
Method Article
High-Density DNA and RNA microarrays - Photolithographic Synthesis, Hybridization and Preparation of Large Nucleic Acid Libraries
In This Article
Summary
In this article, we present and discuss new developments in the synthesis and applications of nucleic acid microarrays fabricated in situ. Specifically, we show how the protocols for DNA synthesis can be extended to RNA and how microarrays can be used to create retrievable nucleic acid libraries.
Abstract
Photolithography is a powerful technique for the synthesis of DNA oligonucleotides on glass slides, as it combines the efficiency of phosphoramidite coupling reactions with the precision and density of UV light reflected from micrometer-sized mirrors. Photolithography yields microarrays that can accommodate from hundreds of thousands up to several million different DNA sequences, 100-nt or longer, in only a few hours. With this very large sequence space, microarrays are ideal platforms for exploring the mechanisms of nucleic acid·ligand interactions, which are particularly relevant in the case of RNA. We recently reported on the preparation of a new set of RNA phosphoramidites compatible with in situ photolithography and which were subsequently used to grow RNA oligonucleotides, homopolymers as well as mixed-base sequences. Here, we illustrate in detail the process of RNA microarray fabrication, from the experimental design, to instrumental setup, array synthesis, deprotection and final hybridization assay using a template 25mer sequence containing all four bases as an example. In parallel, we go beyond hybridization-based experiments and exploit microarray photolithography as an inexpensive gateway to complex nucleic acid libraries. To do so, high-density DNA microarrays are fabricated on a base-sensitive monomer that allows the DNA to be conveniently cleaved and retrieved after synthesis and deprotection. The fabrication protocol is optimized so as to limit the number of synthetic errors and to that effect, a layer of β-carotene solution is introduced to absorb UV photons that may otherwise reflect back onto the synthesis substrates. We describe in a step-by-step manner the complete process of library preparation, from design to cleavage and quantification.
Introduction
The practical use of DNA microarrays have traditionally been in the study of the variations in the gene expression levels between two cell populations, using complementary strands and fluorescence as a detection method1. Occasionally, DNA microarrays venture into binding events with non-nucleic acid ligands, such as proteins, with a strategy of systematic sequence permutation that offers a comprehensive overview of the binding landscape2,3,4,5. This approach effectively transforms microarrays from mere hybridization surfaces into platforms with broad sequence coverage, which would be an asset for the study of the richer and more complex world of RNA structure and function. Supported by the extremely efficient phosphoramidite coupling reaction6, in situ synthesized DNA arrays can now also be regarded as a cheap source of DNA7, which is becoming particularly relevant considering the ever-increasing demand for nucleic acid material for gene assembly8,9, DNA-based nanostructures10, information storage or sequencing11,12. Likewise, sequencing technologies are likely to benefit from the development of methods that yield very complex mixtures of RNA oligonucleotides13. In this context, array fabrication protocols that allow for oligonucleotides to be synthesized in situ and at high density are ideally placed to meet the needs of the rapidly expanding field of nucleic acid biotechnology. However, with a field as diverse as biotechnology, the purpose of each application may require that DNA on microarray be produced either at high-throughput or with a very low amount of synthetic errors14,15, or both, requiring a closer look at the synthesis protocols of DNA microarrays which, historically, have been primarily optimized for hybridization assays. Meanwhile, in situ synthesis of RNA microarrays has been found to be a challenging endeavor, with most of the difficulty associated with the protecting group for the 2'-OH function, usually a silyl moiety in standard solid-phase synthesis that is removed with fluorine-based reagents, chemicals that are incompatible with glass or silicon surfaces. Those issues and challenges in DNA and RNA microarray synthesis have lately been the subject of a large body of work, in particular with the photolithography approach16.
Photolithography uses UV light to unblock oligonucleotides before coupling and requires masks to construct a pattern of UV exposure, thereby spatially organizing and controlling the growth of oligonucleotides. Physical masks have been replaced by computer-controlled micromirrors whose tilting selectively reflects UV light onto the microarray substrate17,18,19. As a UV source, we use 365 nm light from a high power LED source20. Current photolithographic setups are equipped with micromirror arrays containing 1024 × 768 mirrors, corresponding to more than 780,000 individually addressable spots ("features") on a small area of just 1.4 cm2, or 1080p with arrays of 1920 × 1080, or >2 million mirrors. Each of the mirrors in the device therefore has direct control over the sequence grown on the corresponding feature. With the exception of UV light, photolithography functions like a solid-phase synthesis technique and adopts the cycle-based phosphoramidite chemistry. Only it requires an entirely different protection strategy for RNA synthesis to succeed. We developed a new series of light-sensitive RNA phosphoramidites bearing hydrazine-labile protecting groups21. These monomers allow for the RNA to be deprotected under mild conditions that do not affect the integrity of the surface. A first deprotection step uses triethylamine to remove the cyanoethyl phosphodiester protecting groups, while hydrazine is used in a second, separate step to remove those at the 2'-OH and exocyclic amine functions. In so doing, RNA oligonucleotides ~30-nt in length and of any sequence can now be synthesized in situ on microarrays22,23. In parallel, we also recently started to address the questions of throughput, quality and speed in DNA and RNA photolithography. We measured coupling efficiencies >99% for all DNA and RNA amidites (Figure 1) and investigated each individual step in the oligonucleotide elongation cycle, from oxidation time, to choice of activator and to optimal UV exposure24,25. We have brought in new light-sensitive 5' protecting groups that can be removed in seconds only, transforming the synthesis of hundreds of thousands of 100mers into a few hours-long process26. We have also doubled the throughput of array fabrication by exposing two substrates simultaneously27. Finally, we have introduced a dT phosphoramidite containing a base-sensitive succinyl group as a convenient way to cleave, collect and analyze DNA and RNA oligonucleotides, which is central to library preparation28.
In spite of the relatively mundane aspect of DNA and RNA solid-phase synthesis, especially for nucleic acid chemists, microarray photolithography remains a non-trivial upgrade requiring a complex setup, careful control and supervision of the process, and separate instructions for post-synthetic handling depending on the nature of the oligonucleotide and the type of application. In this article, we wish to provide a detailed presentation of the entire step-by-step procedure of in situ synthesis of DNA and RNA microarrays by photolithography, from experimental design to data analysis, with an emphasis on the preparation of instruments and consumables. We then describe the post-synthetic deprotection methods that correspond to the intended purpose of microarray fabrication (i.e., either hybridization or the recovery of nucleic acid libraries).
Protocol
1. Microarray design
- Write the sequences to be synthesized in a text editor, 5'→3', one line per sequence. For the quality control 25mer, use the sequence "GTCATCATCATGAACCACCCTGGTC". Use A, C, G and T letters for DNA nucleotides and 5, 6, 7 and 8 numbers for RNA nucleotides.
- In the case of library preparation, add an extra number (i.e., 9) at the 3' end of each sequence. This will correspond to the coupling of the base-sensitive monomer.
- Each sequence must be followed by a comma. Assign a name to each sequence after the comma, and verify that each lane follows the [sequence,#sequence_name] format (without brackets). Save the list of sequences as a .txt file.
- Start MatLab then load the program ChipDesign.m. Run the program.
- In the Property Panel window, load the microarray EntireChip layout file. Load the MilliChip layout file if the aim is to synthesize the entire array at four identical locations.
- In the Property Panel window, under Chip Specification, select Select Container then Synthesis Area. Under Pattern, select the pattern that yields the correct amount of addressable features, counting the number of sequences × number of replicates. For the quality control 25mer, select the 25:36 pattern.
- Under Select Container, select Fiducial. Fiducial features are usually synthesized at the corners of the synthesis area and are used to extract hybridization data. With Fiducial selected, write a sequence (5'→3') that will be synthesized on fiducial features and which can act as a positive control. For the 25mer experiment, use the same DNA sequence.
- Under Sequence, load the text file containing all written sequences. Make sure Randomize is selected. Give a title to the experiment in Project Title and, in Linker Sequence, write TTTTT (corresponding to a T5 linker).
- Press Generate, then find the array design files and masks (Figure 2) under MaskGen_delta_rc1/Designs folder. Make sure it contains a display script, a flow sequence and a design file.
- For library preparation, open the display script. Add an extra line at the bottom of the display script that copies exactly the first line of the script (e.g., display First_Mask.bmp 150). This will remove the terminal photoprotecting group at the 5' end of all oligonucleotides.
- Start the job creator AutoJob program, load a template by clicking on Load Template, then click on Display Script to load the display script file that was generated by MatLab. Press Generate. This step will create a series of instructions, called a jobfile, that will control the communication of the computer with the micromirrors and the DNA synthesizer.
2. Slide preparation and functionalization
- Drill one slide at two positions corresponding to the location of the inlet and outlet tubing on the synthesis cell. Use a 0.9 mm diamond bit on a CNC router to precisely and reliably drill. Rinse the drilled slides with ultrapure water and arrange them in a slide rack.
- Clean the surfaces by sonicating the slides in a water bath containing 5% of an ammonia-based special purpose cleaner for 30 min at 35 °C. Rinse the slide with double-distilled H2O and transfer them in a clean, dry rack.
- Arrange drilled and undrilled microscope slides in a slide rack. In a large graduated cylinder, prepare the functionalization solution by mixing 475 mL of ethanol (EtOH) with 25 mL of ddH2O, 10 g of silanizing reagent (N-(3-triethoxysilylpropyl)-4-hydroxybutyramide) and 1 mL of acetic acid. Stir well until homogeneous then transfer in a suitable, closed container.
- Place the loaded rack in the container, close the lid and let the container rock gently on an orbital shaker for 4 h at room temperature.
- After 4 h, discard the functionalization solution and replace with 500 mL of wash solution, consisting of 475 mL of EtOH, 25 mL of ddH2O and 1 mL of acetic acid. Agitate slowly for 20 min at room temperature, then discard and replace with 500 mL of fresh wash solution.
- After an additional 20 min at room temperature, discard the solution, dry the slides with a stream of argon and cure them in a pre-heated vacuum oven at 120 °C. After 2 h, switch off the oven and the vacuum pump but leave the slides under reduced pressure overnight. Then, bring the oven back to atmospheric pressure and store the slides in a desiccator until further use.
3. Preparation of synthesis reagents and reactants
- Bring the phosphoramidite powders (Figure 1) from their storage temperature (-25 or -45 °C) to room temperature in a desiccator.
- When the phosphoramidites reached room temperature, dissolve the powder with a volume of ultra-dry acetonitrile (<30 ppm H2O) in order to reach 30 mM concentration for standard DNA and RNA phosphoramidites, and 50 mM for the base-sensitive dT monomer (library preparation). Add a small molecular sieves bag to trap any trace of humidity.
- Prepare a solution of 1% (w/w) imidazole in DMSO by dissolving 11 g of imidazole into 1 L of dry DMSO. Shake well until completely dissolved. Attach the solution to the auxiliary port of the DNA synthesizer. This will be the exposure solvent necessary for complete removal of the 5′-photoprotecting group.
- For the synthesis of libraries, prepare a solution of 1% (w/v) β-carotene in dichloromethane by dissolving 100 mg of β-carotene in 10 mL of dichloromethane. Shake well in an amber glass bottle then wrap in aluminum foil.
4. Preparation and monitoring of microarray synthesis.
- Record temperature and humidity in the microarray fabrication room and make sure the DNA synthesizer is under sufficient helium pressure.
- Turn on the UV-LED and its cooling fan. Attach a UV intensity meter at the focal plane of incoming UV light and turn it on (Figure 3A).
- On the computer, start the jobfile/micromirror/synthesis files controller software (named WiCell). Turn on then initialize the micromirror device and load an all-white mask file by right-clicking on DMD, then selecting Load Image.
- Right-click on the UVS icon and select UV Shutter Open. Read the power value (in mW/cm2) on the intensity meter and count 60 s. After 60 s, read the power value again and note down the beginning and end values. Close the shutter by selecting UV Shutter Close and turn off the intensity meter. Calculate the average UV intensity value in mW/cm2.
- Calculate the exposure time necessary to reach a radiant energy of 6 J/cm2 for 5'-NPPOC photodeprotection (DNA and RNA microarrays) and 3 J/cm2 for 5'-BzNPPOC photodeprotection (DNA libraries), simply following the
 relationship.
relationship. - In the DNA synthesizer Workstation software, create a sequence file in the Sequence Editor by copying and pasting the contents of the flow sequence generated by MatLab. Add an extra two washing steps at the 3' end (default cycle letter: "s") to wash the surface of the substrates before moving on to the first coupling. Save and export the sequence file.
- In the Protocol Editor of the Workstation software, create a protocol that contains a dedicated cycle named after each of the letters and numbers present in the sequence file (e.g., if a sequence file contains only letters A, C, G and T, then the protocol file must contain four cycles, named A, C, G and T).
- Set the coupling time for DNA phosphoramidites (cycles A, C, G and T) to 15 s, to 120 s for rU phosphoramidite (cycle 8) and to 300 s for rA, rC and rG phosphoramidites (cycles 5, 6 and 7). For library preparation, set the coupling of the base-sensitive, cleavable dT monomer to 2 × 120 s (Table 1 and Table 2).
- Make sure the wait time between two Event 2 Out communication events in each cycle corresponds to the calculated UV exposure time to reach the necessary radiant energy. As for the sequence file, save and export the protocol file.
- In the WiCell software, load the job, sequence and protocol files in their respective sub-windows then click Send to send the sequence and protocol files to the DNA synthesizer.
- In the sequence file, count the number of couplings for each phosphoramidite.
- To measure the required volume (in µL) of phosphoramidite solution needed to perform each synthesis, multiply the number of couplings by 60. Add 250 µL to each volume for safety and priming of the line on the DNA synthesizer. Use a base volume of 250 µL for phosphoramidites requiring a single coupling only.
- Quickly transfer the solution into the vials on the DNA synthesizer corresponding to the port letter/number in the protocol cycles.
- Prime the ports with the phosphoramidite solutions with 10 prime pulses each in order to fill the lines with reagent. Prime the acetonitrile wash line before attaching the reaction cell.
- To assemble the synthesis cell, place a thick (250 µm) perfluoroelastomer (FFKM) gasket first on the quartz block of the cell. Place a drilled, functionalized microscope slide on top of the first gasket, and verify that the holes on the slides connect with the inlet and outlet tubing of the synthesis cell.
- Place a second, thin (50 µm) polytetrafluoroethylene (PTFE) gasket over the drilled slide, surrounding the two holes. Finally, place a second, functionalized but undrilled slide atop the second gasket (Figure 3B). Place a 4-screw metal frame on top of the assembled double-substrate cell and tighten the screw to the same clamping force (0.45 Nm) using a torque screwdriver.
- Attach the inlet and outlet tubing to the DNA synthesizer. Prime the acetonitrile wash line and verify the proper flow of acetonitrile (ACN) through the substrates. Disassemble and reassemble the cell if any leakage of ACN can be observed at this stage. Measure the volume of ACN at the waste line after going through 7 cycles of ACN priming. This volume should be 2 mL.
- Attach the synthesis cell at the focal plane of incoming UV light. In the case of library preparation, attach an extra inlet and outlet line to the back of the cell and fill the back chamber with the β-carotene solution (2 mL of solution is sufficient). Make sure there is no leakage (Figure 3C).
- Start the synthesis by first clicking on Run in the WiCell software. At the first WAIT command in the job file, press Start on the DNA synthesizer.
- During synthesis, regularly verify that the display of mask files coincides with UV exposure and opening of the shutter.
- After synthesis of regular microarrays, disconnect the cell from the synthesizer, disassemble the cell and use a diamond pen to etch the synthesis number onto the glass slides. Etch the number on the non-synthesized face of each slide. Transfer the slides into 50 mL centrifuge tubes and store in a desiccated area until further use.
- After the synthesis of library microarrays, first drain the β-carotene solution out of the chamber then wash it by flowing 2 × 5 mL of CH2Cl2, then drain. Proceed with disassembling as per step 4.21. The synthesized array may be visible to the naked eye (Figure 4).
5. DNA microarray deprotection
- Fill a staining glass jar with 20 mL of EtOH and 20 mL of ethylenediamine (EDA). Place the DNA-only microarrays vertically in the jar, close the lid, and leave the slides to deprotect for 2 h at room temperature.
CAUTION: EDA is an acutely toxic, corrosive and flammable liquid. Work with gloves in a well-ventilated fume hood. - After 2 h, retrieve the slides using tweezers and rinse them thoroughly with double-distilled H2O.
- Dry the slides in a microarray centrifuge for a few seconds then store in a desiccator.
6. RNA microarray deprotection
- In a 50 mL centrifuge tube, prepare a dry solution of 2:3 triethylamine/ACN (20 mL and 30 mL of each, respectively). Transfer one RNA microarray slide into the centrifuge tube, close the lid then wrap with plastic sealing film. Gently shake the centrifuge tube on an orbital shaker for 1h and 30 min at room temperature.
- After 1h and 30 min, remove the slide, wash with 2 × 20 mL dry ACN then dry in a microarray centrifuge for a few seconds.
CAUTION: Triethylamine is an acutely toxic, corrosive and flammable liquid. Acetonitrile is toxic and flammable. Work with gloves in a well-ventilated fume hood.
NOTE: First deprotection step complete. - Prepare a 0.5 M hydrazine hydrate solution in 3:2 pyridine/acetic acid. First, mix 20 mL of acetic acid and 30 mL of pyridine in a graduated cylinder. Wait for the solution to cool down before adding 1.21 mL of hydrazine hydrate. Transfer around 40 mL of the resulting solution into a 50 mL centrifuge tube.
CAUTION: Hydrazine hydrate is an acutely toxic and corrosive liquid. Pyridine is highly flammable and acutely toxic. Acetic acid is flammable and corrosive. Work with gloves in a well-ventilated fume hood. - After the first deprotection step, transfer the RNA slide into the hydrazine hydrate solution, close the lid and wrap with plastic sealing film. Gently shake the tube on an orbital shaker for 2 h at room temperature. After 2 h, remove the slide, wash with 2 × 20 mL of dry ACN then dry in a microarray centrifuge for a few seconds.
NOTE: Second deprotection step complete. - If the RNA microarray also contains DNA nucleotides, proceed with a third deprotection step. In a 50 mL centrifuge tube, mix 20 mL of EDA with 20 mL of EtOH. Add the DNA/RNA microarray to the 1:1 EDA/EtOH solution and leave at room temperature for 5 min.
- After 5 min, remove the slide, wash with 2 × 20 mL of sterile water then dry in a microarray centrifuge and store in a desiccator.
7. Hybridization with a fluorescently-labelled complementary strand
- Thaw acetylated bovine serum albumin (10 mg/mL) and the Cy3-labelled complementary strand (100 nM) and warm up to room temperature.
- In a 1.5 mL sterile microcentrifuge tube, mix 150 µL of 2x MES buffer (200 mM 2-(N-morpholino)ethanesulfonic acid; 1.8 M NaCl; 40 mM EDTA; 0.02% Tween-20) with 26.7 µL of Cy3-labelled DNA, 13.4 µL of acetylated BSA and 110 µL of sterile H2O. Mix and vortex. Double the volume if both slides are to be used for hybridization.
- Pre-warm a hybridization oven to a temperature below the Tm of the duplex but high enough to ensure good discrimination between full-match and non-match sequences. For the quality control 25mer, set the temperature to 42 °C (Tm of 59 °C).
- Carefully place a self-adhesive 300 µL hybridization chamber over the synthesis area on each slide and pipet in the hybridization solution prepared above. Cover the holes of the chamber with adhesive dots and wrap the entire slide in aluminum foil.
- Place the microarray slide into the hybridization oven, cover and let it gently rotate at the selected hybridization temperature for 2 h.
- After 2 h, detach the slide, remove the aluminum foil and carefully tear off the hybridization chamber. Transfer the slides into a centrifuge tube containing 30 mL of Non-Stringent Wash Buffer (NSWB; 0.9 M NaCl, 0.06 M phosphate, 6 mM EDTA, 0.01% Tween20, pH 7.4) and shake vigorously for 2 min at room temperature.
- Transfer the slide into a centrifuge tube containing 30 mL of Stringent Wash Buffer (SWB; 100 mM MES, 0.1 M NaCl, 0.01% Tween20) and shake vigorously for 1 min.
- Finally, transfer the slide into a centrifuge tube containing 30 mL of Final Wash Buffer (FWB; 0.1x sodium saline citrate) and shake for a few seconds. Dry the slide in a microarray centrifuge.
- Place the dry microarray, synthesis area facing down, in the slide holder of the microarray scanner. For Cy3-labelled duplexes, scan at 5 µm resolution with a 532 nm excitation wavelength, a 575 nm filter and a photomultiplier of 350. Save the high resolution scan as a .tif image file (Figure 5A).
8. Data extraction and analysis
- Prior to data extraction, rotate the saved array scan in an image editor so as to situate the longest chain of fiducial features at the top-left corner of the scan. Save the rotated image.
- Start NimbleScan, then press File | Open and load the array scan. Then, by clicking Browse in the Design file subsection, load the .ndf design file that was automatically generated during the design of the microarray experiment. Then click Open.
- In View, click Auto Contrast/Brightness. Click on the icon Manually align above the scan. Place four square-shaped markers at the four corners of the scan, then click the now green icon again. Extract the hybridization data by clicking on Analysis, Reports then Probe Report.
- Open the .probe report file in a spreadsheet editor. Keep columns B and I and discard the rest before proceeding with calculating average values and standard deviation from the extracted data (Figure 5B).
9. Library deprotection, cleavage and recovery
- To deprotect and cleave DNA libraries, prepare a solution of 1:1 dry EDA/toluene in a 50 mL centrifuge tube. Immerse the slide into the cleavage solution, close the slide and wrap with plastic sealing film, then gently rotate in an orbital shaker for 2 h at room temperature.
- After 2 h, remove the slide and wash with 2 × 20 mL of scrupulously dry ACN. Remove the slide and let it air dry.
- With a pipette, apply 100 µL of sterile H2O over the now discernable synthesis area. Pipet the solution up and down a few times before transferring it into a 1.5 mL microcentrifuge tube. Repeat the process and combine the microarray eluate into the same tube (Figure 6).
- Evaporate the 2 × 100 µL of chip eluate to dryness then redissolve into 10 µL of nuclease-free H2O. Measure the absorbance on a UV-Vis spectrophotometer.
- Desalt the DNA library on a 10 µL pipette tip equipped with C18 resin. First, transform the chip eluate into a 0.1 M triethylammonium acetate (TEAA) buffered solution.
- Wet the resin by aspiring 3 x 10 µL of H2O/ACN 1:1, and equilibrate the resin by washing with 3 x 10 µL of 0.1 M TEAA buffer. Bind the DNA by pipetting the chip eluate 10 times up and down through the resin. Wash the resin with 3 x 10 µL of 0.1 M TEAA buffer, 3 x 10 µL of H2O.
- Elute the desalted DNA from the tip by washing the resin with 10 µL of H2O/ACN 1:1. Dry the desalted solution down and redissolve into 10 µL of sterile H2O. Measure the absorbance on a UV-Vis spectrophotometer (Figure 7). Evaporate the library to dryness and store at -20 °C until further use.
Results
DNA and RNA microarray hybridization
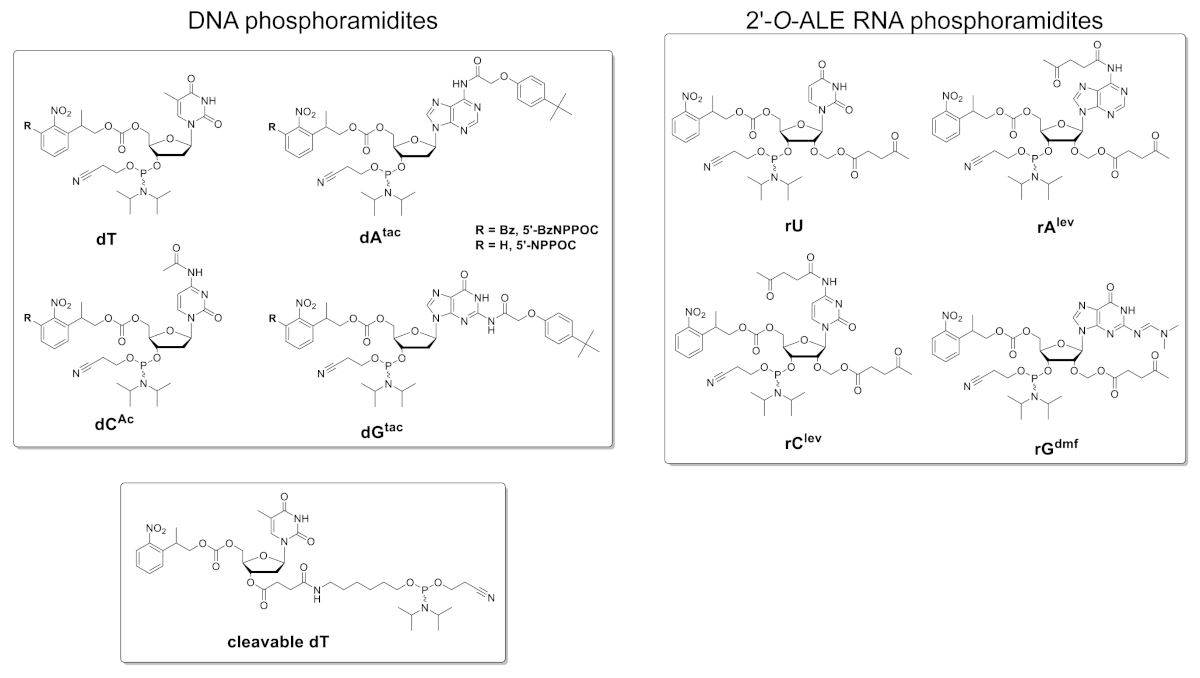
Figure 5 shows the results of a hybridization assay performed on a microarray containing the DNA and RNA versions of a 25mer sequence (5'-GTCATCATCATGAACCACCCTGGTC-3' in DNA form). The scan in Figure 5A appears in a greenscale format corresponding to the excitation/emission spectrum of Cy3 fluorescence, with fluorescence intensity recorded in arbitrary units between 0 and 65536. The array design followed the 25:36 feature layout described in the protocol section. The scan is shown after proper orientation of the array, with the top left corner populated with the longest chain of fiducial features. Here, fiducial features contain the DNA version of the 25mer and should, in principle, always give a positive fluorescence signal in order to perform scan alignment and data extraction. The hybridized microarray should appear uniformly bright, with the edges of the synthesis area being however usually brighter than the center (up to 50% brighter). The large amount of sequence replicates, randomly distributed throughout the area, reduces the impact of spatial artefacts. Here, each sequence (DNA and RNA) was synthesized at 2,000 random locations. There is typically low fluorescence noise (background), <50 a.u., which leads to a signal/noise ratio in the order of 200:1 to 800:1 in hybridization assays. After data extraction, fluorescence intensities are averaged out and plotted ±SD.
There is significant variability in absolute fluorescence values between experiments. Here, we show the results for three independent syntheses using the same fabrication parameters and the same post-synthetic handling. The 25mer DNA, when hybridized to its complementary Cy3-labelled DNA strand, will yield fluorescence signals ranging anywhere from 20,000 to 30,000, very rarely above or below. The 25mer RNA, when hybridized to the same Cy3-labelled DNA complement, will give fluorescence intensities on the corresponding features ranging from 15,000 to 20,000. However, fluorescence intensity of the RNA/DNA duplexes will occasionally drop below 8,000, when the corresponding DNA/DNA duplexes will still fluoresce within the 20,000-30,000 range. In such cases, the results for RNA may be regarded as sub-optimal. A synthesis or hybridization failure, either for DNA or RNA, will be immediately noticeable during scanning from the obvious lack of fluorescence. There are multiple opportunities for the RNA synthesis to either fail or partially succeed and they will be outlined in the discussion part.
Library deprotection, cleavage and recovery
Depending on the complexity and the density of the library, the shape and outline of the synthesized array can be seen without magnification but under proper lighting (Figure 4), with the DNA still in protected form. After deprotection with EDA/toluene, and before the addition of water in order to collect the cleaved library, the synthesis area containing the now deprotected oligonucleotides may stand out as a hydrophilic zone, when the rest of the glass slide will appear covered with a turbid hydrophobic layer. The direct observation of the synthesis area depends on the total area used to synthesize oligonucleotides: greater use of the synthesis area will correspond to a greater chance of clear distinction between hydrophilic and hydrophobic regions on the surface. Conversely, libraries synthesized using fewer mirrors and with smaller features may not be immediately observable.
Similarly, the amount of recovered DNA after desalting is directly proportional to the total area used for synthesis. If all features are used for oligonucleotide synthesis, the cleavage and recovery procedure should yield between 25 and 30 pmol of DNA. A 10% use of the synthesis area will therefore afford only around 3 pmol of DNA.

Figure 1. Chemical structures of the DNA and RNA phosphoramidites used in oligonucleotide synthesis by microarray photolithography. Standard nitrophenylpropyloxycarbonyl (NPPOC) photosensitive protecting groups at the 5'-OH are used in regular DNA and RNA microarray synthesis for hybridization purposes. For the synthesis of complex DNA libraries, the more photolabile benzyl-NPPOC (BzNPPOC) are preferred at the 5'-OH, as BzNPPOC is removed twice as fast as NPPOC, which significantly reduces total microarray synthesis time. DNA oligonucleotides for libraries also require the coupling of a cleavable dT monomer at the 3' end. This monomer, which carries a succinyl ester function, will be cleaved during deprotection, allowing for the DNA to be collected from the microchip. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. Example of a mask as an image file sent to the micromirror device during UV exposure. The white pixels correspond to mirrors which will be tilted in the "ON" position, reflecting UV light onto the synthesis cell. Black pixels correspond to "OFF" mirrors, where the UV light will be reflected away from the cell. White pixels will therefore allow for the coupling of the next incoming phosphoramidite on the oligonucleotides found at the corresponding features on the glass substrates. Oligonucleotides synthesized on the features whose corresponding mirrors are, in this mask file, black pixels will however remain inert during the next coupling event. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Photographs of the microarray photolithography optical and synthesis setup. (A) Optical circuit for UV exposure. UV light from the UV-LED is first homogenized through a rectangular-cross-section light-pipe then reflects onto the micromirrors. Micromirrors which have been tilted into an "OFF" position will reflect UV-light away from the synthesis cell, but micromirrors in the "ON" position will reflect light onto the synthesis cell, situated at the focal plane, by first passing through a 1:1 Offner Relay imaging system. (B) The synthesis cell, once assembled, consists of a drilled slide placed first onto the quartz block of the cell, separated by a thick PTFE gasket (not shown). A second, non-drilled slide is then positioned over the drilled slide, separated by a thin PTFE gasket. A metal frame (not shown) holds the assembly together. (C). For library preparation, once the synthesis cell is attached at the focal plane of incoming UV light, the chamber located between the quartz block and the drilled slide is filled with a 1% solution of β-carotene in CH2Cl2. To do so, an additional inlet and outlet tubing is attached to the quartz block and the orange solution flows from the rightmost to the leftmost position. The flow of reagents and solvents for the synthesis is shown in white arrows. Please click here to view a larger version of this figure.
{kind=link}

Figure 4. The synthesis area is usually visible to the naked eye. Here, a DNA library can be seen on the glass surface right after synthesis, with the DNA still in protected form. Please click here to view a larger version of this figure.
{kind=link}

Figure 5. Hybridization assays to the 25mer DNA and RNA sequences synthesized in situ on microarrays. (A) Fluorescence scan of the entire hybridized DNA and RNA microarray. 25mer DNA and RNA oligonucleotides are hybridized to their Cy3-labelled complementary strands. The array was scanned with a laser at a 532 nm excitation wavelength, at 5 µm resolution. (B) Fluorescence intensities (arbitrary units) of the DNA:DNA and RNA:DNA duplexes in three separate experiments. The light green data for in situ synthesized RNA oligonucleotides can be considered suboptimal, when compared to the fluorescence intensity of the corresponding DNA sequences. Error bars are SD. Please click here to view a larger version of this figure.
{kind=link}

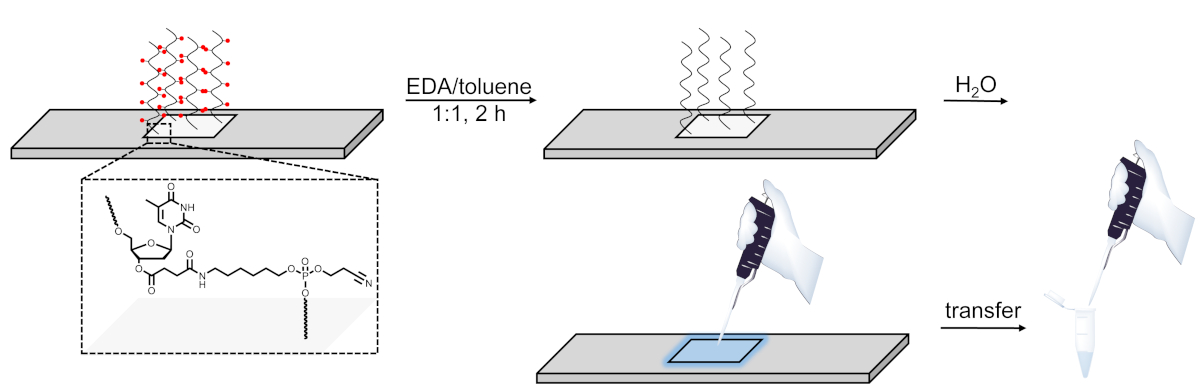
Figure 6. Schematic representation of the deprotection, cleavage and recovery procedure for DNA libraries synthesized on microarrays. DNA sequences are grown on a base-sensitive cleavable dT nucleoside (shown in the zoomed area). After synthesis, deprotection of the DNA oligonucleotides (base protecting groups are represented as red spheres) in EDA/toluene leaves the deprotected material electrostatically bound to the surface and can then be pipetted out by applying a small amount of water onto the synthesized area. Please click here to view a larger version of this figure.
{kind=link}

Figure 7. Representative absorbance spectrum (220 - 350 nm) of a cleaved, desalted DNA library containing 4,000 different sequences, 100-nt in length. A total of 940 ng of DNA was isolated from a single array synthesis, corresponding to 30 pmol of DNA total, or 15 pmol per glass substrate. Please click here to view a larger version of this figure.
{kind=link}

Table 1. Representative cycle protocol for coupling/oxidation/photodeprotection of 5'-BzNPPOC-dA, assuming the corresponding phosphoramidite was loaded on port "A". Coupling time (in seconds) is shown at the "couple monomer" line. The UV photodeprotection time, here corresponding to a radiant energy of 3 J/cm2 (BzNPPOC photochemistry), is calculated as the time elapsed between the two "Event 2 Out" communication signals.

Table 2. Representative cycle protocol for coupling/oxidation/photodeprotection of 5'-NPPOC-rA, assuming the corresponding RNA phosphoramidite was loaded on port "A". Coupling time (in seconds) is shown at the "couple monomer" line. The UV photodeprotection time, here corresponding to a radiant energy of 6 J/cm2 (NPPOC photochemistry), is calculated as the time elapsed between the two "Event 2 Out" communication signals.
Discussion
Solid-phase DNA and RNA synthesis is the bread and butter of every nucleic acid chemistry lab, and although the addition of the photolithography component is admittedly a complex operation, microarray fabrication mediated by UV light is also a very reliable process. It is, in addition, the only available method for in situ RNA synthesis on microarrays. Still, as in any multi-stage experimental procedure, there is ample room for human error.
Perhaps the most critical step is the coupling of a phosphoramidite, as it needs to be a constantly high-yielding chemical reaction in order to afford oligonucleotides with few synthetic errors. In our microarray synthesis protocol, phosphoramidite coupling is even more pivotal to overall synthesis quality since the fabrication process bypasses capping and prevents oligonucleotide purification. Stepwise coupling efficiencies above 99% have been calculated for all photosensitive DNA and RNA phosphoramidites, even for very short coupling times (15 s)24 but lower coupling yields can occasionally occur, particularly in the case of dG amidites. The stability of the solubilized phosphoramidites at room temperature has been investigated before and was shown to depend on the nature of the nucleobase, with guanosine phosphoramidites prone to extensive degradation in only a matter of days29,30. But when stored at -25 °C, dG phosphoramidites dissolved in ACN as 30 mM solution were found to be stable for several weeks. The relative instability of dG phosphoramidite solutions at room temperature does however mean that they should not be kept attached to the DNA synthesizer for several days.
For RNA phosphoramidites, the coupling yield is very dependent on phosphoramidite quality (which can be assessed by 31P NMR spectroscopy) and coupling time. Coupling times of 5 minutes for rA, rG, rC and 2 min for rU appear necessary. Indeed, we found that shortening the condensation time to 2 min for all RNA phosphoramidites led to significantly lower hybridization signals.
The DNA synthesizer itself, as well as the reagents and solvents, certainly needs to be as clean as possible in order to achieve the highest yield of oligonucleotide synthesis. However, insoluble material, salts or particles, can accumulate over time in the lines and tubing of the delivery system, leading to a gradual decrease in consumption of reagents and reactants. Where a general cleaning of the synthesizer does not resolve a low output volume, an increase in the number of pulses can be an alternative solution. Particularly useful in the case of low phosphoramidite consumption, the line in the coupling protocol corresponding to the pumping of a mix of phosphoramidite and activator (third line of the coupling subsection in Table 1 and Table 2) can be modified, from 6 to 9 pulses without any appreciable negative effect on synthesis quality. Furthermore, the number of pulses of activator needed to bring the amidite/activator mix to the synthesis substrate (currently 6, fourth line in the coupling subsection, see Table 1 and Table 2) depends on the DNA synthesizer itself as well as on tubing length in the synthesis cell. This number can be adjusted after replacing the phosphoramidite with a colored solution and counting the number of pulses needed to push the colored mix to the glass substrate for coupling.
The method described herein allows for DNA and RNA synthesis to proceed simultaneously, on the same microarray. Hybrids of DNA and RNA may also be prepared without any change to the array fabrication protocols, and as long as the three-step deprotection protocol is followed. However, it should be noted that RNA-only microarrays only require a two-step deprotection: a decyanoethylation first with Et3N followed by hydroxyl and base deprotection with hydrazine. DNA nucleobases were found to be incompletely deprotected under those conditions, and need the additional step in EDA in order to effect the complete removal of phenoxyacetyl (Pac) groups. This extra treatment with EDA is shorter (5 min) than for the standard deprotection of DNA microarrays31, but it is sufficient to drive it to completion after the triethylamine and hydrazine treatments. In addition, a short reaction time with EDA limits the exposure of a fully-deprotected RNA oligonucleotide to basic conditions.
An advantage of in situ RNA array synthesis over alternative methods like spotting or DNA transcription32,33,34 is the ability to store the synthesized RNA microchip in protected form until use, thus avoiding the risk of potential RNA degradation. Post-synthetic procedures for RNA does, on the other hand, imply that consumables and reagents are kept sterile and that handling is performed under RNase-free conditions. Of note, we found that the addition of RNase inhibitor to the hybridization mix did not yield stronger hybridization signals for the RNA features.
The synthesis of DNA libraries on a base-sensitive monomer is more complex than the synthesis of a few control sequences on a surface, and as such is certainly more prone to design errors. Yet, assuming that the sequence design (i.e., the nature and the number of sequences) is correct, transforming this list into a collection of exposure masks and an ordered series of coupling cycles remains a straightforward process. However, important variations from standard microarray synthesis exist and are critical to a successful fabrication of a high-density library array.
First, a base-sensitive dT monomer is coupled as the first phosphoramidite after synthesis of the linker. The coupling yield of this monomer (Figure 1) was found to be relatively low, around 85%28, which is why efforts are made to improve its incorporation rate, either by increasing its concentration in ACN from 30 mM to 50 mM, or by repeating the coupling step: two consecutive coupling reactions using fresh monomers, or two separate but consecutive coupling cycles.
The second change is the addition of a β-carotene solution in the back chamber of the synthesis cell, which conveniently absorbs 365 nm light. This is an important modification of the photolithography setup as it prevents UV light from reflecting back onto the array substrate. Indeed, after traversing the interstitial medium between the substrates, incoming UV light exits through the drilled slide and reaches the quartz block of the cell. The Fresnel equations predict that ~4% of perpendicularly incident UV light will reflect from each of the three downstream air-glass interfaces (exit side of the 2nd substrate and both sides of the quartz block) and back onto the synthesis substrate, leading to unintended exposure of photoprotected oligonucleotides. Diffraction and scattering also contribute to "off-target" photodeprotection and, therefore, to nucleotide insertion, which directly affects the error rate of synthesis, but these contributions are much smaller than reflections and can mainly be addressed by reducing synthesis density (leaving gaps between features). We have found that the level of β-carotene solution in the lower chamber of the cell tends to slightly decrease only during the first minutes of array synthesis, and therefore needs to be monitored and readjusted.
Finally, the third change is the deprotection solution, replacing EtOH for toluene, which keeps the cleaved DNA library bound to the surface, presumably through electrostatic interactions. Applying a small amount of water to the synthesis area after ACN washing allows for the library to be conveniently collected. The process is however only successful if the water content in EDA and toluene is minimal, rendering the nucleic acid entirely insoluble in the deprotection cocktail. Alternatively, DNA libraries may be cleaved off the chip using ammonia9,10,14,35, then further deprotected by heating the DNA-containing aqueous ammonia solution to 55 °C overnight. The recovery of DNA libraries using ammonia is however not compatible with RNA. RNA oligonucleotides on a base-cleavable substrate can be eluted from the surface using the same EDA/toluene procedure described above, but only at the penultimate stage after the Et3N and hydrazine two-step deprotection strategy28.
Alternative strategies to recover oligonucleotide pools from microarrays without the need for a specific basic treatment exist, are in principle compatible with photolithography and rely on the use of enzymes. For instance, a single deoxyuracil nucleotide can be the target of the uracil-DNA glycosylase (UDG) and excised from the rest of the DNA sequence, or a single RNA unit can be recognized by RNase H type 2 enzymes and the phosphodiester bond 5' to the RNA cleaved, releasing the 5' DNA part23.
We now have a powerful, reliable and high-density method for the synthesis of DNA, RNA, and hybrid DNA/RNA microarrays. These can not only serve as platforms for hybridization or binding assays36, but they also represent a fast and inexpensive way to produce complex nucleic acid libraries. For DNA-based digital data storage, microarray photolithography may become a potential solution to the "writing" bottleneck (i.e., to the encoding of information by synthesis). The success in digital encoding on DNA and in de novo gene assembly depends on sequence fidelity which, at the synthesis level, translates into the error rate. Synthetic and optical errors in our current array fabrication protocols will be discussed and reported on elsewhere. In parallel, efforts are now underway to further increase fabrication scale and throughput.
Disclosures
The authors certify they have no affiliation with any for-profit organization.
Acknowledgements
This work was supported by the Austrian Science Fund (FWF grants P23797, P27275 and P30596) and the Swiss National Science Foundation (Grant #PBBEP2_146174).
Materials
| Name | Company | Catalog Number | Comments |
| Slide functionalization | |||
| Acetic acid >99.8% | Sigma | 33209 | For RNA deprotection |
| CNC router | Stepcraft | 300 CK | |
| Ethanol absolute | VWR | 1.07017.2511 | For deprotection and functionalization |
| N-(3-triethoxysilylpropyl)-4-hydroxybutyramide | Gelest | SIT8189.5 | Silanizing reagent |
| Nexterion Glass D microscope slides | Schott | 1095568 | |
| Polymax 1040 | Heidolph | Orbital shaker | |
| Proclean 507 Ultrasonic water bath | Ulsonix | To clean slides after drilling | |
| Tickopur RW 77 Special Purpose Cleaner | Sigma | Z860086 | To clean slides after drilling |
| Microarray synthesis | |||
| 0.25 M dicyanoimidazole in ACN | Biosolve | 0004712402BS | Activator |
| 0.7 XGA DMD | Texas Instruments | Digital Micromirror Device | |
| 20 mM I2 in pyridine/H2O/THF | Sigma | L860020-06 | Oxidizer |
| 250 μm thick Chemraz 584 perfluoroelastomer | FFKM | Lower teflon gasket | |
| 2'-O-ALE RNA phosphoramidites | ChemGenes | ||
| 365 nm high-power UV-LED | Nichia | NVSU333A | |
| 5'BzNPPOC DNA phosphoramidites | Orgentis | ||
| 5'NPPOC DNA phosphoramidites | FlexGen | ||
| Acetonitrile | Biosolve | 0001205402BS | For DNA synthesis |
| Amidite Diluent for DNA synthesis | Sigma | L010010 | For dissolving phosphoramidites |
| Cleavable dT | ChemGenes | Base-sensitive monomer for library preparation | |
| DMSO | Biosolve | 0004474701BS | As exposure solvent |
| DNA and RNA microarray deprotection | |||
| Ethylenediamine >99.5% | Sigma | 3550 | For deprotection |
| Expedite 8909 | PerSeptive Biosystems | DNA synthesizer | |
| Hydrazine hydrate 50-60% hydrazine | Sigma | 225819 | For RNA deprotection |
| Imidazole | Sigma | 56750 | |
| Industrial Strength lower-density PTFE tape | Gasoila | Thin, upper teflon gasket | |
| Pyridine >99% | Sigma | P57506 | For RNA deprotection |
| Triethylamine >99% | Sigma | T0886 | For RNA deprotection |
| β-carotene | Sigma | C9750 | For library preparation |
| Hybridization and scanning | |||
| 20x Sodium Saline Citrate | Roth | 1054.1 | |
| 5'Cy3-labelled complementary strand | Eurogentec | For duplex hybridization | |
| Biopur Safe-Lock microcentrifuge tube | Eppendorf | ||
| BSA (10 mg/mL) | Promega | R3961 | |
| EDTA molecular biology grade | Promega | H5031 | |
| GenePix 4100A | Molecular Devices | Microarray scanner | |
| Hybridization oven | Boekel Scientific | 230500 | |
| MES monohydrate | Sigma | 69889 | |
| MES sodium | Sigma | M3058 | |
| NaCl >99.5% | Sigma | 71376 | |
| SecureSeal SA200 hybridization chamber | Grace BioLabs | 623503 | |
| Spectrafuge mini | Labnet | C1301 | Microarray centrifuge |
| Tween-20 molecular biology grade | Sigma | P9416 | |
| Data extraction | |||
| Excel | Microsoft | For data extraction | |
| MatLab | MathWorks | Microarray design | |
| NimbleScan 2.1 | Roche NimbleGen | ||
| Desalting and quantification | |||
| NanoDrop One Spectrophotometer | Thermo Scientific | ||
| Toluene | Merck | ||
| ZipTip C18 | Millipore | ZTC18s008 | Desalting pipet tips |
References
- Bumgarner, R. . Current protocols in molecular biology. 101, 22 (2013).
- Berger, M. F., Bulyk, M. L. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nature Protocols. 4 (3), 393-411 (2009).
- Tietjen, J. R., Donato, L. J., Bhimisaria, D., Ansari, A. Z. Sequence-specificity and energy landscapes of DNA-binding molecules. Methods in enzymology. 497, 3-30 (2011).
- Katilius, E., Flores, C., Woodbury, N. W. Exploring the sequence space of a DNA aptamer using microarrays. Nucleic Acids Research. 35 (22), 7626-7635 (2007).
- Franssen-van Hal, N. L. W., et al. Optimized Light-Directed Synthesis of Aptamer Microarrays. Analytical Chemistry. 85 (12), 5950-5957 (2013).
- Matteucci, M. D., Caruthers, M. H. Nucleotide Chemistry .4. Synthesis of Deoxyoligonucleotides on a Polymer Support. Journal of the American Chemical Society. 103 (11), 3185-3191 (1981).
- Eroshenko, N., Kosuri, S., Marblestone, A. H., Conway, N., Church, G. M. Gene Assembly from Chip-Synthesized Oligonucleotides. Current Protocols in Chemical Biology. 2012, (2012).
- Kosuri, S., et al. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nature Biotechnology. 28 (12), 1295 (2010).
- Richmond, K. E., et al. Amplification and assembly of chip-eluted DNA (AACED): a method for high-throughput gene synthesis. Nucleic Acids Research. 32 (17), 5011-5018 (2004).
- Schmidt, T. L., et al. Scalable amplification of strand subsets from chip-synthesized oligonucleotide libraries. Nature Communications. 6, (2015).
- Grass, R. N., Heckel, R., Puddu, M., Paunescu, D., Stark, W. J. Robust Chemical Preservation of Digital Information on DNA in Silica with Error-Correcting Codes. Angewandte Chemie International Edition. 54 (8), 2552-2555 (2015).
- Erlich, Y., Zielinski, D. DNA Fountain enables a robust and efficient storage architecture. Science. 355 (6328), 950-953 (2017).
- Garalde, D. R., et al. Highly parallel direct RNA sequencing on an array of nanopores. Nature Methods. 15 (3), 201-206 (2018).
- Cleary, M. A., et al. Production of complex nucleic acid libraries using highly parallel in situ oligonucleotide synthesis. Nature Methods. 1 (3), 241-248 (2004).
- LeProust, E. M., et al. Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Research. 38 (8), 2522-2540 (2010).
- Lietard, J., Damha, M. J., Somoza, M. M., Fernández-Lucas, J. . Enzymatic and Chemical Synthesis of Nucleic Acid Derivatives. , (2018).
- Fodor, S. P. A., et al. Spatially Addressable Parallel Chemical Synthesis. Science. 251 (4995), 767-773 (1991).
- Singh-Gasson, S., et al. Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array. Nature Biotechnology. 17 (10), 974-978 (1999).
- Pease, A. C., et al. Light-Generated Oligonucleotide Arrays for Rapid DNA-Sequence Analysis. Proceedings of the National Academy of Sciences of the United States of America. 91 (11), 5022-5026 (1994).
- Holz, K., Lietard, J., Somoza, M. M. High-Power 365 nm UV LED Mercury Arc Lamp Replacement for Photochemistry and Chemical Photolithography. ACS Sustainable Chemistry & Engineering. 5 (1), 828-834 (2017).
- Lackey, J. G., Mitra, D., Somoza, M. M., Cerrina, F., Damha, M. J. Acetal Levulinyl Ester (ALE) Groups for 2′-Hydroxyl Protection of Ribonucleosides in the Synthesis of Oligoribonucleotides on Glass and Microarrays. Journal of the American Chemical Society. 131 (24), 8496-8502 (2009).
- Lackey, J. G., Somoza, M. M., Mitra, D., Cerrina, F., Damha, M. J. In-situ chemical synthesis of rU-DNA chimeras on chips and enzymatic recognition. Chimica Oggi-Chemistry Today. 27 (6), 30-33 (2009).
- Lietard, J., Ameur, D., Damha, M., Somoza, M. M. High-density RNA microarrays synthesized in situ by photolithography. Angewandte Chemie International Edition. 57 (46), 15257-15261 (2018).
- Agbavwe, C., et al. Efficiency, Error and Yield in Light-Directed Maskless Synthesis of DNA Microarrays. Journal of Nanobiotechnology. 9, (2011).
- Sack, M., et al. Express photolithographic DNA microarray synthesis with optimized chemistry and high-efficiency photolabile groups. Journal of Nanobiotechnology. 14, (2016).
- Kretschy, N., Holik, A. K., Somoza, V., Stengele, K. P., Somoza, M. M. Next-Generation o-Nitrobenzyl Photolabile Groups for Light-Directed Chemistry and Microarray Synthesis. Angewandte Chemie International Edition. 54 (29), 8555-8559 (2015).
- Sack, M., Kretschy, N., Rohm, B., Somoza, V., Somoza, M. M. Simultaneous Light-Directed Synthesis of Mirror-Image Microarrays in a Photochemical Reaction Cell with Flare Suppression. Analytical Chemistry. 85 (18), 8513-8517 (2013).
- Lietard, J., et al. Base-cleavable microarrays for the characterization of DNA and RNA oligonucleotides synthesized in situ by photolithography. Chemical Communications. 50 (85), 12903-12906 (2014).
- Krotz, A. H., et al. Solution stability and degradation pathway of deoxyribonucleoside phosphoramidites in acetonitrile. Nucleosides Nucleotides & Nucleic Acids. 23 (5), 767-775 (2004).
- Hargreaves, J. S., Kaiser, R., Wolber, P. K. The Degradation of Dg Phosphoramidites in Solution. Nucleosides Nucleotides & Nucleic Acids. 34 (10), 691-707 (2015).
- McGall, G. H., et al. The efficiency of light-directed synthesis of DNA arrays on glass substrates. Journal of the American Chemical Society. 119 (22), 5081-5090 (1997).
- Collett, J. R., et al. Functional RNA microarrays for high-throughput screening of antiprotein aptamers. Analytical Biochemistry. 338 (1), 113-123 (2005).
- Buenrostro, J. D., et al. Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nature Biotechnology. 32 (6), 562-568 (2014).
- Wu, C. -. H., Holden, M. T., Smith, L. M. Enzymatic Fabrication of High-Density RNA Arrays. Angewandte Chemie International Edition. 53 (49), 13514-13517 (2014).
- Tian, J., et al. Accurate multiplex gene synthesis from programmable DNA microchips. Nature. 432 (7020), 1050-1054 (2004).
- Lietard, J., et al. Mapping the affinity landscape of Thrombin-binding aptamers on 2'F-ANA/DNA chimeric G-Quadruplex microarrays. Nucleic Acids Research. 45 (4), 1619-1632 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved