
Method Article
High-Density DNA und RNA Mikroarrays - Photolithographische Synthese, Hybridisierung und Vorbereitung von großen Nukleinsäurebibliotheken
In diesem Artikel
Zusammenfassung
In diesem Artikel stellen wir neue Entwicklungen in der Synthese und Anwendung von Nukleinsäure-Mikroarrays vor, die vor Ort hergestellt werden. Insbesondere zeigen wir, wie die Protokolle für die DNA-Synthese auf RNA erweitert werden können und wie Mikroarrays verwendet werden können, um wiederholbare Nukleinsäurebibliotheken zu erstellen.
Zusammenfassung
Die Photolithographie ist eine leistungsstarke Technik zur Synthese von DNA-Oligonukleotiden auf Glasdias, da sie die Effizienz von Phosphoramidit-Kopplungsreaktionen mit der Präzision und Dichte von UV-Licht kombiniert, das von mikrometergroßen Spiegeln reflektiert wird. Die Photolithographie liefert Mikroarrays, die in nur wenigen Stunden von Hunderttausenden bis zu mehreren Millionen verschiedenen DNA-Sequenzen, 100 nt oder länger, aufnehmen können. Mit diesem sehr großen Sequenzraum sind Mikroarrays ideale Plattformen, um die Mechanismen von Nukleinsäure-Ligand-Wechselwirkungen zu erforschen, die besonders bei RNA relevant sind. Kürzlich berichteten wir über die Herstellung eines neuen Satzes von RNA-Phosphoramiditen, die mit der In-situ-Photolithographie kompatibel sind und anschließend zum Anbau von RNA-Oligonukleotiden, Homopolymeren sowie gemischten Basissequenzen verwendet wurden. Hier veranschaulichen wir detailliert den Prozess der RNA-Mikroarray-Fertigung, vom experimentellen Design bis zum Instrumental-Setup, Array-Synthese, Deprotection und Final-Hybridisierungs-Assay anhand einer Vorlage 25mer-Sequenz, die alle vier Basen als Beispiel enthält. Parallel dazu gehen wir über hybridisierungsbasierte Experimente hinaus und nutzen die Mikroarray-Photolithographie als kostengünstiges Tor zu komplexen Nukleinsäurebibliotheken. Dazu werden DNA-Mikroarrays mit hoher Dichte auf einem basisempfindlichen Monomer hergestellt, das es ermöglicht, die DNA bequem zu spalten und nach Synthese und Deprotection zu bergen. Das Fertigungsprotokoll ist so optimiert, dass die Anzahl der synthetischen Fehler begrenzt wird, und zu diesem Zweck wird eine Schicht von Carotin-Lösung eingeführt, um UV-Photonen zu absorbieren, die sonst auf die Synthesesubstrate zurückfallen könnten. Schritt für Schritt beschreiben wir den gesamten Prozess der Bibliotheksvorbereitung, vom Design bis zur Spaltung und Quantifizierung.
Einleitung
Die praktische Anwendung von DNA-Mikroarrays wurde traditionell in der Untersuchung der Variationen der Genexpressionsniveaus zwischen zwei Zellpopulationen unter Verwendung komplementärer Stränge und Fluoreszenz als Nachweismethode1verwendet. Gelegentlich wagen sich DNA-Mikroarrays in Bindungsereignisse mit Nicht-Nukleinsäure-Liganden, wie Proteinen, mit einer Strategie der systematischen Sequenzpermutation, die einen umfassenden Überblick über die Bindungslandschaft bietet2,3, 4,5. Dieser Ansatz wandelt Mikroarrays effektiv von reinen Hybridisierungsflächen in Plattformen mit breiter Sequenzabdeckung um, was ein Vorteil für die Erforschung der reicheren und komplexeren Welt der RNA-Struktur und -Funktion wäre. Unterstützt durch die extrem effiziente Phosphoramidit-Kopplungsreaktion6können in situ synthetisierte DNA-Arrays nun auch als billige Quelle von DNA7angesehen werden, die angesichts der stetig steigenden Nachfrage nach Nukleinsäurematerial für genassembly8,9, DNA-basierte Nanostrukturen10, Informationsspeicherung oder Sequenzierung11,12. Ebenso dürften Sequenzierungstechnologien von der Entwicklung von Methoden profitieren, die sehr komplexe Mischungen von RNA-Oligonukleotiden ergeben13. In diesem Zusammenhang sind Array-Fertigungsprotokolle, die es ermöglichen, Oligonukleotide vor Ort und bei hoher Dichte zu synthetisiert, ideal positioniert, um den Bedürfnissen des schnell wachsenden Feldes der Nukleinsäurebiotechnologie gerecht zu werden. Bei einem so unterschiedlichen Feld wie der Biotechnologie kann der Zweck jeder Anwendung jedoch erfordern, dass DNA auf Mikroarray entweder bei hohem Durchsatz oder mit einer sehr geringen Menge an synthetischen Fehlern14,15oder beides hergestellt wird, was eine näherer Betrachtung der Syntheseprotokolle von DNA-Mikroarrays, die historisch gesehen in erster Linie für Hybridisierungs-Assays optimiert wurden. In der Zwischenzeit hat sich die In-situ-Synthese von RNA-Mikroarrays als ein schwieriges Unterfangen erwiesen, wobei die meisten Der Mitschwierigkeiten, die mit der Schutzgruppe für die 2'-OH-Funktion verbunden sind, in der Regel ein Silylmoiety in der Standard-Festphasensynthese, das mit reagenzienauf Fluor, Chemikalien, die mit Glas- oder Siliziumoberflächen nicht kompatibel sind. Diese Probleme und Herausforderungen in der DNA- und RNA-Mikroarray-Synthese waren in letzter Zeit Gegenstand einer vielzahln Arbeit, insbesondere mit dem Photolithographie-Ansatz16.
Die Photolithographie verwendet UV-Licht, um Oligonukleotide vor der Kopplung zu entsperren und erfordert Masken, um ein Muster der UV-Exposition zu konstruieren, wodurch das Wachstum von Oligonukleotiden räumlich organisiert und gesteuert wird. Physikalische Masken wurden durch computergesteuerte Mikrospiegel ersetzt, deren Neigung selektiv UV-Licht auf das Mikroarraysubstratreflektiert 17,18,19. Als UV-Quelle verwenden wir 365 nm Licht aus einer Hochleistungs-LED-Quelle20. Aktuelle photolithographische Setups sind mit Mikrospiegel-Arrays mit 1024 x 768 Spiegeln ausgestattet, was mehr als 780.000 individuell adressierbaren Spots ("Features") auf einer kleinen Fläche von nur 1,4cm2oder 1080p mit Arrays von 1920 x 1080 entspricht, oder >2 Millionen Spiegel. Jeder der Spiegel im Gerät hat somit eine direkte Kontrolle über die Sequenz, die auf dem entsprechenden Merkmal gewachsen ist. Mit Ausnahme von UV-Licht funktioniert die Photolithographie wie eine Festphasensynthesetechnik und übernimmt die zyklusbasierte Phosphoramiditchemie. Nur erfordert es eine völlig andere Schutzstrategie für die RNA-Synthese, um erfolgreich zu sein. Wir haben eine neue Serie von lichtempfindlichen RNA-Phosphoramiditen entwickelt, die Hydrazin-Labile-Schutzgruppentragen 21. Diese Monomere ermöglichen es, die RNA unter milden Bedingungen zu degeschützt zu machen, die die Integrität der Oberfläche nicht beeinträchtigen. Ein erster Deprotection-Schritt verwendet Triethylamin, um die Cyanoethylphosphodiester Schutzgruppen zu entfernen, während Hydrazin in einem zweiten, separaten Schritt verwendet wird, um diejenigen an den 2'-OH- und exozyklischen Aminfunktionen zu entfernen. Dabei können RNA-Oligonukleotide in der Länge von 30 nt und jeder Sequenz nun in situ auf mikroarrays22,23synthetisiert werden. Parallel dazu haben wir vor kurzem auch begonnen, uns mit den Fragen des Durchsatzes, der Qualität und der Geschwindigkeit in der DNA- und RNA-Photolithographie zu befassen. Wir haben die Kopplungseffizienzvon >99% für alle DNA- und RNA-Amiditen gemessen (Abbildung 1) und jeden einzelnen Schritt im Oligonukleotid-Dehnungszyklus untersucht, von der Oxidationszeit über die Wahl des Aktivators bis hin zur optimalen UV-Exposition24 , 25. Wir haben neue lichtempfindliche 5' Schutzgruppen eingeführt, die nur in Sekundenschnelle entfernt werden können, wodurch die Synthese von Hunderttausenden von 100 Mers in einen wenigen Stunden langen Prozess umgewandelt wird26. Wir haben auch den Durchsatz der Array-Fertigung verdoppelt, indem wir zwei Substrate gleichzeitig freisetzen27. Schließlich haben wir ein dT-Phosphoramidit eingeführt, das eine grundempfindliche Succinylgruppe enthält, um DNA- und RNA-Oligonukleotide zu spalten, zu sammeln und zu analysieren, was für die Bibliotheksvorbereitung von zentraler Bedeutung ist28.
Trotz des relativ banalen Aspekts der DNA- und RNA-Festphasensynthese, insbesondere für Nukleinsäurechemiker, bleibt die Mikroarray-Photolithographie ein nicht triviales Upgrade, das eine komplexe Einrichtung, eine sorgfältige Kontrolle und Überwachung des Prozesses erfordert, und separate Anweisungen für die postsynthetische Handhabung in Abhängigkeit von der Art des Oligonukleotids und der Art der Anwendung. In diesem Artikel möchten wir eine detaillierte Darstellung des gesamten Schrittweise-Verfahrens der In-situ-Synthese von DNA- und RNA-Mikroarrays durch Photolithographie, vom experimentellen Design bis zur Datenanalyse, mit Schwerpunkt auf der Vorbereitung von Instrumenten und Verbrauchsmaterialien. Wir beschreiben dann die postsynthetischen Deprotection-Methoden, die dem beabsichtigten Zweck der Mikroarray-Fertigung entsprechen (d. h. entweder Hybridisierung oder die Wiederherstellung von Nukleinsäurebibliotheken).
Protokoll
1. Microarray-Design
- Schreiben Sie die zu synthetismisierenden Sequenzen in einen Texteditor, 5'3', eine Zeile pro Sequenz. Für die Qualitätskontrolle 25mer verwenden Sie die Sequenz "GTCATCATCATGAACCACCCTGGTC". Verwenden Sie A-, C-, G- und T-Buchstaben für DNA-Nukleotide und 5, 6, 7 und 8 Zahlen für RNA-Nukleotide.
- Fügen Sie im Falle der Bibliotheksvorbereitung eine zusätzliche Zahl (d. h. 9) am Ende jeder Sequenz hinzu. Dies entspricht der Kopplung des grundempfindlichen Monomers.
- Auf jede Sequenz muss ein Komma folgen. Weisen Sie jeder Sequenz nach dem Komma einen Namen zu, und stellen Sie sicher, dass jede Spur dem Format [Sequenz, #sequence_name] folgt (ohne Klammern). Speichern Sie die Liste der Sequenzen als .txt-Datei.
- Starten Sie MatLab und laden Sie dann das Programm ChipDesign.m. Führen Sie das Programm aus.
- Laden Sie im Fenster Eigenschaftenbereich die Layoutdatei microarray EntireChip. Laden Sie die MilliChip-Layoutdatei, wenn das gesamte Array an vier identischen Stellen synthetisiert werden soll.
- Wählen Sie im Fenster Eigenschaftenbereich unter Chipspezifikationdie Option Container als Synthesebereich auswählen aus. Wählen Sie unter Musterdas Muster aus, das die richtige Menge an adressierbaren Features liefert, wobei Sie die Anzahl der Sequenzen und die Anzahl der Replikationen zählen. Wählen Sie für die Qualitätskontrolle 25mer das Muster 25:36 aus.
- Wählen Sie unter Container auswählenaus, wählen Sie Fiducialaus. Fiducial Features werden in der Regel an den Ecken des Synthesebereichs synthetisiert und werden verwendet, um Hybridisierungsdaten zu extrahieren. Wenn Fiducial ausgewählt ist, schreiben Sie eine Sequenz (5'3'), die auf treuer Weise synthetisiert wird und als positive Kontrolle fungieren kann. Verwenden Sie für das 25mer-Experiment dieselbe DNA-Sequenz.
- Laden Sie unter Sequenzdie Textdatei, die alle geschriebenen Sequenzen enthält. Stellen Sie sicher, dass Randomize ausgewählt ist. Geben Sie dem Experiment einen Titel in Project Title und schreiben Sie in Linker Sequence TTTTT (entsprechend einem T 5-Linker).
- Drücken Sie Generieren, und suchen Sie dann die Arrayentwurfsdateien und -masken (Abbildung 2) unter MaskGen_delta_rc1/Designs. Stellen Sie sicher, dass es ein Anzeigeskript, eine Flowsequenz und eine Entwurfsdateienthält.
- Öffnen Sie zur Bibliotheksvorbereitung das Anzeigeskript. Fügen Sie am unteren Rand des Anzeigeskripts eine zusätzliche Zeile hinzu, die genau die erste Zeile des Skripts kopiert (z. B. First_Mask.bmp 150 ). Dadurch wird die Terminal-Fotoschutzgruppe am 5' Ende aller Oligonukleotide entfernt.
- Starten Sie das AutoJob-Programm, laden Sie eine Vorlage, indem Sie auf Vorlage laden klicken, und klicken Sie dann auf Skript anzeigen, um die von MatLab generierte Anzeigeskriptdatei zu laden. Drücken Sie Generieren. Dieser Schritt wird eine Reihe von Anweisungen erstellen, eine Jobdatei genannt, die die Kommunikation des Computers mit den Mikrospiegeln und dem DNA-Synthesizer steuern.
2. Folienvorbereitung und Funktionalisierung
- Bohren Sie eine Rutsche an zwei Positionen, die der Position des Ein- und Auslassschlauchs auf der Synthesezelle entsprechen. Verwenden Sie ein 0,9-mm-Diamantbit auf einem CNC-Fräser, um präzise und zuverlässig zu bohren. Spülen Sie die gebohrten Dias mit Reinstwasser und ordnen Sie sie in einem Schiebeträger an.
- Reinigen Sie die Oberflächen, indem Sie die Dias in einem Wasserbad beschallen, das 5 % eines Speziellreinigers auf Ammoniakbasis für 30 min bei 35 °C enthält. Spülen Sie die Rutsche mit doppelt destilliertem H2O ab und geben Sie sie in ein sauberes, trockenes Rack.
- Ordnen Sie gebohrte und unrillierte Mikroskopschlitten in einem Dia-Rack an. In einem großen abgestuften Zylinder die Funktionalisierungslösung vorbereiten, indem Sie 475 ml Ethanol (EtOH) mit 25 ml ddH2O, 10 g Silanisiagereagenz (N -(3-Triethoxysilylpropyl)-4-Hydroxybutyramid) und 1 ml Essigsäure mischen. Gut umrühren, bis er homogen ist und dann in einen geeigneten, geschlossenen Behälter geben.
- Legen Sie das beladene Rack in den Behälter, schließen Sie den Deckel und lassen Sie den Behälter sanft auf einem Orbital-Shaker für 4 h bei Raumtemperatur schaukeln.
- Nach 4 h die Funktionalisierungslösung entsorgen und durch 500 ml Waschlösung ersetzen, bestehend aus 475 ml EtOH, 25 ml ddH2O und 1 ml Essigsäure. Langsam 20 min bei Raumtemperatur aufräumen, dann entsorgen und mit 500 ml Frischwaschlösung ersetzen.
- Nach weiteren 20 min bei Raumtemperatur die Lösung entsorgen, die Dias mit einem Argonstrom trocknen und in einem vorgeheizten Vakuumofen bei 120 °C aushärten. Nach 2 H den Ofen und die Vakuumpumpe ausschalten, aber die Dias über Nacht unter reduziertem Druck lassen. Dann den Ofen wieder in den atmosphärischen Druck bringen und die Dias bis zur weiteren Verwendung in einem Trockenschrank aufbewahren.
3. Herstellung von Synthesereagenzien und Reaktanten
- Bringen Sie die Phosphoramiditpulver (Abbildung 1) von ihrer Lagertemperatur (-25 oder -45 °C) auf Raumtemperatur in einem Trockenhaus.
- Wenn die Phosphoramidite Raumtemperatur erreicht enden, lösen Sie das Pulver mit einem Volumen von ultratrockenem Acetonitril (<30 ppm H2O) auf, um eine Konzentration von 30 mM für Standard-DNA- und RNA-Phosphoramidite zu erreichen, und 50 mM für das basisempfindliche dT-Monomer ( Bibliotheksvorbereitung). Fügen Sie eine kleine molekulare Siebe Tasche, um jede Spur von Feuchtigkeit zu fangen.
- Bereiten Sie eine Lösung von 1% (w/w) Imidazol in DMSO vor, indem Sie 11 g Imidazol in 1 L trockenes DMSO auflösen. Gut schütteln, bis er vollständig aufgelöst ist. Befestigen Sie die Lösung am Hilfsanschluss des DNA-Synthesizers. Dies wird das Expositionslösungsmittel sein, das für die vollständige Entfernung der 5-Foto-Schutzgruppe erforderlich ist.
- Für die Synthese von Bibliotheken, bereiten Sie eine Lösung von 1% (w/v) -Carotin in Dichlormethan durch Auflösung von 100 mg -Carotin in 10 ml Dichlormethan. Gut in einer Bernsteinglasflasche schütteln und dann in Aluminiumfolie wickeln.
4. Vorbereitung und Überwachung der Mikroarraysynthese.
- Zeichnen Sie Temperatur und Luftfeuchtigkeit im Mikroarray-Fertigungsraum auf und stellen Sie sicher, dass der DNA-Synthesizer unter ausreichend Heliumdruck steht.
- Schalten Sie die UV-LED und den Lüfter ein. Befestigen Sie ein UV-Intensitätsmessgerät an der Brennebene des eingehenden UV-Lichts und schalten Sie es ein (Abbildung 3A).
- Starten Sie auf dem Computer die Controller-Software "Jobfile/Micromirror/Synthesis files" (WiCell ). Aktivieren Sie dann das Micromirror-Gerät und laden Sie eine vollweiße Maskendatei, indem Sie mit der rechten Maustaste auf DMDklicken, und wählen Sie dann Bild laden aus.
- Klicken Sie mit der rechten Maustaste auf das UVS-Symbol und wählen Sie UV Shutter Open. Lesen Sie den Leistungswert (in mW/cm2) auf dem Intensitätsmesser und zählen Sie 60 s. Lesen Sie nach 60 s den Leistungswert erneut und notieren Sie sich die Anfangs- und Endwerte. Schließen Sie den Verschluss, indem Sie UV Shutter Close auswählen, und schalten Sie das Intensitätsmessgerät aus. Berechnen Sie den durchschnittlichen UV-Intensitätswert in mW/cm2.
- Berechnen Sie die Belichtungszeit, die erforderlich ist, um eine Strahlungsenergie von 6 J/cm2 für 5'-NPPOC Photodeprotection (DNA- und RNA-Mikroarrays) und 3 J/cm2 für 5'-BzNPPOC Photodeprotection (DNA-Bibliotheken) zu erreichen, einfach nach der
 Beziehung.
Beziehung. - Erstellen Sie in der Software DNA-Synthesizer Workstation eine Sequenzdatei im Sequenz-Editor, indem Sie den Inhalt der von MatLab generierten Flow-Sequenz kopieren und einfügen. Fügen Sie am Ende der 3 eine zusätzliche Zwei-Waschschritte hinzu (Standard-Zyklusbuchstabe: "s"), um die Oberfläche der Substrate zu waschen, bevor Sie zur ersten Kupplung übergehen. Speichern und exportieren Sie die Sequenzdatei.
- Erstellen Sie im Protokoll-Editor der Workstation-Software ein Protokoll, das einen dedizierten Zyklus enthält, der nach jedem der Buchstaben und Zahlen in der Sequenzdatei benannt ist (z. B. wenn eine Sequenzdatei nur die Buchstaben A, C, G und T enthält, muss die Protokolldatei vier Zyklen mit den Namen A, C, G und T enthalten).
- Stellen Sie die Kopplungszeit für DNA-Phosphoramidite (Zyklen A, C, G und T) auf 15 s, für rU-Phosphoramidit (Zyklus 8) und auf 300 s für rA-, rC- und rG-Phosphoramidite (Zyklen 5, 6 und 7) fest. Zur Bibliotheksvorbereitung die Kopplung des grundempfindlichen, kleavablen dT-Monomers auf 2 x 120 s (Tabelle 1 und Tabelle 2) einstellen.
- Stellen Sie sicher, dass die Wartezeit zwischen zwei Event 2 Out-Kommunikationsereignissen in jedem Zyklus der berechneten UV-Belichtungszeit entspricht, um die erforderliche Strahlungsenergie zu erreichen. Wie für die Sequenzdatei, speichern und exportieren Sie die Protokolldatei.
- Laden Sie in der WiCell-Software die Auftrags-, Sequenz- und Protokolldateien in ihre jeweiligen Unterfenster und klicken Sie dann auf Senden, um die Sequenz- und Protokolldateien an den DNA-Synthesizer zu senden.
- Zählen Sie in der Sequenzdatei die Anzahl der Kupplungen für jeden Phosphoramidit.
- Um das für jede Synthese erforderliche Volumen (in L) der Phosphoramiditlösung zu messen, multiplizieren Sie die Anzahl der Kupplungen mit 60. Fügen Sie jedem Volumen 250 L hinzu, um die Linie auf dem DNA-Synthesizer zu erhöhen und zu grundieren. Verwenden Sie ein Grundvolumen von 250 l für Phosphoramidite, die nur eine einzige Kopplung benötigen.
- Übertragen Sie die Lösung schnell in die Durchstechflaschen auf dem DNA-Synthesizer, der dem Portbuchstaben/der Portnummer in den Protokollzyklen entspricht.
- Primieren Sie die Ports mit den Phosphoramidit-Lösungen mit jeweils 10 Prime-Impulsen, um die Leitungen mit Reagenz zu füllen. Prime die Acetonitril-Waschlinie vor der Befestigung der Reaktionszelle.
- Um die Synthesezelle zusammenzusetzen, legen Sie zunächst eine dicke (250 m) Perfluorelastomerdichtung (FFKM) auf den Quarzblock der Zelle. Legen Sie einen gebohrten, funktionalisierten Mikroskopschlitten auf die erste Dichtung und stellen Sie sicher, dass sich die Löcher auf den Dias mit dem Ein- und Auslassschlauch der Synthesezelle verbinden.
- Legen Sie eine zweite, dünne (50 m) Polytetrafluorethylendichtung (PTFE) über den gebohrten Schlitten, der die beiden Löcher umgibt. Platzieren Sie schließlich eine zweite, funktionalisierte, aber unrillierte Rutsche auf der zweiten Dichtung (Abbildung 3B). Legen Sie einen 4-Schrauben-Metallrahmen auf die montierte Doppelsubstratzelle und ziehen Sie die Schraube mit einem Drehmomentschrauber auf die gleiche Schließkraft (0,45 Nm) fest.
- Befestigen Sie den Einlass und den Auslassschlauch am DNA-Synthesizer. Prime die Acetonitril-Waschlinie und überprüfen Sie den richtigen Fluss von Acetonitril (ACN) durch die Substrate. Zerlegen und wieder zusammensetzen die Zelle, wenn ein Leck von ACN in diesem Stadium beobachtet werden kann. Messen Sie das VOLUMEN von ACN an der Abfallleitung, nachdem Sie 7 Zyklen ACN-Grundierung durchlaufen haben. Dieses Volumen sollte 2 ml betragen.
- Befestigen Sie die Synthesezelle an der Brennebene des eingehenden UV-Lichts. Bei der Bibliotheksvorbereitung eine zusätzliche Ein- und Auslassleitung an der Rückseite der Zelle befestigen und die hintere Kammer mit der Lösung a-carotin füllen (2 ml Lösung sind ausreichend). Stellen Sie sicher, dass es keine Leckage gibt (Abbildung 3C).
- Starten Sie die Synthese, indem Sie zuerst in der WiCell-Software auf Ausführen klicken. Drücken Sie beim ersten WAIT-Befehl in der Auftragsdatei auf den DNA-Synthesizer starten.
- Während der Synthese, regelmäßig überprüfen, ob die Anzeige von Maskendateien mit UV-Belichtung und Öffnung des Verschlusses übereinstimmt.
- Nach der Synthese von regulären Mikroarrays trennen Sie die Zelle vom Synthesizer, zerlegen Sie die Zelle und verwenden Sie einen Diamantstift, um die Synthesenummer auf die Glasgleitetzujehinaus zu ätzen. Ätzen Sie die Zahl auf der nicht synthetisierten Fläche jeder Folie. Die Dias in 50 ml Zentrifugenrohre übertragen und bis zur weiteren Verwendung in einem ausgetrockneten Bereich lagern.
- Nach der Synthese von Bibliotheksmikroarrays zuerst die Carotin-Lösung aus der Kammer abtropfen lassen und dann durch Fließen von 2 x 5 ml CH2Cl2abtropfen lassen. Fahren Sie mit der Demontage gemäß Schritt 4.21 fort. Das synthetisierte Array kann mit bloßem Auge sichtbar sein (Abbildung 4).
5. DNA-Mikroarray-Deprotection
- Füllen Sie ein Glasmitglas mit 20 ml EtOH und 20 ml Ethylendiamin (EDA). Legen Sie die dna-only-Mikroarrays vertikal in das Glas, schließen Sie den Deckel und lassen Sie die Dias für 2 h bei Raumtemperatur zu detekten.
VORSICHT: EDA ist eine akut giftige, korrosive und brennbare Flüssigkeit. Arbeiten Sie mit Handschuhen in einer gut belüfteten Dunstabzugshaube. - Nach 2 h die Dias mit einer Pinzette abholen und gründlich mit doppelt destilliertem H2O abspülen.
- Trocknen Sie die Dias in einer Mikroarray-Zentrifuge für ein paar Sekunden dann in einem Austrocknungsgerät lagern.
6. RNA-Mikroarray-Deprotection
- In einem 50 ml Zentrifugenrohr eine trockene Lösung von 2:3 Triethylamin/ACN (jeweils 20 ml bzw. 30 ml) zubereiten. Übertragen Sie ein RNA-Mikroarray-Dia in das Zentrifugenrohr, schließen Sie den Deckel und wickeln Sie ihn dann mit Kunststoff-Dichtungsfolie. Schütteln Sie das Zentrifugenrohr auf einem Orbital-Shaker 1h und 30 min bei Raumtemperatur vorsichtig.
- Nach 1h und 30 min den Schlitten entfernen, mit 2 x 20 ml trockenem ACN waschen und dann in einer Mikroarrayzentrifuge für ein paar Sekunden trocknen.
VORSICHT: Triethylamin ist eine akut giftige, korrosive und brennbare Flüssigkeit. Acetotril ist giftig und entzündlich. Arbeiten Sie mit Handschuhen in einer gut belüfteten Dunstabzugshaube.
HINWEIS: Erster Deschutzschritt abgeschlossen. - Bereiten Sie eine 0,5 M Hydrazinhydratlösung in 3:2 Pyridin/Essigsäure vor. Mischen Sie zunächst 20 ml Essigsäure und 30 ml Pyridin in einem abgestuften Zylinder. Warten Sie, bis die Lösung abgekühlt ist, bevor Sie 1,21 ml Hydrazinhydrat hinzufügen. Rund 40 ml der resultierenden Lösung in ein 50 ml Zentrifugenrohr übertragen.
VORSICHT: Hydrazinhydrat ist eine akut giftige und korrosive Flüssigkeit. Pyridin ist leicht entzündlich und akut giftig. Essigsäure ist entzündlich und ätzend. Arbeiten Sie mit Handschuhen in einer gut belüfteten Dunstabzugshaube. - Nach dem ersten Deprotection-Schritt die RNA-Rutsche in die Hydrazinhydratlösung übertragen, den Deckel schließen und mit Kunststoff-Dichtungsfolie umwickeln. Schütteln Sie das Rohr vorsichtig auf einem Orbital-Shaker für 2 h bei Raumtemperatur. Nach 2 h den Schlitten entfernen, mit 2 x 20 ml trockenem ACN waschen und dann in einer Mikroarrayzentrifuge für einige Sekunden trocknen.
HINWEIS: Zweiter Deprotection-Schritt abgeschlossen. - Wenn das RNA-Mikroarray auch DNA-Nukleotide enthält, fahren Sie mit einem dritten Deprotection-Schritt fort. In einem 50 ml Zentrifugenrohr 20 ml EDA mit 20 ml EtOH mischen. Fügen Sie das DNA/RNA-Mikroarray in die 1:1 EDA/EtOH-Lösung ein und lassen Sie es bei Raumtemperatur 5 min.
- Nach 5 min den Schlitten entfernen, mit 2 x 20 ml sterilem Wasser waschen, dann in einer Mikroarrayzentrifuge trocknen und in einem Trockenschrank lagern.
7. Hybridisierung mit einem fluoreszierend gekennzeichneten Komplementärstrang
- Acetyliertes Rinderserumalbumin (10 mg/ml) und den Cy3-gekennzeichneten Komplementärstrang (100 nM) auftauen und auf Raumtemperatur erwärmen.
- In einem 1,5 ml sterilen Mikrozentrifugenrohr Mischen Sie 150 l 2x MES-Puffer(200 mM 2-(N-morpholino)ethanesulfonsäure; 1,8 M NaCl; 40 mM EDTA; 0,02% Tween-20) mit 26,7 l Cy3-markierte DNA, 13,4 l acetylierte BSA und 110 l sterile H2O und wirbel. Verdoppeln Sie das Volumen, wenn beide Folien für die Hybridisierung verwendet werden sollen.
- Vorerwärmen Sie einen Hybridisierungsofen auf eine Temperatur unter dem Tm des Duplex, aber hoch genug, um eine gute Diskriminierung zwischen Full-Match- und Non-Match-Sequenzen zu gewährleisten. Für die Qualitätskontrolle 25mer die Temperatur auf42 °C (Tm von 59 °C) einstellen.
- Legen Sie vorsichtig eine selbstklebende 300-L-Hybridisierungskammer über den Synthesebereich auf jedem Schlitten und jeder Pipette in die oben vorbereitete Hybridisierungslösung. Bedecken Sie die Löcher der Kammer mit Klebepunkten und wickeln Sie die gesamte Rutsche in Aluminiumfolie.
- Legen Sie den Mikroarray-Schlitten in den Hybridisierungsofen, decken Sie ihn ab und lassen Sie es bei der gewählten Hybridisierungstemperatur für 2 h sanft drehen.
- Nach 2 h die Rutsche lösen, die Aluminiumfolie entfernen und die Hybridisierungskammer vorsichtig abreißen. Die Dias in ein Zentrifugenrohr mit 30 ml Nicht-Stringent Wash Buffer (NSWB; 0,9 M NaCl, 0,06 M Phosphat, 6 mM EDTA, 0,01% Tween20, pH 7,4) übertragen und bei Raumtemperatur 2 min kräftig schütteln.
- Das Dia in ein Zentrifugenrohr mit 30 ml Stringent Wash Buffer (SWB; 100 mM MES, 0,1 M NaCl, 0,01% Tween20) geben und 1 min kräftig schütteln.
- Schließlich das Dia in ein Zentrifugenrohr mit 30 ml Endwaschpuffer (FWB; 0,1x Natriumkochincitrat) übertragen und für einige Sekunden schütteln. Trocknen Sie den Schlitten in einer Mikroarray-Zentrifuge.
- Platzieren Sie das trockene Mikroarray, den Synthesebereich nach unten, in den Diahalter des Mikroarray-Scanners. Bei Cy3-gekennzeichneten Duplexen können Sie mit einer Auflösung von 532 nm, einem 575 nm Filter und einem Photomultiplier von 350 mit einer Auflösung von 5m scannen. Speichern Sie den hochauflösenden Scan als .tif-Bilddatei (Abbildung 5A).
8. Datenextraktion und -analyse
- Drehen Sie vor der Datenextraktion den gespeicherten Array-Scan in einem Bildeditor, um die längste Kette von Fiducial-Features in der oberen linken Ecke des Scans zu verorten. Speichern Sie das gedrehte Bild.
- Starten Sie NimbleScan, und drücken Sie dann Datei | Öffnen und laden Sie den Array-Scan. Wenn Sie dann im Unterabschnitt Designdatei auf Durchsuchen klicken, laden Sie die . ndf-Entwurfsdatei, die während des Entwurfs des Microarray-Experiments automatisch generiert wurde. Klicken Sie dann auf Öffnen.
- Klicken Sie in der Ansichtauf Auto-Kontrast/Helligkeit. Klicken Sie auf das Symbol Manuell über dem Scan ausrichten. Platzieren Sie vier quadratische Markierungen an den vier Ecken des Scans, und klicken Sie dann erneut auf das jetzt grüne Symbol. Extrahieren Sie die Hybridisierungsdaten, indem Sie auf Analyse, Berichte und Probebericht klicken.
- Öffnen Sie die . Prüfpunktdatei in einem Tabellenkalkulationseditor. Bewahren Sie die Spalten B und I auf, und verwerfen Sie den Rest, bevor Sie mit der Berechnung der Durchschnittswerte und der Standardabweichung von den extrahierten Daten fortfahren (Abbildung 5B).
9. Bibliotheksdeschutz, Spaltung und Wiederherstellung
- Um DNA-Bibliotheken zu deprotecten und zu spalten, bereiten Sie eine Lösung von 1:1 trockenem EDA/Toluin in einem 50 ml Zentrifugenrohr vor. Tauchen Sie das Dia in die Dekolletélösung ein, schließen Sie den Schlitten und wickeln Sie ihn mit Kunststoff-Dichtungsfolie, und drehen Sie dann vorsichtig in einem Orbital-Shaker für 2 h bei Raumtemperatur.
- Nach 2 h den Schlitten entfernen und mit 2 x 20 ml sorgfältig trockenem ACN waschen. Entfernen Sie den Schlitten und lassen Sie ihn an der Luft trocknen.
- Mit einer Pipette 100 l sterilesH2 O über den nun erkennbaren Synthesebereich auftragen. Pipet die Lösung ein paar Mal nach oben und unten, bevor sie in ein 1,5 ml Mikrozentrifugenrohr übertragen wird. Wiederholen Sie den Vorgang und kombinieren Sie das Mikroarray-Eluat in ein und dasselbe Rohr (Abbildung 6).
- Verdampfen Sie die 2 x 100 l Chip-Eluat bis zur Trockenheit und lösen Siesie dann wieder in 10 l nukleasefreies H2 O auf. Messen Sie die Absorption auf einem UV-Vis-Spektralphotometer.
- Entalt die DNA-Bibliothek auf einer 10 L Pipettenspitze mit C18-Harz ausgestattet. Verwandeln Sie zunächst das Spaneluat in eine 0,1 M Triethylammoniumacetat (TEAA) gepufferte Lösung.
- Befeuchten Sie das Harz, indem Sie 3 x 10 l H2O/ACN 1:1 anstreben, und gleichnden Sie das Harz durch Waschen mit 3 x 10 l 0,1 M TEAA Puffer. Binden Sie die DNA, indem Sie den Chip zehnmal durch das Harz nach oben und unten pfeifen. Waschen Sie das Harz mit 3 x 10 l 0,1 M TEAA Puffer, 3 x 10 l H2O.
- Elute die entsalzte DNA von der Spitze, indem sie das Harz mit 10 l H2O/ACN 1:1 waschen. Trocknen Sie die entsalzte Lösung nach unten und lösenSie sie wieder in 10 l steriles H2 O auf. Messen Sie die Absorption auf einem UV-Vis-Spektralphotometer (Abbildung 7). Verdampfen Sie die Bibliothek bis zur Trockenheit und lagern Sie sie bei -20 °C bis zur weiteren Verwendung.
Ergebnisse
DNA- und RNA-Mikroarray-Hybridisierung
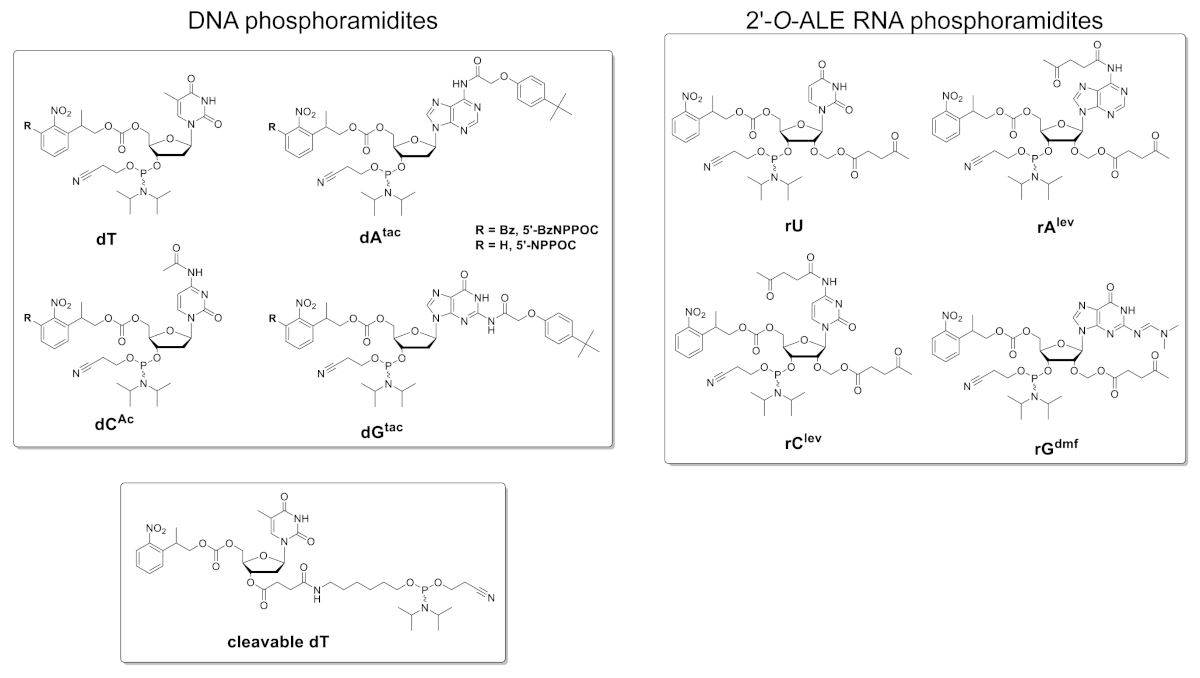
Abbildung 5 zeigt die Ergebnisse eines Hybridisierungstests, der an einem Mikroarray durchgeführt wurde, das die DNA- und RNA-Versionen einer 25mer-Sequenz enthält (5'-GTCATCATCATGAACCACCCTGGTC-3' in DNA-Form). Der Scan in Abbildung 5A erscheint in einem grünen Format, das dem Anregungs-/Emissionsspektrum der Cy3-Fluoreszenz entspricht, wobei die Fluoreszenzintensität in beliebigen Einheiten zwischen 0 und 65536 aufgezeichnet wird. Der Arrayentwurf folgte dem im Protokollabschnitt beschriebenen 25:36-Feature-Layout. Der Scan wird nach der richtigen Ausrichtung des Arrays angezeigt, wobei die obere linke Ecke mit der längsten Kette von Fiducial-Features gefüllt ist. Hier enthalten die handelsgebundenen Merkmale die DNA-Version des 25mer und sollten grundsätzlich immer ein positives Fluoreszenzsignal geben, um Scanausrichtung und Datenextraktion durchzuführen. Das hybridisierte Mikroarray sollte gleichmäßig hell erscheinen, wobei die Ränder des Synthesebereichs jedoch in der Regel heller als das Zentrum (bis zu 50% heller) sind. Die große Anzahl von Sequenzreplikationen, die zufällig über das Gebiet verteilt sind, reduziert die Auswirkungen räumlicher Artefakte. Hier wurde jede Sequenz (DNA und RNA) an 2.000 zufälligen Stellen synthetisiert. Es gibt typischerweise niedrige Fluoreszenzrauschen (Hintergrund), <50 a.u., was zu einem Signal-Rausch-Verhältnis in der Größenordnung von 200:1 bis 800:1 in Hybridisierungstests führt. Nach der Datenextraktion werden die Fluoreszenzintensitäten gemittelt und mit sD geplottet.
Die absoluten Fluoreszenzwerte zwischen den Experimenten sind signifikant variabel. Hier zeigen wir die Ergebnisse für drei unabhängige Synthesen mit den gleichen Fertigungsparametern und der gleichen postsynthetischen Handhabung. Die 25mer-DNA, wenn sie mit ihrem komplementären Cy3-markierten DNA-Strang hybridisiert wird, liefert Fluoreszenzsignale von 20.000 bis 30.000, sehr selten über oder unter. Die 25mer-RNA, wenn sie mit der gleichen Cy3-markierten DNA-Ergänzung hybridisiert wird, wird Fluoreszenzintensitäten auf die entsprechenden Merkmale von 15.000 bis 20.000 geben. Die Fluoreszenzintensität der RNA/DNA-Duplexe wird jedoch gelegentlich unter 8.000 sinken, wenn die entsprechenden DNA/DNA-Duplexe noch im Bereich von 20.000-30.000 fluoreszieren. In solchen Fällen können die Ergebnisse für RNA als suboptimal angesehen werden. Ein Synthese- oder Hybridisierungsversagen, entweder für DNA oder RNA, wird sofort beim Scannen aus dem offensichtlichen Mangel an Fluoreszenz auffallen. Es gibt mehrere Möglichkeiten für die RNA-Synthese, entweder zu scheitern oder teilweise erfolgreich zu sein, und sie werden im Diskussionsteil skizziert.
Bibliotheksdeschutz, Spaltung und Wiederherstellung
Je nach Komplexität und Dichte der Bibliothek können die Form und umriss des synthetisierten Arrays ohne Vergrößerung, aber unter richtiger Beleuchtung (Abbildung 4) gesehen werden, wobei die DNA noch in geschützter Form ist. Nach dem Deschutz mit EDA/Toluol und vor dem Zusatz von Wasser, um die spaltete Bibliothek zu sammeln, kann der Synthesebereich, der die jetzt degeschützten Oligonukleotide enthält, als hydrophile Zone auffallen, wenn der Rest der Glasrutsche erscheint mit einer trüben hydrophoben Schicht bedeckt. Die direkte Beobachtung des Synthesebereichs hängt von der Gesamtfläche ab, die zur Synthese von Oligonukleotiden verwendet wird: Eine stärkere Nutzung des Synthesebereichs wird einer größeren Chance einer klaren Unterscheidung zwischen hydrophilen und hydrophoben Regionen an der Oberfläche entsprechen. Umgekehrt sind Bibliotheken, die mit weniger Spiegeln und mit kleineren Features synthetisiert werden, möglicherweise nicht sofort beobachtbar.
In ähnlicher Weise ist die Menge der wiedergewonnenen DNA nach der Entsalzung direkt proportional zur Gesamtfläche, die für die Synthese verwendet wird. Wenn alle Merkmale für die Oligonukleotid-Synthese verwendet werden, sollte das Spaltungs- und Rückgewinnungsverfahren zwischen 25 und 30 pmol DNA ergeben. Eine 10%ige Nutzung des Synthesebereichs wird daher nur etwa 3 pmol DNA ermöglichen.

Abbildung 1 . Chemische Strukturen der DNA- und RNA-Phosphoramidite, die bei der Oligonukleotidsynthese durch Mikroarray-Photolithographie verwendet werden. Standard-Nitrophenylpropyloxycarbonyl (NPPOC) lichtempfindliche Schutzgruppen am 5'-OH werden in der regulären DNA- und RNA-Mikroarraysynthese zur Hybridisierung eingesetzt. Für die Synthese komplexer DNA-Bibliotheken wird am 5'-OH das photolabile Benzyl-NPPOC (BzNPPOC) bevorzugt, da BzNPPOC doppelt so schnell entfernt wird wie NPPOC, was die Gesamt-Mikroarraysynthesezeit deutlich reduziert. DNA-Oligonukleotide für Bibliotheken erfordern auch die Kopplung eines cleavablen dT-Monomers am 3'-Ende. Dieses Monomer, das eine Succinylester-Funktion trägt, wird während des Deschutzes zerklaut tappt, so dass die DNA aus dem Mikrochip gesammelt werden kann. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2 . Beispiel für eine Maske als Bilddatei, die während der UV-Bestrahlung an das Mikrospiegelgerät gesendet wird. Die weißen Pixel entsprechen Spiegeln, die in der Position "ON" gekippt werden und UV-Licht auf die Synthesezelle reflektieren. Schwarze Pixel entsprechen" OFF"-Spiegeln, bei denen das UV-Licht von der Zelle weg reflektiert wird. Weiße Pixel ermöglichen daher die Kopplung des nächsten eingehenden Phosphoramidites auf die Oligonukleotide, die an den entsprechenden Merkmalen auf den Glassubstraten zu finden sind. Oligonukleotide synthetisiert auf den Features, deren entsprechende Spiegel sind, in dieser Maskendatei, schwarze Pixel bleiben jedoch während des nächsten Kopplungsereignisses inert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3 . Fotos der Mikroarray Photolithographie optische und Synthese-Setup. (A) Optischer Schaltkreis für UV-Belichtung. UV-Licht der UV-LED wird zunächst durch ein rechteckig-querschnittsförmiges Lichtrohr homogenisiert und dann auf die Mikrospiegel reflektiert. Mikrospiegel, die in eine "OFF"-Position gekippt wurden, reflektieren UV-Licht von der Synthesezelle weg, aber Mikrospiegel in der Position "ON" reflektieren Licht auf die Synthesezelle, die sich auf der Brennebene befindet, indem sie zuerst ein 1:1 Offner-Relais passieren. Bildgebungssystem. (B) Die Synthesezelle besteht nach der Montage aus einem bohrenden Schlitten, der zuerst auf den Quarzblock der Zelle gelegt wird und durch eine dicke PTFE-Dichtung getrennt ist (nicht dargestellt). Ein zweiter, nicht gebohrter Schlitten wird dann über dem gebohrten Schlitten positioniert, der durch eine dünne PTFE-Dichtung getrennt ist. Ein Metallrahmen (nicht dargestellt) hält die Baugruppe zusammen. (C). Zur Bibliotheksvorbereitung wird die Kammer zwischen Quarzblock und Bohrschlitten nach der Befestigung der Synthesezelle an der Brennebene des einfallenden UV-Lichts mit einer 1%-Lösung von .-carotin in CH2Cl2gefüllt. Dazu wird ein zusätzlicher Ein- und Auslassschlauch am Quarzblock befestigt und die orangenlösung fließt von der rechten bis zur linken Position. Der Fluss von Reagenzien und Lösungsmitteln für die Synthese wird in weißen Pfeilen dargestellt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4 . Der Synthesebereich ist in der Regel mit bloßem Augesichtbar. Hier ist direkt nach der Synthese eine DNA-Bibliothek auf der Glasoberfläche zu sehen, wobei die DNA noch in geschützter Form ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5 . Hybridisierungs-Assays zu den 25mer-DNA- und RNA-Sequenzen, die in situ auf Mikroarrays synthetisiert werden. (A) Fluoreszenzscan des gesamten hybridisierten DNA- und RNA-Mikroarrays. 25mer-DNA- und RNA-Oligonukleotide werden zu ihren Cy3-markierten Komplementärsträngen hybridisiert. Das Array wurde mit einem Laser mit einer Anregungswellenlänge von 532 nm mit einer Auflösung von 5 m gescannt. (B) Fluoreszenzintensitäten (willkürliche Einheiten) der DNA:DNA und RNA:DNA-Duplexe in drei separaten Experimenten. Die hellgrünen Daten für in situ synthetisierte RNA-Oligonukleotide können im Vergleich zur Fluoreszenzintensität der entsprechenden DNA-Sequenzen als suboptimal betrachtet werden. Fehlerleisten sind SD. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

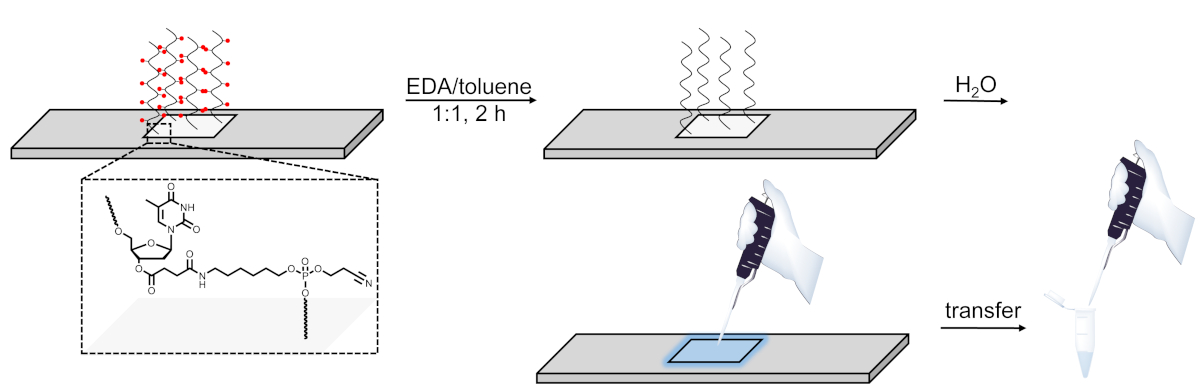
Abbildung 6 . Schematische Darstellung des Deprotection-, Spaltungs- und Wiederherstellungsverfahrens für DNA-Bibliotheken, die auf Mikroarrays synthetisiert werden. DNA-Sequenzen werden auf einem basisempfindlichen, spaltbaren dT-Nukleosid (im vergrößerten Bereich dargestellt) angebaut. Nach der Synthese lässt der Deschutz der DNA-Oligonukleotide (Basisschutzgruppen werden als rote Kugeln dargestellt) in EDA/Toluol das degeschützte Material elektrostatisch an die Oberfläche gebunden und kann dann durch Auftragen einer kleinen Menge Wasser auf den synthetisierten Bereich. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 7 . Repräsentatives Absorptionsspektrum (220 - 350 nm) einer geklammerten, entsalzten DNA-Bibliothek mit 4.000 verschiedenen Sequenzen, 100 nt lang. Insgesamt wurden 940 ng DNA aus einer einzigen Arraysynthese isoliert, was 30 pmol DNA-Gesamtmenge oder 15 pmol pro Glassubstrat entspricht. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Tabelle 1. Repräsentatives Zyklusprotokoll für Kopplung/Oxidation/Photodeprotection von 5'-BzNPPOC-dA unter der Annahme, dass der entsprechende Phosphoramidit auf Port "A" geladen wurde. Die Kopplungszeit (in Sekunden) wird in der Linie "Paarmonomer" angezeigt. Die UV-Photodeprotektionszeit, hier einer Strahlungsenergie von 3 J/cm2 (BzNPPOC Photochemie) entspricht, wird als Zeit zwischen den beiden "Event 2 Out"-Kommunikationssignalen berechnet.

Tabelle 2. Repräsentatives Zyklusprotokoll für Kopplung/Oxidation/Photodeprotection von 5'-NPPOC-rA unter der Annahme, dass der entsprechende RNA-Phosphoramidit auf Port "A" geladen wurde. Die Kopplungszeit (in Sekunden) wird in der Linie "Paarmonomer" angezeigt. Die UV-Photodeschutzzeit, hier einer Strahlungsenergie von 6 J/cm2 (NPPOC Photochemie) entspricht, wird als Zeit zwischen den beiden "Event 2 Out"-Kommunikationssignalen berechnet.
Diskussion
Die Festphasen-DNA- und RNA-Synthese ist das Brot und die Butter jedes Nukleinsäure-Chemielabors, und obwohl die Zugabe der Photolithographiekomponente zugegebenermaßen eine komplexe Operation ist, ist die Mikroarray-Fertigung, die durch UV-Licht vermittelt wird, auch ein sehr zuverlässiger Prozess. . Darüber hinaus ist es die einzige verfügbare Methode für die In-situ-RNA-Synthese auf Mikroarrays. Dennoch gibt es, wie bei jedem mehrstufigen experimentellen Verfahren, genügend Raum für menschliches Versagen.
Der vielleicht wichtigste Schritt ist die Kopplung eines Phosphoramidites, da es sich um eine konstant ertragreiche chemische Reaktion handelt, um sich Oligonukleotide mit wenigen synthetischen Fehlern leisten zu können. In unserem Mikroarray-Syntheseprotokoll ist die Phosphoramiditkopplung für die Gesamtsynthesequalität noch wichtiger, da der Herstellungsprozess die Deckelung umgeht und die Oligonukleotidreinigung verhindert. Für alle lichtempfindlichen DNA- und RNA-Phosphoramidite wurden stufenweise Kopplungseffizienzen von über 99% berechnet, selbst für sehr kurze Kopplungszeiten (15 s)24, aber gelegentlich können geringere Kopplungsausbeuten auftreten, insbesondere bei dG-Amiditen. Die Stabilität der löslichen Phosphoramidite bei Raumtemperatur wurde bereits untersucht und war nachweislich von der Art der Nukleobase abhängig, wobei Guanosinphosphoramidite in nur wenigen Tagen zu einem umfangreichen Abbau neigen29, 30. Bei -25 °C wurden jedoch in ACN als 30 mM Lösung gelöste dG-Phosphoramidite für mehrere Wochen als stabil befunden. Die relative Instabilität von dG-Phosphoramiditlösungen bei Raumtemperatur bedeutet jedoch, dass sie nicht mehrere Tage am DNA-Synthesizer befestigt werden sollten.
Bei RNA-Phosphoramiditen ist die Kopplungsausbeute stark von der Phosphoramiditqualität (die durch 31P NMR-Spektroskopie beurteilt werden kann) und der Kopplungszeit abhängig. Kupplungszeiten von 5 Minuten für rA, rG, rC und 2 min für rU erscheinen notwendig. Tatsächlich fanden wir heraus, dass die Verkürzung der Kondensationszeit auf 2 min für alle RNA-Phosphoramidite zu deutlich niedrigeren Hybridisierungssignalen führte.
Der DNA-Synthesizer selbst, sowie die Reagenzien und Lösungsmittel, müssen sicherlich so sauber wie möglich sein, um die höchste Ausbeute an Oligonukleotid-Synthese zu erreichen. Unlösliches Material, Salze oder Partikel können sich jedoch im Laufe der Zeit in den Leitungen und Schläuchen des Fördersystems ansammeln, was zu einer allmählichen Abnahme des Verbrauchs von Reagenzien und Reaktanten führt. Wenn eine allgemeine Reinigung des Synthesizers ein geringes Ausgangsvolumen nicht auflöst, kann eine Erhöhung der Anzahl der Impulse eine alternative Lösung sein. Besonders nützlich bei geringem Phosphoramiditverbrauch kann die Leitung im Kopplungsprotokoll, die dem Pumpen eines Gemischs aus Phosphoramidit und Aktivator entspricht (dritte Linie des Kupplungsunterabschnitts in Tabelle 1 und Tabelle 2) von 6 auf 9 Impulse ohne nennenswerte negative Auswirkungen auf die Synthesequalität. Darüber hinaus hängt die Anzahl der Impulse des Aktivators, die benötigt werden, um den Amidit-Aktivator-Mix auf das Synthesesubstrat zu bringen (derzeit 6, vierte Zeile im Kopplungsunterabschnitt, siehe Tabelle 1 und Tabelle2), sowohl vom DNA-Synthesizer selbst als auch von Rohrlänge in der Synthesezelle. Diese Zahl kann angepasst werden, nachdem der Phosphoramidit durch eine farbige Lösung ersetzt und die Anzahl der Impulse gezählt wurde, die benötigt werden, um die farbige Mischung zur Kopplung auf das Glassubstrat zu schieben.
Die hier beschriebene Methode ermöglicht es, dass die DNA- und RNA-Synthese gleichzeitig auf demselben Mikroarray verläuft. Hybriden von DNA und RNA können auch ohne Änderung der Array-Fertigungsprotokolle hergestellt werden, und solange das dreistufige Deprotection-Protokoll befolgt wird. Es sollte jedoch beachtet werden, dass reine RNA-Mikroarrays nur einen zweistufigen Deprotection erfordern: eine Decyanoethylierung zuerst mit Et3N, gefolgt von Hydroxyl- und Basenschutz mit Hydrazin. DNA-Nukleobasen wurden unter diesen Bedingungen als unvollständig degeschützt festgestellt und benötigen den zusätzlichen Schritt in EDA, um die vollständige Entfernung von Phenoxyacetylgruppen (Pac) zu bewirken. Diese zusätzliche Behandlung mit EDA ist kürzer (5 min) als für den Standard-Deprotection von DNA-Mikroarrays31, aber es reicht aus, um es nach den Triethylamin- und Hydrazin-Behandlungen zum Abschluss zu bringen. Darüber hinaus begrenzt eine kurze Reaktionszeit mit EDA die Exposition eines vollständig degeschützten RNA-Oligonukleotids an Grundbedingungen.
Ein Vorteil der In-situ-RNA-Array-Synthese gegenüber alternativen Methoden wie Spotting oder DNA-Transkription32,33,34 ist die Möglichkeit, den synthetisierten RNA-Mikrochip bis zur Anwendung in geschützter Form zu speichern und so die Risiko eines möglichen RNA-Abbaus. Postsynthetische Verfahren für RNA implizieren hingegen, dass Verbrauchsmaterialien und Reagenzien steril gehalten werden und dass die Handhabung unter RNase-freien Bedingungen erfolgt. Bemerkenswert erweise haben wir festgestellt, dass die Zugabe von RNase-Inhibitoren zum Hybridisierungsmix keine stärkeren Hybridisierungssignale für die RNA-Features lieferte.
Die Synthese von DNA-Bibliotheken auf einem basisempfindlichen Monomer ist komplexer als die Synthese einiger Kontrollsequenzen auf einer Oberfläche und als solche sicherlich anfälliger für Konstruktionsfehler. Geht man jedoch davon aus, dass der Sequenzentwurf (d. h. die Art und die Anzahl der Sequenzen) korrekt ist, bleibt die Umwandlung dieser Liste in eine Sammlung von Belichtungsmasken und einer geordneten Reihe von Kopplungszyklen ein einfacher Prozess. Es gibt jedoch wichtige Variationen von der Standard-Mikroarray-Synthese und ist entscheidend für eine erfolgreiche Herstellung eines Bibliotheksarrays mit hoher Dichte.
Zunächst wird ein grundempfindliches dT-Monomer als erstes Phosphoramidit nach der Synthese des Linkers gekoppelt. Die Kopplungsausbeute dieses Monomers (Abbildung 1) wurde als relativ gering befunden, etwa 85%28, weshalb Anstrengungen unternommen werden, um seine Einbaurate zu verbessern, entweder durch Erhöhung der AcN-Konzentration von 30 mM auf 50 mM oder durch Wiederholung der Kupplungsschritt: zwei aufeinander folgende Kupplungsreaktionen mit frischen Monomeren oder zwei separate, aber aufeinander folgende Kupplungszyklen.
Die zweite Änderung ist die Zugabe einer Carotin-Lösung in der Hinterkammer der Synthesezelle, die bequem 365 nm Licht absorbiert. Dies ist eine wichtige Änderung der Photolithographie-Einrichtung, da es verhindert, dass UV-Licht wieder auf das Array-Substrat reflektiert wird. Tatsächlich tritt nach dem Durchlaufen des interstitiellen Mediums zwischen den Substraten einfallendes UV-Licht durch den gebohrten Schlitten aus und erreicht den Quarzblock der Zelle. Die Fresnel-Gleichungen sagen voraus, dass 4 % des senkrecht einfallenden UV-Lichts von jeder der drei nachgeschalteten Luft-Glas-Schnittstellen (Ausgangsseite des 2. Substrats und beidseitig des Quarzblocks) und zurück auf das Synthesesubstrat reflektieren werden. zu unbeabsichtigter Exposition von photogeschützten Oligonukleotiden führen. Beugung und Streuung tragen auch zum "Off-Target"-Photodeprotection und damit zur Nukleotideinfügung bei, was sich direkt auf die Fehlerrate der Synthese auswirkt, aber diese Beiträge sind viel kleiner als Reflexionen und können hauptsächlich durch Reflexionen angegangen werden. Verringerung der Synthesedichte (Lücken zwischen den Merkmalen). Wir haben festgestellt, dass der Gehalt an Carotin-Lösung in der unteren Kammer der Zelle nur in den ersten Minuten der Arraysynthese leicht abnimmt und daher überwacht und nachjustiert werden muss.
Schließlich ist die dritte Änderung die Deprotection-Lösung, die EtOH durch Toluen ersetzt, das die geklammerte DNA-Bibliothek an die Oberfläche gebunden hält, vermutlich durch elektrostatische Wechselwirkungen. Wenn Sie nach dem ACN-Waschen eine kleine Menge Wasser auf den Synthesebereich auftragen, können Sie die Bibliothek bequem sammeln. Der Prozess ist jedoch nur erfolgreich, wenn der Wassergehalt in EDA und Toluen minimal ist, wodurch die Nukleinsäure im Deprotection-Cocktail völlig unlöslich wird. Alternativ können DNA-Bibliotheken mit Ammoniak 9,10,14,35vom Chip abgeklaut und dann durch Erhitzen der DNA-haltigen wässrigen Ammoniaklösung über Nacht weiter auf 55 °C erhitzt werden. Die Wiederherstellung von DNA-Bibliotheken mit Ammoniak ist jedoch nicht mit RNA kompatibel. RNA-Oligonukleotide auf einem basissturierbaren Substrat können von der Oberfläche mit dem oben beschriebenen EDA/Toluol-Verfahren eluiert werden, jedoch nur in der vorletzten Phase nach der zweistufigen Deprotection-Strategie Et3N und Hydrazin28.
Alternative Strategien zur Rückgewinnung von Oligonukleotidbecken aus Mikroarrays ohne die Notwendigkeit einer spezifischen Grundbehandlung existieren, sind grundsätzlich mit der Photolithographie kompatibel und setzen auf den Einsatz von Enzymen. Zum Beispiel kann ein einzelnes Deoxyuracil-Nukleotid das Ziel der Uracil-DNA-Glykosomalase (UDG) sein und aus dem Rest der DNA-Sequenz entfernt werden, oder eine einzelne RNA-Einheit kann durch RNase H Typ 2-Enzyme erkannt werden und die Phosphodiesterbindung 5' an die RNA , die 5' DNA Teil23.
Wir haben jetzt eine leistungsfähige, zuverlässige und hochdichte Methode für die Synthese von DNA, RNA und hybriden DNA/RNA-Mikroarrays. Diese können nicht nur als Plattformen für Hybridisierung oder Bindungstests36dienen, sondern stellen auch eine schnelle und kostengünstige Möglichkeit dar, komplexe Nukleinsäurebibliotheken herzustellen. Für die DNA-basierte digitale Datenspeicherung kann die Mikroarray-Photolithographie zu einer potenziellen Lösung für den "Schreib"-Engpass (d. h. zur Kodierung von Informationen durch Synthese) werden. Der Erfolg bei der digitalen Codierung auf DNA und in der de novo Genassembly hängt von der Sequenztreue ab, die sich auf Syntheseebene in der Fehlerrate niederschlägt. Synthetische und optische Fehler in unseren aktuellen Array-Fertigungsprotokollen werden an anderer Stelle diskutiert und berichtet. Parallel dazu werden derzeit Anstrengungen unternommen, um den Fertigungsumfang und den Durchsatz weiter zu erhöhen.
Offenlegungen
Die Autoren bescheinigen, dass sie keine Verbindung zu einer gewinnorientierten Organisation haben.
Danksagungen
Diese Arbeit wurde vom Wissenschaftsfonds (FWF-Stipendien P23797, P27275 und P30596) und dem Schweizerischen Nationalfonds (Grant #PBBEP2_146174) unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Slide functionalization | |||
| Acetic acid >99.8% | Sigma | 33209 | For RNA deprotection |
| CNC router | Stepcraft | 300 CK | |
| Ethanol absolute | VWR | 1.07017.2511 | For deprotection and functionalization |
| N-(3-triethoxysilylpropyl)-4-hydroxybutyramide | Gelest | SIT8189.5 | Silanizing reagent |
| Nexterion Glass D microscope slides | Schott | 1095568 | |
| Polymax 1040 | Heidolph | Orbital shaker | |
| Proclean 507 Ultrasonic water bath | Ulsonix | To clean slides after drilling | |
| Tickopur RW 77 Special Purpose Cleaner | Sigma | Z860086 | To clean slides after drilling |
| Microarray synthesis | |||
| 0.25 M dicyanoimidazole in ACN | Biosolve | 0004712402BS | Activator |
| 0.7 XGA DMD | Texas Instruments | Digital Micromirror Device | |
| 20 mM I2 in pyridine/H2O/THF | Sigma | L860020-06 | Oxidizer |
| 250 μm thick Chemraz 584 perfluoroelastomer | FFKM | Lower teflon gasket | |
| 2'-O-ALE RNA phosphoramidites | ChemGenes | ||
| 365 nm high-power UV-LED | Nichia | NVSU333A | |
| 5'BzNPPOC DNA phosphoramidites | Orgentis | ||
| 5'NPPOC DNA phosphoramidites | FlexGen | ||
| Acetonitrile | Biosolve | 0001205402BS | For DNA synthesis |
| Amidite Diluent for DNA synthesis | Sigma | L010010 | For dissolving phosphoramidites |
| Cleavable dT | ChemGenes | Base-sensitive monomer for library preparation | |
| DMSO | Biosolve | 0004474701BS | As exposure solvent |
| DNA and RNA microarray deprotection | |||
| Ethylenediamine >99.5% | Sigma | 3550 | For deprotection |
| Expedite 8909 | PerSeptive Biosystems | DNA synthesizer | |
| Hydrazine hydrate 50-60% hydrazine | Sigma | 225819 | For RNA deprotection |
| Imidazole | Sigma | 56750 | |
| Industrial Strength lower-density PTFE tape | Gasoila | Thin, upper teflon gasket | |
| Pyridine >99% | Sigma | P57506 | For RNA deprotection |
| Triethylamine >99% | Sigma | T0886 | For RNA deprotection |
| β-carotene | Sigma | C9750 | For library preparation |
| Hybridization and scanning | |||
| 20x Sodium Saline Citrate | Roth | 1054.1 | |
| 5'Cy3-labelled complementary strand | Eurogentec | For duplex hybridization | |
| Biopur Safe-Lock microcentrifuge tube | Eppendorf | ||
| BSA (10 mg/mL) | Promega | R3961 | |
| EDTA molecular biology grade | Promega | H5031 | |
| GenePix 4100A | Molecular Devices | Microarray scanner | |
| Hybridization oven | Boekel Scientific | 230500 | |
| MES monohydrate | Sigma | 69889 | |
| MES sodium | Sigma | M3058 | |
| NaCl >99.5% | Sigma | 71376 | |
| SecureSeal SA200 hybridization chamber | Grace BioLabs | 623503 | |
| Spectrafuge mini | Labnet | C1301 | Microarray centrifuge |
| Tween-20 molecular biology grade | Sigma | P9416 | |
| Data extraction | |||
| Excel | Microsoft | For data extraction | |
| MatLab | MathWorks | Microarray design | |
| NimbleScan 2.1 | Roche NimbleGen | ||
| Desalting and quantification | |||
| NanoDrop One Spectrophotometer | Thermo Scientific | ||
| Toluene | Merck | ||
| ZipTip C18 | Millipore | ZTC18s008 | Desalting pipet tips |
Referenzen
- Bumgarner, R. Current protocols in molecular biology. 101, 22(2013).
- Berger, M. F., Bulyk, M. L. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nature Protocols. 4 (3), 393-411 (2009).
- Tietjen, J. R., Donato, L. J., Bhimisaria, D., Ansari, A. Z. Sequence-specificity and energy landscapes of DNA-binding molecules. Methods in enzymology. 497, 3-30 (2011).
- Katilius, E., Flores, C., Woodbury, N. W. Exploring the sequence space of a DNA aptamer using microarrays. Nucleic Acids Research. 35 (22), 7626-7635 (2007).
- Franssen-van Hal, N. L. W., et al. Optimized Light-Directed Synthesis of Aptamer Microarrays. Analytical Chemistry. 85 (12), 5950-5957 (2013).
- Matteucci, M. D., Caruthers, M. H. Nucleotide Chemistry .4. Synthesis of Deoxyoligonucleotides on a Polymer Support. Journal of the American Chemical Society. 103 (11), 3185-3191 (1981).
- Eroshenko, N., Kosuri, S., Marblestone, A. H., Conway, N., Church, G. M. Gene Assembly from Chip-Synthesized Oligonucleotides. Current Protocols in Chemical Biology. 2012, (2012).
- Kosuri, S., et al. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nature Biotechnology. 28 (12), 1295(2010).
- Richmond, K. E., et al. Amplification and assembly of chip-eluted DNA (AACED): a method for high-throughput gene synthesis. Nucleic Acids Research. 32 (17), 5011-5018 (2004).
- Schmidt, T. L., et al. Scalable amplification of strand subsets from chip-synthesized oligonucleotide libraries. Nature Communications. 6, (2015).
- Grass, R. N., Heckel, R., Puddu, M., Paunescu, D., Stark, W. J. Robust Chemical Preservation of Digital Information on DNA in Silica with Error-Correcting Codes. Angewandte Chemie International Edition. 54 (8), 2552-2555 (2015).
- Erlich, Y., Zielinski, D. DNA Fountain enables a robust and efficient storage architecture. Science. 355 (6328), 950-953 (2017).
- Garalde, D. R., et al. Highly parallel direct RNA sequencing on an array of nanopores. Nature Methods. 15 (3), 201-206 (2018).
- Cleary, M. A., et al. Production of complex nucleic acid libraries using highly parallel in situ oligonucleotide synthesis. Nature Methods. 1 (3), 241-248 (2004).
- LeProust, E. M., et al. Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Research. 38 (8), 2522-2540 (2010).
- Lietard, J., Damha, M. J., Somoza, M. M. Enzymatic and Chemical Synthesis of Nucleic Acid Derivatives. Fernández-Lucas, J. , Wiley Online Books. (2018).
- Fodor, S. P. A., et al. Spatially Addressable Parallel Chemical Synthesis. Science. 251 (4995), 767-773 (1991).
- Singh-Gasson, S., et al. Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array. Nature Biotechnology. 17 (10), 974-978 (1999).
- Pease, A. C., et al. Light-Generated Oligonucleotide Arrays for Rapid DNA-Sequence Analysis. Proceedings of the National Academy of Sciences of the United States of America. 91 (11), 5022-5026 (1994).
- Holz, K., Lietard, J., Somoza, M. M. High-Power 365 nm UV LED Mercury Arc Lamp Replacement for Photochemistry and Chemical Photolithography. ACS Sustainable Chemistry & Engineering. 5 (1), 828-834 (2017).
- Lackey, J. G., Mitra, D., Somoza, M. M., Cerrina, F., Damha, M. J. Acetal Levulinyl Ester (ALE) Groups for 2′-Hydroxyl Protection of Ribonucleosides in the Synthesis of Oligoribonucleotides on Glass and Microarrays. Journal of the American Chemical Society. 131 (24), 8496-8502 (2009).
- Lackey, J. G., Somoza, M. M., Mitra, D., Cerrina, F., Damha, M. J. In-situ chemical synthesis of rU-DNA chimeras on chips and enzymatic recognition. Chimica Oggi-Chemistry Today. 27 (6), 30-33 (2009).
- Lietard, J., Ameur, D., Damha, M., Somoza, M. M. High-density RNA microarrays synthesized in situ by photolithography. Angewandte Chemie International Edition. 57 (46), 15257-15261 (2018).
- Agbavwe, C., et al. Efficiency, Error and Yield in Light-Directed Maskless Synthesis of DNA Microarrays. Journal of Nanobiotechnology. 9, (2011).
- Sack, M., et al. Express photolithographic DNA microarray synthesis with optimized chemistry and high-efficiency photolabile groups. Journal of Nanobiotechnology. 14, (2016).
- Kretschy, N., Holik, A. K., Somoza, V., Stengele, K. P., Somoza, M. M. Next-Generation o-Nitrobenzyl Photolabile Groups for Light-Directed Chemistry and Microarray Synthesis. Angewandte Chemie International Edition. 54 (29), 8555-8559 (2015).
- Sack, M., Kretschy, N., Rohm, B., Somoza, V., Somoza, M. M. Simultaneous Light-Directed Synthesis of Mirror-Image Microarrays in a Photochemical Reaction Cell with Flare Suppression. Analytical Chemistry. 85 (18), 8513-8517 (2013).
- Lietard, J., et al. Base-cleavable microarrays for the characterization of DNA and RNA oligonucleotides synthesized in situ by photolithography. Chemical Communications. 50 (85), 12903-12906 (2014).
- Krotz, A. H., et al. Solution stability and degradation pathway of deoxyribonucleoside phosphoramidites in acetonitrile. Nucleosides Nucleotides & Nucleic Acids. 23 (5), 767-775 (2004).
- Hargreaves, J. S., Kaiser, R., Wolber, P. K. The Degradation of Dg Phosphoramidites in Solution. Nucleosides Nucleotides & Nucleic Acids. 34 (10), 691-707 (2015).
- McGall, G. H., et al. The efficiency of light-directed synthesis of DNA arrays on glass substrates. Journal of the American Chemical Society. 119 (22), 5081-5090 (1997).
- Collett, J. R., et al. Functional RNA microarrays for high-throughput screening of antiprotein aptamers. Analytical Biochemistry. 338 (1), 113-123 (2005).
- Buenrostro, J. D., et al. Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nature Biotechnology. 32 (6), 562-568 (2014).
- Wu, C. -H., Holden, M. T., Smith, L. M. Enzymatic Fabrication of High-Density RNA Arrays. Angewandte Chemie International Edition. 53 (49), 13514-13517 (2014).
- Tian, J., et al. Accurate multiplex gene synthesis from programmable DNA microchips. Nature. 432 (7020), 1050-1054 (2004).
- Lietard, J., et al. Mapping the affinity landscape of Thrombin-binding aptamers on 2'F-ANA/DNA chimeric G-Quadruplex microarrays. Nucleic Acids Research. 45 (4), 1619-1632 (2017).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten