
Method Article
RNA-Pulldown-Verfahren, RNA-Ziele eine lange nicht-kodierende RNA zu identifizieren
In diesem Artikel
Zusammenfassung
Diese RNA Pulldown-Methode ermöglicht es, die RNA-Ziele eine lange nicht-kodierende RNA (LncRNA) zu identifizieren. Basierend auf der Hybridisierung von hausgemachten, gestaltete Gegenstrang DNA-Oligonukleotid Sonden spezifisch für diese LncRNA in einem entsprechend festen Gewebe oder Zell-Linie, ermöglicht es effizient die Erfassung aller RNA-Ziele von der LncRNA.
Zusammenfassung
Lange nicht-kodierende RNA (LncRNA), die Sequenzen von mehr als 200 Nukleotiden ohne einen definierten Leseraster sind, gehören zu den regulatorischen nicht-kodierender RNA Familie. Obwohl ihre biologischen Funktionen weitgehend unbekannt bleiben, die Zahl dieser LncRNAs stetig gestiegen und es wird jetzt geschätzt, dass der Mensch mehr als 10.000 solcher Protokolle können. Einige davon sind bekanntermaßen wichtige regulatorische Signalwege der Genexpression beteiligt sein, die auf transkriptioneller Ebene, sondern auch auf verschiedenen Stufen der RNA co- und Posttranskriptionale Reifung stattfinden. In den letztgenannten Fällen haben RNAs, die durch das LncRNA ausgerichtet sind, identifiziert werden. Das ist der Grund, warum ist es sinnvoll, eine Methode, die es ermöglicht die Identifizierung der RNAs verbunden direkt oder indirekt mit einer LncRNA von Interesse zu entwickeln.
Dieses Protokoll, das von zuvor veröffentlichten Protokolle, so dass die Isolierung des LncRNA zusammen mit den zugehörigen Chromatin-Sequenzen inspiriert wurde, wurde angepasst um die Isolierung der zugehörigen RNAs zu ermöglichen. Wir festgestellt, dass zwei Schritte entscheidend für die Effizienz dieses Protokolls sind. Das erste ist das Design von spezifischen Anti-Sense DNA-Oligonukleotid Sonden zu hybridisieren, die LncRNA von Interesse. Zu diesem Zweck die LncRNA Sekundärstruktur wurde von Bioinformatik vorausgesagt und Gegenstrang Oligonukleotid Sonden wurden entwickelt, mit einer starken Affinität für Regionen, in denen eine geringe Wahrscheinlichkeit der internen Basenpaarung angezeigt. Der zweite entscheidende Schritt des Verfahrens stützt sich auf das Fixativ Bedingungen des Gewebes oder kultivierten Zellen, die das Netzwerk zwischen allen molekularen Partnern zu erhalten. Gepaart mit hohem Durchsatz RNA Sequenzierung, bieten diese RNA Pulldown-Protokoll der gesamten RNA Interaktom des LncRNA von Interesse.
Einleitung
Das übergeordnete Ziel des hier beschriebenen Verfahrens ist RNA Molekulare Partner eine lange nichtcodierender RNA (LncRNA) zu identifizieren. LncRNA Sequenzen von mehr als 200 Nukleotide ohne einen definierten Leseraster entsprechen. Einige von ihnen haben gezeigt, dass Ausdruck Genregulation, nicht nur auf transkriptioneller Ebene, sondern auch auf verschiedenen Stufen der RNA mit- und Posttranskriptionale Reifung beteiligt sein. In den letztgenannten Fällen sind Molekulare Partner von der LncRNA RNAs, die identifiziert werden. Entscheidend für die Entwicklung wäre eine Methode ermöglicht die Identifizierung der RNAs verbunden direkt oder indirekt mit einer LncRNA von Interesse.
Zuvor veröffentlichten Methoden, wie Chromatin Isolierung von RNA-Reinigung (ChIRP)1,2 und erfassen Hybridisierung Analyse der RNA Ziele (Diagramm)3,4, erlauben Hochdurchsatz-Entdeckung RNA-gebundene Proteine und genomische Bindungsstellen für einen bestimmten LncRNA. In diesen zwei Methoden, die LncRNA von Interesse war zuerst zu biotinylierte komplementären Oligonukleotiden hybridisiert, und die Anlage war dann isoliert mit Streptavidin-Perlen. Der Hauptunterschied zwischen diesen beiden Techniken bezieht sich auf das Design der Sonden, die auf LncRNAs. Zwitschern, die Strategie von RNA-Fisch, bestand aus einen Pool von kurzen komplementären DNA-Oligonukleotid Sonden decken die gesamte Länge der LncRNA Gestaltung inspiriert. Im Gegensatz dazu im Diagramm angepasst die Autoren einen RNase H Zuordnung Assay auf LncRNAs Websites zur Hybridisierung zu untersuchen.
Das Verfahren hier vorgeschlagene Design Gegenstrang DNA biotinylierte Oligonukleotid Sonden verwendet, die Bioinformatik Modellierung von LncRNA Sekundärstruktur5 markieren Sie mit einer starken Affinität für Regionen-Sonden, die eine geringe Wahrscheinlichkeit der internen anzeigen Base pairing. Dieses Verfahren hat den Vorteil, dass weniger teuer als solche auf Basis von Pools von Fliesen Oligonukleotid Sonden2 und weniger zeitaufwändig als solche auf Basis von RNAse-H Empfindlichkeit4.
Da gibt es eine wachsende Zahl von Beweisen für posttranskriptionelle Genregulation durch LncRNAs6, ist es sehr nützlich, um einen Ansatz ermöglicht die Erfassung von RNAs, die Ziele von einem LncRNA zu entwickeln. Darüber hinaus wurde der Ansatz für die meisten Anwendungen verwendbar, in kultivierten Zellen und Gewebe-Extrakte optimiert.
Protokoll
Alle Verfahren wurden unter strikter Einhaltung der Europäischen Wirtschaftsgemeinschaft für die Pflege und Verwendung von Labortieren (86/609/EWG und 2010/63/UE) und unter einer Lizenz gewährt, D. Becquet (Préfecture des Bouches-du-Rhône, Genehmigung Nr. 13-002) durchgeführt.
1. Probe Design
-
Mit der primären Sequenz des LncRNA von Interesse, generieren Sie die Sekundärstruktur auf der "RNAstructure Webserver"5.
Hinweis: Auf dieser Seite können verschiedene Algorithmen verwendet werden. Die drei Vorhersage-Tools, die die besten Ergebnisse zu geben, denn die Sonde Designs sind: "Fold" (niedrigste freie Energiestruktur), "MaxExpect" (sehr wahrscheinlich Basenpaare) und "Probnot" (wahrscheinlich Basenpaare, einschließlich der Pseudoknots). Diese drei Analysen können durchgeführt und verglichen werden. Anderen Webservern, wie die Wiener RNA Websuite7, können auch verwendet werden.- Wählen Sie die Regionen, die eine geringe Wahrscheinlichkeit der internen Basenpaarung anzeigen und Gegenstrang Oligonukleotid Sonden von 25 Basen mit einer starken Affinität für diese Regionen zu entwerfen.
Hinweis: Der GC Inhalt diese Sonden sollte zwischen 40 und 60 % bestehen. Mit einer Ausrichtung Suchwerkzeug (Blast), stellen Sie sicher, dass die ausgewählten Gegenstrang Oligonukleotid Sonden Nukleotidsequenzen in anderen RNA ausgedrückt in der ausgewählten zellensystem nicht kennen.
- Wählen Sie die Regionen, die eine geringe Wahrscheinlichkeit der internen Basenpaarung anzeigen und Gegenstrang Oligonukleotid Sonden von 25 Basen mit einer starken Affinität für diese Regionen zu entwerfen.
- Design auch eine unspezifische DNA-Oligonukleotid Sonde, von 25 Basen die weder Affinität für die LncRNA von Interesse, noch für andere RNA-Sequenzen im Genom der Interesse zeigt.
- Bestellen Sie die Sonden mit Biotin am 3'-Ende.
Hinweis: Um die sterische Behinderung zu reduzieren, muss der Abstand zwischen Oligonukleotid und Biotin mit einem Triethyleneglycerol-Abstandhalter erhöht werden. Für ein optimales Ergebnis und die Spezifität der Ergebnisse des Pull-Down beurteilen zu können, es wird empfohlen, 3 verschiedene Design Gegenstrang Oligonukleotid Sonden und dann experimentell ihre Effizienz zu vergleichen.
2. Vernetzung
-
Kultivierten Zellen Vernetzung
- Kultur der GH4C1 Sommatolactotroph Hypophysen Zellen in Hams F10 Medium mit 15 % Pferd Serum und 2 % fetalen Kälberserum ergänzt. Wachsen Sie die Zellen bis zum Zusammenfluss in 78,5 cm2 Kultur Platten. Dies entspricht ca. 1 x 107 Zellen.
- Entfernen Sie das Zellkulturmedium aus einem konfluierende GH4C1 78,5 cm2 Kultur Teller, dann Spülen mit 1 x die mittlere Lautstärke von Phosphat gepufferte Kochsalzlösung (PBS)
- Befestigen Sie die Zellen mit einer 1 % igen Paraformaldehyd Lösung mit PBS-Puffer (10 mL für eine 78,5 cm2 Gerichte); Diese Lösung muss von einer 4 % Paraformaldehyd Vorratslösung frisch zubereitet werden. Vernetzen Sie unter Agitation für 10 min bei Raumtemperatur (RT).
Achtung: Paraformaldehyd (PFA) ist giftig und muss mit Vorsicht behandelt werden. - Paraformaldehyd-Aktion zu stillen, durch Hinzufügen von 1/10 Volumen von Glycin 1,25 M (1 mL pro 10 mL der Lösung Paraformaldehyd); 5 min bei RT agitieren
- Verwerfen Sie die Medien durch streben und spülen zwei Zeiten (5 min) mit 1 X das mittlere Volumen der PBS.
- Fügen Sie ein Volumen von PBS entspricht 1/10 des Volumens der Medien, sammeln Zellen mit einer Zelle Schaber und übertragen Sie dann zu einem Zentrifugenröhrchen.
- Mit 510 g bei 4 ° C für 5 min drehen.
- Entfernen Sie so viel überstand wie möglich.
- Speichern von Pellets auf unbestimmte Zeit bei Bedarf bei-80 ° C.
-
Vernetzung von Gewebe
- Setzen Sie 5 mg des frisch gewonnenen Maus Hypophyse Gewebe in einer Lösung von 1 % Paraformaldehyd in PBS (ca. 10 x das Volumen des Gewebes) verdünnt, agitieren für 10 min bei RT
- Stillen Sie die Paraformaldehyd Aktion durch Hinzufügen einer 1,25 M Glycin-Lösung (1 mL pro 10 mL der Lösung Paraformaldehyd) zu und rühren Sie 5 min bei RT
- Verwerfen Sie die Medien durch Absaugen und spülen Sie zweimal mit PBS (ca. 10 x das Volumen des Gewebes). Entfernen Sie so viel überstand wie möglich.
- Speichern der vernetzten Gewebes auf unbestimmte Zeit bei-80 ° C.
(3) Zellen oder Gewebe Lyse
- Bereiten Sie die Lyse-Puffer (50 mM Tris-HCl pH 7,0, 10 mM EDTA, 1 % SDS ergänzt mit 200 U/mL einer RNAse-Inhibitor-Lösung und einem Cocktail von Proteasen Inhibitor 5 µL/mL).
- Um lysierten Proben ohne vorherige Auftauen zu erhalten, wieder aussetzen der Zelle Pellets oder vernetzten Gewebe mit dieser Puffer (ca. 1 mL pro 100 mg Pellet Zelle oder eines Gewebes). Ein Zelle Pellet 1 x 10-7 -Zellen gewonnenen ergeben sich eine lysierten Probe enthält etwa 20 mg Protein.
Hinweis: Je nach Gewebe verwendet, sollte ein Schritt von mechanischen Störungen hinzugefügt werden. In diesem Fall ist es wichtig, die Erwärmung der Proben während diesen zusätzlichen Schritt zu vermeiden.
(4) Beschallung
-
Optimierung der Beschallung Bedingungen
- Programm der sonikator mit 2 bis 5 Reihe von 30 s ein und 30 s aus.
- Durchführen Sie Tests, um Beschallung Bedingungen auf verdünnten lysierten Proben (Verdünnungsfaktor ½ oder ¼ entspricht ungefähr 10 oder 5 mg Protein) zu optimieren. Ort verdünnt lysierten Proben im Wasserbad 4 ° C und starten Sie die Anwendung von Ultraschall-Serie.
- RNAs zu reinigen, mit einem RNA-Reinigung-Kit oder mit einer RNA-Isolierung-Reagenz (z.B.Trizol).
- Laden Sie die Gesamtheit der gereinigten RNA auf einem 1 % Agarose-Gelelektrophorese in TBE-Puffer, überprüfen Sie die Länge der RNA-Fragmente. Diese Länge sollte zwischen 200 und 800 bp liegen.
Hinweis: Abhängig von der RNA-Fragment-Größe kann die Gegenstrang Oligonukleotid Sonden Effizienz variieren. Es wird dann empfohlen, um die Effizienz der Sonden unter verschiedenen Bedingungen der Zellulite überprüfen.
-
Beschallung der lysierten Proben
- Ort lysiert Proben entsprechend 20 mg von Proteinen (nach Schritt 3.2 erhalten) in die 4 ° C Wasserbad und die Beschallung-Serie zu starten, wie in Schritt 4.1 optimiert.
- Unmittelbar nach der Beschallung, Zentrifuge für 5 min bei 12.000 g bei 4 ° C. Übertragen Sie Überstände in neue Zentrifuge Rohre.
Hinweis: Um Homogenität zu gewährleisten, replizieren Überstände gebündelt und werden bei diesem Schritt umverteilt.
(5) RNA-Pulldown
-
Tag 1 – Hybridisierung Schritt
- 2 Bände der Hybridisierung Puffer (50 mM Tris-HCl pH 7,0, 750 mM NaCl, 1 mM EDTA, 1 % SDS, 15 % Formamid, aus dem Stegreif hinzugefügt) hinzufügen, Überstände nach der Beschallung Schritt gesammelt wurden. Vortex.
- 20 µL jeder Probe in ein Zentrifugenröhrchen (eingabesamples) übertragen und speichern bei-20 ° C.
- Jede Probe fügen Sie 100 Pmol biotinylierte Oligonukleotid Sonden (spezifische oder unspezifische; siehe Tabelle 1 hinzu). Inkubieren Sie 4 bis 6 h unter mäßiger Erregung auf ein Rohr Rotator bei RT
- Fügen Sie 50 µL der magnetischen Streptavidin Perlen mit 200 U/mL einer RNAse-Inhibitor-Lösung und einem Cocktail von Proteasen Inhibitor 5 µL/mL ergänzt.
- Über Nacht unter mäßiger Erregung auf den Rohr-Rotator bei RT inkubieren
-
Tag 2 – RNA Isolierung Schritt
- Nutzen Sie magnetische Support um Perlen aus Zelle lysate zu trennen, den überstand zu verwerfen und die Perlen mit 900 µL Waschpuffer waschen (SDS 0,5 %, SSC 2 X). Wiederholen Sie, dazwischen 5 mal 5 min Unruhe auf den Rotator bei RT
- Nach dem letzten Waschen, Dekantieren ein letztes Mal und 95 µL Proteinease K-Puffer (10 mM Tris-HCl pH 7,0, 100 mM NaCl, 1 mM EDTA, 0,5 % SDS) hinzufügen und 5 µL Proteinase K (20 mg/mL) zu Proben.
- Tauen Sie auf dem Eis eingabesamples (20 μl auf) und fügen Sie 75 µL Proteinease K-Puffer und 5 µL Proteinase-K (20 mg/mL).
- Inkubieren Sie alle Proben mit Proteinase K 45 min bei 50 ° C dann 10 min bei 95 ° c
- Kühlen Sie die Probe für 3 min auf Eis, bevor die Perlen von RNAs mit magnetischen Unterstützung trennt. Halten Sie den überstand und entsorgen Sie die Perlen.
- Reinigen Sie RNAs mit einem RNS Reinigung Kit, die einen DNA-Verdauung Schritt umfassen sollte. Shop-RNAs bei-80 ° C.
- Durchführen Sie reversen Transkription qPCR (RT-qPCR) mit einem RT-qPCR spezifische Primer (Tabelle 1) mit anschließender Kit.
- Erstellen Sie zwei DNA-Bibliotheken entspricht den zwei RNA-Pools mit jeweils spezifischen Neat1 Sonden (Tabelle 1) erhalten. Durchführen Sie Sequenzierung auf einem Next Generation Sequencing-System.
Ergebnisse
Mehrere Studien haben gezeigt, dass LncRNAs in fast allen wichtigen biologischen Verfahren eine wesentliche Rolle spielen und dass diese Rolle durch die Kontrolle der Genexpression auftreten, sowohl auf die transkriptionelle und posttranskriptionelle Ebene erreicht wird zeigen in diesem Fall, dass RNAs das Ziel von LncRNAs6sein können.
Die LncRNA nuklearen angereicherten reichlich Transkript 1 (Neat1) ist in verschiedenen Neuropathologies als Frontotemporale Demenz, Amyotrophe Lateralsklerose oder Epilepsie8,9,10verwickelt, und ist auch misreguliert in verschiedenen Krebsarten11,12.
Diese LncRNA ist auch bekannt, der strukturellen Komponente des bestimmten nuklearen Einrichtungen, die Paraspeckles und posttranskriptionelle Circadiane Regulierung der Gen-Expression-13beteiligt sein. Paraspeckles, die findet man in jedem Zellkern und werden gebildet, um nicht nur Neat1, die notwendig ist für ihre Ausbildung, sondern auch um mehrere RNA-bindende Proteine (RBP)14, sind in der Tat bekannt, RNA Ziele innerhalb des Zellkerns15 behalten zu können . Die Bildung des Paraspeckles erfolgt durch die Vereinigung der verschiedenen Komponenten. Diese Formation zeigte sich ein circadiane rhythmisches Muster eine rhythmische nuklearen Aufbewahrung von RNA Ziele13fahren angezeigt. Die nukleare Beibehaltung der RNA Ziele von Paraspeckles kann auftreten durch Bindung an RBP oder direkt durch RNA/RNA Verein, aber das Ausmaß der RNAs, die gezielt durch Paraspeckles ermittelt werden musste. Um die RNA zu identifizieren gezielt direkt oder indirekt durch Neat1, eine RNA Pulldown-Protokoll wurde entwickelt, die erlaubt, die Isolierung und Identifizierung aller Neat1 RNA-Ziele in kultivierten Zellen als auch in Gewebeproben (siehe Abbildung 1 für eine grafische Präsentation der Technik).
Das Protokoll wurde auch erfolgreich zur Identifizierung der RNA Ziele von einem anderen LncRNA Metastasierung assoziiert Lunge Adenokarzinom Transkript 1 (Malat1) eingesetzt. Malat1 ist ein hoch konservierte und ausdrücklichen LncRNA in nuklearen Flecken zusammen mit mehreren RNA splicing-Faktoren gefunden. Malat1 ist bekannt, in der Verordnung das Spleißen von mehreren im Entstehen begriffenen Pre-mRNA16,17einbezogen.
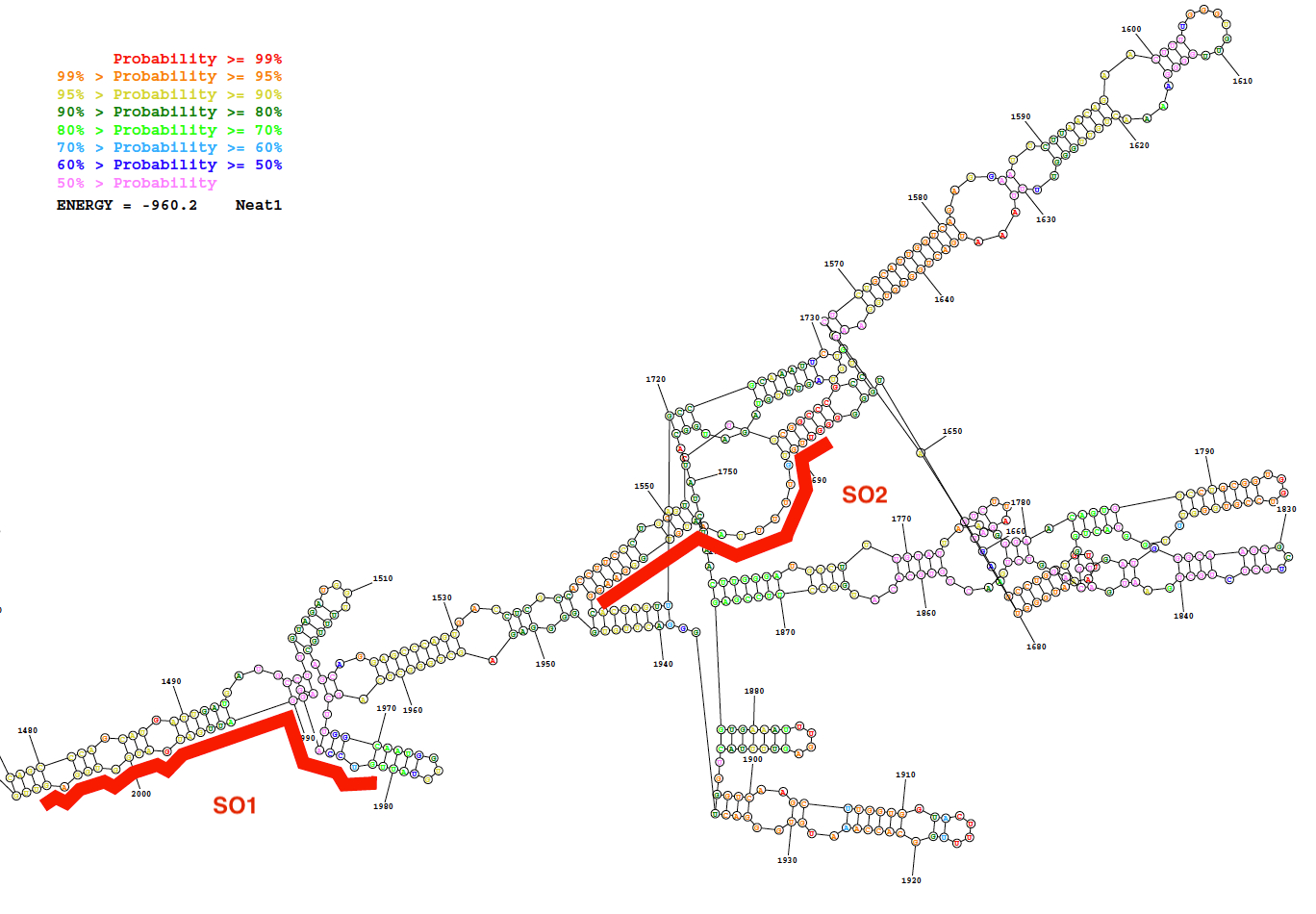
(SO) spezifische und unspezifische Oligonukleotid (NSO) Sonden wurden mit der Sonde Designstrategie beschrieben hier erzeugt. Diese Strategie basiert auf die Auswahl der Regionen, in denen eine geringe Wahrscheinlichkeit der internen Basis Paarung wie vorhergesagt durch die Sekundärstruktur der LncRNA angezeigt und das Design von spezifischen Sonden mit einer starken Affinität für diese Regionen. Als ein repräsentatives Ergebnis dieser Bioinformatik Vorhersagen, ein Bild von den vorhergesagten Sekundärstruktur aus einer Folge von Neat1 (Nukleotide 1.480 bis 2.000) zusammen mit der Position von zwei entwickelt, so dass Sonden in Abbildung 2gegeben sind.
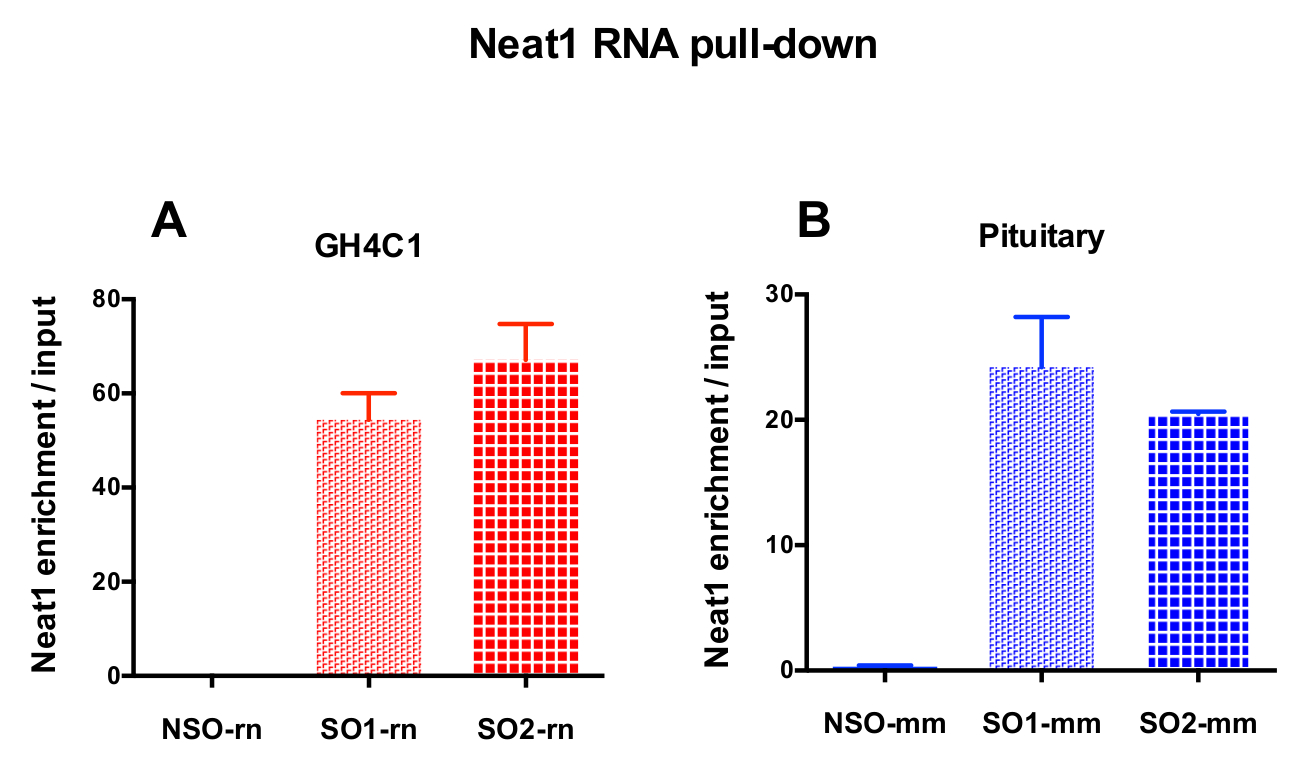
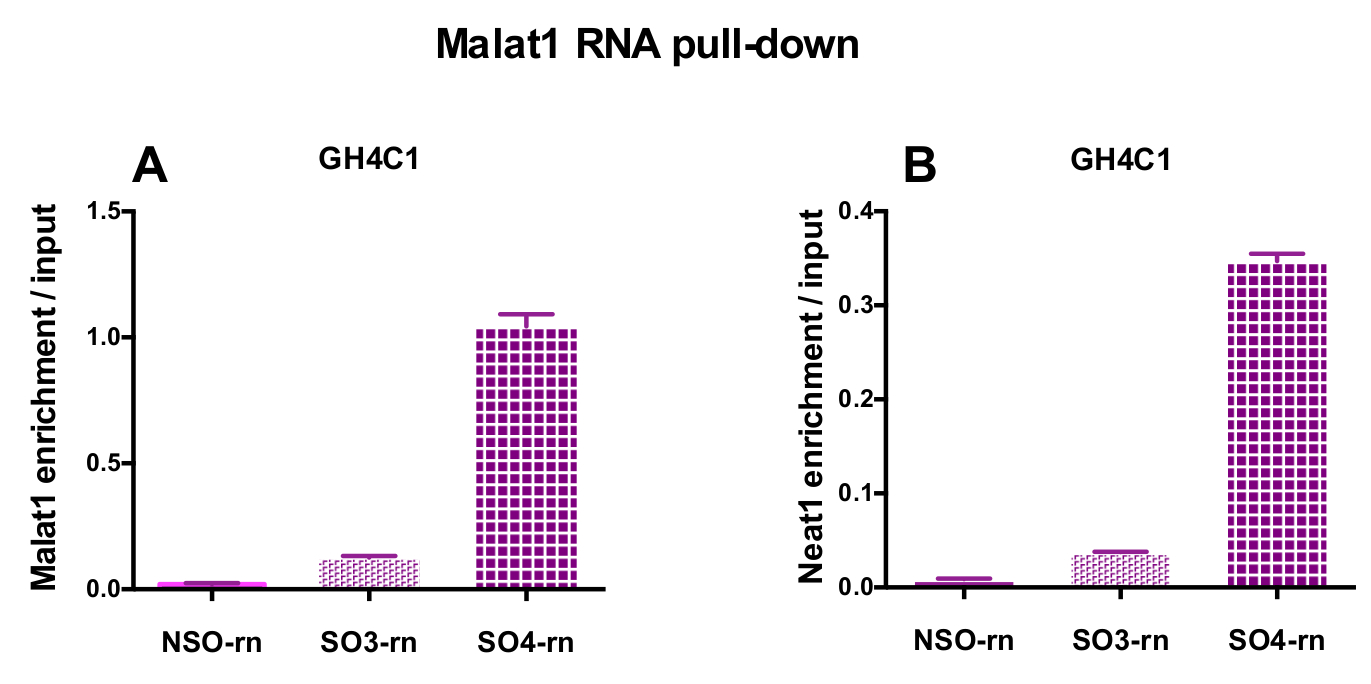
Die entworfenen Sonden wurden angewiesen, Neat1 oder Malat1 für GH4C1, die kultivierten Zellen und Maus Neat1 für Hypophyse Gewebe-Extrakte (Tabelle 1) Ratte. Die relative Anreicherung in Neat1 oder Malat1 wurde für die unspezifischen und spezifischen Sonden im Verhältnis zu der eingabesamples berechnet. Abbildung 3 zeigt die Effizienz der spezifischen Sonden zu Pulldown-Neat1 bei der Ratte GH4C1 Hypophyse Zelllinie (Abbildung 3A) und in der Maus Hypophyse Gewebe extrahiert (Abb. 3 b). Wenn die Sonde Design-Protokoll verwenden, um spezifische Oligonukleotid generieren (damit) Sonden an Malat1 weitergeleitet, verworfen eine effiziente Sonde erhalten wurde, während ein anderer nicht effizient genug war und wurde (Abb. 4A).
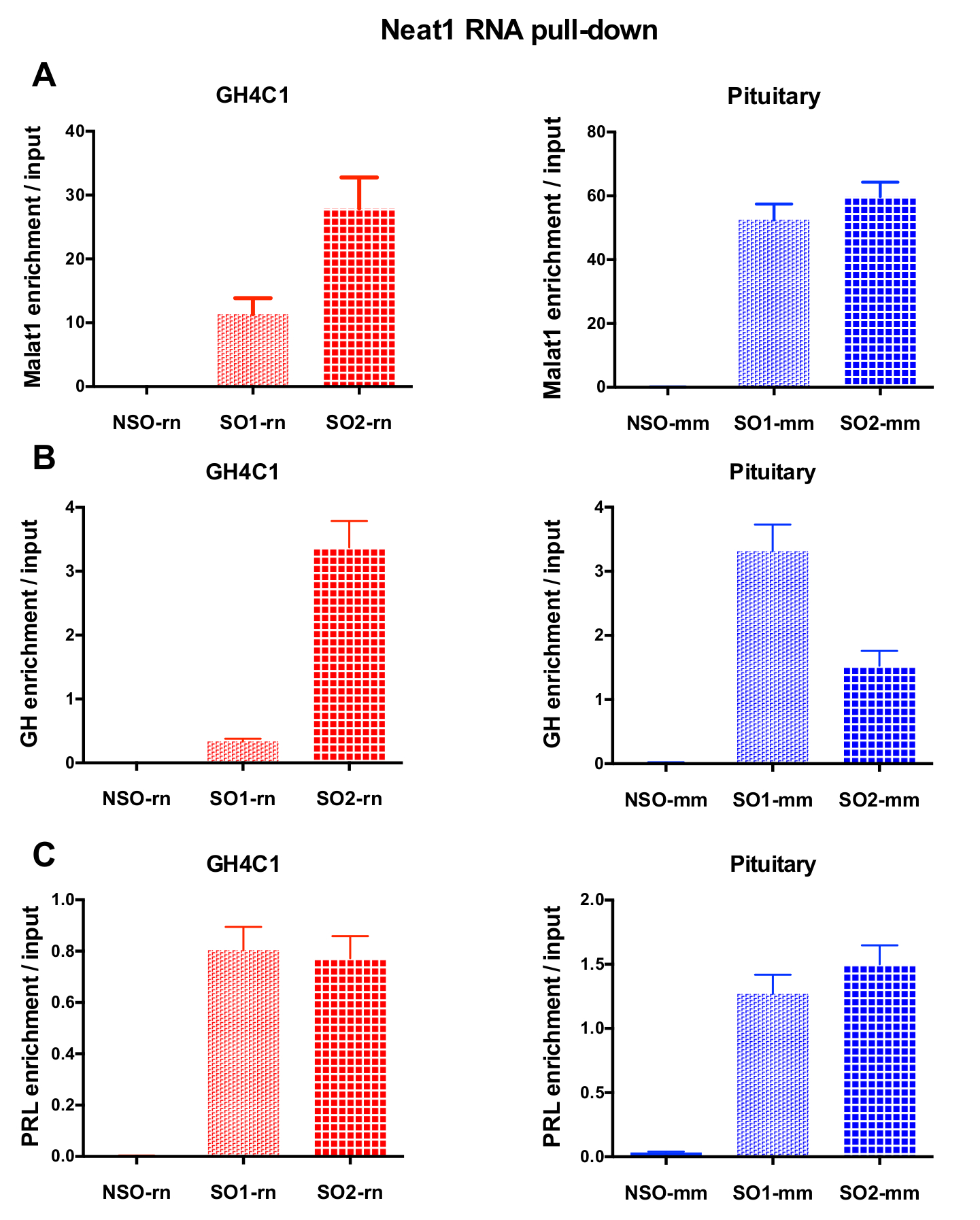
Nach einer RNA Pulldown-Verfahren, gefolgt von RT-qPCR Experimente, einige RNAs bewertet mit spezifischen Primern (Tabelle 1) erwiesen sich als Neat1 oder Malat1 GH4C1 auszugsweise zugeordnet werden. Der RNAs, die im Zusammenhang mit Neat1 in GH4C1 zellextrakte zeigten auch auszugsweise Hypophyse Gewebe mit Neat1 in Verbindung gebracht werden. In der Tat wurde nach Neat1 RNA Pulldown-Malat1 gefunden, von Neat1 in der GH4C1-Zell-Linie und auszugsweise Hypophyse Maus Gewebe (Abb. 5A) ausgerichtet werden. Umgekehrt war Neat1 wesentlich bereichert, nachdem Malat1 RNA Pulldown-Menü mit einer speziellen Sonde in den GH4C1 Zellen (Abbildung 4 b) durchgeführt. Durch die enge Beziehung zwischen den beiden LncRNAs hervorheben, diese Ergebnisse stehen im Einklang mit der möglichen ko-Rolle der Neat1 und Malat1, vorgeschlagen von Malat1-Knockout-Mäusen, die Variationen an Neat1 RNA Ausdruck18, 19. die Transkripte der beiden wichtigsten Hypophyse Hormone, Wachstumshormon (Gh) (Abb. 5 b) und Prolaktin (Prl) (Abbildung 5) wurden wesentlich bereichert nach Neat1 RNA Pulldown-Menü mit speziellen Sonden in GH4C1 Zellen und Hypophyse extrahiert, was auf eine mögliche Regulierung der beiden Hormone durch Neat1. Vergleicht man die zwei spezifischen Sonden verwendet, schien es, dass ihre Effizienz, je nach RNA Ziel betrachtet (Abbildung 5 b und Abbildung 5 variieren kann). Diese Ergebnisse unterstreichen die Notwendigkeit der Gestaltung mehrere spezifische Sonden, um die Anzeige nicht nur den besten Wirkungsgrad bei der Anreicherung von Pulldown-LncRNA, sondern auch die beste Effizienz bei der Anreicherung der RNA Ziele auswählen.
Die RNA-Pull-Down-Methode kann auch RNA Hochdurchsatz-Sequenzierung erhalten die umfassende Liste der RNA Ziele von LncRNA Interesse13folgen. Eine RNA-Seq-Analyse auf GH4C1 Hypophysen Zellen nach Neat1 RNA Pulldown-Menü mit zwei spezifischen Sonden, die oben beschriebenen durchgeführt wurde. Es sei darauf hingewiesen, dass eine Negativkontrolle mit einem NSO auch RNA-Seq-Analyse unterzogen werden könnte wenn das Niveau der RNA wiederhergestellt, nachdem das RNA-Pulldown-Menü mit NSO ausreicht, um den Bau von Bibliotheken ermöglichen. Dies war nicht der Fall in früheren Erfahrung13. Bibliotheken, die erzeugt wurden, nach der Verwendung von spezifischen Sonden wurden analysiert mit Tophat/Manschettenknöpfe pipeline-20 und nur Abschriften mit Werten des Fragments pro Kilobase pro Million des zugeordneten lautet (FPKM) höher als 1 berücksichtigt wurden. Die Listen mit den zwei spezifischen Sonden an Neat1 gerichtet erhalten (Tabelle 1) wurden gekreuzt, um die Spezifität der Ergebnisse zu bewerten. 4.268 Gene fanden sich verbunden mit Paraspeckles, die 28 % der zum Ausdruck gebrachten Abschriften in GH4C1 Zellen13vertreten. In Übereinstimmung mit erzielten Ergebnisse mit qPCR Analyse (Abbildung 5A-C), die Transkripte der Gh, Prl und Malat1 fanden sich Neat1 zugeordnet werden. Die RNA-Pull-Down-Methode wurde daher erwies sich ein effizientes Werkzeug, um die Interaktion zwischen LncRNAs und ihre RNA Ziele erkunden werden.

Abbildung 1: grafische Darstellung der RNA pull-down Verfahren. Am ersten Tag wurden Zellen oder Geweben vernetzt mit Paraformaldehyd, lysiert und vor dem Schritt der Hybridisierung, der durchgeführt wurde, indem man spezifische Sonden biotinylierte beschallt. Magnetische Streptavidin Perlen wurden dann hinzugefügt, um bestimmtes Material vom Rest der Zelle lysate zu trennen. Am zweiten Tag waren Perlen isoliert durch einen Magneten und mehrmals gewaschen. Ein de-Vernetzung Schritt ermöglichte Wiederherstellung von RNAs, die gereinigt und für RT-qPCR oder RNA-Seq-Analyse verwendet wurden. Bitte klicken Sie hier für eine größere Version dieser Figur.
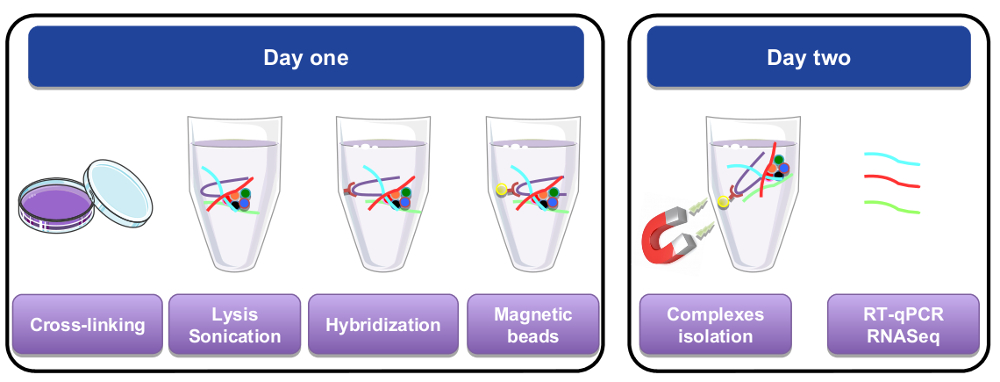{kind=link}

Abbildung 2: sekundäre Struktur aus mehreren Neat1 (Nukleotid 1.480 bis 2.000) wie vorhergesagt von Bioinformatik-Ressource (RNAstructure-Webserver; niedrigste freie Energie-Struktur). Die Struktur ist je nach Grad der Wahrscheinlichkeit der Basenpaarung gefärbt. Die zwei Oligonukleotide Sonden (SO1 und SO2) in rot sind entlang der Neat1 RNA-Struktur aufgestellt. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 3: qPCR Validierung der Neat1 Bereicherung gegenüber Eingang. qPCR Validierung der Neat1 Bereicherung gegen Eingabe nach Neat1 RNA herunterziehen von zwei verschiedenen spezifischen Sonden (SO1-Rn und SO2-Rn für GH4C1 Zellen und 1 mm und SO2-mm für Hypophyse Gewebe) im Vergleich zu einer unspezifischen (NSO-Rn für GH4C1 Zellen und NSO-mm für Hypophyse Gewebe) in GH4C1 rattenzellen (A) und Maus Hypophyse Gewebe extrahiert (B). Ergebnisse sind Mittelwert ± SEM in 3 bis 10 Experimenten gewonnen. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 4: qPCR Validierung von Malat1 und Neat1 Bereicherung gegen Eingabe nach Malat1 RNA herunterziehen. qPCR Validierung des Malat1 (A) und Neat1 (B) Anreicherung versus Eingabe nach Malat1 RNA durch zwei verschiedene spezifische Sonden (SO3-Rn und SO4-Rn) im Vergleich zu einer unspezifischen herunterziehen einer (NSO-Rn) in GH4C1 rattenzellen. Ergebnisse sind Mittelwert ± SEM in 3 Experimente erhalten. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}

Abbildung 5: qPCR Validierung von Malat1, Gh, Prl Bereicherung gegen Eingabe nach Neat1 RNA herunterziehen. qPCR Validierung des Malat1 (A), Gh (B), Prl (C) Anreicherung versus Eingabe nach Neat1 RNA pull-down mit verschiedenen spezifischen Sonden im Vergleich zu einer unspezifischen GH4C1 Ratte Zellen und Maus Hypophyse Gewebe extrahiert. Ergebnisse sind Mittelwert ± SEM in 3 bis 8 Experimente erhalten. Bitte klicken Sie hier für eine größere Version dieser Figur.
{kind=link}
| SONDE NAMEN | Sequenzen | |
| NSO-Rn | TAAAATACCATTTGATGTTTGAAATTAT | |
| SO1-Rn | CTCCACCATCATCAATCCTCTGGAC | |
| SO2-Rn | GCCTTCCCACATTTAAAAACACAAC | |
| SO3-Rn | AACTCGTGGCTCAAGTGAGGTGACA | |
| SO4-Rn | AAGACTCTCAGGCTCCTGCTCATTC | |
| NSO-mm | GTTTGTGGTTTAACAGTGGGAAGGC | |
| SO1-mm | GCCTTCCCACTGTTAAACCACAAAC | |
| SO2-mm | CTCACCCGCACCCCGACTCCTTCAA | |
| qPCR Primer: | ||
| Rattus norvegicus | ||
| Neat1 | AAGGCACGAGTTAGCCGCAAAT | |
| TGTGCACAGTCAGACCTGTCATTC | ||
| Malat1 | GAAGGCGTGTACTGCTATGCTGTT | |
| TCTCCTGAGGTGACTGTGAACCAA | ||
| GH1 | CCGCGTCTATGAGAAACTGAAGGA | |
| GGTTTGCTTGAGGATCTGCCCAAT | ||
| PRL | TGAACCTGATCCTCAGTTTGGT | |
| AGCTGCTTGTTTTGTTCCTCAA | ||
| Mus musculus | ||
| Neat1 | TGGGCCCTGGGTCATCTTACTAGATA | |
| CACAGCTGTTCCAATGAGCGATCT | ||
| GH1 | CTCGGACCGTGTCTATGAGAAACTGA | |
| TTTGCTTGAGGATCTGCCCAACAC | ||
| PRL | TGAACCTGATCCTCAGTTTGGT | |
| AGCTGCTTGTTTTGTTCCTCAA | ||
Tabelle 1: Sequenzen von DNA-Oligonukleotid Sonden und qPCR Primer
Diskussion
Lange Forensisches RNAs (LncRNAs) durch ihre Anzahl und Vielfalt ein großes Forschungsfeld dar, die meisten ihrer Rollen sind noch zu entdecken. Viele von diesen LncRNAs haben eine nukleare Lokalisierung und unter ihnen sind einige an rechtlichen Wege der Genexpression durch transkriptionelle oder posttranskritionelle Mechanismen beteiligt. Eines der aktuellen Herausforderungen in diesem Bereich ist die Relevanz dieser LncRNAs in Posttranskriptionale Verarbeitung von RNAs zu verstehen. Zu diesem Zweck müssen RNAs gezielt durch LncRNAs identifiziert werden. Inspiriert von früheren Studien konzentrierte sich auf die Vereinigung von LncRNAs mit Chromatin, entwickelten wir ein Verfahren, das von RNAs, die einem LncRNA zugeordnet werden können. Der Erfolg dieses Protokolls, namens RNA Pulldown-richtet sich vor allem auf zwei entscheidende Schritte, nämlich die Gestaltung der Gegenstrang DNA-Oligonukleotid Sonden, die speziell und ausschließlich mit der LncRNA von Interesse, und die Bedingungen des Gewebes zu hybridisieren oder Zelle-Fixierung, die die Integrität des Netzwerks zwischen allen molekularen Partnern zu erhalten.
Zuvor veröffentlichten Protokollen vorgesehenen Verfahren um eine LncRNA zusammen mit seinen zugehörigen Chromatin-Sequenzen (ChIRP1,2, Tabelle3,4) zu isolieren. In diesen Protokollen wurden verschiedene Strategien Design beschäftigt, die dem Gegenstrang DNA biotinylierte Oligonukleotid Sonden. In der ChIRP-Prozedur verwendet die Autoren einen Pool von DNA biotinylierte Oligonukleotid Sonden umfasst die gesamte Länge des LncRNA von Interesse nach Ausschluss aller überflüssig und nicht-spezifischen Sonden1,2. Protokoll der CHART die Autoren identifiziert die Bereiche der LncRNA, die zugänglicher für die Hybridisierung sind und Capture-Oligonukleotide, die auf diesen Gebieten entwickelt. Diese Regionen wurden aufgrund ihrer Empfindlichkeit RNase-H ausgewählt. In der Tat, mit der Eigenschaft der RNAse-H, hydrolyseneigung RNAs an Standorten der RNA-DNA-Bindung, der Oligonukleotide, die an zugänglichen Seiten in den LncRNA hybridisieren RNA-DNA-Hybriden zu produzieren und zu enzymatische Spaltung von der LncRNA führen. Die Autoren drei von diesen Kandidaten Erfassung Oligonukleotide ausgewählt und in ein cocktail3,4verwendet.
Das Verfahren zum Design der Gegenstrang DNA biotinylierte Oligonukleotid Sonden war nah an, die in das Diagramm-Protokoll verwendet, aber die Hybridisierung verfügbaren Regionen von der gewünschten LncRNA auf der Grundlage ihrer RNAse-H-Empfindlichkeit nicht ausgewählt wurden, sondern nach deren geringe Wahrscheinlichkeit der internen Basenpaarung wie von Bioinformatik Modellierung von LncRNA Sekundärstruktur bestimmt. Es muss festgestellt werden, dass verschiedene Sekundärstrukturen werden mit verschiedenen Algorithmen vorausgesagt werden, Sonden ausgewählt werden sollten diejenigen, die verfügbaren Sequenzen der LncRNA in der größten Zahl der Sekundärstrukturen vorhergesagt hybridisieren sein. Die gleichen Ergebnisse wurden erzielt mit entweder ein Cocktail aus drei entworfen, spezifische Sonden oder einer einzigen Sonde einzeln. Dies veranlasste die Verwendung von zwei separaten, spezifischen Sonden und die Berücksichtigung der positiven Ergebnisse als jene, die für diese zwei Sonden sind. Endlich, es wird daher empfohlen, am Anfang der Entwicklung der Methode, für ein optimales Ergebnis und Sonden, die Spezifität der Ergebnisse das Pulldown-Menü, 3 verschiedene Anti-Sense-Oligonukleotid entwerfen bewerten zu können und dann zu vergleichen experimentell ihre Effizienz, zumal Sonde Effizienz durch Zelle lysate Vorbereitung verändert werden kann. Dennoch, das Verfahren der Sonde Design basierend auf Bioinformatik Modellierung von LncRNA sekundäre Struktur verwendeten wir preiswerter als die basierend auf Pools von Fliesen Oligonukleotid Sonden2blieb, und das war weniger zeitaufwändig als die Methode basiert RNAse-H Empfindlichkeit4.
Eine negative Kontrolle sollte auch mit negativen erfassen Oligonukleotid entweder die Sinne DNA biotinylierte Oligonukleotid Sonden durchgeführt werden oder Rührei Oligonukleotid Sonden oder gegen eine unabhängige RNA-Oligonukleotide. Wegen der Existenz der natürlichen antisense Transkripte LncRNAs möglicherweise Verwendung von Sinn Oligonukleotid Sonden manchmal unzureichend. Unabhängig von der Oligonukleotid-Sonde für die negativ-Kontrolle ausgewählt ist es notwendig, durch Explosion zu überprüfen, die es nicht mit einem bekannten RNA hybridisieren und im Auge zu behalten, die diese Oligonukleotid, noch un-kommentierte LncRNA hybridisieren kann.
Die Zelle Lysates verwendet diese RNA Pulldown-Experimente stammen aus 106 bis 107 Zellen bei der Arbeit mit kultivierten Zellen und von 1 bis 10 mg, bei der Arbeit mit Gewebe. Die Vorbereitung der Zelle Lysates muss angepasst werden, je nach Gewebe oder Zellen verwendet mit zwei Hauptschritte, die optimiert werden müssen: nämlich die Vernetzung Schritt, die Bildung von kovalenten Bindungen zwischen den LncRNA und ihren molekularen Partnern ermöglicht und die Beschallung-Schritt, der die Viskosität verringert durch Schreddern Chromatin.
Das Ziel der Vernetzung Schritt soll sicherstellen, dass alle RNA-Ziele für den LncRNA geschlossen bleiben, durch Induktion der Bildung eines Netzwerkes zwischen allen Partnern der molekularen. Ein Paraformaldehyd Behandlungsschritt, die bilden kovalente Bindungen zwischen den LncRNA und ihren Partnern kann das Netzwerk sein netzartige. Im Diagramm Protokoll, es wurde vorgeschlagen, wenn arbeiten mit nuklearen LncRNA, um eine erste Behandlung mit Paraformaldehyd durchzuführen, im großen und ganzen lysate Zelle und eine zweite Behandlung am isoliert Nukleinsäuren Bruchteil3,4. Wir beobachteten, dass dieser zusätzliche Schritt die Effizienz der Sonden, wahrscheinlich verringert durch die Reduzierung der LncRNA Zugänglichkeit in den Zellen. Daher der Grad der Vernetzung von Paraformaldehyd muss unter Berücksichtigung der Zelle angepasst oder Gewebetyp verwendet, die Lokalisierung von LncRNA von Interesse, und die Effizienz der entworfenen Sonden.
Während der Lyse der Zellen, das Chromatin ist in der lysate freigegeben und erhöht seine Viskosität; Es ist dann notwendig, um das Chromatin durch Ultraschallbehandlung zu erhöhen die Fließfähigkeit der Proben und erleichtern damit die Zugänglichkeit des Oligonukleotids Zerkleinern, die LncRNA von Interesse-Sonden. Jedoch wird Beschallung auch Schreddern der RNAs extrahiert mit der LncRNA von Interesse. Es ist dann wichtig, die beschallungsdauer so minimieren, dass es effizient reduziert die Viskosität von der lysate, erlaubt es auch, dass die Einholung der RNA Fragmente mit einer Länge zwischen 200-800 BP. Hinweis enthalten, die die beschallungsdauer hoch werden abhängig von der Menge und der Art des Gewebes oder kultivierten Zellen verwendet.
Abschließend ermöglicht die hier beschriebene Vorgehensweise in 2-3 Tagen die Erfassung der RNA Ziele der gewünschten LncRNA. Gepaart mit RT-qPCR, werden diese Methoden ermöglichen eine Verordnung für eine mRNA und bestimmte Zuordnung durch die gewünschte LncRNA als Kandidat Ansatz suchen. Für eine genomweite Ansatz können RNA Pulldown-Experimente von Hochdurchsatz-RNA-Sequenzierung ermöglicht den Abruf von allen RNAs, verbunden mit der gewünschten LncRNA analysiert werden. Was die analytische Strategie gewählt, sollte das RNA-Pulldown-Verfahren neuen wichtigen Erkenntnisse über RNA-Verordnung von LncRNAs vermitteln.
Offenlegungen
Die Autoren haben nichts preisgeben.
Danksagungen
Diese Arbeit wurde unterstützt von der Universität Aix-Marseille und CNRS und finanziert durch einen Zuschuss von Pfizer Laboratorien.
Materialien
| Name | Company | Catalog Number | Comments |
| Bioruptor Plus | Diagenode | B01020001 | Sonicator |
| Dynabeads My One | Thermo-Fisher | 65001 | Magnetic streptavidin beads |
| Formamide | Thermo-Fisher | 15515-026 | |
| Gel electrophoresis apparatus | Advance | Mupid-One | Gel electrophoresis apparatus |
| Proteinase K | Sigma | P2308 | |
| RNA XS purification kit | Macherey-Nagel | 740902 | RNA purificationkit |
| RNAseOUT | Thermo-Fisher | 10777-019 | RNAse inhibitor |
| Trizol | Thermo-Fisher | 15596018 | RNA purification |
| Tube Rotator | Stuart | SB2 | Eppendorf tube rotator |
| RNA to DNA | Thermo-Fisher | 4387405 | Reverse transcription kit |
| iTaq Universal SYBR Green Supermix | BioRad | 1725124 | qPCR reagent |
| Applied 7500 Fast | Thermo-Fisher | 4351107 | qPCR apparatus |
| Illumina TruSeq Stranded mRNA Sample Preparation kit | Illumina | 20020594 | DNA library construction kit |
| Illumina NextSeq 500 | Illumina | SY-415-1002 | NGS system |
Referenzen
- Chu, C., Qu, K., Zhong, F. L., Artandi, S. E., Chang, H. Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. , 667-678 (2011).
- Chu, C., Quinn, J., Chang, H. Y. Chromatin isolation by RNA purification (ChIRP). J Vis Exp. , e3912 (2012).
- Simon, M. D., Wang, C. I., Kharchenko, P. V., West, J. A., Chapman, B. A., Alekseyenko, A. A., Borowsky, M. L., Kuroda, M. I., Kingston, R. E. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A. , 20497-20502 (2011).
- Simon, M. D. Capture hybridization analysis of RNA targets (CHART). Curr Protoc Mol Biol. , (2013).
- Reuter, J. S., Mathews, D. H. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, 129 (2010).
- Sun, X., Haider Ali, M. S. S., Moran, M. The role of interactions of long non-coding RNAs and heterogeneous nuclear ribonucleoproteins in regulating cellular functions. Biochem J. , 2925-2935 (2017).
- Gruber, A. R., Lorenz, R., Bernhart, S. H., Neuböck, R., Hofacker, I. L. The Vienna RNA websuite. Nucleic Acids Res. , W70-W74 (2008).
- Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., König, J., Hortobágyi, T., Nishimura, A. L., Zupunski, V., Patani, R., Chandran, S., Rot, G., Zupan, B., Shaw, C. E., Ule, J. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. , 452-458 (2011).
- Riva, P., Ratti, A., Venturin, M. The long non-coding RNAs in neurodegenerative diseases: novel mechanisms of pathogenesis. Curr Alzheimer Res. (27338628), (2016).
- Barry, G., Briggs, J. A., Hwang, D. W., Nayler, S. P., Fortuna, P. R., Jonkhout, N., Dachet, F., Maag, J. L., Mestdagh, P., Singh, E. M., Avesson, L., Kaczorowski, D. C., Ozturk, E., Jones, N. C., Vetter, I., Arriola-Martinez, L., Hu, J., Franco, G. R., Warn, V. M., Gong, A., Dinger, M. E., Rigo, F., Lipovich, L., Morris, M. J., O'Brien, T. J., Lee, D. S., Loeb, J. A., Blackshaw, S., Mattick, J. S., Wolvetang, E. J. The long non-coding RNA NEAT1 is responsive to neuronal activity and is associated with hyperexcitability states. Sci Rep. , 40127 (2017).
- Adriaens, C., Standaert, L., Barra, J., Latil, M., Verfaillie, A., Kalev, P., Boeckx, B., Wijnhoven, P. W., Radaelli, E., Vermi, W., Leucci, E., Lapouge, G., Beck, B., van den Oord, J., Nakagawa, S., Hirose, T., Sablina, A. A., Lambrechts, D., Aerts, S., Blanpain, C., Marine, J. C. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat Med. , (2016).
- Fang, J., Qiao, F., Tu, J., Xu, J., Ding, F., Liu, Y., Akuo, B. A., Hu, J., Shao, S. High expression of long non-coding RNA NEAT1 indicates poor prognosis of human cancer. Oncotarget. , (2017).
- Torres, M., Becquet, D., Blanchard, M. P., Guillen, S., Boyer, B., Moreno, M., Franc, J. L., François-Bellan, A. M. Circadian RNA expression elicited by 3'-UTR IRAlu-paraspeckle associated elements. Elife. , (2016).
- Chen, L. L., DeCerbo, J. N., Carmichael, G. G. Alu element-mediated gene silencing. EMBO J. , 1694-1705 (2008).
- Tripathi, V., Ellis, J. D., Shen, Z., Song, D. Y., Pan, Q., Watt, A. T., Freier, S. M., Bennett, C. F., Sharma, A., Bubulya, P. A., Blencowe, B. J., Prasanth, S. G., Prasanth, K. V. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. , 925-938 (2010).
- Engreitz, J. M., Sirokman, K., McDonel, P., Shishkin, A. A., Surka, C., Russell, P., Grossman, S. R., Chow, A. Y., Guttman, M., Lander, E. S. RNA-RNA Interactions Enable Specific Targeting of Noncoding RNAs to Nascent Pre-mRNAs and Chromatin Sites. Cell. , 188-199 (2014).
- Nakagawa, S., Ip, J. Y., Shioi, G., Tripathi, V., Zong, X., Hirose, T., Prasanth, K. V. Malat1 is not an essential component of nuclear speckles in mice. RNA. , (2012).
- Zhang, B., Arun, G., Mao, Y. S., Lazar, Z., Hung, G., Bhattacharjee, G., Xiao, X., Booth, C. J., Wu, J., Zhang, C., Spector, D. L. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. , 111-123 (2012).
- Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., Pimentel, H., Salzberg, S. L., Rinn, J. L., Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. , 562-578 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten