
Method Article
Amélioration des cartes de densité en supprimant la majorité des particules dans les piles finales de microscopie électronique cryogénique à particule unique
* Ces auteurs ont contribué à parts égales
Dans cet article
Résumé
Une méthode avancée de sélection de particules pour la cryo-EM, à savoir CryoSieve, améliore la résolution de la carte de densité en supprimant la majorité des particules dans les piles finales, comme le démontre son application sur un ensemble de données du monde réel.
Résumé
Au cours de la dernière décennie, les progrès technologiques et méthodologiques dans le domaine de la microscopie électronique cryogénique (cryo-EM) et de l’analyse de particules uniques (SPA) ont considérablement amélioré notre capacité d’examen structurel à haute résolution des macromolécules biologiques. Cette avancée a inauguré une nouvelle ère de connaissances moléculaires, remplaçant la cristallographie aux rayons X comme méthode dominante et fournissant des réponses à des questions de longue date en biologie. Étant donné que la cryo-EM ne dépend pas de la cristallisation, ce qui est une limitation importante de la cristallographie aux rayons X, elle capture des particules de qualité variable. Par conséquent, la sélection des particules est cruciale, car la qualité des particules sélectionnées influence directement la résolution de la carte de densité reconstruite. Une approche itérative innovante pour la sélection des particules, appelée CryoSieve, améliore considérablement la qualité des cartes de densité reconstruites en réduisant efficacement le nombre de particules dans la pile finale. Des preuves expérimentales montrent que cette méthode peut éliminer la majorité des particules dans les piles finales, ce qui entraîne une amélioration notable de la qualité des cartes de densité. Cet article décrit le flux de travail détaillé de cette approche et présente son application sur un ensemble de données du monde réel.
Introduction
L’analyse de particules uniques (SPA) par microscopie électronique cryogénique (cryo-EM) est devenue une méthode dominante pour déterminer des cartes de densité tridimensionnelles à haute résolution de macromolécules biologiques. Grâce à une série d’innovations technologiques 1,2,3,4,5,6, appelées révolution de résolution 7, la cryo-EM a la capacité de déterminer les structures de macromolécules biologiques avec une résolution atomique allant jusqu’à un rythme sans précédent. Cette percée marque le début d’une nouvelle ère dans les connaissances moléculaires, dépassant la cristallographie aux rayons X en tant que technique prédominante et répondant à des questions biologiques de longue date.
La Cryo-EM SPA s’écarte de la cristallographie aux rayons X en ne nécessitant pas la cristallisation de macromolécules biologiques. Au lieu de cela, une solution contenant les macromolécules biologiques cibles est rapidement congelée dans de la glace vitrée. Il est ensuite imagé à l’aide d’un faisceau d’électrons pour produire une série de micrographies, en contournant le besoin de cristallisation8. Par la suite, des algorithmes de sélection de particules sont utilisés pour extraire des particules brutes individuelles de ces micrographies 4,9,10,11,12. Comme la cryo-EM ne dépend pas de la cristallisation, il est naturel que les particules extraites soient principalement endommagées ou dans des états conformationnels indésirables, ce qui nécessite plusieurs cycles de sélection de particules pour obtenir une carte de densité à haute résolution. Dans le traitement d’images cryo-EM SPA, la sélection des particules est donc cruciale pour obtenir des cartes de densité à haute résolution13.
Dans la cryo-EM SPA, les méthodes standard de sélection des particules comprennent la classification bidimensionnelle (2D) et tridimensionnelle (3D)14. La classification 2D catégorise les particules en un nombre prédéfini de groupes, ce qui permet d’obtenir une image moyenne et une résolution 2D estimée pour chaque classe. Les chercheurs peuvent ensuite inspecter visuellement ces classes, en supprimant les particules des groupes à faible résolution pour utiliser les autres dans des reconstructions visant à atteindre une résolution plus élevée. Une fois que les poses des particules auront été établies à l’aide d’algorithmes de raffinement, les chercheurs procéderont à la classification 3D, en regroupant les particules en plusieurs classes. Cela permet une inspection visuelle de la carte de densité reconstruite pour chaque classe, ce qui permet d’exclure les particules indésirables, telles que celles provenant de conformations indésirables. Après plusieurs cycles de classification, on obtient un dernier empilement comprenant des particules de qualité relativement élevée. Ces empilements finaux jouent un rôle déterminant dans la production de cartes de densité à résolution atomique ou quasi atomique.
Zhu et ses collègues ont démontré qu’une sélection supplémentaire de particules peut être effectuée sur ces empilements finaux15. CryoSieve15, une méthode itérative innovante pour la sélection des particules, peut être appliquée pour améliorer la qualité de la carte de densité finale en réduisant considérablement le nombre de particules. Bien que d’autres critères et logiciels de tri des particules, tels que la méthode de corrélation croisée normalisée (NCC)16, l’approche de cohérence de graphe angulaire (AGC)17 et la classification de non-alignement5, soient actuellement utilisés sur le terrain, il a été démontré que cette méthode surpasse ces algorithmes en termes d’efficacité.
Dans cette étude, nous présentons un guide détaillé de l’ensemble du processus. À titre d’étude de cas, nous avons appliqué cette nouvelle méthode à l’ensemble de données du trimère d’hémagglutinine de la grippe (entrée EMPIAR : 10097)18, qui comprend 130 000 particules dans sa pile finale. Notre procédure a réussi à éliminer environ 73,8 % des particules de la dernière pile de cet ensemble de données, améliorant ainsi la résolution de la carte de densité reconstruite de 4,11 Å à 3,62 Å. En plus du trimère de l’hémagglutinine de la grippe, les résultats de plusieurs ensembles de données sont présentés dans une publication antérieure15, mettant en évidence une variété de résolutions et de poids moléculaires de biomolécules.
Protocole
1. L’installation
- Vérification et configuration de l’environnement d’accélération GPU
- Ouvrez le terminal et entrez la commande : nvidia-smi. Assurez-vous que la commande affiche correctement toutes les informations sur la ou les cartes GPU et que la version CUDA est supérieure à 10.2. Exécutez la commande : conda -V pour vérifier si Conda est installé (Figure supplémentaire 1).
- Configurer l’environnement virtuel
- Entrez la commande suivante pour configurer l’environnement virtuel, en remplaçant CRYOSIEVE_ENV par le nom d’environnement souhaité : conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge. Patientez quelques minutes jusqu’à ce que l’environnement soit correctement configuré (Figure supplémentaire 2).
REMARQUE : Les utilisateurs ont la possibilité de modifier le nom de l’environnement selon leurs besoins. La commande fournie est spécifique à CUDA 10.2. Si vous souhaitez une autre version de CUDA, ajustez le numéro de version de cudatoolkit.
- Entrez la commande suivante pour configurer l’environnement virtuel, en remplaçant CRYOSIEVE_ENV par le nom d’environnement souhaité : conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge. Patientez quelques minutes jusqu’à ce que l’environnement soit correctement configuré (Figure supplémentaire 2).
- Installer CryoSieve
- Activez l’environnement en exécutant la commande : conda activate CRYOSIEVE_ENV. Installez le logiciel en exécutant : pip install cryosieve ou conda install -c mxhulab cryosieve (Figure supplémentaire 3). Entrez dans le cryotamis -h et assurez-vous que les informations d’aide s’affichent correctement (Figure supplémentaire 4).
2. Tamis des particules
- Récupérer les données
- Téléchargez l’ensemble de données de l’empilement final EMPIAR-10097 sur EMPIAR (voir Tableau des matériaux). Téléchargez le fichier star, le fichier mask (mask.mrc) et le modèle initial (pour l’étape de réestimation ; initial.mrc) depuis Github (voir Table des matériaux). Placez tous ces fichiers dans un dossier (Figure supplémentaire 5).
REMARQUE : Le référentiel de https://github.com/mxhulab/cryosieve-demos utilise Git Large File Storage (Git LFS). L’installation de Git LFS est essentielle pour cloner l’ensemble du dépôt. Vous pouvez également accéder au fichier via le lien GitHub et cliquer sur le bouton Télécharger le fichier brut pour télécharger un fichier individuel.
- Téléchargez l’ensemble de données de l’empilement final EMPIAR-10097 sur EMPIAR (voir Tableau des matériaux). Téléchargez le fichier star, le fichier mask (mask.mrc) et le modèle initial (pour l’étape de réestimation ; initial.mrc) depuis Github (voir Table des matériaux). Placez tous ces fichiers dans un dossier (Figure supplémentaire 5).
- Tamisage des particules de processus
- Ouvrez le terminal et utilisez la commande : cd FILEPATH pour accéder au dossier où se trouve l’ensemble de données. Activez l’environnement Conda en : conda activate CRYOSIEVE_ENV.
- Entrez la commande suivante pour démarrer notre expérience de tamisage de particules : cryosieve --reconstruct_software relion_reconstruct --postprocess_software relion_postprocess --i T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star --o output/ --mask mask.mrc --angpix 1.3099979 --num_iters 10 --frequency_start 40 --frequency_end 3 --retention_ratio 0.8 --sym C3 --num_gpus 1 --balance (Figure supplémentaire 5). Pendant l’exécution, le terminal affichera les journaux de sortie pour chaque itération.
REMARQUE : Des instructions détaillées pour chaque option se trouvent dans le Fichier supplémentaire 1. Le temps de traitement et les exigences minimales pour l’exécution sont détaillés dans le fichier supplémentaire 2. T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star a été affiné par CryoSPARC à partir de T40_HA_130K-Equalized_run-data.star (téléchargé à partir d’EMPIAR) afin d’atténuer les effets provoqués par les progrès des techniques d’estimation de l’orientation.
3. Trouver l’itération optimale
- Vérifier les résolutions
- Utilisez la commande : grep « + FINAL RESOLUTION : » output/_postprocess*.txt pour imprimer les résultats de résolution pour les 10 itérations de tamisage (Figure 1). Étant donné que l’empilement de particules filtré dans la 7eitération a la résolution la plus élevée avec le moins de particules, il est probable qu’il fournira le résultat optimal.
REMARQUE : Pour éviter le transfert involontaire d’informations des particules mises au rebut vers les particules conservées15 et pour s’assurer que l’empilement de particules après la 7e itération est effectivement optimal, les utilisateurs doivent exécuter une étape de réestimation pour les itérations voisines. Dans ce protocole, les itérations 4, 5, 6, 7 et 8 sont soumises à une vérification.
- Utilisez la commande : grep « + FINAL RESOLUTION : » output/_postprocess*.txt pour imprimer les résultats de résolution pour les 10 itérations de tamisage (Figure 1). Étant donné que l’empilement de particules filtré dans la 7eitération a la résolution la plus élevée avec le moins de particules, il est probable qu’il fournira le résultat optimal.
- Importer des particules tamisées
- Ouvrez l’interface Web de CryoSPARC et procédez comme suit : Entrez dans un espace de travail et cliquez sur le bouton Builder en haut à droite du panneau. Dans le panneau, sélectionnez et cliquez sur l’option Importer la pile de particules . Dans la section Paramètres du panneau Importation de pile de particules, spécifiez le méta-chemin d’accès aux particules en tant que fichier _iter{n}.star situé dans le dossier de sortie des résultats terminés et le chemin d’accès aux données de particules vers le dossier où le fichier mrcs est stocké. Cliquez sur le bouton File d’attente de la tâche , puis sur le bouton File d’attente pour lancer le processus. Utilisez la même méthode pour importer les itérations restantes qui doivent être réestimées (Figure supplémentaire 6A).
- Importer le modèle initial
- Cliquez sur le bouton Builder en haut à droite du panneau. Dans le panneau, sélectionnez et cliquez sur l’option Importer des volumes 3D .
- Spécifiez le chemin d’accès aux données du volume en tant que fichier initial.mrc. Cliquez sur le bouton File d’attente de la tâche , puis sur le bouton File d’attente pour lancer le processus (Figure supplémentaire 6B).
REMARQUE : Le modèle initial peut également être généré par reconstruction ab initio (Fichier supplémentaire 3).
- Raffinement homogène (travail de construction)
- Cliquez sur le bouton Builder en haut à droite du panneau. Dans le panneau, sélectionnez et cliquez sur l’option Affinement homogène .
REMARQUE : Un raffinement non uniforme s’applique également.
- Cliquez sur le bouton Builder en haut à droite du panneau. Dans le panneau, sélectionnez et cliquez sur l’option Affinement homogène .
- Raffinement homogène (importation de particules)
- Dans le panneau principal de gauche, ouvrez la tâche d’importation de la pile de particules de la 5eitération (ou de l’itération souhaitée). Faites glisser le module de particules importées depuis le côté droit du panneau principal et déposez-le dans la section Piles de particules du constructeur sur la droite. Fermez la tâche Importer la pile de particules en cliquant sur le X rouge dans le coin supérieur droit du panneau principal.
- Ouvrez la tâche d’importation de volumes 3D. Faites glisser le module de volumes importés depuis le côté droit du panneau principal et déposez-le dans la section Volume initial du constructeur à droite.
- Raffinement homogène (Modifier les paramètres)
- Sous le pli Paramètres, localisez l’option Symétrie et réglez-la sur C3. Trouvez l’option Forcer le fractionnement GS et désactivez-la. Cliquez sur le bouton File d’attente de la tâche , puis sur le bouton File d’attente pour lancer l’affinement homogène. Effectuez un raffinement homogène pour les itérations restantes à l’aide de la même méthode (Figure supplémentaire 6C-D).
REMARQUE : L’option Forcer la réédition GS est essentielle. La désactivation de cette option garantit que CryoSPARC conserve la division de référence donnée par la lime étoile, évitant ainsi le surapprentissage. Une justification détaillée de la désactivation de Force Re-do GS Split se trouve dans le Fichier supplémentaire 4.
- Sous le pli Paramètres, localisez l’option Symétrie et réglez-la sur C3. Trouvez l’option Forcer le fractionnement GS et désactivez-la. Cliquez sur le bouton File d’attente de la tâche , puis sur le bouton File d’attente pour lancer l’affinement homogène. Effectuez un raffinement homogène pour les itérations restantes à l’aide de la même méthode (Figure supplémentaire 6C-D).
- Attendez la fin de l’exécution de tous les travaux pour obtenir les résultats. Sur la base des résultats, il est confirmé que l’empilement de particules filtré dans la 6eitération est le résultat optimal réel.
REMARQUE : Il est normal que les résultats obtenus présentent des écarts aléatoires mineurs par rapport aux résultats fournis dans ce protocole. Ces écarts n’affectent pas la conclusion globale.
Résultats
Dans ce protocole, nous avons utilisé l’ensemble de données sur le trimère de l’hémagglutinine de la grippe (entrée EMPIAR : 10097) comme démonstration de l’efficacité de ce processus. En raison de l’orientation préférée de l’échantillon, l’acquisition des données a nécessité une inclinaison à 40°. La protéine présente une symétrie C3 et a un poids moléculaire de 150 kDa.
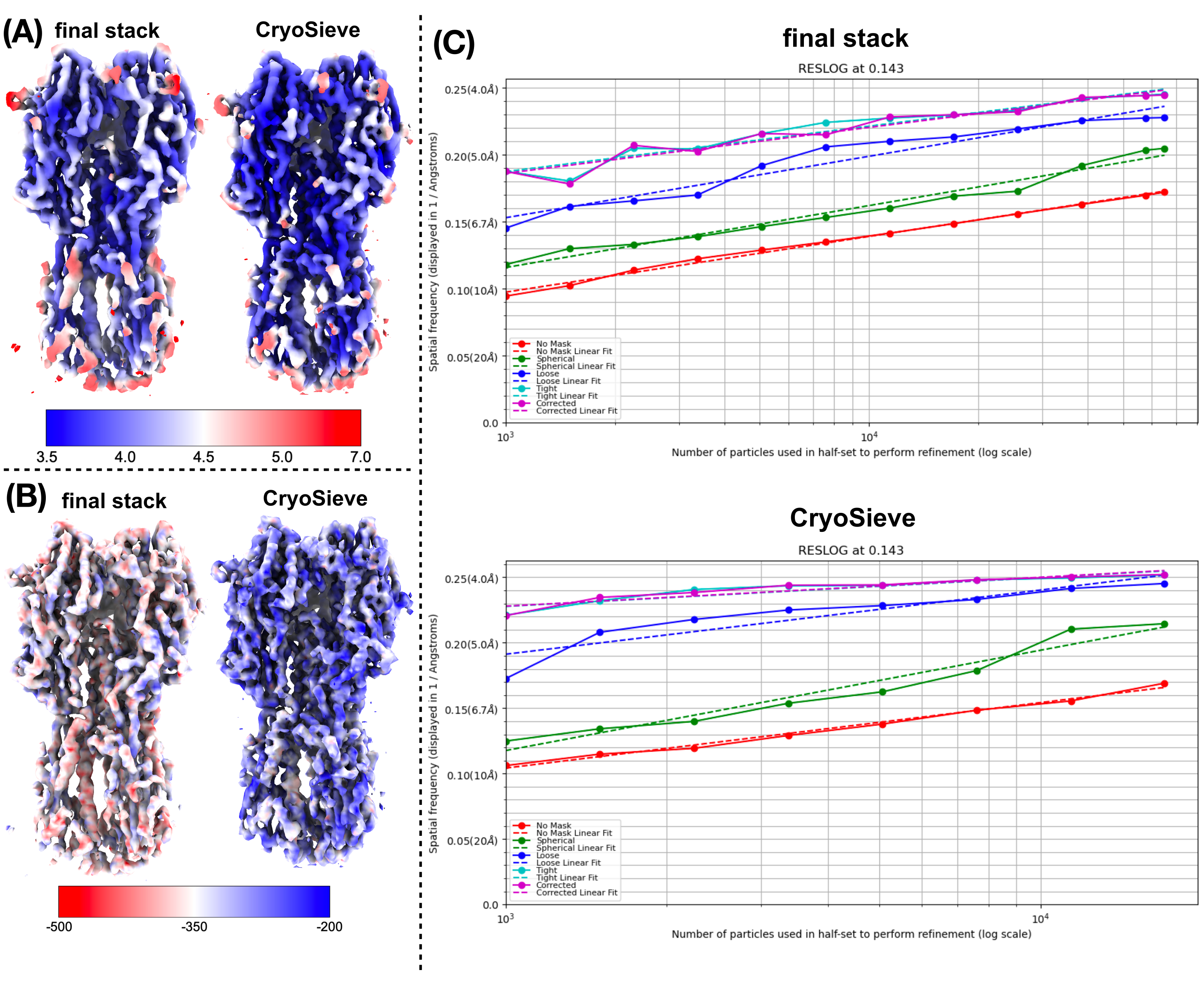
Nous avons mis en œuvre le protocole décrit précédemment pour traiter l’empilement final de particules. Il a progressivement éliminé 20 % des particules à chaque itération, ce qui a donné un taux de rétention de 80,0 %, 64,0 %, 51,2 %, etc. Comme le montrent les figures 1 et 2, la résolution des particules retenues s’est initialement améliorée, mais a finalement diminué. Parmi les itérations, la 6e itération a été identifiée comme le sous-ensemble le plus optimal, contenant le moins de particules tout en atteignant la résolution la plus élevée. Notre algorithme a réussi à identifier un sous-ensemble de particules ne représentant que 26,2 % de l’empilement d’origine, ce qui a permis d’améliorer la résolution de 4,19 Å à 3,62 Å (réestimée par CryoSPARC), comme le montre la figure 2. De plus, les cartes de densité avant et après l’utilisation de CryoSieve ont été comparées dans la figure 3. La courbe de corrélation de l’enveloppe de Fourier (FSC) et les demi-cartes de la courbe FSC des cartes de densité reconstruites avant et après la méthode sont également illustrées (figure 3A-B). Les cartes de densité brutes et les cartes de densité nettes obtenues ont également été comparées, avec le niveau de contour équivalent appliqué (figure 3C). Les chaînes latérales de cartes de densité nettes ont été comparées, montrant l’amélioration des cartes de densité reconstruites. Le facteur B de Rosenthal-Henderson estimé a également été adopté pour le critère de la qualité des particules19. Après avoir éliminé la majorité des particules de la pile finale, le facteur B de Rosenthal-Henderson est passé de 226,9 Å2 à 146,2 Å2 (Figure 3D). La résolution locale, le facteur Blocal 20 et le ResLog21 ont également été utilisés pour la comparaison, indiquant que CryoSieve améliore effectivement à la fois la qualité des cartes de densité et des particules (Figure 4).

Figure 1 : Résolutions de chaque itération. Les résolutions qui ont été signalées sont mises en évidence dans des encadrés rouges. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Résolutions de chaque itération. Les résolutions identifiées par des tâches d’affinement homogènes sont mises en évidence dans des cases rouges. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Cartes de densité. (A) Comparaison de la courbe FSC modèle-carte des cartes de densité reconstruites avant et après l’utilisation de CryoSieve. L’axe des y représente le FSC, tandis que l’axe des x représente la résolution. La ligne pointillée rouge marque le seuil de 0,5 pour le FSC. La ligne pointillée verticale illustre la résolution des cartes de densité obtenues sous un seuil de 0,5. (B) Des demi-cartes de la courbe FSC ont été obtenues à partir de cartes de densité reconstruites avant et après l’utilisation de CryoSieve via CryoSPARC. L’axe des y représente le FSC, tandis que l’axe des x représente la résolution. (C) Des cartes de densité brutes et des cartes de densité nettes ont été présentées à la fois pour les particules retenues par CryoSieve et pour l’ensemble complet des particules dans les piles finales. Le niveau de contour équivalent de 0,65 a été appliqué pour les cartes de densité brutes. Le niveau de contour équivalent de 0,84 a été appliqué pour les cartes de densité nettes. Des cartes de densité précises ont été obtenues directement par CryoSPARC. Les cartes de densité nettes ont été post-traitées automatiquement, d’abord pondérées par FSC (sur la base des FSC fournies par CryoSPARC). Ensuite, le facteur B a été affûté à l’aide des facteurs B auto-déterminés (232,0 Å2 pour toutes les particules de la pile finale et 160,8 Å2 pour CryoSieve). Les chaînes latérales des cartes de densité nettes ont été comparées, en incorporant des modèles atomiques comme référence. Les flèches rouges mettent en évidence les régions améliorées. (D) Le facteur B de Rosenthal-Henderson estimé a été montré à la fois pour les particules retenues par CryoSieve et pour l’ensemble complet des particules dans les cheminées finales. L’axe des y représente le nombre de particules utilisées, et l’axe des x représente l’inverse du carré de la résolution. En se déplaçant de haut en bas, chaque point représente la moitié des particules du précédent. Les résolutions ont été déterminées par le raffinement. Les facteurs B ont été déterminés à l’aide d’une approximation des points mesurés par les moindres carrés, comme le montrent les courbes d’ajustement. Les facteurs B estimés de Rosenthal et Henderson sont indiqués dans les légendes : l’orange représente les particules retenues par CryoSieve, tandis que le bleu désigne toutes les particules de la pile finale. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Comparaison de diverses mesures de cartes de densité. (A) Comparaison de cartes de résolution locale avant et après l’utilisation de CryoSieve obtenues par CryoSPARC. La résolution locale varie entre 7 Å (rouge) et 3,5 Å (bleu). (B) Comparaison des cartes de densité avant et après l’utilisation de CryoSieve, colorées avec la carte locale du facteur B obtenue par LocBFactor en utilisant une plage de résolution de [20-3,5] Å. (C), Comparaison des parcelles ResLog avant et après l’utilisation de CryoSieve obtenue par CryoSPARC. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Figure supplémentaire 1 : Utilisation des commandes nvidia-smi et conda -V pour vérifier les conditions préalables. Si les conditions préalables sont remplies, la saisie de la commande nvidia-smi affichera la version du pilote GPU, la version CUDA et l’état des cartes GPU. De même, la saisie de la commande conda -V devrait afficher correctement la version installée de Conda. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 2 : Le processus de création de nouveaux environnements d’accélération GPU. L’écran affiche la sortie générée par la commande utilisée pour créer l’environnement Conda. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 3 : Installation de CryoSieve dans l’environnement d’accélération GPU. Après avoir activé l’environnement Conda nouvellement créé, l’écran affiche le résultat de l’exécution de la commande d’installation de CryoSieve à l’aide de Pip. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 4 : Informations d’aide. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 5 : Processus en cours. Lors de l’exécution de CryoSieve via la ligne de commande, l’écran affiche alors des informations concernant le processus en cours. Veuillez cliquer ici pour télécharger ce fichier.
Figure supplémentaire 6 : La configuration des tâches de CryoSPARC. (A) Importer la pile de particules. (B) Importer des volumes 3D. (C-D) Raffinement homogène. Veuillez cliquer ici pour télécharger ce fichier.
Dossier supplémentaire 1 : Options de CryoSieve. Veuillez cliquer ici pour télécharger ce fichier.
Dossier supplémentaire 2 : Temps de traitement et exigence minimale pour faire fonctionner Cryosieve. Veuillez cliquer ici pour télécharger ce fichier.
Dossier supplémentaire 3 : Génération du modèle initial par CryoSPARC. Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 4 : Justification de l’inactivation de la réactivation de la réactivation de la division GS. Veuillez cliquer ici pour télécharger ce fichier.
Dossier supplémentaire 5 : Options de cryosieve-csrefine. Veuillez cliquer ici pour télécharger ce fichier.
Dossier supplémentaire 6 : Options du cryotamiseur-csrhbfactor. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
La cryo-EM est une technique essentielle pour élucider les structures des molécules biologiques. Dans ce processus, après la collecte des données par microscopie, l’extraction des particules à partir des micrographies est essentielle, suivie de leur classification en plusieurs étapes pour compiler la pile finale. Un défi courant est la prédominance de particules endommagées ou non conformées de manière indésirable, ce qui souligne la nécessité d’une sélection répétée des particules pour obtenir des cartes de densité à haute résolution. La sélection des particules est donc une étape critique de la cryo-EM SPA pour obtenir des cartes de densité de haute qualité. Les techniques de sélection de particules existantes comprennent l’algorithme de validation statistique sans inclinaison22, l’approche basée sur le score z23 et la méthode d’estimation de la précision angulaire24.
CryoSieve apparaît comme un outil précieux dans ce contexte, capable d’éliminer un nombre important de particules étrangères de la pile finale. Cette réduction améliore non seulement l’efficacité de calcul de la reconstruction, mais rationalise également le processus. Il offre une suite complète pour la sélection des particules, où l’ampleur des rejets de particules et l’amélioration conséquente de la résolution dépendent en grande partie de la qualité initiale des données et des méthodologies employées dans le traitement des données.
Dans ce manuscrit, nous avons présenté un flux complet de criblage de particules à l’aide de l’ensemble de données de cas réels de trimère d’hémagglutinine de grippe (entrée EMPIAR : 10097). Les étapes couvertes et discutées ici peuvent être résumées comme le tamisage des particules et la réestimation de la pose. Le volume final reconstruit en 3D a atteint une résolution de 3,62 Å, et les chaînes latérales en hélices alpha étaient plus claires dans le volume post-traité par rapport à la carte de densité publiée.
CryoSieve est une méthode open-source qui est disponible sur GitHub (https://github.com/mxhulab/cryosieve). Un tutoriel détaillé peut également être trouvé sur sa page d’accueil. Les utilisateurs peuvent l’installer et l’utiliser en suivant le tutoriel. De plus, deux modules, cryosieve-csrefine et cryosieve-csrhbfactor, sont fournis. Le module cryosieve-csrefine est spécialement conçu pour automatiser l’exécution séquentielle de diverses opérations au sein de CryoSPARC (Fichier supplémentaire 5). Ces opérations comprennent l’importation d’empilements de particules et la réalisation de travaux de raffinement ab initio, homogènes ou non uniformes. D’autre part, le module cryosieve-csrhbfactor est conçu pour automatiser la détermination du facteur B de Rosenthal-Henderson en exploitant les capacités de cryosieve-csrefine (Fichier supplémentaire 6).
À l’heure actuelle, l’application de cette méthode est limitée aux scénarios de conformation unique. Par conséquent, dans les cas où les particules représentent plusieurs conformations, leurs capacités sont limitées. Il est conseillé aux utilisateurs de s’engager d’abord dans la classification 3D pour séparer les particules de conformations disparates avant de l’utiliser pour une sélection raffinée des particules. De plus, bien que la méthode démontre sa capacité à filtrer plus de 50 % des particules de la pile finale, l’origine de ces particules rejetées et les raisons sous-jacentes de leur contribution négligeable à la qualité de la reconstruction restent incertaines. Cette lacune dans la compréhension nécessite des recherches supplémentaires pour aborder de manière exhaustive et éventuellement rectifier cette limitation.
Il existe trois méthodes possibles de tri ou de tamisage des particules. Tout d’abord, cisTEM4 peut rapporter un score pour chaque image de particule après raffinement 3D. Les utilisateurs pouvaient trier les particules à l’aide du score cisTEM pour éliminer les particules. L’approche AGC (Angular Graph Consistency)17 est également une méthode pour éliminer les particules mal alignées. De plus, la classification de non-alignement5 est une méthode traditionnelle d’élimination des particules à l’aide de la classification 3D. Nous avons comparé la qualité des particules retenues par ces méthodes avec CryoSieve et avons constaté que les particules retenues de CryoSieve sont de meilleure qualité15. La méthode présentée ici surpasse considérablement les méthodes alternatives et permet d’obtenir le plus petit nombre de particules à la même résolution.
Comme le démontre le résultat, la majorité des particules d’une pile finale cryo-EM ne contribuent pas à la reconstruction de la carte de densité. En d’autres termes, parmi toutes les particules recueillies lors de l’acquisition de l’image, seules quelques-unes, à savoir le sous-ensemble le plus fin, contribuent réellement à la reconstruction finale. Par conséquent, le rapport entre ce dernier sous-ensemble et le nombre total de particules collectées pourrait servir de mesure quantitative pour évaluer la qualité de l’échantillon. Plus ce rapport est élevé, meilleure est la qualité de l’échantillon. Malgré les progrès techniques qui ont rendu la cryo-EM plus accessible aux biologistes structurels, la préparation des échantillons reste un goulot d’étranglement majeur dans le flux de travail. Les scientifiques et les ingénieurs concentrent donc leurs efforts sur ce défi25. Dans l’analyse des particules uniques (SPA), la préparation des échantillons se compose de deux étapes cruciales : l’optimisation des échantillons et la préparation de la grille. La première consiste à purifier l’échantillon tout en maintenant son état biochimique optimal. Ce dernier implique la préparation de l’échantillon pour l’analyse au microscope, y compris le traitement chimique ou plasma de la grille, le dépôt de l’échantillon et la vitrification. De nombreuses techniques ont été proposées pour traiter l’instabilité macromoléculaire, mais l’efficacité d’une approche par rapport à une autre dépend des caractéristiques de l’échantillon25,26. À l’heure actuelle, les résultats de la préparation du réseau sont fortement influencés par l’expertise et l’expérience de l’utilisateur, ce qui peut rendre le processus long et difficile27,28. Les nombreuses variables rencontrées dans la préparation de l’échantillon et de la grille posent des défis pour établir des relations de cause à effet, car les chercheurs ne peuvent évaluer l’échantillon au niveau moléculaire qu’à l’aide du microscope. En conséquence, les statistiques quantitatives provenant de comparaisons de différents protocoles de préparation d’échantillons et de grilles font toujours défaut, et une approche systématique est nécessaire pour étudier les tendances et comprendre les mécanismes fondamentaux du comportement de l’échantillon29.
Déclarations de divulgation
Tous les autres auteurs ne déclarent aucun intérêt concurrent.
Remerciements
Ce travail a été soutenu par l’Académie de recherche et de traduction de Shenzhen (à M.H.), le Centre d’innovation avancée pour la biologie structurale (à M.H.), le Beijing Frontier Research Center for Biological Structure (à M.H.), le National Key R&D Program of China (n° 2021YFA1001300) (à C.B.), la National Natural Science Foundation of China (n° 12271291) (à C.B.), et la Fondation nationale des sciences naturelles de Chine (n° 12071244) (à Z.S.).
matériels
| Name | Company | Catalog Number | Comments |
| CryoSPARC | Structura Biotechnology Inc. Toronto, Canada | CryoSPARC (Cryo-EM Single Particle Ab-Initio Reconstruction and Classification) is a state of the art HPC software solution for complete processing of single-particle cryo-electron microscopy (cryo-EM) data. CryoSPARC is useful for solving cryo-EM structures of membrane proteins, viruses, complexes, flexible molecules, small particles, phase plate data and negative stain data. | |
| EMPIAR-10097 Dataset | https://ftp.ebi.ac.uk/empiar/world_availability/10097/data/Particle-Stack/T40_HA_130K-Equalized-Particle-Stack.mrcs | This dataset comprises single-particle cryo-EM data of the Influenza Hemagglutinin trimer, characterized by its highly preferred orientation, collected using a 40-degree tilted collection strategy. | |
| initial.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| mask.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| RELION | 4.0-beta-2 | RELION (REgularised LIkelihood OptimisatioN) is an open-source software for cryo-electron microscopy (cryo-EM) data processing, particularly for refining macromolecular structures. Utilizing a Bayesian approach, it excels in separating signal from noise, enabling high-resolution structure determination. RELION supports single-particle analysis, tomography, and sub-tomogram averaging, and has become widely used in structural biology due to its effectiveness and user-friendly interface. | |
| T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | Metadata file for the final stack of particles from EMPIAR-10097 |
Références
- Bai, X. C., Fernandez, I. S., Mcmullan, G., Scheres, S. H. Ribosome structures to near-atomic resolution from thirty thousand cryo-em particles. elife. 2, 00461(2013).
- Campbell, M. G., et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 20 (11), 1823-1828 (2012).
- Li, X., et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-em. Nat Meth. 10 (6), 584-590 (2013).
- Grant, T., Rohou, A., Grigorieff, N. Cis tem, user-friendly software for single-particle image processing. eLife. 7, e35383(2018).
- Scheres, S. H. Relion: Implementation of a bayesian approach to cryo-em structure determination. J Str Biol. 180 (3), 519-530 (2012).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. Cryosparc: Algorithms for rapid unsupervised cryo-em structure determination. Nat Meth. 14 (3), 290-296 (2017).
- Kühlbrandt, W. The resolution revolution. Science. 343 (6178), 1443-1444 (2014).
- Dubochet, J., et al. Cryo-electron microscopy of vitrified specimens. Quart Rev Biophys. 21 (2), 129-228 (1988).
- Wagner, T., et al. Sphire-cryolo is a fast and accurate fully automated particle picker for cryo-EM. Comm Biol. 2 (1), 218(2019).
- Bepler, T., et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Meth. 16 (11), 1153-1160 (2019).
- Wang, F., et al. Deeppicker: A deep learning approach for fully automated particle picking in cryo-em. J Str Biol. 195 (3), 325-336 (2016).
- Heimowitz, A., Andén, J., Singer, A. Apple picker: Automatic particle picking, a low-effort cryo-em framework. J Str Biol. 204 (2), 215-227 (2018).
- Glaeser, R. M. How good can single-particle cryo-em become? What remains before it approaches its physical limits. Ann Rev Biophys. 48, 45-61 (2019).
- Diiorio, M. C., Kulczyk, A. W. A robust single-particle cryo-electron microscopy (cryo-em) processing workflow with cryosparc, relion, and scipion. J Vis Exp. (179), e63387(2022).
- Zhu, J., et al. A minority of final stacks yields superior amplitude in single-particle cryo-em. Nat Comm. 14 (1), 7822(2023).
- Zhou, Y., Moscovich, A., Bendory, T., Bartesaghi, A. Unsupervised particle sorting for high-resolution single-particle cryo-em. Inv Probl. 36 (4), 044002(2020).
- Méndez, J., Garduno, E., Carazo, J. M., Sorzano, C. O. S. Identification of incorrectly oriented particles in cryo-em single particle analysis. J Str Biol. 213 (3), 107771(2021).
- Tan, Y. Z., et al. Addressing preferred specimen orientation in single-particle cryo-em through tilting. Nat Meth. 14 (8), 793-796 (2017).
- Rosenthal, P. B., Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 333 (4), 721-745 (2003).
- Kaur, S., et al. Local computational methods to improve the interpretability and analysis of cryo-em maps. Nat Comm. 12 (1), 1240(2021).
- Stagg, S. M., Noble, A. J., Spilman, M., Chapman, M. S. Reslog plots as an empirical metric of the quality of cryo-em reconstructions. J Str Biol. 185 (3), 418-426 (2014).
- Vargas, J., Otón, J., Marabini, R., Carazo, J. M., Sorzano, C. Particle alignment reliability in single particle electron cryomicroscopy: A general approach. Sci Rep. 6 (1), 21626(2016).
- Vargas, J., et al. Particle quality assessment and sorting for automatic and semiautomatic particle-picking techniques. J Str Biol. 183 (3), 342-353 (2013).
- Vargas, J., Melero, R., Gomez-Blanco, J., Carazo, J. -M., Sorzano, C. O. S. Quantitative analysis of 3d alignment quality: Its impact on soft-validation, particle pruning and homogeneity analysis. Sci Rep. 7 (1), 6307(2017).
- Carragher, B., et al. Current outcomes when optimizing 'standard'sample preparation for single-particle cryo-em. J Microsc. 276 (1), 39-45 (2019).
- Drulyte, I., et al. Approaches to altering particle distributions in cryo-electron microscopy sample preparation. Acta Crystallographica Sec D: Str Biol. 74 (6), 560-571 (2018).
- Glaeser, R. M. How good can cryo-em become. Nat Meth. 13 (1), 28-32 (2016).
- Kim, L. Y., et al. Benchmarking cryo-em single particle analysis workflow. Front Mol Biosci. 5, 50(2018).
- Weissenberger, G., Henderikx, R. J., Peters, P. J. Understanding the invisible hands of sample preparation for cryo-em. Nat Meth. 18 (5), 463-471 (2021).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.