtenidos tengan pequeñas desviaciones aleatorias de los resultados proporcionados en este protocolo. Estas desviaciones no afectan a la conclusión general.
Resultados
En este protocolo, utilizamos el conjunto de datos del trímero de hemaglutinina de la influenza (entrada EMPIAR: 10097) como demostración de la eficacia de este proceso. Debido a la orientación preferida de la muestra, la adquisición de datos requirió una inclinación de 40°. La proteína exhibe simetría C3 y tiene un peso molecular de 150 kDa.
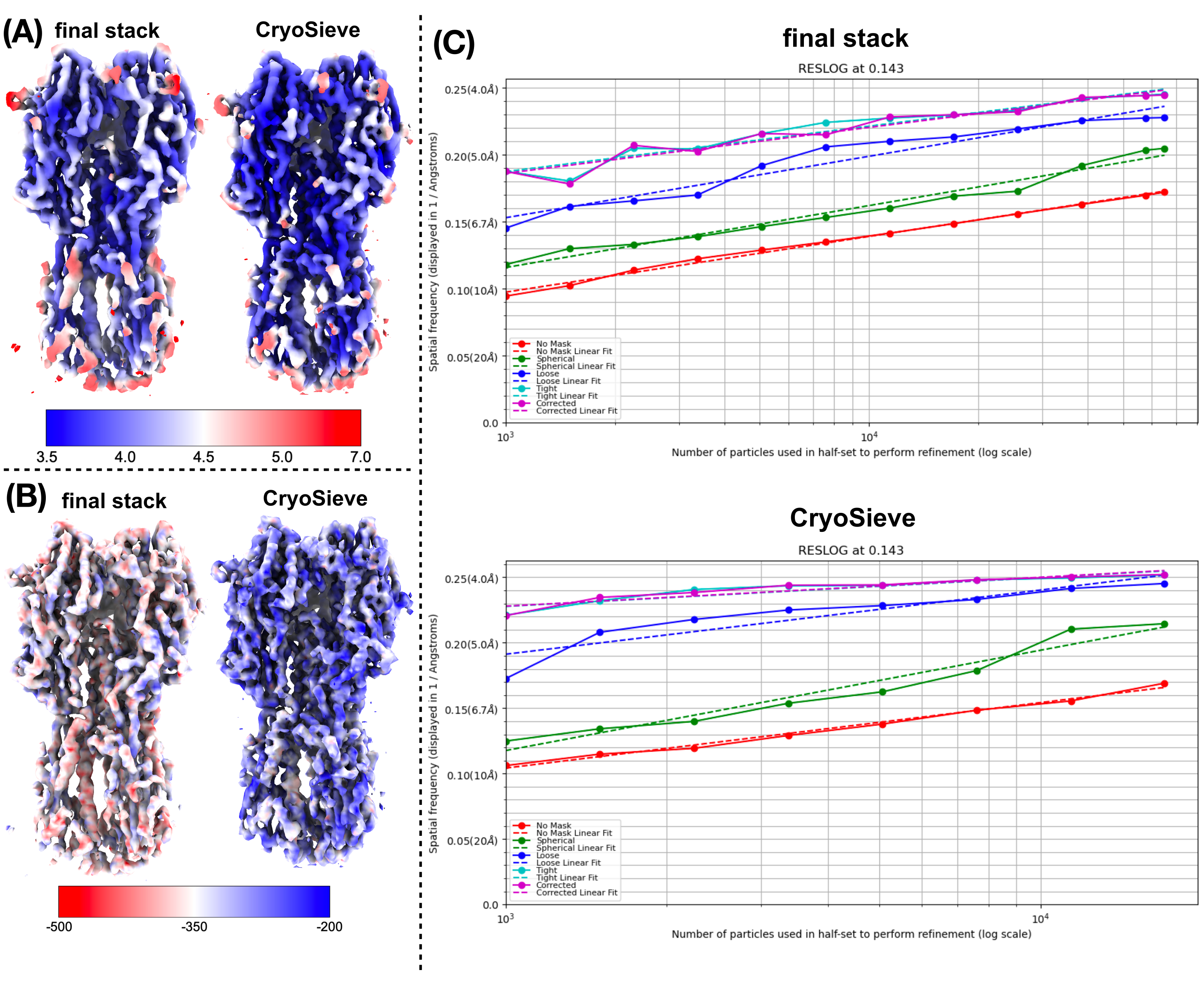
Hemos implementado el protocolo descrito anteriormente para procesar la pila final de partículas. Eliminó progresivamente el 20% de las partículas en cada iteración, lo que resultó en una tasa de retención del 80,0 %, 64,0 %, 51,2 %, etc. Como se muestra en la Figura 1 y la Figura 2, la resolución de las partículas retenidas inicialmente mejoró, pero finalmente disminuyó. Entre las iteraciones, lasexta iteración se identificó como el subconjunto más óptimo, que contiene la menor cantidad de partículas pero logra la resolución más alta. Nuestro algoritmo identificó con éxito un subconjunto de partículas que comprenden solo el 26,2% de la pila original, lo que resultó en una resolución mejorada de 4,19 Å a 3,62 Å (reestimada por CryoSPARC), como se muestra en la Figura 2. Además, se compararon los mapas de densidad antes y después del uso de CryoSieve en la Figura 3. También se muestra la curva de correlación de capas de Fourier (FSC) entre modelos y semimapas de la curva FSC de los mapas de densidad reconstruidos antes y después del método (Figura 3A-B). También se compararon los mapas de densidad brutos y los mapas de densidad nítidos obtenidos, con el nivel de contorno equivalente aplicado (Figura 3C). Se compararon las cadenas laterales de los mapas de densidad nítidos, mostrando la mejora de los mapas de densidad reconstruidos. También se adoptó el factor B estimado de Rosenthal-Henderson para los criterios de calidad de partícula19. Después de eliminar la mayoría de las partículas en la pila final, el factor B de Rosenthal-Henderson aumentó de 226,9 Å2 a 146,2 Å2 (Figura 3D). La resolución local, el factor B local20 y ResLog21 también se utilizaron para la comparación, lo que indica que CryoSieve mejora tanto la calidad de los mapas de densidad como la de las partículas (Figura 4).

Figura 1: Resoluciones de cada iteración. Las resoluciones que se han notificado se resaltan en cuadros rojos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Resoluciones de cada iteración. Las resoluciones identificadas por trabajos de refinamiento homogéneos se resaltan en cuadros rojos. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Mapas de densidad. (A) Comparación de la curva FSC de modelo a mapa de mapas de densidad reconstruidos antes y después de usar CryoSieve. El eje Y representa FSC, mientras que el eje X representa la resolución. La línea discontinua roja marca el umbral de 0,5 para el FSC. La línea discontinua vertical ilustra la resolución de los mapas de densidad obtenidos bajo un umbral de 0,5. (B) Los medios mapas de la curva FSC se obtuvieron a partir de mapas de densidad reconstruidos antes y después de usar CryoSieve a través de CryoSPARC. El eje Y representa FSC, mientras que el eje X representa la resolución. (C) Se mostraron mapas de densidad en bruto y mapas de densidad nítidos tanto para las partículas retenidas por Cryosieve como para el conjunto completo de partículas en las pilas finales. Se aplicó un nivel de contorno equivalente de 0,65 para los mapas de densidad sin procesar. Se aplicó un nivel de contorno equivalente de 0,84 para mapas de densidad nítidos. Los mapas de densidad nítidos se obtuvieron directamente con CryoSPARC. Los mapas de densidad nítidos se autoprocesaron posteriormente, primero ponderados por FSC (basados en FSC proporcionados por CryoSPARC). A continuación, el factor B se afinó utilizando los factores B autodeterminados (232,0 Å2 para todas las partículas de la pila final y 160,8 Å2 para CryoSieve). Se compararon las cadenas laterales en los mapas de densidad nítidos, incorporando modelos atómicos como referencia. Las flechas rojas resaltan las regiones mejoradas. (D) El factor B estimado de Rosenthal-Henderson se mostró tanto para las partículas retenidas por Cryosieve como para el conjunto completo de partículas en las pilas finales. El eje y representa el número de partículas utilizadas, y el eje x representa el recíproco del cuadrado de la resolución. Moviéndose de arriba a abajo, cada punto representa la mitad de las partículas del anterior. Las resoluciones fueron determinadas por el refinamiento. Los factores B se determinaron utilizando una aproximación por mínimos cuadrados de los puntos medidos, como se muestra en las curvas de ajuste. Los factores B estimados de Rosenthal y Henderson se indican en las leyendas: el naranja representa las partículas retenidas por CryoSieve, mientras que el azul denota todas las partículas en la pila final. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Comparación de varias métricas de mapas de densidad. (A) Comparación de mapas de resolución local antes y después de usar CryoSieve obtenidos por CryoSPARC. La resolución local oscila entre 7 Å (rojo) y 3,5 Å (azul). (B) Comparación de mapas de densidad antes y después de usar CryoSieve, coloreados con el mapa del factor B local obtenido por LocBFactor usando un rango de resolución de [20-3.5] Å. (C), Comparación de gráficos de ResLog antes y después de usar CryoSieve obtenidos por CryoSPARC. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Figura complementaria 1: Uso de los comandos nvidia-smi y conda -V para verificar los requisitos previos. Si se cumplen los requisitos previos, al escribir el comando nvidia-smi se mostrará la versión del controlador de GPU, la versión de CUDA y el estado de las tarjetas de GPU. Del mismo modo, al escribir el comando conda -V se debería mostrar correctamente la versión instalada de Conda. Haga clic aquí para descargar este archivo.
Figura complementaria 2: El proceso de creación de nuevos entornos de aceleración de GPU. La pantalla muestra la salida generada por el comando utilizado para crear el entorno Conda. Haga clic aquí para descargar este archivo.
Figura complementaria 3: Instalación de CryoSieve en el entorno de aceleración de GPU. Después de activar el entorno Conda recién creado, la pantalla muestra el resultado de ejecutar el comando para instalar CryoSieve usando Pip. Por favor, haga clic aquí para descargar este archivo.
Figura complementaria 4: Información de ayuda. Haga clic aquí para descargar este archivo.
Figura complementaria 5: Proceso en ejecución. Al ejecutar CryoSieve a través de la línea de comandos, la pantalla muestra información sobre el proceso en ejecución. Haga clic aquí para descargar este archivo.
Figura complementaria 6: Configuración de los trabajos de CryoSPARC. (A) Importar pila de partículas. (B) Importar volúmenes 3D. (C-D) Refinamiento homogéneo. Haga clic aquí para descargar este archivo.
Ficha complementaria 1: Opciones de CryoSieve. Haga clic aquí para descargar este archivo.
Archivo complementario 2: Tiempo de procesamiento y requisito mínimo para ejecutar Cryosieve. Haga clic aquí para descargar este archivo.
Archivo complementario 3: Generación del modelo inicial por CryoSPARC. Haga clic aquí para descargar este archivo.
Archivo complementario 4: Justificación para deshabilitar la reactivación de la rehacer forzada la división GS. Haga clic aquí para descargar este archivo.
Fichero complementario 5: Opciones de cryosieve-csrefine. Haga clic aquí para descargar este archivo.
Archivo complementario 6: Opciones de cryosieve-csrhbfactor. Haga clic aquí para descargar este archivo.
Discusión
Cryo-EM se presenta como una técnica fundamental para dilucidar las estructuras de las moléculas biológicas. En este proceso, después de la recopilación de datos mediante microscopía, es esencial la extracción de partículas de las micrografías, seguida de su clasificación en múltiples etapas para compilar la pila final. Un desafío común es el predominio de partículas dañadas o no deseadas, lo que subraya la necesidad de una selección repetida de partículas para obtener mapas de densidad de alta resolución. Esto hace que la selección de partículas sea un paso crítico en la SPA crio-EM para lograr mapas de densidad de alta calidad. Las técnicas de selección de partículas existentes incluyen el algoritmo de validación estadística sin inclinación22, el enfoque basado en la puntuación z23 y el método de estimación de precisión angular24.
CryoSieve emerge como una herramienta valiosa en este contexto, experta en eliminar un número significativo de partículas extrañas de la pila final. Esta reducción no solo mejora la eficiencia computacional de la reconstrucción, sino que también agiliza el proceso. Ofrece un conjunto completo para la selección de partículas, donde el grado de descarte de partículas y la consiguiente mejora en la resolución dependen en gran medida de la calidad inicial de los datos y las metodologías empleadas en el procesamiento de datos.
En este manuscrito, hemos presentado un flujo de trabajo completo de tamizado de partículas utilizando el conjunto de datos de casos reales del trímero de hemaglutinina de influenza (entrada EMPIAR: 10097). Los pasos cubiertos y discutidos aquí se pueden resumir como tamizado de partículas y reestimación de planteas. El volumen final reconstruido en 3D alcanzó una resolución de 3,62 Å, y las cadenas laterales en las hélices alfa fueron más claras en el volumen postprocesado en comparación con el mapa de densidad publicado.
CryoSieve es un método de código abierto que está disponible en GitHub (https://github.com/mxhulab/cryosieve). También se puede encontrar un tutorial detallado en su página de inicio. Los usuarios pueden instalarlo y usarlo siguiendo el tutorial. Además, se proporcionan dos módulos, cryosieve-csrefine y cryosieve-csrhbfactor. El módulo cryosieve-csrefine está diseñado específicamente para automatizar la ejecución secuencial de varias operaciones dentro de CryoSPARC (Archivo complementario 5). Estas operaciones incluyen la importación de pilas de partículas y la realización de trabajos de refinamiento ab initio, homogéneos o no uniformes. Por otro lado, el módulo cryosieve-csrhbfactor está diseñado para automatizar la determinación del factor B de Rosenthal-Henderson aprovechando las capacidades de cryosieve-csrefine (Archivo suplementario 6).
En la actualidad, la aplicación de este método se limita a escenarios de conformación única. En consecuencia, en los casos en que las partículas representan múltiples conformaciones, sus capacidades son limitadas. Se aconseja a los usuarios que inicialmente participen en la clasificación 3D para segregar partículas de conformaciones dispares antes de emplearla para una selección refinada de partículas. Además, aunque el método demuestra ser competente para filtrar más del 50% de las partículas de la pila final, los orígenes de estas partículas desechadas y las razones subyacentes de su insignificante contribución a la calidad de la reconstrucción siguen sin estar claros. Esta brecha en la comprensión requiere investigación adicional para abordar de manera integral y potencialmente rectificar esta limitación.
Existen tres posibles métodos de clasificación de partículas o tamizado de partículas. En primer lugar, cisTEM4 puede informar de una puntuación para cada imagen de partícula después del refinamiento 3D. Los usuarios podían clasificar las partículas utilizando la puntuación cisTEM para descartar las partículas. El enfoque de consistencia de grafo angular (AGC)17 también es un método para descartar partículas desalineadas. Además, la clasificación de no alineación5 es una forma tradicional de descartar partículas mediante la clasificación 3D. Comparamos la calidad de las partículas retenidas por estos métodos con CryoSieve y encontramos que las partículas retenidas de CryoSieve son de mayor calidad15. El método presentado aquí supera significativamente a los métodos alternativos y logra el menor número de partículas con la misma resolución.
Como se demuestra en el resultado, la mayoría de las partículas en una pila final de crio-EM no contribuyen a la reconstrucción del mapa de densidad. En otras palabras, entre todas las partículas recogidas durante la adquisición de imágenes, solo unas pocas, es decir, el subconjunto más fino, contribuyen realmente a la reconstrucción final. En consecuencia, la relación entre este último subconjunto y el número total de partículas recolectadas podría servir como una métrica cuantitativa para evaluar la calidad de la muestra. Cuanto mayor sea esta proporción, mejor será la calidad de la muestra. A pesar de los avances técnicos que han hecho que la crio-EM sea más accesible para los biólogos estructurales, la preparación de muestras sigue siendo un cuello de botella importante en el flujo de trabajo. Por lo tanto, los científicos e ingenieros están centrando sus esfuerzos en este desafío25. En el análisis de una sola partícula (SPA), la preparación de la muestra consta de dos pasos cruciales: la optimización de la muestra y la preparación de la cuadrícula. El primero consiste en purificar la muestra manteniendo su estado bioquímico óptimo. Este último implica la preparación de la muestra para su análisis en el microscopio, incluido el tratamiento químico o con plasma de la rejilla, la deposición de la muestra y la vitrificación. Se han propuesto numerosas técnicas para abordar la inestabilidad macromolecular, pero la eficacia de un enfoque sobre otro depende de las características de la muestra25,26. En la actualidad, los resultados de la preparación de la red están muy influenciados por los conocimientos y la experiencia del usuario, lo que puede hacer que el proceso sea lento y desafiante27,28. Las numerosas variables encontradas en la preparación de muestras y cuadrículas plantean desafíos para establecer relaciones de causa y efecto, ya que los investigadores solo pueden evaluar la muestra a nivel molecular usando el microscopio. Como resultado, aún faltan estadísticas cuantitativas a partir de comparaciones de diferentes protocolos de preparación de muestras y cuadrículas, y es necesario un enfoque sistemático para investigar las tendencias y comprender los mecanismos fundamentales del comportamiento de la muestra29.
Divulgaciones
Todos los demás autores declaran no tener intereses contrapuestos.
Agradecimientos
Este trabajo fue apoyado por la Academia de Investigación y Traducción de Shenzhen (a M.H.), el Centro de Innovación Avanzada para la Biología Estructural (a M.H.), el Centro de Investigación de la Frontera de Beijing para la Estructura Biológica (a M.H.), el Programa Nacional Clave de Investigación y Desarrollo de China (No.2021YFA1001300) (a C.B.), la Fundación Nacional de Ciencias Naturales de China (No.12271291) (a C.B.), y la Fundación Nacional de Ciencias Naturales de China (No.12071244) (a Z.S.).
Materiales
| Name | Company | Catalog Number | Comments |
| CryoSPARC | Structura Biotechnology Inc. Toronto, Canada | CryoSPARC (Cryo-EM Single Particle Ab-Initio Reconstruction and Classification) is a state of the art HPC software solution for complete processing of single-particle cryo-electron microscopy (cryo-EM) data. CryoSPARC is useful for solving cryo-EM structures of membrane proteins, viruses, complexes, flexible molecules, small particles, phase plate data and negative stain data. | |
| EMPIAR-10097 Dataset | https://ftp.ebi.ac.uk/empiar/world_availability/10097/data/Particle-Stack/T40_HA_130K-Equalized-Particle-Stack.mrcs | This dataset comprises single-particle cryo-EM data of the Influenza Hemagglutinin trimer, characterized by its highly preferred orientation, collected using a 40-degree tilted collection strategy. | |
| initial.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| mask.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| RELION | 4.0-beta-2 | RELION (REgularised LIkelihood OptimisatioN) is an open-source software for cryo-electron microscopy (cryo-EM) data processing, particularly for refining macromolecular structures. Utilizing a Bayesian approach, it excels in separating signal from noise, enabling high-resolution structure determination. RELION supports single-particle analysis, tomography, and sub-tomogram averaging, and has become widely used in structural biology due to its effectiveness and user-friendly interface. | |
| T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | Metadata file for the final stack of particles from EMPIAR-10097 |
Referencias
- Bai, X. C., Fernandez, I. S., Mcmullan, G., Scheres, S. H. Ribosome structures to near-atomic resolution from thirty thousand cryo-em particles. elife. 2, 00461(2013).
- Campbell, M. G., et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 20 (11), 1823-1828 (2012).
- Li, X., et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-em. Nat Meth. 10 (6), 584-590 (2013).
- Grant, T., Rohou, A., Grigorieff, N. Cis tem, user-friendly software for single-particle image processing. eLife. 7, e35383(2018).
- Scheres, S. H. Relion: Implementation of a bayesian approach to cryo-em structure determination. J Str Biol. 180 (3), 519-530 (2012).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. Cryosparc: Algorithms for rapid unsupervised cryo-em structure determination. Nat Meth. 14 (3), 290-296 (2017).
- Kühlbrandt, W. The resolution revolution. Science. 343 (6178), 1443-1444 (2014).
- Dubochet, J., et al. Cryo-electron microscopy of vitrified specimens. Quart Rev Biophys. 21 (2), 129-228 (1988).
- Wagner, T., et al. Sphire-cryolo is a fast and accurate fully automated particle picker for cryo-EM. Comm Biol. 2 (1), 218(2019).
- Bepler, T., et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Meth. 16 (11), 1153-1160 (2019).
- Wang, F., et al. Deeppicker: A deep learning approach for fully automated particle picking in cryo-em. J Str Biol. 195 (3), 325-336 (2016).
- Heimowitz, A., Andén, J., Singer, A. Apple picker: Automatic particle picking, a low-effort cryo-em framework. J Str Biol. 204 (2), 215-227 (2018).
- Glaeser, R. M. How good can single-particle cryo-em become? What remains before it approaches its physical limits. Ann Rev Biophys. 48, 45-61 (2019).
- Diiorio, M. C., Kulczyk, A. W. A robust single-particle cryo-electron microscopy (cryo-em) processing workflow with cryosparc, relion, and scipion. J Vis Exp. (179), e63387(2022).
- Zhu, J., et al. A minority of final stacks yields superior amplitude in single-particle cryo-em. Nat Comm. 14 (1), 7822(2023).
- Zhou, Y., Moscovich, A., Bendory, T., Bartesaghi, A. Unsupervised particle sorting for high-resolution single-particle cryo-em. Inv Probl. 36 (4), 044002(2020).
- Méndez, J., Garduno, E., Carazo, J. M., Sorzano, C. O. S. Identification of incorrectly oriented particles in cryo-em single particle analysis. J Str Biol. 213 (3), 107771(2021).
- Tan, Y. Z., et al. Addressing preferred specimen orientation in single-particle cryo-em through tilting. Nat Meth. 14 (8), 793-796 (2017).
- Rosenthal, P. B., Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 333 (4), 721-745 (2003).
- Kaur, S., et al. Local computational methods to improve the interpretability and analysis of cryo-em maps. Nat Comm. 12 (1), 1240(2021).
- Stagg, S. M., Noble, A. J., Spilman, M., Chapman, M. S. Reslog plots as an empirical metric of the quality of cryo-em reconstructions. J Str Biol. 185 (3), 418-426 (2014).
- Vargas, J., Otón, J., Marabini, R., Carazo, J. M., Sorzano, C. Particle alignment reliability in single particle electron cryomicroscopy: A general approach. Sci Rep. 6 (1), 21626(2016).
- Vargas, J., et al. Particle quality assessment and sorting for automatic and semiautomatic particle-picking techniques. J Str Biol. 183 (3), 342-353 (2013).
- Vargas, J., Melero, R., Gomez-Blanco, J., Carazo, J. -M., Sorzano, C. O. S. Quantitative analysis of 3d alignment quality: Its impact on soft-validation, particle pruning and homogeneity analysis. Sci Rep. 7 (1), 6307(2017).
- Carragher, B., et al. Current outcomes when optimizing 'standard'sample preparation for single-particle cryo-em. J Microsc. 276 (1), 39-45 (2019).
- Drulyte, I., et al. Approaches to altering particle distributions in cryo-electron microscopy sample preparation. Acta Crystallographica Sec D: Str Biol. 74 (6), 560-571 (2018).
- Glaeser, R. M. How good can cryo-em become. Nat Meth. 13 (1), 28-32 (2016).
- Kim, L. Y., et al. Benchmarking cryo-em single particle analysis workflow. Front Mol Biosci. 5, 50(2018).
- Weissenberger, G., Henderikx, R. J., Peters, P. J. Understanding the invisible hands of sample preparation for cryo-em. Nat Meth. 18 (5), 463-471 (2021).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados