
Method Article
RNA Pull-down Procedure to Identify RNA Targets of a Long Non-coding RNA
In This Article
Summary
This RNA pull-down method allows identifying the RNA targets of a long non-coding RNA (lncRNA). Based on the hybridization of home-made, designed anti-sense DNA oligonucleotide probes specific to this lncRNA in an appropriately fixed tissue or cell line, it efficiently allows the capture of all RNA targets of the lncRNA.
Abstract
Long non-coding RNA (lncRNA), which are sequences of more than 200 nucleotides without a defined reading frame, belong to the regulatory non-coding RNA's family. Although their biological functions remain largely unknown, the number of these lncRNAs has steadily increased and it is now estimated that humans may have more than 10,000 such transcripts. Some of these are known to be involved in important regulatory pathways of gene expression which take place at the transcriptional level, but also at different steps of RNA co- and post-transcriptional maturation. In the latter cases, RNAs that are targeted by the lncRNA have to be identified. That's the reason why it is useful to develop a method enabling the identification of RNAs associated directly or indirectly with a lncRNA of interest.
This protocol, which was inspired by previously published protocols allowing the isolation of a lncRNA together with its associated chromatin sequences, was adapted to permit the isolation of associated RNAs. We determined that two steps are critical for the efficiency of this protocol. The first is the design of specific anti-sense DNA oligonucleotide probes able to hybridize to the lncRNA of interest. To this end, the lncRNA secondary structure was predicted by bioinformatics and anti-sense oligonucleotide probes were designed with a strong affinity for regions that display a low probability of internal base pairing. The second crucial step of the procedure relies on the fixative conditions of the tissue or cultured cells that have to preserve the network between all molecular partners. Coupled with high throughput RNA sequencing, this RNA pull-down protocol can provide the whole RNA interactome of a lncRNA of interest.
Introduction
The overall goal of the method described here is to identify RNA molecular partners of a long noncoding RNA (lncRNA). LncRNA correspond to sequences of more than 200 nucleotides without a defined reading frame. Some of them have been shown to be involved in gene expression regulation, not only at the transcriptional level, but also at different steps of RNA co- and post-transcriptional maturation. In the latter cases, molecular partners of the lncRNA are RNAs that are to be identified. A method enabling the identification of RNAs associated directly or indirectly with a lncRNA of interest would then be essential to develop.
Previously published methods, such as Chromatin Isolation by RNA Purification (ChIRP)1,2 and Capture Hybridization Analysis of RNA Targets (CHART)3,4, allow high-throughput discovery of RNA-bound proteins and genomic binding sites of a specific lncRNA. In these two methods, the lncRNA of interest was first hybridized to biotinylated complementary oligonucleotides, and the complex was then isolated using streptavidin beads. The main difference between these two techniques is related to the design of probes that target lncRNAs. ChIRP, the strategy inspired by RNA FISH, consisted of designing a pool of short complementary DNA oligonucleotide probes to tile the entire length of the lncRNA. By contrast, in CHART, the authors adapted an RNase H mapping assay on lncRNAs to probe sites available to hybridization.
The procedure proposed here to design the anti-sense DNA biotinylated oligonucleotide probes uses bioinformatics modeling of lncRNA secondary structure5 to select probes with a strong affinity for regions that display a low probability of internal base pairing. This procedure has the advantage of being less expensive than those based on pools of tiling oligonucleotide probes2 and less time consuming than those based on RNAse-H sensitivity4.
As there is a growing body of evidence for post-transcriptional gene regulation by lncRNAs6, it is very useful to develop an approach enabling the capture of RNAs that are targets of a lncRNA. Furthermore, to be usable for most applications, the approach was optimized both in cultured cells and tissue extracts.
Protocol
All procedures were performed in strict accordance with European Economic Community for the care and use of laboratory animals (86/609/EEC and 2010/63/UE) and under a license granted to D. Becquet (Préfecture des Bouches-du-Rhône, Authorization no. 13-002).
1. Probe Design
- Using the primary sequence of the lncRNA of interest, generate its secondary structure on the "RNAstructure Webserver"5.
NOTE: On this site, different algorithms can be used. The three prediction tools that give the best results for the probe designs are: "Fold" (lowest free energy structure), "MaxExpect" (highly probable base pairs), and "Probnot" (probable base pairs including pseudoknots). These three analyses can be performed and compared. Other web servers, such as the Vienna RNA websuite7, can also be used.- Select regions that display a low probability of internal base pairing and design anti-sense oligonucleotide probes of 25 bases with a strong affinity for these regions.
NOTE: The GC contents of these probes should be comprised between 40 and 60%. Using an alignment search tool (Blast), make sure that the selected anti-sense oligonucleotide probes do not recognize nucleotide sequences in other RNA expressed in the chosen cell system.
- Select regions that display a low probability of internal base pairing and design anti-sense oligonucleotide probes of 25 bases with a strong affinity for these regions.
- Design also a non-specific DNA oligonucleotide probe of 25 bases which displays neither affinity for the lncRNA of interest nor for other RNA sequences in the genome of interest.
- Order the probes with biotin at the 3'-end.
NOTE: In order to reduce the steric hindrance, the distance between oligonucleotide and Biotin has to be increased with a triethyleneglycerol spacer. For an optimal result, and to be able to assess the specificity of results of the pull-down, it is recommended to design 3 different anti-sense oligonucleotide probes and then to experimentally compare their efficiency.
2. Cross-linking
- Cultured cell cross-linking
- Culture the GH4C1 sommatolactotroph pituitary cells in Ham's F10 medium supplemented with 15% horse serum and 2% fetal calf serum. Grow the cells until confluence in 78.5 cm2 culture plates. This corresponds approximately to 1 x 107 cells.
- Remove the cell culture medium from a confluent GH4C1 78.5 cm2 culture plate, then rinse with 1x the medium volume of phosphate buffered saline (PBS)
- Fix the cells with a 1% paraformaldehyde solution in PBS (10 mL for a 78.5 cm2 dishes); this solution must be freshly prepared from a 4% paraformaldehyde stock solution. Cross-link under agitation for 10 min at room temperature (RT).
CAUTION: Paraformaldehyde (PFA) is toxic, and must be handled with caution. - Quench the paraformaldehyde action by adding 1/10 volume of glycine 1.25 M (1 mL per 10 mL of paraformaldehyde solution); agitate 5 min at RT.
- Discard the media by aspiration and rinse two times (5 min) with 1X the medium volume of PBS.
- Add a volume of PBS corresponding to 1/10th of the volume of media, collect cells with a cell scraper, and then transfer to a centrifuge tube.
- Spin at 510 g at 4 °C for 5 min.
- Remove as much supernatant as possible.
- Store pellets indefinitely as needed at -80 °C.
- Tissue Cross-linking
- Put 5 mg of freshly obtained mouse pituitary gland tissue in a solution of 1% paraformaldehyde diluted in PBS (approximately 10x the volume of the tissue), agitate for 10 min at RT.
- Quench the paraformaldehyde action by adding a 1.25M glycine solution (1 mL per 10 mL of paraformaldehyde solution) and agitate 5 min at RT.
- Discard the media by aspiration and rinse two times with PBS (approximately 10x the volume of the tissue). Remove as much supernatant as possible.
- Store the cross-linked tissue indefinitely at -80 °C.
3. Cell or Tissue Lysis
- Prepare the Lysis Buffer (50 mM Tris-HCl pH 7.0, 10 mM EDTA, 1% SDS supplemented with 200 U/mL of a RNAse inhibitor solution, and a cocktail of proteases inhibitor 5 µL/mL).
- To obtain lysed samples without prior thawing, re-suspend the cell pellets or cross-linked tissues with this buffer (approximately 1 mL per 100 mg of cell pellet or tissue). A cell pellet obtained from 1 x 107 cells give rise to a lysed sample containing roughly 20 mg of protein.
NOTE: Depending on the tissue used, a step of mechanical disruption should be added. In this case, it is important to avoid heating of the samples during this additional step.
4. Sonication
- Optimization of the sonication conditions
- Program the sonicator with 2 to 5 series of 30 s ON and 30 s OFF.
- Perform tests to optimize sonication conditions on diluted lysed samples (dilution factor ½ or ¼ corresponding to approximately 10 or 5 mg of protein). Place diluted lysed samples in the 4 °C water bath and start the sonication series.
- Purify RNAs either with a RNA purification kit or with an RNA isolation reagent (e.g., Trizol).
- Load the totality of purified RNA on a 1% agarose gel electrophoresis in TBE buffer, check the length of RNA fragments. This length should range between 200 and 800 bp.
NOTE: Depending on the RNA fragment size, the anti-sense oligonucleotide probes' efficiency can vary. It is then recommended to check the efficiency of the probes under different conditions of sonication.
- Sonication of the lysed samples
- Place lysed samples corresponding to 20 mg of proteins (obtained after step 3.2) in the 4 °C water bath and start the sonication series as optimized in step 4.1.
- Immediately after sonication, centrifuge for 5 min at 12,000 g at 4 °C. Transfer supernatants in new centrifuge tubes.
NOTE: To ensure homogeneity, replicate supernatants can be pooled and redistributed at this step.
5. RNA Pull-down
- Day 1 – hybridization step
- Add 2 volumes of hybridization buffer (50 mM Tris-HCl pH 7.0, 750 mM NaCl, 1 mM EDTA, 1% SDS, 15% Formamide added extemporaneously) to supernatants collected after the sonication step. Vortex.
- Transfer 20 µL of each sample in a centrifuge tube (input samples) and store at -20 °C.
- Add 100 pmol of biotinylated oligonucleotide probes (specific or non-specific; see Table 1) to each sample. Incubate 4 to 6 h under moderate agitation on a tube rotator at RT.
- Add 50 µL of magnetic streptavidin beads supplemented with 200 U/mL of a RNAse inhibitor solution and a cocktail of proteases inhibitor 5 µL/mL.
- Incubate overnight under moderate agitation on the tube rotator at RT.
- Day 2 – RNA isolation step
- Use magnetic support to separate beads from cell lysate, discard the supernatant, and wash the beads with 900 µL of wash buffer (SDS 0.5%, SSC 2x). Repeat 5 times interspersed with 5 min agitation on the rotator at RT.
- After the last wash, decant one last time and add 95 µL of Proteinease K buffer (10 mM Tris-HCl pH 7.0, 100 mM NaCl, 1 mM EDTA, 0.5% SDS) and 5 µL of proteinase-K (20 mg/mL) to the samples.
- On ice, thaw the input samples (20 μL) and add 75 µL of Proteinease K buffer and 5 µL of proteinase-K (20 mg/mL).
- Incubate all samples with proteinase K for 45 min at 50 °C then 10 min at 95 °C.
- Chill the sample on ice for 3 min before separating the beads from RNAs with the magnetic support. Keep the supernatant and discard the beads.
- Purify RNAs with a RNA purification kit, which should include a DNA digestion step. Store RNAs at -80 °C.
- Perform reverse transcription qPCR (RT-qPCR) using a RT kit followed by qPCR using specific primers (Table 1).
- Construct two DNA libraries corresponding to the two RNA pools obtained with each of the specific Neat1 probes (Table 1). Perform sequencing on a next-generation sequencing system.
Results
Several recent studies have shown that lncRNAs play an essential role in almost every important biological process and that this role is achieved through the control of gene expression occurring both at the transcriptional and the post-transcriptional levels showing in this latter case that RNAs may be the target of lncRNAs6.
The lncRNA Nuclear enriched abundant transcript 1 (Neat1) is implicated in different neuropathologies as frontotemporal dementia, amyotrophic lateral sclerosis, or epilepsy8,9,10, and is also misregulated in different cancers11,12.
This lncRNA is also known to be the structural component of specific nuclear bodies, the paraspeckles, and to be involved in post-transcriptional circadian regulation of gene expression13. Paraspeckles that are found in every cell nucleus and are formed around not only Neat1, which is necessary for their formation, but also around several RNA binding proteins (RBP)14, are indeed known to be able to retain RNA targets within the nucleus15. The formation of paraspeckles is achieved through the association of the different components. This formation was shown to display a circadian rhythmic pattern driving a rhythmic nuclear retention of RNA targets13. The nuclear retention of RNA targets by paraspeckles may occur through binding to RBP or directly through RNA/RNA association, but the extent of RNAs targeted by paraspeckles had to be determined. To identify the RNA targeted directly or indirectly by Neat1, an RNA pull-down protocol was designed that allows the isolation and the identification of all Neat1 RNA targets in cultured cells as well as in tissue samples (see Figure 1 for a graphical presentation of the technique).
The protocol was also successfully applied to the identification of RNA targets of an another lncRNA Metastasis Associated Lung Adenocarcinoma Transcript 1 (Malat1). Malat1 is a highly conserved and expressed lncRNA found in nuclear speckles together with several RNA splicing factors. Malat1 is known to be involved in the regulation of the splicing of several nascent pre-mRNA16,17.
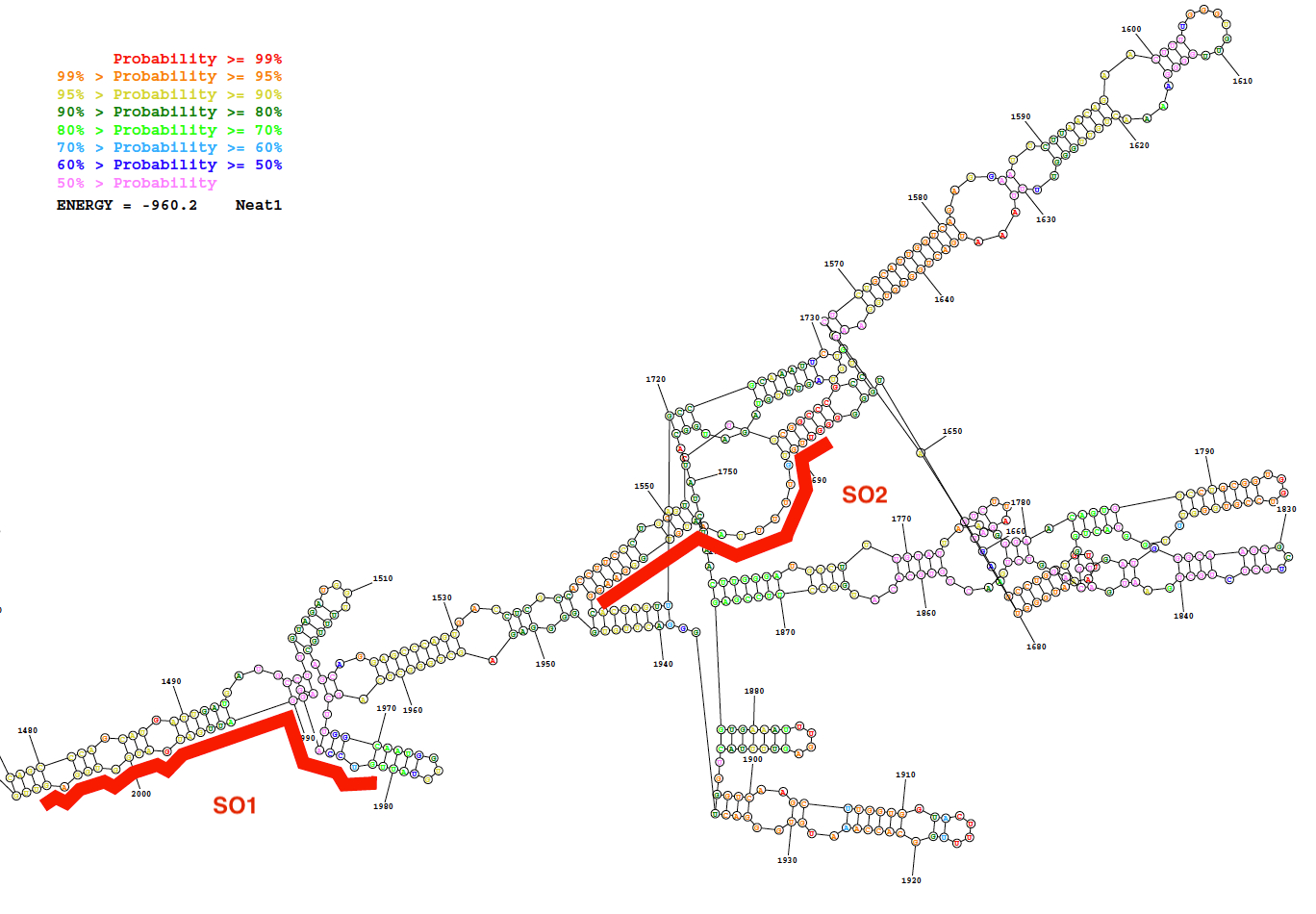
Specific (SO) and non-specific oligonucleotide (NSO) probes were generated using the probe design strategy described here. This strategy relies on the selection of regions that display a low probability of internal base pairing as predicted by the secondary structure of the lncRNA and on the design of specific probes with a strong affinity for these regions. As a representative result of these bioinformatics predictions, a picture of the predicted secondary structure of a sequence of Neat1 (nucleotides 1,480 to 2,000) together with the position of two designed SO probes are given in Figure 2.
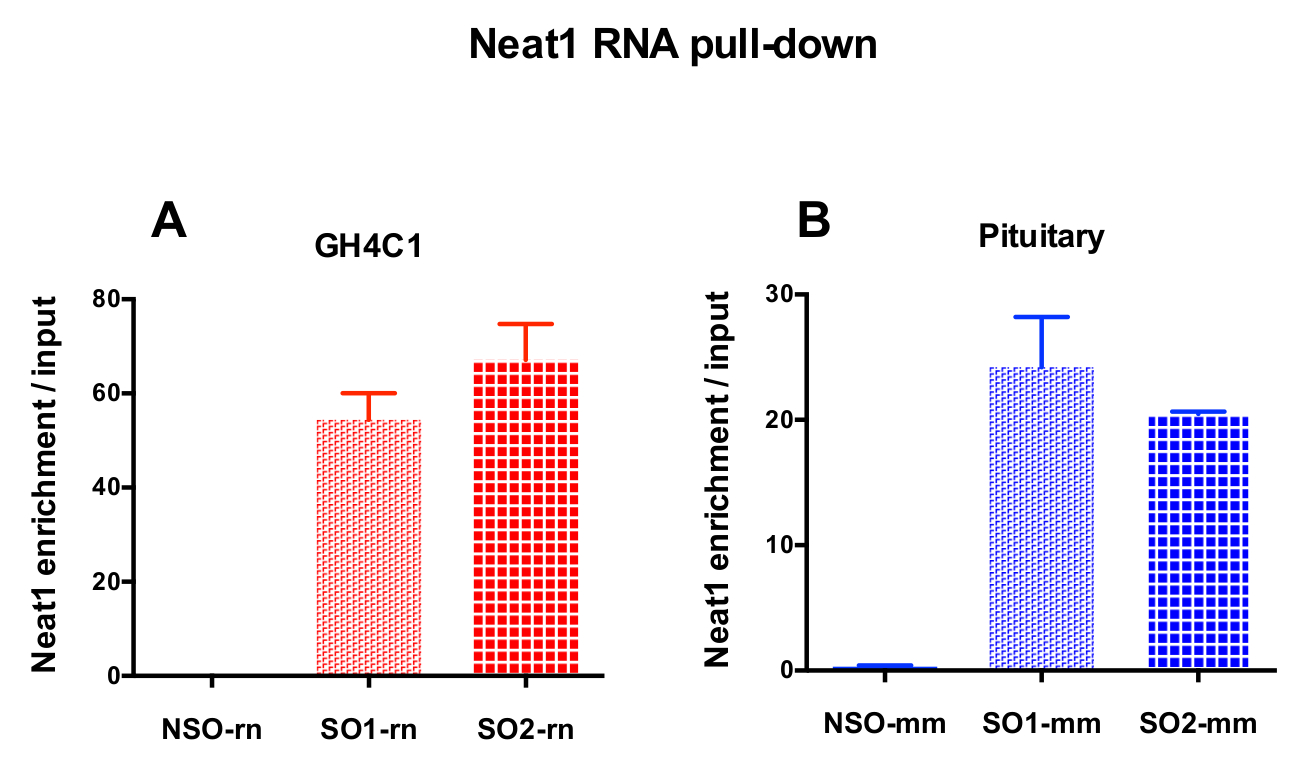
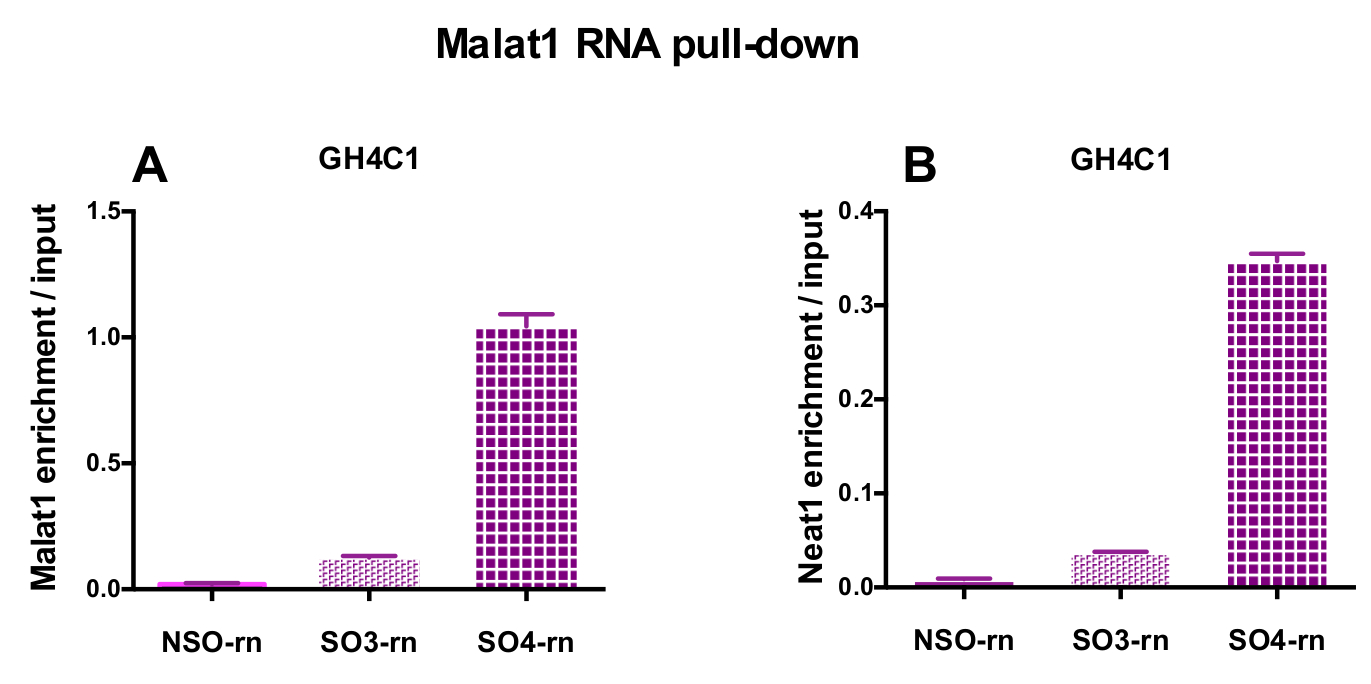
The designed probes were directed to rat Neat1 or Malat1 for GH4C1 cultured cells and to mouse Neat1 for pituitary tissue extracts (Table 1). The relative enrichment in Neat1 or Malat1 was calculated for non-specific and specific probes relative to the input samples. Figure 3 shows the efficiency of the specific probes to pull-down Neat1 in the rat GH4C1 pituitary cell line (Figure 3A) and in mouse pituitary tissue extracts (Figure 3B). When using the probe design protocol to generate specific oligonucleotide (SO) probes directed to Malat1, one efficient probe was obtained while another was not efficient enough and was discarded (Figure 4A).
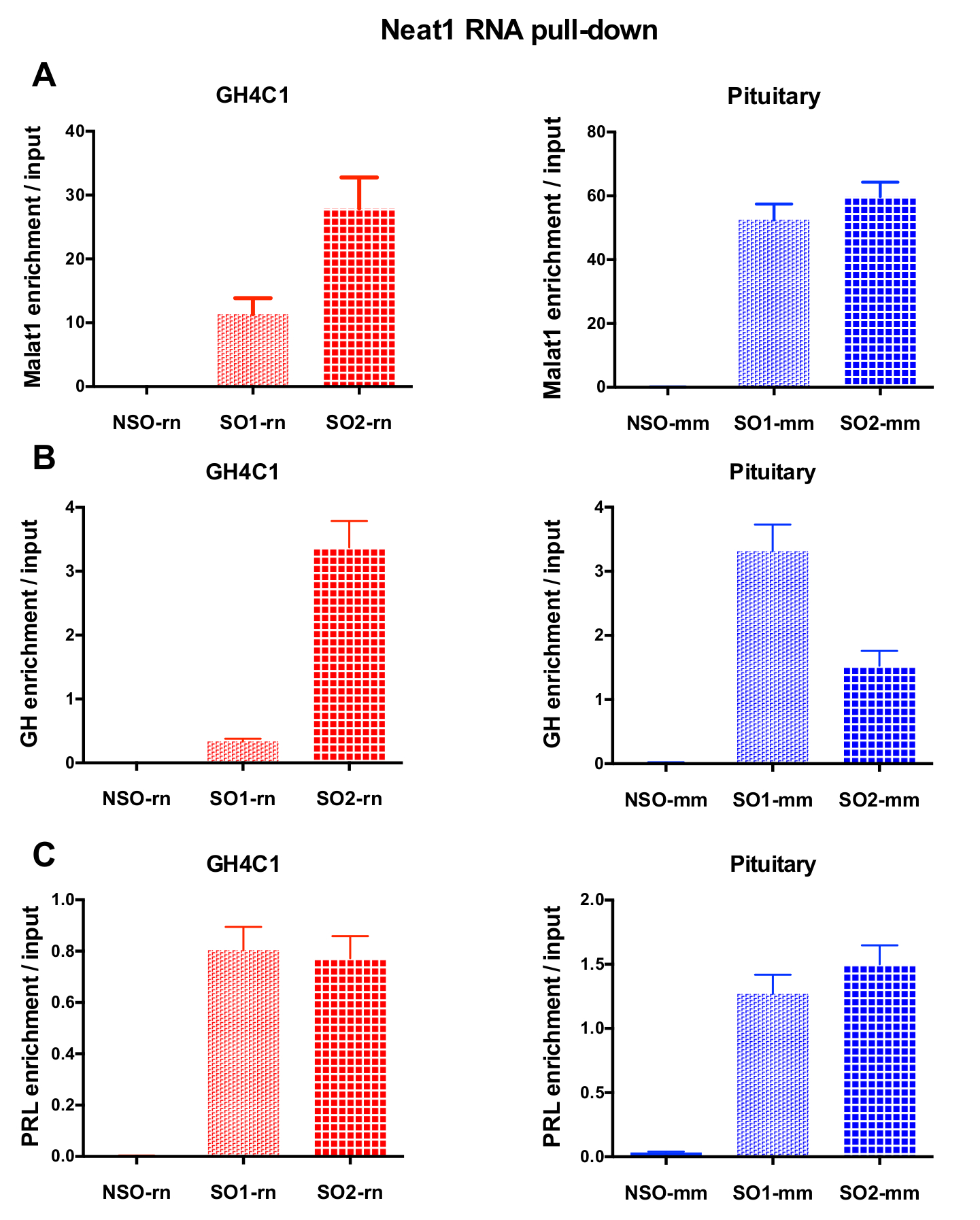
After an RNA pull-down procedure followed by RT-qPCR experiments, some RNAs assessed with specific primers (Table 1) were shown to be associated with Neat1 or Malat1 in GH4C1 extracts. The RNAs associated with Neat1 in GH4C1 cell extracts were also shown to be associated with Neat1 in pituitary tissue extracts. Indeed, after Neat1 RNA pull-down, Malat1 was found to be targeted by Neat1 both in the GH4C1 cell line and in pituitary mouse tissue extracts (Figure 5A). Reciprocally, Neat1 was significantly enriched after Malat1 RNA pull-down performed with a specific probe in GH4C1 cells (Figure 4B). By highlighting the close relationship between the two lncRNAs, these results are consistent with the potential co-regulatory role of Neat1 and Malat1 suggested by Malat1 knockout mice that display variations in Neat1 RNA expression18,19. The transcripts of two main pituitary hormones, growth hormone (Gh) (Figure 5B) and prolactin (Prl) (Figure 5C) were significantly enriched following Neat1 RNA pull-down with specific probes in both GH4C1 cells and pituitary extracts, suggesting a possible regulation of the two hormones by Neat1. When comparing the two specific probes used, it appeared that their efficiency could vary depending on the RNA target considered (Figure 5B and Figure 5C). These results highlight the necessity of designing several specific probes in order to select those displaying not only the best efficiency in the enrichment of the pull-down lncRNA, but also the best efficiency in the enrichment of its RNA targets.
The RNA pull-down method can also be followed by RNA high-throughput sequencing to obtain the comprehensive list of RNA targets of a lncRNA of interest13. An RNA-seq analysis on GH4C1 pituitary cells after Neat1 RNA pull-down using the two specific probes described above was performed. It should be noted that a negative control using a NSO could also be subjected to RNA-seq analysis, if the level of RNA recovered after the RNA pull-down with NSO is sufficient to allow the construction of libraries. This was not the case in previous experience13. Libraries that were generated after use of specific probes were analyzed using Tophat/cufflinks pipeline20 and only transcripts with values of fragment per kilobase per million of mapped reads (FPKM) higher than 1 were taken into account. The lists obtained with the two specific probes directed to Neat1 (Table 1) were crossed to assess the specificity of the results. 4,268 genes were found associated with paraspeckles, which represented 28% of expressed transcripts in GH4C1 cells13. Consistent with results obtained using qPCR analysis (Figure 5A-C), the transcripts of Gh, Prl, and Malat1 were found to be associated with Neat1. The RNA pull-down method has therefore been proved to be an efficient tool to explore the interaction between lncRNAs and their RNA targets.

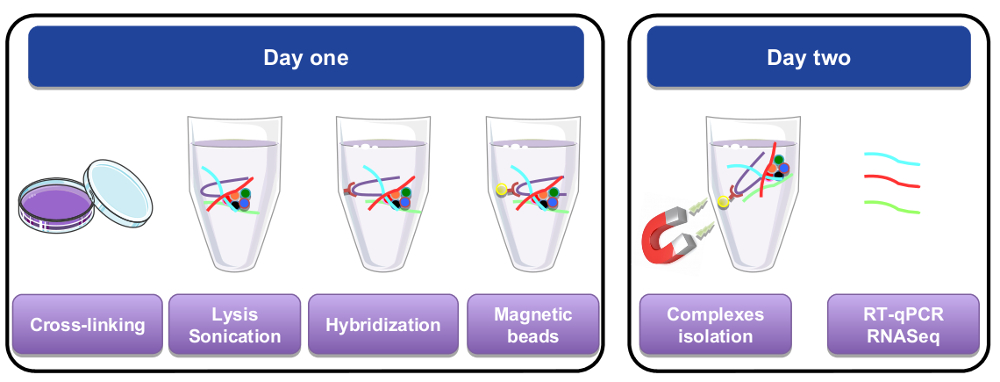
Figure 1: Graphical representation of the RNA pull down procedure. On the first day, cells or tissues were cross-linked with paraformaldehyde, lysed, and sonicated before the hybridization step that was performed by adding biotinylated specific probes. Magnetic streptavidin beads were then added to separate specific material from the rest of the cell lysate. On the second day, beads were isolated by a magnet and washed several times. A de-crosslinking step allowed recovery of RNAs that were purified and used for RT-qPCR or RNA-seq analysis. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Secondary structure of a Neat1 sequence (nucleotide 1,480 to 2,000) as predicted by bioinformatics resource (the RNAstructure webserver; lowest free energy structure). The structure is colored according to degree of probability of base pairing. The two oligonucleotides probes (SO1 and SO2) in red are positioned along the Neat1 RNA structure. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: qPCR validation of Neat1 enrichment versus input. qPCR validation of Neat1 enrichment versus input after Neat1 RNA pull down by two different specific probes (SO1-rn and SO2-rn for GH4C1 cells and SO1-mm and SO2-mm for pituitary tissue) as compared to a non-specific one (NSO-rn for GH4C1 cells and NSO-mm for pituitary tissue) in GH4C1 rat cells (A) and in mouse pituitary tissue extracts (B). Results are mean ± SEM obtained in 3 to 10 experiments. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: qPCR validation of Malat1 and Neat1 enrichment versus input after Malat1 RNA pull down. qPCR validation of Malat1 (A) and Neat1 (B) enrichment versus input after Malat1 RNA pull down by two different specific probes (SO3-rn and SO4-rn) as compared to a non-specific one (NSO-rn) in GH4C1 rat cells. Results are mean ± SEM obtained in 3 experiments. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: qPCR validation of Malat1, Gh, Prl enrichment versus input after Neat1 RNA pull down. qPCR validation of Malat1 (A), Gh (B), Prl (C) enrichment versus input after Neat1 RNA pull down using different specific probes as compared to a non-specific one in GH4C1 rat cells and mouse pituitary tissue extracts. Results are mean ± SEM obtained in 3 to 8 experiments. Please click here to view a larger version of this figure.
{kind=link}
| PROBE NAMES | Sequences | |
| NSO-Rn | TAAAATACCATTTGATGTTTGAAATTAT | |
| SO1-Rn | CTCCACCATCATCAATCCTCTGGAC | |
| SO2-Rn | GCCTTCCCACATTTAAAAACACAAC | |
| SO3-Rn | AACTCGTGGCTCAAGTGAGGTGACA | |
| SO4-Rn | AAGACTCTCAGGCTCCTGCTCATTC | |
| NSO-mm | GTTTGTGGTTTAACAGTGGGAAGGC | |
| SO1-mm | GCCTTCCCACTGTTAAACCACAAAC | |
| SO2-mm | CTCACCCGCACCCCGACTCCTTCAA | |
| qPCR PRIMERS : | ||
| Rattus norvegicus | ||
| Neat1 | AAGGCACGAGTTAGCCGCAAAT | |
| TGTGCACAGTCAGACCTGTCATTC | ||
| Malat1 | GAAGGCGTGTACTGCTATGCTGTT | |
| TCTCCTGAGGTGACTGTGAACCAA | ||
| Gh1 | CCGCGTCTATGAGAAACTGAAGGA | |
| GGTTTGCTTGAGGATCTGCCCAAT | ||
| Prl | TGAACCTGATCCTCAGTTTGGT | |
| AGCTGCTTGTTTTGTTCCTCAA | ||
| Mus musculus | ||
| Neat1 | TGGGCCCTGGGTCATCTTACTAGATA | |
| CACAGCTGTTCCAATGAGCGATCT | ||
| Gh1 | CTCGGACCGTGTCTATGAGAAACTGA | |
| TTTGCTTGAGGATCTGCCCAACAC | ||
| Prl | TGAACCTGATCCTCAGTTTGGT | |
| AGCTGCTTGTTTTGTTCCTCAA | ||
Table 1: Sequences of DNA oligonucleotide probes and qPCR primers
Discussion
Long noncoding RNAs (lncRNAs) by their number and diversity represent a large field of research and most of their roles are still to be discovered. Many of these lncRNAs have a nuclear localization and among them, some are implicated in regulatory pathways of gene expression through transcriptional or posttranscriptional mechanisms. One of the current challenges in this field is to understand the relevance of those lncRNAs in post-transcriptional processing of RNAs. For this purpose, RNAs targeted by lncRNAs have to be identified. Inspired by previous studies focused on association of lncRNAs with chromatin, we developed a procedure that allows the identification of RNAs associated with a lncRNA. The success of this protocol, named RNA pull-down, is mainly dependent on two crucial steps, namely the design of anti-sense DNA oligonucleotide probes that have to specifically and exclusively hybridize with the lncRNA of interest, and the conditions of tissue or cell fixation that have to preserve the integrity of the network between all molecular partners.
Previously published protocols provided procedures to isolate a lncRNA together with its associated chromatin sequences (ChIRP1,2, CHART3,4). In those protocols, different strategies were employed to design the anti-sense DNA biotinylated oligonucleotide probes. In the ChIRP procedure, the authors used a pool of DNA biotinylated oligonucleotide probes encompassing the entire length of the lncRNA of interest after exclusion of all redundant and non-specific probes1,2. In the CHART protocol, the authors identified the regions of the lncRNA that are more accessible for hybridization and designed capture oligonucleotides that target these regions. These regions were selected on the basis of their RNase-H sensitivity. Indeed, using the property of RNAse-H to hydrolyze RNAs at sites of RNA-DNA binding, the oligonucleotides that hybridize to accessible sites in the lncRNA produce RNA-DNA hybrids and lead to enzymatic cleavage of the lncRNA. The authors selected three of these candidate capture oligonucleotides and used them in a cocktail3,4.
The procedure we used to design the anti-sense DNA biotinylated oligonucleotide probes was close to that used in the CHART protocol, but the hybridization-available regions of the desired lncRNA were not selected on the basis of their RNAse-H sensitivity, but according to their low probability of internal base pairing as determined by bioinformatics modeling of lncRNA secondary structure. It must be noticed that different secondary structures will be predicted using different algorithms, and probes to be selected should be those that hybridize to available sequences of the lncRNA in the largest number of secondary structures predicted. The same results were obtained using either a cocktail of three designed, specific probes or a single probe individually. This prompted the use of two separate, specific probes and the consideration of positive results as those that are common to these two probes. Finally, it is therefore recommended at the beginning of the development of the method, for an optimal result and to be able to assess the specificity of results of the pull-down, to design 3 different anti-sense oligonucleotide probes and then to compare experimentally their efficiency, especially since probe efficiency may be altered by cell lysate preparation. Nevertheless, the procedure of probe design based on bioinformatics modeling of lncRNA secondary structure we used remained less expensive than that based on pools of tiling oligonucleotide probes2, and this was less time consuming than the method based on RNAse-H sensitivity4.
A negative control should also be performed using as negative capture oligonucleotide either the sense DNA biotinylated oligonucleotide probes or scrambled oligonucleotide probes, or oligonucleotides directed against an unrelated RNA. Because of the existence of natural antisense transcripts lncRNAs, use of sense oligonucleotide probes may sometimes be inadequate. Irrespective of the oligonucleotide probe selected for the negative control, it is necessary to check by blast that it does not hybridize with a known RNA and to keep in mind that this oligonucleotide can hybridize to a still un-annotated lncRNA.
The cell lysates used in these RNA pull-down experiments were obtained from 106 to 107 cells when working with cultured cells, and from 1 to 10 mg when working with tissue. The preparation of cell lysates needs to be adapted according to the tissue or cell type used with two main steps that have to be optimized: namely, the cross-linking step that allows formation of covalent bonds between the lncRNA and its molecular partners, and the sonication step that reduces the viscosity by shredding chromatin.
The aim of the cross-linking step is to ensure that all RNA targets remain closed to the lncRNA by inducing the formation of a network between all molecular partners. A paraformaldehyde treatment step that will form covalent bonds between the lncRNA and its partners allows the network to be reticulated. In the CHART protocol, it was suggested if working with nuclear lncRNA, to perform a first treatment with paraformaldehyde on the whole cell lysate and a second treatment on the isolated nucleic fraction3,4. We observed that this supplementary step decreased the efficiency of the probes, probably by reducing the lncRNA accessibility in the cells. Hence, the degree of reticulation by paraformaldehyde has to be adapted taking into account the cell or tissue type used, the localization of the lncRNA of interest, and the efficiency of the designed probes.
While lysing the cells, the chromatin is released in the lysate and increases its viscosity; it is then necessary to shred the chromatin by sonication to increase the fluidity of the samples and hence facilitate the accessibility of the oligonucleotide probes to the lncRNA of interest. However, sonication will also shred the RNAs extracted with the lncRNA of interest. It is then important to minimize the sonication time in such a way that while it efficiently reduces the viscosity of the lysate, it also allows the obtainment of RNA fragments with a length comprised between 200-800 bp. Note that the sonication time will be highly dependent upon both the quantity and the type of tissue or cultured cells used.
In conclusion, the procedure described here enables in 2-3 days the capture of the RNA targets of a desired lncRNA. Coupled with RT-qPCR, these methods will allow looking for a specific association and regulation of an mRNA by the desired lncRNA as a candidate approach. For a genome-wide approach, RNA pull-down experiments can be analyzed by high-throughput RNA-sequencing allowing the retrieval of all RNAs associated with the desired lncRNA. Whatever the analytical strategy chosen, the RNA pull-down procedure should provide new significant knowledge on RNA regulation by lncRNAs.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by Aix-Marseille University and CNRS and funded by a grant from Pfizer Laboratories.
Materials
| Name | Company | Catalog Number | Comments |
| Bioruptor Plus | Diagenode | B01020001 | Sonicator |
| Dynabeads My One | Thermo-Fisher | 65001 | Magnetic streptavidin beads |
| Formamide | Thermo-Fisher | 15515-026 | |
| Gel electrophoresis apparatus | Advance | Mupid-One | Gel electrophoresis apparatus |
| Proteinase K | Sigma | P2308 | |
| RNA XS purification kit | Macherey-Nagel | 740902 | RNA purificationkit |
| RNAseOUT | Thermo-Fisher | 10777-019 | RNAse inhibitor |
| Trizol | Thermo-Fisher | 15596018 | RNA purification |
| Tube Rotator | Stuart | SB2 | Eppendorf tube rotator |
| RNA to DNA | Thermo-Fisher | 4387405 | Reverse transcription kit |
| iTaq Universal SYBR Green Supermix | BioRad | 1725124 | qPCR reagent |
| Applied 7500 Fast | Thermo-Fisher | 4351107 | qPCR apparatus |
| Illumina TruSeq Stranded mRNA Sample Preparation kit | Illumina | 20020594 | DNA library construction kit |
| Illumina NextSeq 500 | Illumina | SY-415-1002 | NGS system |
References
- Chu, C., Qu, K., Zhong, F. L., Artandi, S. E., Chang, H. Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. , 667-678 (2011).
- Chu, C., Quinn, J., Chang, H. Y. Chromatin isolation by RNA purification (ChIRP). J Vis Exp. , e3912 (2012).
- Simon, M. D., Wang, C. I., Kharchenko, P. V., West, J. A., Chapman, B. A., Alekseyenko, A. A., Borowsky, M. L., Kuroda, M. I., Kingston, R. E. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A. , 20497-20502 (2011).
- Simon, M. D. Capture hybridization analysis of RNA targets (CHART). Curr Protoc Mol Biol. , (2013).
- Reuter, J. S., Mathews, D. H. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 11, 129 (2010).
- Sun, X., Haider Ali, M. S. S., Moran, M. The role of interactions of long non-coding RNAs and heterogeneous nuclear ribonucleoproteins in regulating cellular functions. Biochem J. , 2925-2935 (2017).
- Gruber, A. R., Lorenz, R., Bernhart, S. H., Neuböck, R., Hofacker, I. L. The Vienna RNA websuite. Nucleic Acids Res. , W70-W74 (2008).
- Tollervey, J. R., Curk, T., Rogelj, B., Briese, M., Cereda, M., Kayikci, M., König, J., Hortobágyi, T., Nishimura, A. L., Zupunski, V., Patani, R., Chandran, S., Rot, G., Zupan, B., Shaw, C. E., Ule, J. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. , 452-458 (2011).
- Riva, P., Ratti, A., Venturin, M. The long non-coding RNAs in neurodegenerative diseases: novel mechanisms of pathogenesis. Curr Alzheimer Res. (27338628), (2016).
- Barry, G., Briggs, J. A., Hwang, D. W., Nayler, S. P., Fortuna, P. R., Jonkhout, N., Dachet, F., Maag, J. L., Mestdagh, P., Singh, E. M., Avesson, L., Kaczorowski, D. C., Ozturk, E., Jones, N. C., Vetter, I., Arriola-Martinez, L., Hu, J., Franco, G. R., Warn, V. M., Gong, A., Dinger, M. E., Rigo, F., Lipovich, L., Morris, M. J., O'Brien, T. J., Lee, D. S., Loeb, J. A., Blackshaw, S., Mattick, J. S., Wolvetang, E. J. The long non-coding RNA NEAT1 is responsive to neuronal activity and is associated with hyperexcitability states. Sci Rep. , 40127 (2017).
- Adriaens, C., Standaert, L., Barra, J., Latil, M., Verfaillie, A., Kalev, P., Boeckx, B., Wijnhoven, P. W., Radaelli, E., Vermi, W., Leucci, E., Lapouge, G., Beck, B., van den Oord, J., Nakagawa, S., Hirose, T., Sablina, A. A., Lambrechts, D., Aerts, S., Blanpain, C., Marine, J. C. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat Med. , (2016).
- Fang, J., Qiao, F., Tu, J., Xu, J., Ding, F., Liu, Y., Akuo, B. A., Hu, J., Shao, S. High expression of long non-coding RNA NEAT1 indicates poor prognosis of human cancer. Oncotarget. , (2017).
- Torres, M., Becquet, D., Blanchard, M. P., Guillen, S., Boyer, B., Moreno, M., Franc, J. L., François-Bellan, A. M. Circadian RNA expression elicited by 3'-UTR IRAlu-paraspeckle associated elements. Elife. , (2016).
- Chen, L. L., DeCerbo, J. N., Carmichael, G. G. Alu element-mediated gene silencing. EMBO J. , 1694-1705 (2008).
- Tripathi, V., Ellis, J. D., Shen, Z., Song, D. Y., Pan, Q., Watt, A. T., Freier, S. M., Bennett, C. F., Sharma, A., Bubulya, P. A., Blencowe, B. J., Prasanth, S. G., Prasanth, K. V. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. , 925-938 (2010).
- Engreitz, J. M., Sirokman, K., McDonel, P., Shishkin, A. A., Surka, C., Russell, P., Grossman, S. R., Chow, A. Y., Guttman, M., Lander, E. S. RNA-RNA Interactions Enable Specific Targeting of Noncoding RNAs to Nascent Pre-mRNAs and Chromatin Sites. Cell. , 188-199 (2014).
- Nakagawa, S., Ip, J. Y., Shioi, G., Tripathi, V., Zong, X., Hirose, T., Prasanth, K. V. Malat1 is not an essential component of nuclear speckles in mice. RNA. , (2012).
- Zhang, B., Arun, G., Mao, Y. S., Lazar, Z., Hung, G., Bhattacharjee, G., Xiao, X., Booth, C. J., Wu, J., Zhang, C., Spector, D. L. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. , 111-123 (2012).
- Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., Pimentel, H., Salzberg, S. L., Rinn, J. L., Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. , 562-578 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved