
Method Article
Verbesserung der Dichtekarten durch Entfernen der Mehrheit der Partikel in den finalen Stacks der kryogenen Elektronenmikroskopie mit Einzelpartikeln
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Eine fortschrittliche Partikelauswahlmethode für Kryo-EM, nämlich CryoSieve, verbessert die Auflösung der Dichtekarte, indem sie einen Großteil der Partikel in den endgültigen Stapeln entfernt, wie ihre Anwendung auf einem realen Datensatz zeigt.
Zusammenfassung
In den letzten zehn Jahren haben technologische und methodische Fortschritte auf dem Gebiet der kryogenen Elektronenmikroskopie (Kryo-EM) und der Einzelteilchenanalyse (SPA) unsere Fähigkeit zur hochauflösenden Strukturuntersuchung biologischer Makromoleküle erheblich verbessert. Dieser Fortschritt hat eine neue Ära molekularer Erkenntnisse eingeläutet, die die Röntgenkristallographie als dominierende Methode abgelöst und Antworten auf langjährige Fragen in der Biologie gegeben hat. Da die Kryo-EM nicht von der Kristallisation abhängt, die eine wesentliche Einschränkung der Röntgenkristallographie darstellt, werden Partikel unterschiedlicher Qualität eingefangen. Daher ist die Auswahl der Partikel entscheidend, da die Qualität der ausgewählten Partikel die Auflösung der rekonstruierten Dichtekarte direkt beeinflusst. Ein innovativer iterativer Ansatz für die Partikelauswahl, genannt CryoSieve, verbessert die Qualität der rekonstruierten Dichtekarten erheblich, indem er die Anzahl der Partikel im endgültigen Stapel effektiv reduziert. Experimentelle Beweise zeigen, dass diese Methode die Mehrheit der Partikel in den endgültigen Stapeln eliminieren kann, was zu einer bemerkenswerten Verbesserung der Qualität der Dichtekarten führt. In diesem Artikel wird der detaillierte Workflow dieses Ansatzes beschrieben und seine Anwendung auf ein reales Dataset vorgestellt.
Einleitung
Kryogene Elektronenmikroskopie (Kryo-EM) Die Einzelpartikelanalyse (SPA) hat sich zu einer dominierenden Methode zur Bestimmung hochauflösender dreidimensionaler Dichtekarten biologischer Makromoleküle entwickelt. Aufgrund einer Reihe von technologischen Innovationen 1,2,3,4,5,6, genannt Auflösungsrevolution 7, ist die Kryo-EM in der Lage, die Strukturen biologischer Makromoleküle mit bis zu atomarer Auflösung in einer noch nie dagewesenen Geschwindigkeit zu bestimmen. Dieser Durchbruch markiert den Beginn einer neuen Ära molekularer Erkenntnisse, die die Röntgenkristallographie als vorherrschende Technik ablöste und langjährige biologische Fragen beantwortete.
Die Kryo-EM SPA unterscheidet sich von der Röntgenkristallographie, da sie keine Kristallisation biologischer Makromoleküle erfordert. Stattdessen wird eine Lösung, die die biologischen Zielmakromoleküle enthält, schnell in glasartigem Eis eingefroren. Es wird dann mit einem Elektronenstrahl abgebildet, um eine Reihe von Mikroskopaufnahmen zu erstellen, wobei die Notwendigkeit einer Kristallisationumgeht 8. Anschließend werden Particle-Picking-Algorithmen verwendet, um einzelne Rohpartikel aus diesen Mikroaufnahmen zu extrahieren 4,9,10,11,12. Da die Kryo-EM nicht von der Kristallisation abhängt, ist es natürlich, dass extrahierte Partikel überwiegend beschädigt sind oder sich in unerwünschten Konformationszuständen befinden, was mehrere Partikelauswahlrunden erforderlich macht, um eine hochauflösende Dichtekarte zu erhalten. In der Kryo-EM-SPA-Bildverarbeitung ist die Partikelselektion daher entscheidend für die Erstellung hochauflösender Dichtekarten13.
Zu den Standardmethoden der Partikelauswahl bei der Kryo-EM gehören die zweidimensionale (2D) und die dreidimensionale (3D) Klassifizierung14. Die 2D-Klassifizierung kategorisiert Partikel in eine vordefinierte Anzahl von Gruppen, was zu einem durchschnittlichen Bild und einer geschätzten 2D-Auflösung für jede Klasse führt. Forscher können diese Klassen dann visuell inspizieren und Partikel aus Gruppen mit niedrigerer Auflösung entfernen, um die verbleibenden in Rekonstruktionen zu verwenden, um eine höhere Auflösung zu erreichen. Sobald die Partikelposen mithilfe von Verfeinerungsalgorithmen festgelegt sind, fahren die Forscher mit der 3D-Klassifizierung fort und gruppieren die Partikel in mehrere Klassen. Dies ermöglicht eine visuelle Inspektion der rekonstruierten Dichtekarte für jede Klasse, wodurch unerwünschte Partikel, wie z. B. solche aus unerwünschten Konformationen, ausgeschlossen werden können. Nach mehreren Klassifizierungsrunden wird ein endgültiger Stapel erhalten, der aus relativ hochwertigen Partikeln besteht. Diese finalen Stacks sind maßgeblich an der Erstellung von Dichtekarten mit atomarer oder nahezu atomarer Auflösung beteiligt.
Zhu und ihre Kollegen haben gezeigt, dass auf diesen finalen Stacks15 eine weitere Partikelselektion durchgeführt werden kann. CryoSieve15, eine innovative iterative Methode zur Partikelselektion, kann eingesetzt werden, um die Qualität der endgültigen Dichtekarte zu verbessern, indem die Anzahl der Partikel erheblich reduziert wird. Während andere Kriterien und Software für die Partikelsortierung, wie z. B. die normierte Kreuzkorrelationsmethode (NCC)16, der Angular Graph Consistency (AGC)-Ansatz17 und die Non-Alignment-Klassifizierung5, derzeit in diesem Bereich verwendet werden, hat sich gezeigt, dass diese Methode diese Algorithmen in Bezug auf die Wirksamkeit übertrifft.
In dieser Studie stellen wir Ihnen einen detaillierten Leitfaden für den gesamten Prozess vor. Als Fallstudie haben wir diese neue Methode auf den Datensatz des Influenza-Hämagglutinin-Trimers (EMPIAR-Eintrag: 10097)18 angewendet, der 130.000 Partikel in seinem endgültigen Stapel enthält. Unser Verfahren verwarf erfolgreich etwa 73,8% der Partikel aus dem letzten Stapel dieses Datensatzes, wodurch die Auflösung der rekonstruierten Dichtekarte von 4,11 Å auf 3,62 Å verbessert wurde. Zusätzlich zum Influenza-Hämagglutinin-Trimer werden in der früheren Veröffentlichung15 Ergebnisse aus mehreren Datensätzen vorgestellt, die eine Vielzahl von Auflösungen und Molekulargewichten von Biomolekülen zeigen.
Protokoll
1. Einbau
- Überprüfen und Konfigurieren der GPU-Beschleunigungsumgebung
- Öffnen Sie das Terminal und geben Sie den Befehl nvidia-smi ein. Stellen Sie sicher, dass der Befehl erfolgreich alle Informationen zu den GPU-Karten anzeigt und die CUDA-Version höher als 10.2 ist. Führen Sie den Befehl conda -V aus, um zu überprüfen, ob Conda installiert ist (Ergänzende Abbildung 1).
- Konfigurieren der virtuellen Umgebung
- Geben Sie den folgenden Befehl ein, um die virtuelle Umgebung einzurichten, und ersetzen Sie CRYOSIEVE_ENV durch den gewünschten Umgebungsnamen: conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge. Warten Sie einige Minuten, bis die Umgebung erfolgreich konfiguriert ist (ergänzende Abbildung 2).
HINWEIS: Benutzer haben die Flexibilität, den Umgebungsnamen nach Bedarf zu ändern. Der bereitgestellte Befehl ist spezifisch für CUDA 10.2. Wenn eine andere CUDA-Version gewünscht wird, passen Sie die Versionsnummer für cudatoolkit an.
- Geben Sie den folgenden Befehl ein, um die virtuelle Umgebung einzurichten, und ersetzen Sie CRYOSIEVE_ENV durch den gewünschten Umgebungsnamen: conda create -n CRYOSIEVE_ENV python=3.8 cudatoolkit=10.2 cupy=10.0 pytorch=1.10 -c pytorch -c conda-forge. Warten Sie einige Minuten, bis die Umgebung erfolgreich konfiguriert ist (ergänzende Abbildung 2).
- Installieren Sie CryoSieve
- Aktivieren Sie die Umgebung, indem Sie den Befehl conda activate CRYOSIEVE_ENV ausführen. Installieren Sie die Software, indem Sie pip install cryosieve oder conda install -c mxhulab cryosieve ausführen (Ergänzende Abbildung 3). Geben Sie cryosieve -h ein und stellen Sie sicher, dass die Hilfeinformationen korrekt angezeigt werden (Ergänzende Abbildung 4).
2. Partikel-Siebung
- Abrufen der Daten
- Laden Sie den EMPIAR-10097 final stack Datensatz von EMPIAR herunter (siehe Materialtabelle). Laden Sie die Sterndatei, die Maskendatei (mask.mrc) und das ursprüngliche Modell (für den Schritt der Neuschätzung; initial.mrc) von Github herunter (siehe Materialtabelle). Platzieren Sie alle diese Dateien zusammen in einem Ordner (Ergänzende Abbildung 5).
HINWEIS: Das Repository in https://github.com/mxhulab/cryosieve-demos verwendet Git Large File Storage (Git LFS). Die Installation von Git LFS ist für das Klonen des gesamten Repositorys unerlässlich. Alternativ können Sie über den GitHub-Link auf die Datei zugreifen und auf die Schaltfläche Rohdatei herunterladen klicken, um eine einzelne Datei herunterzuladen.
- Laden Sie den EMPIAR-10097 final stack Datensatz von EMPIAR herunter (siehe Materialtabelle). Laden Sie die Sterndatei, die Maskendatei (mask.mrc) und das ursprüngliche Modell (für den Schritt der Neuschätzung; initial.mrc) von Github herunter (siehe Materialtabelle). Platzieren Sie alle diese Dateien zusammen in einem Ordner (Ergänzende Abbildung 5).
- Prozess-Partikelsiebung
- Öffnen Sie das Terminal und verwenden Sie den Befehl: cd FILEPATH, um zu dem Ordner zu navigieren, in dem sich das Dataset befindet. Aktivieren Sie die Conda-Umgebung durch: conda activate CRYOSIEVE_ENV.
- Geben Sie den folgenden Befehl ein, um unser Teilchensiebexperiment zu starten: cryosieve --reconstruct_software relion_reconstruct --postprocess_software relion_postprocess --i T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star --o output/ --mask mask.mrc --angpix 1.3099979 --num_iters 10 --frequency_start 40 --frequency_end 3 --retention_ratio 0.8 --sym C3 --num_gpus 1 --balance (Ergänzende Abbildung 5). Während der Ausführung zeigt das Terminal die Ausgabeprotokolle für jede Iteration an.
HINWEIS: Detaillierte Anweisungen für jede Option finden Sie in der Zusatzdatei 1. Die Bearbeitungszeit und die Mindestanforderungen für die Ausführung sind in der Zusatzdatei 2 aufgeführt. T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star wurde von CryoSPARC von T40_HA_130K-Equalized_run-data.star (heruntergeladen von EMPIAR) verfeinert, um die Auswirkungen zu mildern, die durch Fortschritte bei der Orientierungsschätzung verursacht wurden.
3. Die optimale Iteration finden
- Überprüfen der Auflösungen
- Verwenden Sie den Befehl: grep "+ FINAL RESOLUTION:" output/_postprocess*.txt, um die Auflösungsergebnisse für die 10 Iterationen der Siebung zu drucken (Abbildung 1). Da der in der 7. Iteration gefilterte Partikelstapel die höchste Auflösung mit den wenigsten Partikeln aufweist, ist es wahrscheinlich, dass er das optimale Ergebnis liefert.
HINWEIS: Um eine unbeabsichtigte Informationsübertragung von verworfenen zu zurückgehaltenen Partikeln15 zu vermeiden und sicherzustellen, dass der Partikelstapel nach der 7. Iteration tatsächlich optimal ist, müssen Benutzer einen Neuschätzungsschritt für benachbarte Iterationen ausführen. In diesem Protokoll werden die Iterationen 4, 5, 6, 7 und 8 einer Verifizierung unterzogen.
- Verwenden Sie den Befehl: grep "+ FINAL RESOLUTION:" output/_postprocess*.txt, um die Auflösungsergebnisse für die 10 Iterationen der Siebung zu drucken (Abbildung 1). Da der in der 7. Iteration gefilterte Partikelstapel die höchste Auflösung mit den wenigsten Partikeln aufweist, ist es wahrscheinlich, dass er das optimale Ergebnis liefert.
- Importiert gesiebte Partikel
- Öffnen Sie die CryoSPARC-Weboberfläche und gehen Sie folgendermaßen vor: Betreten Sie einen Arbeitsbereich und klicken Sie auf die Schaltfläche Builder oben rechts im Panel. Wählen Sie im Bedienfeld die Option Partikelstapel importieren aus und klicken Sie darauf. Geben Sie im Abschnitt "Parameter" des Bedienfelds "Partikelstapelimport" den Partikelmetapfad als _iter{n}.star-Datei an, die sich im Ausgabeordner der abgeschlossenen Ergebnisse befindet, sowie den Partikeldatenpfad zu dem Ordner, in dem die MRCS-Datei gespeichert ist. Klicken Sie auf die Schaltfläche "Auftrag in die Warteschlange" und dann auf die Schaltfläche "Warteschlange", um den Vorgang zu starten. Verwenden Sie die gleiche Methode, um die verbleibenden Iterationen zu importieren, die neu geschätzt werden müssen (Ergänzende Abbildung 6A).
- Importieren des ursprünglichen Modells
- Klicken Sie auf die Schaltfläche Builder oben rechts im Panel. Wählen Sie im Bedienfeld die Option 3D-Volumen importieren aus und klicken Sie darauf.
- Geben Sie den Datenpfad des Volumes als Datei initial.mrc an. Klicken Sie auf die Schaltfläche "Job in die Warteschlange" und dann auf die Schaltfläche "Warteschlange", um den Vorgang zu starten (Ergänzende Abbildung 6B).
HINWEIS: Das ursprüngliche Modell kann auch durch Ab-initio-Rekonstruktion generiert werden (Ergänzungsdatei 3).
- Homogene Verfeinerung (Bauauftrag)
- Klicken Sie auf die Schaltfläche Builder oben rechts im Panel. Wählen Sie im Bedienfeld die Option Homogene Verfeinerung aus und klicken Sie darauf.
HINWEIS: Eine ungleichmäßige Verfeinerung ist ebenfalls möglich.
- Klicken Sie auf die Schaltfläche Builder oben rechts im Panel. Wählen Sie im Bedienfeld die Option Homogene Verfeinerung aus und klicken Sie darauf.
- Homogene Veredelung (Import von Partikeln)
- Öffnen Sie im Hauptfenster auf der linken Seite den Auftrag zum Importieren des Partikelstapels der 5. Iteration (oder der gewünschten Iteration). Ziehen Sie das importierte Partikelmodul von der rechten Seite des Hauptfensters und legen Sie es im Abschnitt "Partikelstapel" des Builders auf der rechten Seite ab. Schließen Sie den Auftrag "Partikelstapel importieren", indem Sie auf das rote X in der oberen rechten Ecke des Hauptfensters klicken.
- Öffnen Sie den Auftrag zum Importieren von 3D-Volumen. Ziehen Sie das importierte Volumes-Modul von der rechten Seite des Hauptfensters und legen Sie es im Abschnitt "Initial volume" des Builders auf der rechten Seite ab.
- Homogene Verfeinerung (Ändern Sie die Parameter)
- Suchen Sie unter der Parameterfalte die Option Symmetrie, und legen Sie sie auf C3 fest. Suchen Sie die Option Erneutes Wiederholen der GS-Teilung erzwingen und deaktivieren Sie sie. Klicken Sie auf die Schaltfläche "Auftrag in die Warteschlange" und dann auf die Schaltfläche "Warteschlange", um die homogene Verfeinerung zu starten. Führen Sie die homogene Verfeinerung für die verbleibenden Iterationen mit derselben Methode durch (Ergänzende Abbildung 6C-D).
HINWEIS: Die Option Wiederholung des GS-Splits erzwingen ist entscheidend. Das Deaktivieren dieser Option stellt sicher, dass CryoSPARC die Goldstandard-Aufteilung beibehält, die durch die Sterndatei gegeben ist, wodurch eine Überanpassung vermieden wird. Eine detaillierte Begründung für die Deaktivierung von Force Re-do GS Split finden Sie in der Zusatzdatei 4.
- Suchen Sie unter der Parameterfalte die Option Symmetrie, und legen Sie sie auf C3 fest. Suchen Sie die Option Erneutes Wiederholen der GS-Teilung erzwingen und deaktivieren Sie sie. Klicken Sie auf die Schaltfläche "Auftrag in die Warteschlange" und dann auf die Schaltfläche "Warteschlange", um die homogene Verfeinerung zu starten. Führen Sie die homogene Verfeinerung für die verbleibenden Iterationen mit derselben Methode durch (Ergänzende Abbildung 6C-D).
- Warten Sie, bis alle Aufträge ausgeführt wurden, um die Ergebnisse zu erhalten. Basierend auf den Ergebnissen wird bestätigt, dass der in der 6. Iteration gefilterte Partikelstapel das tatsächlich optimale Ergebnis ist.
HINWEIS: Es ist normal, dass die erzielten Ergebnisse geringfügige zufällige Abweichungen von den in diesem Protokoll angegebenen Ergebnissen aufweisen. Diese Abweichungen haben keinen Einfluss auf die Gesamtschlussfolgerung.
Ergebnisse
In diesem Protokoll haben wir den Influenza-Hämagglutinin-Trimer-Datensatz (EMPIAR-Eintrag: 10097) verwendet, um die Wirksamkeit dieses Prozesses zu demonstrieren. Aufgrund der bevorzugten Ausrichtung der Probe musste die Datenerfassung um 40° gekippt werden. Das Protein weist eine C3-Symmetrie auf und hat ein Molekulargewicht von 150 kDa.
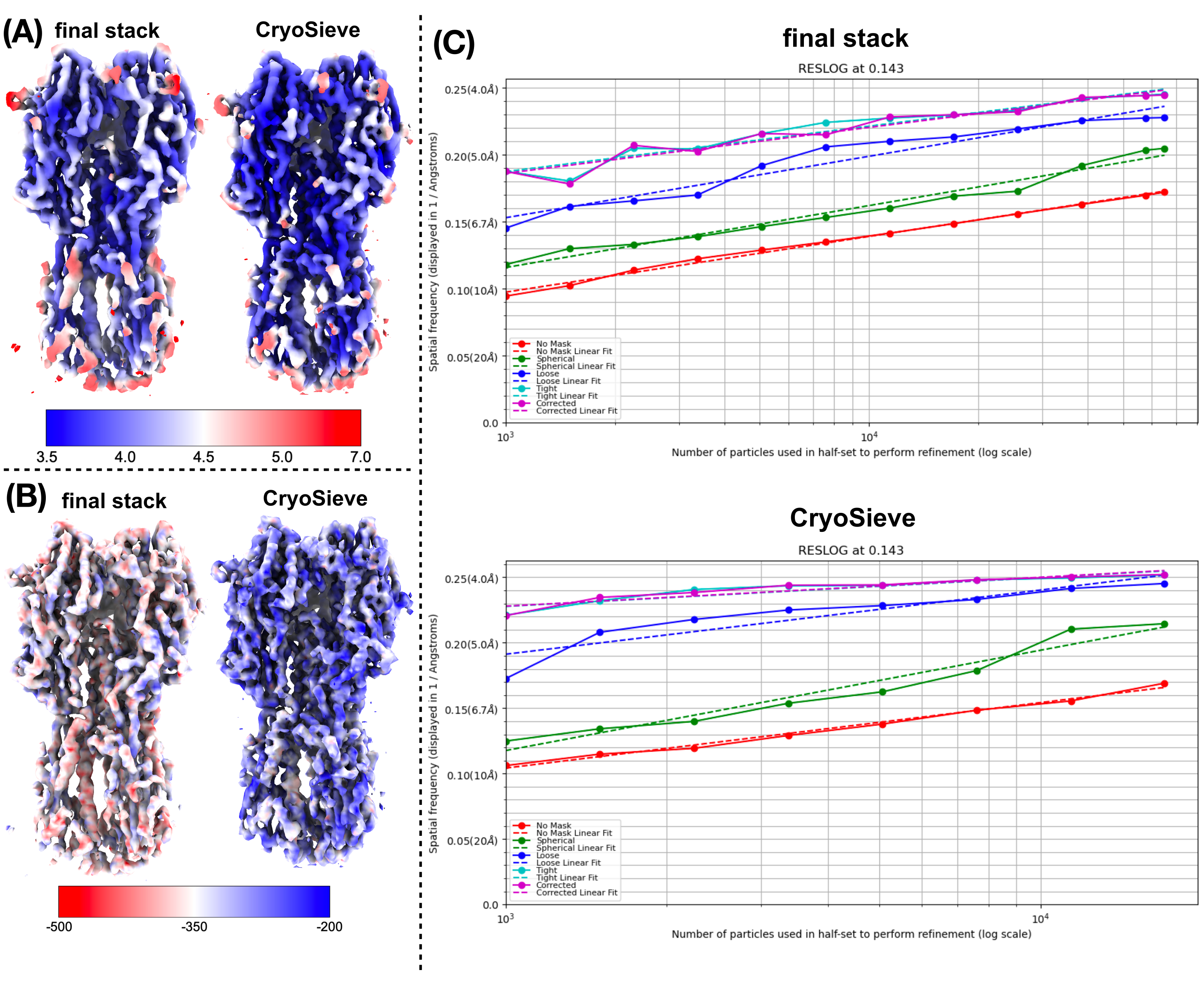
Wir haben das zuvor beschriebene Protokoll implementiert, um den endgültigen Partikelstapel zu verarbeiten. In jeder Iteration wurden nach und nach 20 % der Partikel entfernt, was zu einem Rückhalteverhältnis von 80,0 %, 64,0 %, 51,2 % usw. führte. Wie in Abbildung 1 und Abbildung 2 dargestellt, verbesserte sich die Auflösung der zurückgehaltenen Teilchen zunächst, nahm aber schließlich ab. Unter den Iterationen wurde die 6. Iteration als die optimalste Teilmenge identifiziert, die die wenigsten Partikel enthielt und dennoch die höchste Auflösung erreichte. Unser Algorithmus identifizierte erfolgreich eine Untergruppe von Partikeln, die nur 26,2 % des ursprünglichen Stapels ausmachte, was zu einer verbesserten Auflösung von 4,19 Å auf 3,62 Å führte (neu geschätzt durch CryoSPARC), wie in Abbildung 2 dargestellt. Darüber hinaus wurden in Abbildung 3 Dichtekarten vor und nach der Verwendung von CryoSieve verglichen. Die Modell-zu-Kartierung der Fourier-Schalen-Korrelation (FSC)-Kurve und die Halbkarten-FSC-Kurve der rekonstruierten Dichtekarten vor und nach der Methode sind ebenfalls dargestellt (Abbildung 3A-B). Rohe Dichtekarten und erhaltene scharfe Dichtekarten wurden ebenfalls verglichen, wobei das äquivalente Konturniveau angewendet wurde (Abbildung 3C). Die Seitenketten von scharfen Dichtekarten wurden verglichen, was die Verbesserung der rekonstruierten Dichtekarten zeigt. Der geschätzte Rosenthal-Henderson-B-Faktor wurde auch für das Kriterium der Partikelqualität19 verwendet. Nachdem der Großteil der Partikel im finalen Stapel entfernt worden war, stieg der Rosenthal-Henderson B-Faktor von 226,9 Å2 auf 146,2 Å2 (Abbildung 3D). Die lokale Auflösung, der lokale B-Faktor20 und ResLog21 wurden ebenfalls zum Vergleich verwendet, was darauf hindeutet, dass CryoSieve tatsächlich sowohl die Qualität der Dichtekarten als auch der Partikel verbessert (Abbildung 4).

Abbildung 1: Auflösungen der einzelnen Iterationen. Resolutionen, die gemeldet wurden, sind in roten Kästchen hervorgehoben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Auflösungen der einzelnen Iterationen. Auflösungen, die durch homogene Verfeinerungsaufträge gekennzeichnet sind, werden in roten Kästchen hervorgehoben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Dichtekarten. (A) Vergleich der Modell-zu-Karte-FSC-Kurve von rekonstruierten Dichtekarten vor und nach der Verwendung von CryoSieve. Die y-Achse stellt FSC dar, während die x-Achse die Auflösung darstellt. Die rote gestrichelte Linie markiert den Schwellenwert von 0,5 für den FSC. Die vertikale gestrichelte Linie veranschaulicht die Auflösung der Dichtekarten, die unter einem Schwellenwert von 0,5 erhalten wurden. (B) Die halbe FSC-Kurve wurde aus rekonstruierten Dichtekarten vor und nach der Verwendung von CryoSieve über CryoSPARC erhalten. Die y-Achse stellt FSC dar, während die x-Achse die Auflösung darstellt. (C) Rohdichtekarten und scharfe Dichtekarten wurden sowohl für die mit dem CryoSieve zurückgehaltenen Partikel als auch für den vollständigen Satz von Partikeln in den endgültigen Stapeln gezeigt. Für Rohdichtekarten wurde der äquivalente Konturpegel von 0,65 angewendet. Für scharfe Dichtekarten wurde der äquivalente Konturpegel von 0,84 angewendet. Scharfe Dichtekarten wurden direkt mit CryoSPARC erstellt. Die scharfen Dichtekarten wurden automatisch nachbearbeitet, zunächst FSC-gewichtet (basierend auf FSCs, die von CryoSPARC gegeben wurden). Anschließend wurde der B-Faktor mit den automatisch ermittelten B-Faktoren (232,0 Å2 für alle Partikel im finalen Stapel und 160,8 Å2 für CryoSieve) geschärft. Die Seitenketten in den scharfen Dichtekarten wurden verglichen, wobei atomare Modelle als Referenz einbezogen wurden. Rote Pfeile markieren die verbesserten Regionen. (D) Der geschätzte Rosenthal-Henderson-B-Faktor wurde sowohl für die CryoSieve-zurückgehaltenen Partikel als auch für den gesamten Satz von Partikeln in den endgültigen Stapeln gezeigt. Die y-Achse stellt die Anzahl der verwendeten Partikel dar, und die x-Achse stellt den Kehrwert des Quadrats der Auflösung dar. Wenn man sich von oben nach unten bewegt, repräsentiert jeder Punkt die Hälfte der Partikel des vorherigen. Die Vorsätze wurden durch Verfeinerung bestimmt. Die B-Faktoren wurden unter Verwendung einer Approximation der gemessenen Punkte nach den kleinsten Quadraten bestimmt, wie sie durch die Anpassungskurven dargestellt wird. Die geschätzten B-Faktoren von Rosenthal und Henderson sind in den Legenden angegeben: Orange steht für Partikel, die von CryoSieve zurückgehalten werden, während Blau alle Partikel im endgültigen Stapel bezeichnet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Vergleich verschiedener Metriken von Dichtekarten. (A) Vergleich von lokalen Auflösungskarten vor und nach der Verwendung von CryoSieve, die mit CryoSPARC erhalten wurden. Die lokale Auflösung liegt zwischen 7 Å (rot) und 3,5 Å (blau). (B) Vergleich von Dichtekarten vor und nach der Verwendung von CryoSieve, eingefärbt mit der lokalen B-Faktor-Karte, die mit LocBFactor unter Verwendung eines Auflösungsbereichs von [20-3,5] Å erstellt wurde. (C), Vergleich von ResLog-Diagrammen vor und nach der Verwendung von CryoSieve, die mit CryoSPARC erhalten wurden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Ergänzende Abbildung 1: Verwenden der Befehle nvidia-smi und conda -V zur Überprüfung der Voraussetzungen. Wenn die Voraussetzungen erfüllt sind, werden durch Eingabe des Befehls nvidia-smi die GPU-Treiberversion, die CUDA-Version und der Status der GPU-Karten angezeigt. Ebenso sollte bei Eingabe des Befehls conda -V die installierte Version von Conda korrekt angezeigt werden. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung 2: Der Prozess der Erstellung neuer GPU-Beschleunigungsumgebungen. Auf dem Bildschirm wird die Ausgabe angezeigt, die von dem Befehl generiert wurde, der zum Erstellen der Conda-Umgebung verwendet wurde. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung 3: Installation von CryoSieve in der GPU-Beschleunigungsumgebung. Nach der Aktivierung der neu erstellten Conda-Umgebung wird auf dem Bildschirm die Ausgabe angezeigt, die sich aus der Ausführung des Befehls zur Installation von CryoSieve mit Pip ergibt. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung 4: Hilfeinformationen. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung 5: Laufender Prozess. Beim Ausführen von CryoSieve über die Befehlszeile werden auf dem Bildschirm Informationen zum laufenden Prozess angezeigt. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung 6: Die Konfiguration der CryoSPARC-Jobs. (A) Partikelstapel importieren. (B) Importieren von 3D-Volumen. (C-D) Homogene Veredelung. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 1: Optionen von CryoSieve. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 2: Verarbeitungszeit und minimale Anforderungen für den Betrieb von Cryosieve. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 3: Generierung des ersten Modells durch CryoSPARC. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 4: Begründung für die Deaktivierung der Wiederholung der GS-Teilung. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 5: Optionen von cryosieve-csrefine. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Datei 6: Optionen des Kryosieb-csrhb-Faktors. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Die Kryo-EM gilt als zentrale Technik zur Aufklärung der Strukturen biologischer Moleküle. In diesem Prozess ist nach der Datenerfassung mittels Mikroskopie die Extraktion von Partikeln aus Mikroskopbildern unerlässlich, gefolgt von deren Klassifizierung in mehreren Schritten, um den endgültigen Stapel zusammenzustellen. Eine häufige Herausforderung ist das Vorherrschen von beschädigten oder unerwünscht konformen Partikeln, was die Notwendigkeit einer wiederholten Partikelauswahl unterstreicht, um hochauflösende Dichtekarten zu erhalten. Dies macht die Partikelauswahl zu einem entscheidenden Schritt in der Kryo-EM-SPA, um qualitativ hochwertige Dichtekarten zu erhalten. Zu den bestehenden Partikelauswahltechniken gehören der statistische Nicht-Kipp-Validierungsalgorithmus22, der auf Z-Scoresbasierende Ansatz 23 und das Verfahren zur Schätzung der Winkelgenauigkeit24.
CryoSieve erweist sich in diesem Zusammenhang als wertvolles Werkzeug, das in der Lage ist, eine beträchtliche Anzahl von Fremdpartikeln aus dem endgültigen Stapel zu eliminieren. Diese Reduzierung erhöht nicht nur die Recheneffizienz der Rekonstruktion, sondern rationalisiert auch den Prozess. Es bietet eine umfassende Suite für die Partikelauswahl, bei der das Ausmaß des Partikelverwurfs und die daraus resultierende Verbesserung der Auflösung weitgehend von der anfänglichen Datenqualität und den bei der Datenverarbeitung verwendeten Methoden abhängen.
In diesem Manuskript haben wir einen vollständigen Arbeitsablauf der Partikelsiebung unter Verwendung des realen Falldatensatzes von Influenza-Hämagglutinin-Trimer vorgestellt (EMPIAR-Eintrag: 10097). Die hier behandelten und diskutierten Schritte können als Partikelsiebung und Neuschätzung der Pose zusammengefasst werden. Das endgültige 3D-rekonstruierte Volumen erreichte eine Auflösung von 3,62 Å, und die Seitenketten in Alpha-Helices waren im nachbearbeiteten Volumen im Vergleich zur veröffentlichten Dichtekarte klarer.
CryoSieve ist eine Open-Source-Methode, die auf GitHub (https://github.com/mxhulab/cryosieve) verfügbar ist. Ein ausführliches Tutorial finden Sie auch auf der Homepage. Benutzer können es installieren und verwenden, indem sie dem Tutorial folgen. Zusätzlich werden zwei Module, cryosieve-csrefine und cryosieve-csrhbfactor, bereitgestellt. Das Modul cryosieve-csrefine wurde speziell entwickelt, um die sequentielle Ausführung verschiedener Vorgänge innerhalb von CryoSPARC zu automatisieren (Ergänzende Datei 5). Zu diesen Vorgängen gehören das Importieren von Partikelstapeln und das Ausführen von Ab-initio-, homogenen oder ungleichmäßigen Verfeinerungsaufträgen. Auf der anderen Seite wurde das Kryosieve-csrhbfactor-Modul entwickelt, um die Bestimmung des Rosenthal-Henderson-B-Faktors zu automatisieren, indem es die Fähigkeiten von cryosieve-csrefine nutzt (Supplementary File 6).
Derzeit ist die Anwendung dieser Methode auf einzelne Konformationsszenarien beschränkt. Folglich sind in Fällen, in denen Partikel mehrere Konformationen darstellen, ihre Fähigkeiten eingeschränkt. Anwendern wird empfohlen, zunächst eine 3D-Klassifizierung durchzuführen, um Partikel unterschiedlicher Konformationen zu trennen, bevor sie sie für eine verfeinerte Partikelauswahl verwenden. Obwohl die Methode in der Lage ist, über 50 % der Partikel aus dem endgültigen Stapel herauszufiltern, bleiben die Herkunft dieser weggeworfenen Partikel und die zugrunde liegenden Gründe für ihren vernachlässigbaren Beitrag zur Rekonstruktionsqualität unklar. Diese Verständnislücke erfordert zusätzliche Forschung, um diese Einschränkung umfassend anzugehen und möglicherweise zu beheben.
Es gibt drei mögliche Methoden der Partikelsortierung oder Partikelsiebung. Zunächst einmal kann cisTEM4 für jedes einzelne Partikelbild nach der 3D-Verfeinerung eine Punktzahl melden. Benutzer können Partikel mithilfe des cisTEM-Scores sortieren, um Partikel zu verwerfen. Der Winkelgraph-Konsistenz-Ansatz (AGC)17 ist auch ein Verfahren zum Verwerfen falsch ausgerichteter Partikel. Darüber hinaus ist die Nichtausrichtungsklassifizierung5 eine traditionelle Methode, um Partikel mithilfe der 3D-Klassifizierung zu verwerfen. Wir verglichen die Qualität der mit diesen Methoden zurückgehaltenen Partikel mit CryoSieve und stellten fest, dass die zurückgehaltenen Partikel von CryoSieve von höherer Qualität sind15. Das hier vorgestellte Verfahren übertrifft alternative Verfahren deutlich und erreicht die geringste Anzahl von Partikeln bei gleicher Auflösung.
Wie das Ergebnis zeigt, trägt die Mehrheit der Partikel in einem Kryo-EM-Endstapel nicht zur Rekonstruktion der Dichtekarte bei. Mit anderen Worten, unter allen Partikeln, die während der Bildaufnahme gesammelt werden, tragen nur einige wenige, nämlich die feinste Teilmenge, tatsächlich zur endgültigen Rekonstruktion bei. Folglich könnte das Verhältnis dieser letzten Teilmenge zur Gesamtzahl der gesammelten Partikel als quantitative Metrik für die Beurteilung der Probenqualität dienen. Je höher dieses Verhältnis, desto besser ist die Probenqualität. Trotz technischer Fortschritte, die die Kryo-EM für Strukturbiologen zugänglicher gemacht haben, bleibt die Probenvorbereitung ein großer Engpass im Arbeitsablauf. Naturwissenschaftler und Ingenieure konzentrieren sich daher auf diese Herausforderung25. Bei der Einzelpartikelanalytik (SPA) besteht die Probenvorbereitung aus zwei entscheidenden Schritten: der Probenoptimierung und der Gittervorbereitung. Ersteres beinhaltet die Reinigung der Probe unter Beibehaltung ihres optimalen biochemischen Zustands. Letzteres beinhaltet die Vorbereitung der Probe für die Analyse im Mikroskop, einschließlich chemischer oder Plasmabehandlung des Gitters, Probenabscheidung und Vitrifizierung. Zahlreiche Techniken wurden vorgeschlagen, um makromolekulare Instabilität zu beheben, aber die Wirksamkeit eines Ansatzes gegenüber einem anderen hängt von den Eigenschaften der Probe ab25,26. Derzeit werden die Ergebnisse der Netzvorbereitung stark von der Expertise und Erfahrung des Benutzers beeinflusst, was den Prozess zeitaufwändig und herausfordernd machen kann27,28. Die zahlreichen Variablen, die bei der Proben- und Gittervorbereitung auftreten, stellen eine Herausforderung bei der Feststellung von Ursache-Wirkungs-Beziehungen dar, da die Forscher die Probe nur auf molekularer Ebene mit dem Mikroskop beurteilen können. Infolgedessen fehlen noch quantitative Statistiken aus Vergleichen verschiedener Proben- und Gittervorbereitungsprotokolle, und ein systematischer Ansatz ist notwendig, um Trends zu untersuchen und die grundlegenden Mechanismen des Probenverhaltens zu verstehen29.
Offenlegungen
Alle anderen Autoren erklären, dass keine konkurrierenden Interessen bestehen.
Danksagungen
Diese Arbeit wurde unterstützt von der Shenzhen Academy of Research and Translation (an M.H.), dem Advanced Innovation Center for Structural Biology (an M.H.), dem Beijing Frontier Research Center for Biological Structure (an M.H.), dem National Key R&D Program of China (Nr. 2021YFA1001300) (an C.B.), der National Natural Science Foundation of China (Nr. 12271291) (an C.B.), und die National Natural Science Foundation of China (Nr. 12071244) (an Z.S.).
Materialien
| Name | Company | Catalog Number | Comments |
| CryoSPARC | Structura Biotechnology Inc. Toronto, Canada | CryoSPARC (Cryo-EM Single Particle Ab-Initio Reconstruction and Classification) is a state of the art HPC software solution for complete processing of single-particle cryo-electron microscopy (cryo-EM) data. CryoSPARC is useful for solving cryo-EM structures of membrane proteins, viruses, complexes, flexible molecules, small particles, phase plate data and negative stain data. | |
| EMPIAR-10097 Dataset | https://ftp.ebi.ac.uk/empiar/world_availability/10097/data/Particle-Stack/T40_HA_130K-Equalized-Particle-Stack.mrcs | This dataset comprises single-particle cryo-EM data of the Influenza Hemagglutinin trimer, characterized by its highly preferred orientation, collected using a 40-degree tilted collection strategy. | |
| initial.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| mask.mrc | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | ||
| RELION | 4.0-beta-2 | RELION (REgularised LIkelihood OptimisatioN) is an open-source software for cryo-electron microscopy (cryo-EM) data processing, particularly for refining macromolecular structures. Utilizing a Bayesian approach, it excels in separating signal from noise, enabling high-resolution structure determination. RELION supports single-particle analysis, tomography, and sub-tomogram averaging, and has become widely used in structural biology due to its effectiveness and user-friendly interface. | |
| T40_HA_130K-Equalized_run-data_CryoSPARC_refined.star | https://github.com/mxhulab/cryosieve-demos/tree/master/EMPIAR-10097 | Metadata file for the final stack of particles from EMPIAR-10097 |
Referenzen
- Bai, X. C., Fernandez, I. S., Mcmullan, G., Scheres, S. H. Ribosome structures to near-atomic resolution from thirty thousand cryo-em particles. elife. 2, 00461(2013).
- Campbell, M. G., et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 20 (11), 1823-1828 (2012).
- Li, X., et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-em. Nat Meth. 10 (6), 584-590 (2013).
- Grant, T., Rohou, A., Grigorieff, N. Cis tem, user-friendly software for single-particle image processing. eLife. 7, e35383(2018).
- Scheres, S. H. Relion: Implementation of a bayesian approach to cryo-em structure determination. J Str Biol. 180 (3), 519-530 (2012).
- Punjani, A., Rubinstein, J. L., Fleet, D. J., Brubaker, M. A. Cryosparc: Algorithms for rapid unsupervised cryo-em structure determination. Nat Meth. 14 (3), 290-296 (2017).
- Kühlbrandt, W. The resolution revolution. Science. 343 (6178), 1443-1444 (2014).
- Dubochet, J., et al. Cryo-electron microscopy of vitrified specimens. Quart Rev Biophys. 21 (2), 129-228 (1988).
- Wagner, T., et al. Sphire-cryolo is a fast and accurate fully automated particle picker for cryo-EM. Comm Biol. 2 (1), 218(2019).
- Bepler, T., et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Meth. 16 (11), 1153-1160 (2019).
- Wang, F., et al. Deeppicker: A deep learning approach for fully automated particle picking in cryo-em. J Str Biol. 195 (3), 325-336 (2016).
- Heimowitz, A., Andén, J., Singer, A. Apple picker: Automatic particle picking, a low-effort cryo-em framework. J Str Biol. 204 (2), 215-227 (2018).
- Glaeser, R. M. How good can single-particle cryo-em become? What remains before it approaches its physical limits. Ann Rev Biophys. 48, 45-61 (2019).
- Diiorio, M. C., Kulczyk, A. W. A robust single-particle cryo-electron microscopy (cryo-em) processing workflow with cryosparc, relion, and scipion. J Vis Exp. (179), e63387(2022).
- Zhu, J., et al. A minority of final stacks yields superior amplitude in single-particle cryo-em. Nat Comm. 14 (1), 7822(2023).
- Zhou, Y., Moscovich, A., Bendory, T., Bartesaghi, A. Unsupervised particle sorting for high-resolution single-particle cryo-em. Inv Probl. 36 (4), 044002(2020).
- Méndez, J., Garduno, E., Carazo, J. M., Sorzano, C. O. S. Identification of incorrectly oriented particles in cryo-em single particle analysis. J Str Biol. 213 (3), 107771(2021).
- Tan, Y. Z., et al. Addressing preferred specimen orientation in single-particle cryo-em through tilting. Nat Meth. 14 (8), 793-796 (2017).
- Rosenthal, P. B., Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 333 (4), 721-745 (2003).
- Kaur, S., et al. Local computational methods to improve the interpretability and analysis of cryo-em maps. Nat Comm. 12 (1), 1240(2021).
- Stagg, S. M., Noble, A. J., Spilman, M., Chapman, M. S. Reslog plots as an empirical metric of the quality of cryo-em reconstructions. J Str Biol. 185 (3), 418-426 (2014).
- Vargas, J., Otón, J., Marabini, R., Carazo, J. M., Sorzano, C. Particle alignment reliability in single particle electron cryomicroscopy: A general approach. Sci Rep. 6 (1), 21626(2016).
- Vargas, J., et al. Particle quality assessment and sorting for automatic and semiautomatic particle-picking techniques. J Str Biol. 183 (3), 342-353 (2013).
- Vargas, J., Melero, R., Gomez-Blanco, J., Carazo, J. -M., Sorzano, C. O. S. Quantitative analysis of 3d alignment quality: Its impact on soft-validation, particle pruning and homogeneity analysis. Sci Rep. 7 (1), 6307(2017).
- Carragher, B., et al. Current outcomes when optimizing 'standard'sample preparation for single-particle cryo-em. J Microsc. 276 (1), 39-45 (2019).
- Drulyte, I., et al. Approaches to altering particle distributions in cryo-electron microscopy sample preparation. Acta Crystallographica Sec D: Str Biol. 74 (6), 560-571 (2018).
- Glaeser, R. M. How good can cryo-em become. Nat Meth. 13 (1), 28-32 (2016).
- Kim, L. Y., et al. Benchmarking cryo-em single particle analysis workflow. Front Mol Biosci. 5, 50(2018).
- Weissenberger, G., Henderikx, R. J., Peters, P. J. Understanding the invisible hands of sample preparation for cryo-em. Nat Meth. 18 (5), 463-471 (2021).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten