
Method Article
Dissection, Culture and Analysis of Primary Cranial Neural Crest Cells from Mouse for the Study of Neural Crest Cell Delamination and Migration
* Diese Autoren haben gleichermaßen beigetragen
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt die Zerlegung und Kultur von kranialen neuralen Kammzellen aus Mausmodellen, in erster Linie für die Untersuchung der Zellmigration. Wir beschreiben die verwendeten Live-Bildgebungstechniken und die Analyse von Geschwindigkeits- und Zellformänderungen.
Zusammenfassung
In den letzten Jahrzehnten hat es eine erhöhte Verfügbarkeit von genetisch veränderten Mausmodellen gegeben, die verwendet werden, um menschliche Pathologien nachzuahmen. Die Fähigkeit, Zellbewegungen und Differenzierung in vivo zu untersuchen, ist jedoch nach wie vor sehr schwierig. Neurokristalltopathien oder Störungen der neuralen Kammlinie sind aufgrund der mangelnden Zugänglichkeit wichtiger embryonaler Stadien und der Schwierigkeiten bei der Trennung des neuralen Kammmesenchyms vom benachbarten mesodermalen Mesenchym besonders schwierig zu studieren. Hier haben wir uns vorgenommen, ein klar definiertes Routineprotokoll für die Kultur der primären kranialen neuralen Kammzellen zu etablieren. In unserem Ansatz sezieren wir den neuronalen Plattenrand der Maus während der ersten neuralen Kamminduktionsphase. Der neuronale Plattengrenzbereich wird explantiert und kultiviert. Die neuralen Kammzellen bilden sich in einem Epithelblatt, das die neurale Plattengrenze umgibt, und beginnen sich um 24 h nach der Explantation zu delaminieren, indem sie einen epitheliaal-mesenchymalen Übergang (EMT) durchlaufen, um vollständig motile Neuralkammzellen zu werden. Aufgrund unseres zweidimensionalen Kultivierungsansatzes lassen sich die unterschiedlichen Gewebepopulationen (neurale Platte versus Vorwander- und Wandernalkamm) leicht unterscheiden. Mithilfe von Live-Bildgebungsansätzen können wir dann Veränderungen in der neuralen Kamminduktion, EMT und Migrationsverhalten identifizieren. Die Kombination dieser Technik mit genetischen Mutanten wird ein sehr kraftvoller Ansatz sein, um normale und pathologische neuronale Kammzellbiologie zu verstehen.
Einleitung
Die Neuralkamm (NC) Abstammung ist eine transiente, multipotente und wandernde Population von Zellen, die ausschließlich bei Wirbeltieren während der frühen embryonalen Entwicklung1,2erscheint. Neurale Kammderivate sind extrem vielfältig und umfassen Glia, glatte Muskeln, Melanozyten, Neuronen und craniofacial Knochen und Knorpel3,4. Da der neurale Kamm zur Funktion vieler Organsysteme beiträgt, ist diese Abstammung für die menschliche Embryogenese unerlässlich. Aberrante NC-Entwicklung ist in einer Vielzahl der häufigsten menschlichen Geburtsfehler (d.h. Lippen- und Gaumenspalte)5, sowie Erkrankungen wie Hirschsprung-Krankheit (HSCR), Wardensburg-Syndrom (WS), CHARGE-Syndrom und Williams-Syndrom 6 involviert ,7,8,9.
Die NC-Entwicklung wurde in einer Reihe von nicht-mammalianischen Modellsystemen untersucht, einschließlich Xenopus,Küken- und Zebrafischmodellen. Bei Säugetieren hat die Arbeit in Mausmodellen einige der wichtigsten genetischen Ereignisse identifiziert, die der Entwicklung des neuronalen Kamms zugrunde liegen; jedoch war es schwieriger, die Zellbiologie der neuralen Kammmigration zu verfolgen, aufgrund der Unzugänglichkeit des Mausembryons (an anderer Stelle überprüft10,11). Während Studien an Küken, Xenopus und Zebrafischen ein Gen-Regulierungsnetzwerk für NC aufgebaut haben, weisen Funktionsverluststudien in diesen Tiermodellen manchmal keinen vergleichbaren Phänotyp in der Maus auf. Zum Beispiel, in Xenopus, Zebrafisch und Küken, nicht-kanonische Wnt Signalisierung ist einer der zellulären Mechanismen, die die NC ermöglicht, seine Migrationskapazität zu erwerben12,13,14,15 . Bei der Maus scheint der Verlust der nicht-kanonischen Wnt-Signalisierung jedoch keine Auswirkungen auf Migration zu haben16. Da die in vivo NC-Migration über lange Zeiträume bei der Maus schwer zu verfolgen war, ist unklar, ob diese Artenunterschiede unterschiedliche Migrationsweisen oder Unterschiede in der molekularen Regulierung widerspiegeln.
Wie bereits erwähnt, waren NC-Studien an Mäusen sehr anspruchsvoll, da die Ex-utero-Kultur von Embryonen mühsam ist. Darüber hinaus steht der NC ständig in intimem Kontakt mit angrenzenden Geweben wie Mesoderm und Neurectoderm. Die jüngste Verwendung von neuronalen Kamm-spezifischen Cre-Treibern oder exogenen Farbstoffen hat es uns ermöglicht, die wandernde NC fluoreszierend zu kennzeichnen; diese Ansätze sind jedoch noch begrenzt. Trotz mehrerer Berichte, die verschiedene Techniken zur Visualisierung der NC-Migration17,18beschreiben, war es schwierig, diese Techniken in einem einfachen und routinemäßigen Verfahren aufzulösen.
Es ist klar, dass es einen Bedarf an Techniken gibt, die die Handhabung und Charakterisierung von Säugetier-NC ermöglichen. Wir konzentrierten unsere Bemühungen auf die Maus Schädel NC, da es das primäre Modell für die Untersuchung der menschlichen craniofacial Entwicklung und Neurokristalltopathien ist. Wir verfeinerten unseren Ansatz auf der Grundlage mehrerer interessanter Berichte, die die Primärkultur der NC-Zellen19,20,21beschreiben. Hier beschreiben wir ausführlich die optimalen Kulturtechniken für die Explantation primärer NC-Zellen. Wir zeigen die Live-Zell-Bildgebungsmethode und den optimalen Einsatz verschiedener Matrizen zur Beschichtung der Kulturplatten. Unser Protokoll beschreibt, wie die Migration von lebenden NC-Zellen mit einem invertierten Mikroskop erfasst werden kann, das als Richtschnur für den Einsatz mit anderen Mikroskopen gedacht ist, sowie eine detaillierte Zusammenfassung unserer zellulären Analysen.
Das erwartete Ergebnis der Explantation sollte eine schön angelegte Verteilung von Zellen sein, die unter dem Mikroskop klar unterschieden werden, wobei man drei verschiedene Populationen von Zellen sehen kann, die (i) neuronale Platte darstellen, (ii) vorwandernd und, (iii) wandernde neuronale Kammzellen. Wir zeigen, wie das Zellverhalten am Rand der vorwandernden Population von Zellen während des epitheliaal-mesenchymalen Übergangs analysiert wird. Wir konzentrierten uns auch auf die Untersuchung vollständig migrierender Zellen auf Zellgeschwindigkeit, Entfernung und Zellmorphologie.
Protokoll
Alle tierischen Arbeiten wurden vom King es College London Ethical Review Process ethisch genehmigt und wurden in Übereinstimmung mit der BRITISCHEN Home Office Project License P8D5E2773 (KJL) durchgeführt.
1. Herstellung von Reagenzien
- Vorbereiten allgemeiner Lösungen und Werkzeuge, einschließlich steriler Phosphatpufferkochin (PBS), 70% Ethanol, Sezierwerkzeuge (Zangen und Sezierklingen oder sterile Nadeln), Kunststoffplatten oder Glasgärinnen, die mit einer handelsüblichen extrazellulären Matrix beschichtet sind ( ECM-basiertes Hydrogel oder Fibronectin (siehe Materialtabelle) und neuronale Kammmedien (siehe unten).
- Bereiten Sie das neurale Kamm-Basalmedium mit Dulbeccos modifiziertem Eagle-Medium (DMEM, 4500 mg/L Glukose), 15% fetalem Rinderserum (FBS), 0,1 mM minimum essential medium nonessential aminosäures (MEM NEAA 100X), 1 mM Natriumpyruvat, 55 'M-Mercaptoethanol, 100 Einheiten/ml Penicillin, 100 Einheiten/ml Streptomycin und 2 mM L-Glutamin.
- Konditionieren Sie die Medien über Nacht mit wachstumshemmenden STO-Feederzellen21.
- Bereiten Sie STO-Zellen (siehe Tabelle der Materialien) Medien vor, um DMEM zu enthalten, ergänzt durch 10% FBS und 100 U/ml Penicillin, 100 U/ml Streptomycin. STO-Zellen wachsen und erweitern, um in 25 cm2 Mitlohnen, die mit 0,1% Gelatine beschichtet sind, zusammenzufließen. 5000 rad Gammabestrahlung anwenden.
- Samen ca. 3 x 106 wachstumshemmte Zellen auf eine 10 cm2 Schale oder 25 cm2 Kolben (ab Schritt 1.2.1.1). Etwa 10–12 ml neuralem Kamm-Basalmedium hinzufügen und über Nacht inkubieren.
HINWEIS: Saatzellen können verwendet werden, um bedingtes Medium für bis zu 10 Tage zu produzieren. Überprüfen Sie das Erscheinungsbild von Zellen regelmäßig
- Filtern Sie das Medium (0,22 m Porengröße) und ergänzen Sie es mit 25 ng/ml Basis-Fibroblasten-Wachstumsfaktor (bFGF) und 1000 U des Leukämie-Inhibitorfaktors (LIF).
HINWEIS: Bei 4 °C lagern und innerhalb eines Monats verwenden oder bei -20 °C lagern und innerhalb von 3 Monaten verwenden.
- Konditionieren Sie die Medien über Nacht mit wachstumshemmenden STO-Feederzellen21.
- Beschichten Sie die Gewebekulturoberflächen mit extrazellulärer Matrix.
HINWEIS: Je nach der zu stellenden biologischen Frage kann die Matrix auf Glasboden-Kulturgeschirr, Kunststoff-Gewebekulturgeschirr oder Glasabdeckungsschlips beschichtet werden. Siehe unten für unterschiedliche ECM-basierte Hydrogelverdünnungen, die vom Matrixsubstrat abhängen. Fibronectin wurde auf Glasbodenschalen getestet und Deckschschlüpft nur bei den unten angegebenen Konzentrationen. Hierwerden bezeichnen wir die substratbeschichteten Oberflächen als "beschichtete Platten".- Beschichten Sie die Gewebekulturoberflächen mit ECM-basiertem Hydrogel.
HINWEIS: Halten Sie das Substrat kalt bis zur Beschichtung, entweder durch Abkühlen der Medien oder auf Eis halten.- Das Hydrogel über Nacht bei 4 °C auftauen. Fügen Sie 5 ml 10% FBS in DMEM auf 5 ml Hydrogel für ein Endvolumen von 10 ml hinzu (siehe Materialtabelle).
- Machen Sie 0,5–1 ml Aliquots bequem und lagern Sie bei -20 °C.
- Das Hydrogel auf Eis auftauen.
- Verwenden Sie eine Verdünnung des Hydrogels 1:20, um Kunststoff zu beschichten.
- Verwenden Sie eine 1:5 Verdünnung des Hydrogels, um Glasschlitten und Glasboden-Gewebekulturplatten zu beschichten.
HINWEIS: Das Hydrogel in kaltem DMEM verdünnen. - Tragen Sie genügend verdünntes Hydrogel auf, um den gewünschten Bereich auf Platten/Schlitten abzudecken, und brüten Sie 30–45 min bei 37 °C.
- Verwenden Sie beschichtete Platten/Dias sofort oder lagern Sie beschichtete Dias bei 4 °C über Nacht.
- Entfernen Sie Exzesse und spülen Sie Dias mit hoher Glukose DMEM (optional) vor der Verwendung.
- Beschichten Sie die Gewebekulturoberflächen mit Fibronectin.
- Herstellung von Aliquots von 1 mg/ml Fibronectin-Stammlösung und Lagern Sie bei -80 °C. Fibronectin mit Dulbeccos PBS (dPBS) auf eine Endkonzentration von 1 g/ml verdünnen.
- Tragen Sie ausreichend Fibronectin auf, um den gewünschten Bereich abzudecken, und brüten Sie bei Raumtemperatur für 15 min.
- Entfernen Sie Restfibronectin und lassen Sie das Glas für 30-45 min trocknen.
- Spülen Sie Vorbrunnen oder Deckelscheine mit hoher Glukose DMEM (optional) vor gebrauchen.
- Beschichten Sie die Gewebekulturoberflächen mit ECM-basiertem Hydrogel.
2. Tag 1: Zerlegung von Embryonen im Frühstadium
HINWEIS: Verwenden Sie sterile Werkzeuge und sterile Lösungen. Wenn Genotypisierung erforderlich ist, sammeln Sie den Körper des Embryos für die DNA-Extraktion.
- Die Zerlegung der kranialen neuralen Platte ist auf Embryonen bei 8,5 Tagen nach dem Koitum (dpc) beschränkt. Wählen Sie Embryonen im 5-8-Stadium aus. Sezieren Sie die Gebärmutter in PBS und schneiden Sie das Mesometrium, um jeden Embryo zu trennen (Abbildung 1A). Die Muskelwand der Gebärmutter zieht zusammen und das Dezidualgewebe wird sichtbar (Abbildung 1B).
HINWEIS: Bewahren Sie Embryonen in der Gebärmutter in eiskaltem PBS auf, während Sezierungen jeweils einen Embryo durchführen. Bewegen Sie Embryonen mit einer glasigen Pasteurpipette in frische sterile PBS, um die Sichtbarkeit zu verbessern und die Kontamination zu reduzieren. - Schieben Sie Zangen zwischen der Muskelschicht und dem Dezidualgewebe und entfernen Sie die Muskelschicht mit einem zweiten Zangenpaar (Abbildung 1C).
- Mit Zangen, durchbohren Sie das Deciduum an den Rändern des Mesometrial-Pols und mit einem zweiten Paar Zangen reißen, um senkrecht zum Pol zu öffnen.
- Schälen Sie das Dezidualgewebe mit den Zangen zurück, um die Membran des Reichert zu visualisieren.
- Entfernen Sie Reicherts Membran sorgfältig. Der viszerale Dottersack wird sichtbar und der Embryo ist im Inneren zu sehen (Abbildung 1D).
- Entfernen Sie den viszeralen Dottersack und das Amnion (Abbildung 1E) und positionieren Sie den Embryo, um die Kopffalte zu visualisieren (Abbildung 1F).
- Schneiden Sie die Kopffalte über dem Herzen und kratzen Sie das zugrunde liegende Mesoderm mit Zangen und/oder Wimpernwerkzeugen weg, um eine saubere neuronale Platte (NP) zu erhalten (Abbildung 1H).
HINWEIS: Das NP kann ganz oder geteilt auf der Anteroposterior-Achse gehalten werden, so dass jede Seite einzeln plattiert werden kann. Die neurale Plattengrenze kann weiter von der neuralen Platte abgeschnitten werden, um neuronale Beiträge zu den Expflanzen zu minimieren. - Verwenden Sie eine gläserne Pasteurpipette, um die sezierte neurale Platte auf eine hydrogelbeschichtete Schale zu übertragen, die mit konditionierten neuralen Kammmedien gefüllt ist.
- Mischen Sie die Schale vorsichtig, um den NP in der Mitte des Brunnens zu positionieren. Dies ist wichtig für die Maximierung der Phasenqualität für die Live-Zell-Bildgebung (am 2. Tag).
- Über Nacht (oder zum gewünschten Zeitpunkt) bei 37 °C in 5%CO2inkubieren. Neurale Kammzellen sollten sichtbar aus der neuralen Platte wandern.
HINWEIS: Zellen werden in der Regel innerhalb von 6–8 h angefügt. Nachdem die Explantation angefügt wurde, lassen Sie mehr Zeit, um migrierende Zellen zu visualisieren. In der Regel können wir durch 24 h Postexplant drei unterscheidbare Populationen von Zellen finden. Die erste Population, in der Mitte der Explantation, ist die neurale Platte (NP). Die zweite Population, die vorwandernde NC (pNC), umgibt den NP in einem Epithelblatt von Zellen. Die dritte Population, im äußeren Ring, besteht aus wanderndem NC (mNC), die größer sind und vollständig mesenchymal erscheinen (Abbildung 2).
3. Tag 2: Live-Zell-Bildgebung von murinen Kranial-Neuralkammzellen
HINWEIS: Die Bildgebung sollte bei einer 24-Stunden-Post-Explantation durchgeführt werden, um die Migration neuronaler Kammzellen optimal abzubilden und zu quantifizieren. NC-Induktionsmedien müssen nicht vor der Live-Zell-Bildgebung aktualisiert werden. Zugang zu einem invertierten Mikroskop mit motorisierter Bühne und integrierter Umgebungskammer ist erforderlich. Verwenden Sie Multi-Well-Gewebekultur-Gerichte geeignet für die Bildgebung (Tabelle der Materialien).
-
Mikroskop-Setup
- Stellen Sie die Umgebungskammer auf 37 °C und 5%CO2ein.
- Durchbohren Sie ein Loch in den Deckel des Gewebekulturplattendeckels, damit dieCO2-Nadel, die mit der CO2-Befeuchtungskammer verbunden ist, innerhalb der Platte sitzen kann.
- Legen Sie die Gewebekulturschale in den Probenhalter und kleben Sie den Plattendeckel und dieCO2-Nadel herunter, um ein Schütteln während der Multi-Well-Erfassung zu verhindern.
- Schalten Sie den Mikroskop-Controller, den Bühnenregler und die Bildverarbeitungssoftware ein.
- Konzentrieren Sie sich auf die Schädel-NC-Zellen bei 10-facher Vergrößerung (mit passendem Phasenring im ausgewählten Kondensator).
- Stellen Sie einen hochwertigen Phasenkontrast auf dem Mikroskop ein, indem Sie die Feldirismembran, die Blende Irismembran und das Zentrierteleskop anpassen, wie im Mikroskop-Setup-Handbuch angegeben.
-
Phasenkontrast Live-Zell-Bildgebung
- Legen Sie das Verzeichnis oder den Dateispeicherort fest, an dem die Zeitrafferdateien gespeichert werden.
- Legen Sie belichtungszeit, Binning und Kamerabereich fest.
- Legen Sie die Anzahl der Zeitpunkte, die Dauer der Bildgebung und das Zeitintervall zwischen Frames fest.
- Um die NC-Zellmigrationskapazität zu quantifizieren, stellen Sie das Mikroskop auf die 10-fache Vergrößerung ein und nehmen Sie 1 Frame alle 5 min (217 Zeitpunkte über 18 h). Um die Zellmorphologie zu quantifizieren, stellen Sie die Vergrößerung auf 40x, wobei 1 Frame/min (61 Zeitpunkte über 1 h) genommen wird. Um die lamellipodiale Dynamik zu quantifizieren, stellen Sie die Vergrößerung auf 40x oder 60x Vergrößerung ein, wobei 1 Frame alle 10 s (über 10 min) genommen wird.
- Legen Sie für die Multi-Well-Bildgebung die mechanische Stufe so fest, dass sie zwischen ausgewählten XY-Positionen von Interesse bewegt wird. Bestätigen Sie, dass die Kranial-NC-Zellen im Fokus sind und die Stufenpositionen korrekt sind.
- Verwenden Sie den Befehl Acquire, um die Zeitraffer-Bildgebung zu starten.
- Sobald die Zeitraffer-Bildgebung abgeschlossen ist, überprüfen Sie die mehrdimensionalen Daten und exportieren .stk-Dateien zur Analyse.
HINWEIS: .stk ist eine TIFF-Stack-Datei. - Verlassen Sie die Software, fahren Sie den Computer herunter und schalten Sie die Bühne, kamera- und Mikroskop-Controller aus.
4. Bildgebungsanalyse: Quantifizierung der neuralen Kammzellmigration
ANMERKUNG: Um das zelluläre Verhalten besser zu definieren, indem murine kraniale neuronale Kammzellen migriert werden, haben wir eine Reihe quantifizierbarer Migrationsparameter analysiert, die sich speziell auf die Migrationskapazität und die Zellformdynamik konzentrieren. (1) Migration (akkumulierte Entfernung) ist die gesamte Pfadlänge, die von der Zelle genommen wird ( m); (2) Migration (Euklidischer Abstand) ist der geradlinige Abstand zwischen DerAnfangs- und Endposition der Zelle (m); (3) Migration (Zellgeschwindigkeit) ist die entfernungsweise Zelle pro Zeiteinheit (m/min); (4) Zellform (Zellfläche) ist die gesamte von Zelle bedeckte Fläche. Stellen Sie die Pixel-Skala auf Mikron-Skala gemäß dem Bildgebungsmikroskop ein. (A = Apx x Npx, wobei Apx = Pixelbereich und Npx = Anzahl der Pixel. Einheiten: m2; (5) Zellform (Zellzirrularität) ist die Abweichung der Zellform von einem perfekten Kreis, die durch einen Zirkitätswert von 1,0 (4x (A/P2)) angegeben wird, wobei A = Fläche und P = Umfang.
-
Einzelzellverfolgung
HINWEIS: Um die NC-Zellmigration zu messen, werden XY-Koordinaten einzelner Zellen über alle Zeitrafferrahmen hinweg generiert. Dies ermöglicht eine nachträgliche Analyse von Entfernungs-, Geschwindigkeits- und Persistenzmessungen der Zellmigration.- Öffnen Sie ImageJ, und importieren Sie Daten als TIFF-Stack-Dateien.
- Klicken Sie auf Analysieren | Stellen Sie Scale ein, um die .stk-Dateien entsprechend den Mikroskopeinstellungen zu kalibrieren, und arbeiten Sie in Pixel/m.
- Klicken Sie auf Plugins | Tracking | Manuelle sepermnachade, um Image J manuelles Zellverfolgungs-Plugin zu öffnen. Um mit der Zellenverfolgung zu beginnen, wählen Sie Spur hinzufügenaus.
- Verfolgen Sie Zellen durch alle Frames von Zeitrafferfilmen, indem Sie den Kern als Referenzpunkt verwenden.
HINWEIS: 10–20 Zellen sollten pro Explantation verfolgt werden, wobei insgesamt 60 Zellen verfolgt werden sollten (n = 3). Zellen, die im Laufe des Zeitraffers einer Zellteilung unterzogen werden, sollten von der Analyse ausgeschlossen werden. - Speichern und exportieren Sie die Ergebnisse als CSV-Datei. Die Ergebnisse stellen die einzelne Zellspurnummer, Die Slice-Zahl und die XY-Koordinaten über alle Frames hinweg dar.
-
Quantifizierung der Migrationskapazität der neuralen Kammzellen
- Öffnen Sie Einzelzellen-Tracking-Daten (siehe oben). Konvertieren Sie .csv-Dateien in das .txt-Dateiformat.
- Öffnen Sie die Migrationssoftware (Materialtabelle). Klicken Sie auf die Registerkarte Daten importieren, um die Zellverfolgungsdaten als .txt-Datei zu importieren.
- Unter Datensätze | Initialisierung, wählen Sie die Anzahl der zu analysierenden Slices oder Frames aus, und legen Sie die XY-Kalibrierung und das Zeitintervall zwischen Frames fest. Wählen Sie Einstellungen anwenden aus, um die Einstellungen zu speichern.
- Wählen Sie das Diagrammdatensymbol aus, um Flugbahndiagramme zu erstellen. Wählen Sie das Symbol Statistik aus, um Entfernungs- und Geschwindigkeitsmessungen zu quantifizieren.
- Speichern Sie die Trajektorien diagramme als Bitmap -Dateien (.bmp) und Entfernungs- und Geschwindigkeitsmessungen als .txt-Dateien. Wählen Sie das Symbol Daten entfernen aus. Wiederholen Sie diesen Vorgang für andere Zeitrafferdateien.
HINWEIS: Trajektoriediagramme können verwendet werden, um die Direktheit einzelner Zellpfade für eine bestimmte Zellbedingung oder einen bestimmten Zustand im Laufe von Zeitrafferfilmen zu visualisieren (Abbildung 4A). Die in den .txt-Dateien gespeicherten Entfernungs- und Geschwindigkeitsdaten können dann für weitere Analysen verwendet werden.
-
Quantifizierung des neuralen Kammzellbereichs und der Zirkularität
- Öffnen Sie die Zeitraffer-.stk-Dateien in ImageJ und kalibrieren Sie nach Mikroskopeinstellungen, arbeiten Sie in Pixel/m.
- Unter Analysieren | Festlegen von Messungen, klicken Sie auf , um die Zellenformparameter auszuwählen: Zellfläche, Umfang und Formbeschreibung.
- Verwenden Sie das Werkzeug Freihandauswahl, um jede Zelle manuell zu zeichnen, wobei Zellmembrangrenzen als Leitfaden verwendet werden.
- Drücken Sie die Strg + B-Tasten auf der Tastatur, um die Zellumrissüberbeinen auf dem Bild beizubehalten. Wiederholen Sie diesen Vorgang für Zellen über jeden Zeitrafferrahmen.
- Verwenden sie das Bild | Overlay | An ROI Manager, um die Werte zu speichern.
- Nachdem alle Zellen von Interesse pro Frame umrissen wurden, klicken Sie auf Messen. Speichern Sie die Ergebnisse als CSV-Datei.
HINWEIS: 10–20 Zellen pro Film sollten skizziert werden, wobei insgesamt 30–60 Zellen pro Bedingung analysiert werden (n = 3). Zellformdaten (.csv-Dateien) können verwendet werden, um zu quantifizieren, wie sich die Zellformdynamik im Laufe der Zeit ändert (Abbildung 4C) oder wie Morphologie unter verschiedenen Zellbehandlungen verändert werden kann.
Ergebnisse
Mit dem hier gezeigten Verfahren wurden Mausembryonen aus der Gebärmutter seziert und extraembryonale Gewebe entfernt (Abbildung 1A–D). Embryonen wurden inszeniert (nur mit Embryonen bei 5–8.ss), Abbildung 1E,F). Die kraniale neurale Platte wurde dann seziert und das Neuroepithel isoliert. Mesodermale Zellen, die als lose, kreisförmige, mesenchymale Zellen identifiziert wurden, wurden sanft abgebürstet (Abbildung 1G-L). Die vordere Neuralplatte kann ganz explantiert werden, wobei das neurale Kammgewebe seitlich auftaucht und sich radial um das Explant ausdehnt, oder jeder neurale Plattenrand (rechts und links) kann separat explantiert werden. Dies ist besonders nützlich, wenn von genetischen Mutanten explantiert wird.
Innerhalb von 24 h kann eine Region des vorwandernden (epitheliale) kranialen Neuralkamms deutlich um das neurale Plattenexplant herum gesehen werden (Abbildung 2B). Darüber hinaus hat eine Subpopulation von neuralen Kammzellen epitheliale bis mesenchymale Übergänge durchlaufen und erscheinen vollständig mesenchymal (Abbildung 2). So haben wir mehrere konzentrische Ringe von verschiedenen Zellen, mit der neuralen Platte (NP) in der Mitte, dem vorwandernden neuralen Kamm (pNC) im Zwischenkreis und einer Population von wanderndem Neuralkamm (mNC) im Äußeren Ring (Abbildung 2B). Um NC-Zellen nachzuverfolgen, ist es möglich, genetisch veränderte Mausmodelle zu verwenden, wie wir in Abbildung 2Czeigen. In diesem Fall haben wir den neuralen Kamm spezifisch Wnt1::Cre; RosamTmG, was dazu führt, dass NC-Zellen grün markiert werden. Bei diesen Mäusen drücken Zellen Membrantomate (mT, in rot) aus, es sei denn, sie exprimieren Cre Recombinase. Rekombination führt zu Zellen, die membrangrünes fluoreszierendes Protein (GFP, in grün) exdrücken. Die roten Zellen, die in der Mitte des Explantationsmittels dargestellt werden, sind neurale Plattenzellen. Einige dorsale neurale Plattenzellen exprimieren auch GFP; für langfristige Kultur würden wir alle Zellen in der Mitte verbrauchen. Für unsere Zwecke reicht die Reinheit des Explants aus, um die verschiedenen neuralen Kammzellpopulationen zu verfolgen. Wo eine höhere Reinheit des Neuralkamms erforderlich ist, kann diese genetische Etikettierungsstrategie mit fluoreszierender aktivierter Zellsortierung (FACS) kombiniert werden, um die Reinheit der Population zu gewährleisten. Alternativ ist es möglich, die Expflanzen zu fixieren und die NC-Population mit Antikörperkennzeichnung zu identifizieren.
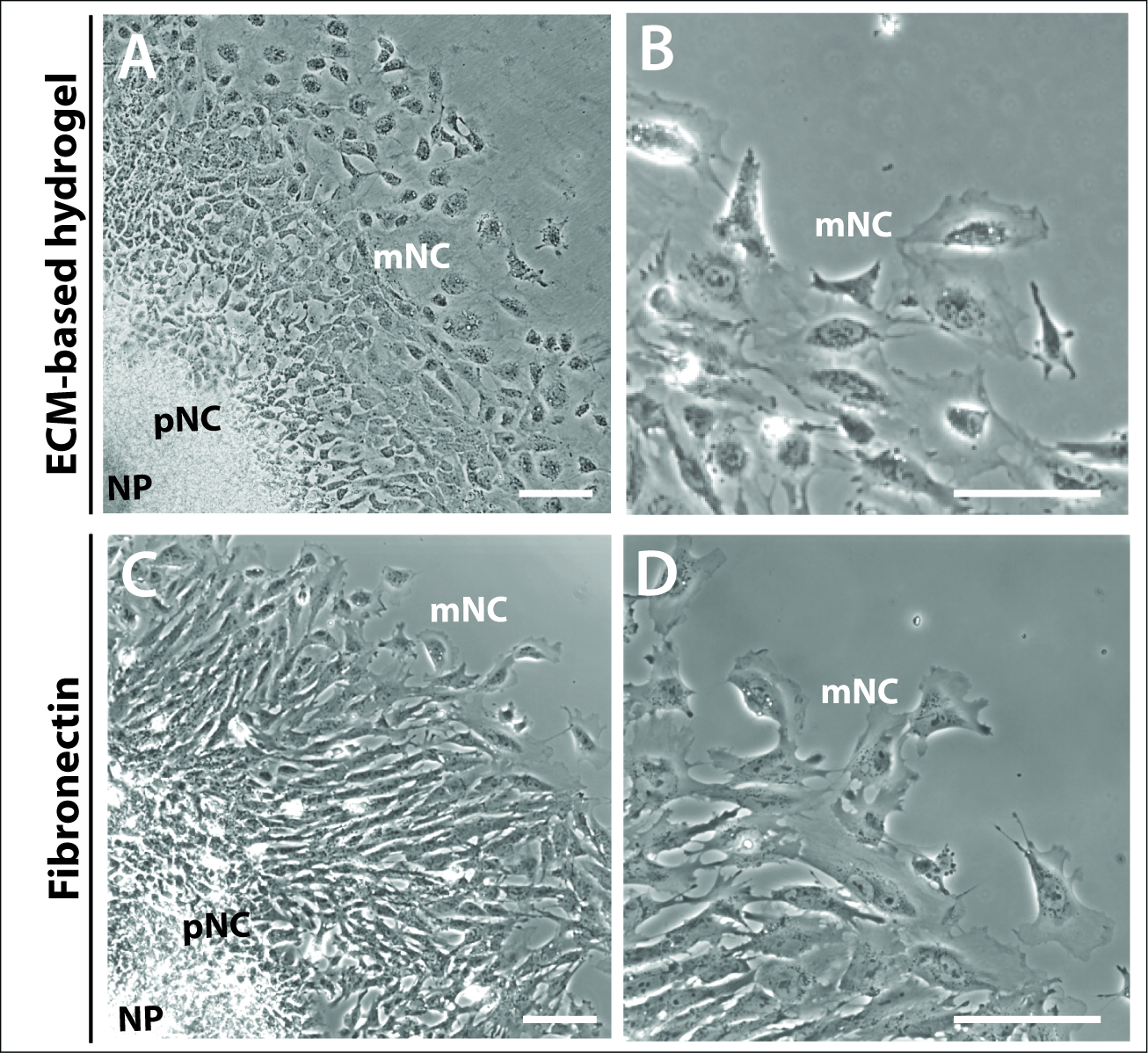
Es zeigte sich auch um 24 h, daß die charakteristischen konzentrischen Ringe von vorwandernden und vollständig wandernden NC-Zellen der Explantationskulturen weder durch Matrixwahl abhängig noch bestimmt waren (Abbildung 3). Explant-Kulturen, die sowohl auf einem ECM-basierten Hydrogel als auch fibronectin plattiert waren, bildeten vergleichbare Explantationsstrukturen, die die drei Zellpopulationen NP, pNC und mNC umfassten (Abbildung 3A,C). Die morphologie der neuralen Kammzellwartologie war auch vergleichbar zwischen denen, die auf dem ECM-basierten Hydrogel und Fibronectin plattiert waren (Abbildung 3B,D). Jedoch, Explanten auf Fibronectin plattiert produziert Zellen mit prominenter Lamellipodie an der Zellvorderkante, scheinbar mehr polarisiert in Richtung der Migration (Abbildung 3B,D).
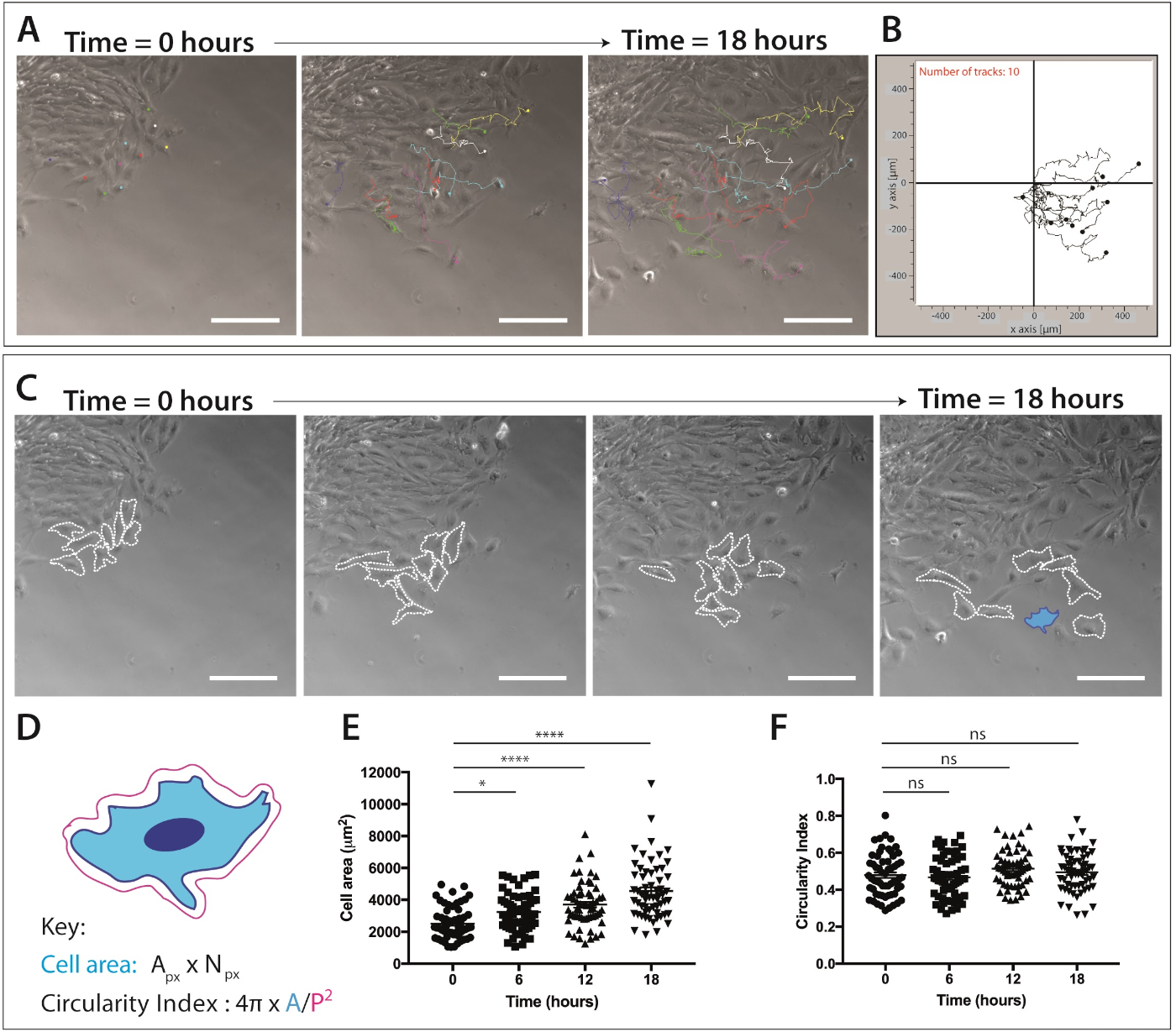
Sobald eine Population von wandernden neuronalen Kammzellen offensichtlich ist, kann die Live-Zell-Bildgebung abgeschlossen werden. Die Zeitraffermikroskopie wird auf die 10-fache Vergrößerung (18 h, 1 Frame/5 min) für die nachfolgende Analyse der NC-Zellmigration eingestellt (Abbildung 4A). ImageJ Manual Tracking Plug-in generiert XY-Koordinaten einzelner Zellen über alle Frames der Zeitrafferfilme (Abbildung 4B). Diese Koordinaten können mit der Migrationssoftware verarbeitet werden. Diese Software ermöglicht die Visualisierung einzelner Zellspuren im Zeitverlauf (Abbildung 4B) und kann zur Quantifizierung der akkumulierten und euklidischen Entfernung sowie der Zellgeschwindigkeit verwendet werden.
Zeitraffer-Bildgebungsdaten liefern auch eine Fülle von Informationen über die Zellmorphologie während der Migration von kranialen neuronalen Kammzellen (Abbildung 4C). Durch die Umrissung einzelner Zellmembranen können Zellflächen- und Umfangsmessungen aus allen Frames der Filme berechnet werden (Abbildung 4C). Diese Messungen ermöglichen die anschließende Quantifizierung der Zellfläche und der Zirkularität (Abbildung 4D). Abbildung 4C zeigt eine Analyse der Zellformveränderungen über 18 h. Beachten Sie, dass zellenförmig, wenn sie vom Explant wegwandern, der Zellbereich signifikant zunimmt, während die Zellzirkularität relativ konstant bleibt (einwegige ANOVA, Tukeys Mehrfachvergleichstest) ( Abbildung 4E,F). Dies deutet darauf hin, dass Zellen, wenn sie von der Epithelkante abweichen und Zell-Zell-Kontakte verlieren, einen erhöhten Zellausbreitungsbereich aufweisen. Die Zellzirkularitätsmaße haben sich im Laufe der Zeit nicht wesentlich geändert; Kurzfristige Veränderungen der Zirkularität können jedoch auftreten, wenn eine erhöhte Anzahl von Zeitpunkten quantifiziert wird. Zellzirkularitätsmessungen können auch interessante Daten zur Zellformdynamik in Gegenwart eines chemotaktischen Hinweises oder unter eingeschränkten Bedingungen liefern.

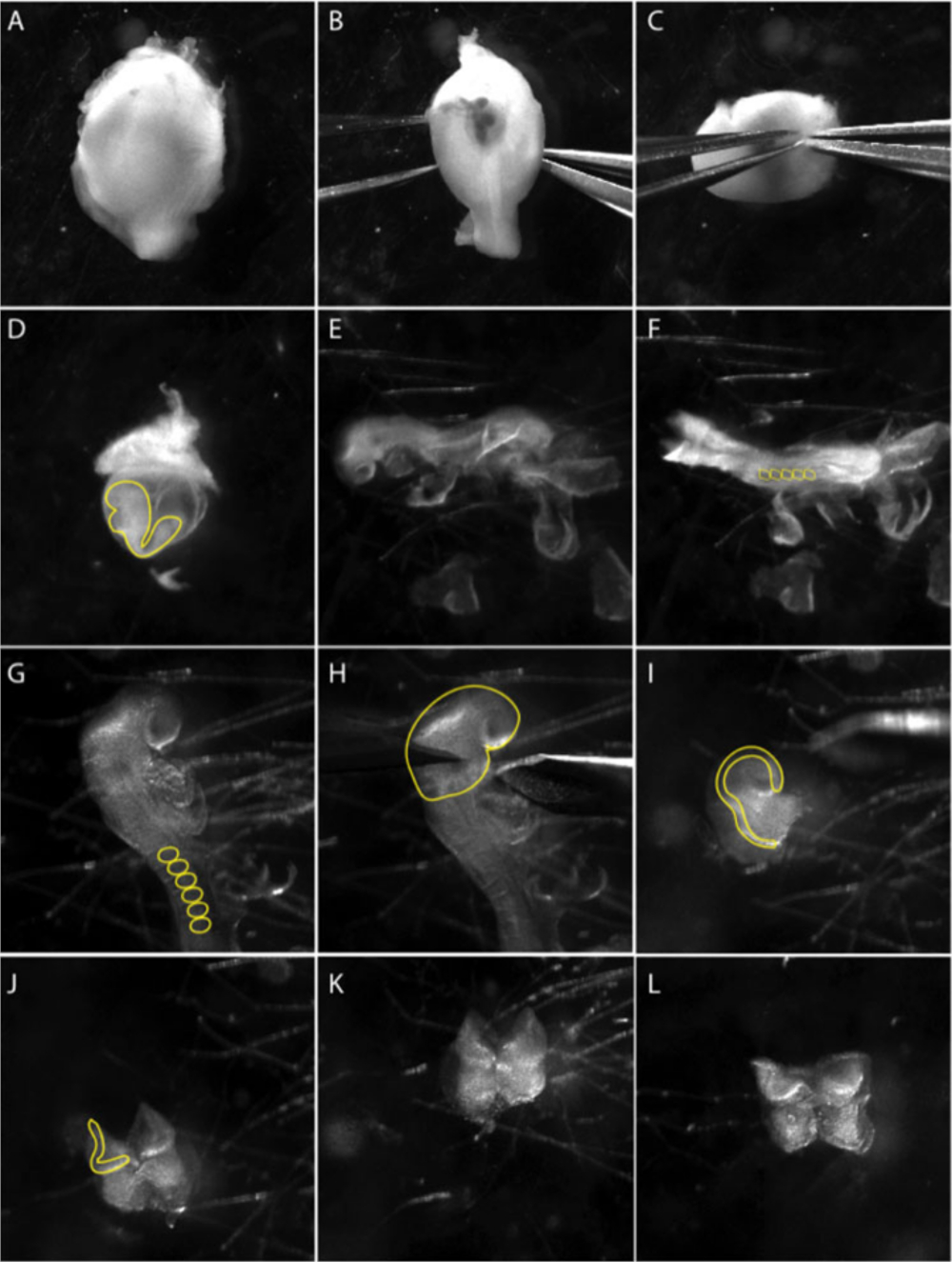
Abbildung 1: Isolierung von kranialen Neuralkammexplanten von einem e8.5-Embryo.
Bilder sind Standbilder aus einem Video, das die Mikro-Sektionstechnik dokumentiert. (A–C) Zerlegung des Embryos aus der Gebärmutter. (B–C) Mit zwei scharfen Zangen, ziehen Sie vorsichtig die Muskelschicht auseinander. Panel (D) zeigt Embryo im viszeralen Dottersack (gelbe Linie). Extrahieren Sie den Embryo aus dem viszeralen Dottersack. (E) Seitliche Ansichten des Embryos im Stadium 8.5 lateral. (F) Dorsale Sicht auf den Embryo im Stadium 8.5. Zählung (ss) zur Bestimmung des Alters der Embryonen; in der Regel 5–8 ss (gelbe Kreise in F). (G) Genauer Blick auf die Schädelregion des Embryos. Extraembryonale Membranen aus dem Schädelbereich entfernen; Die sind mit einer gelben Linie markiert. (H) Unter dem ersten Astbogen (gelbe Linie) werden Die Abschnitte der vorderen neuralen Platte durchgeführt. (I) Seitliche Ansicht der vorderen neuralen Plattensektion. Neuralfalten, bei denen neurale Kammzellen entstehen, sind mit einer gelben Linie markiert. (J–L) Entfernen Sie mesodermales Gewebe (fluffy mesenchymale Zellen), das den vorderen neuronalen Falten zugrunde liegt, so weit wie möglich, bevor Sie NP auf zubereitete Kulturgerichte plattieren. Der Film wurde mit einem Stereomikroskop mit einer Breitfeld-Apochromatischen Linse mit 3,0-Fach-Zoom aufgenommen (siehe Tabelle der Materialien). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Murine Kranial neural kam Kamm Explant.
(A) Schematische Darstellung der dorsalen Ansicht eines e8.5-Mausembryons. Der Schädelbereich des Embryos wird an der gestrichelten Linie geschnitten. Die neurale Plattengrenze (rot hervorgehoben) ist vom umgebenden Mesodermgewebe isoliert und für 24 h kultiviert, damit der kraniale Neuralkamm auswandern kann. Schemaangepasst von22,23. (B) Links: Repräsentatives helles Feldbild eines kranialen Neuralkamms 24 Stunden nach der Beschichtung. Es werden drei Populationen von Zellen beobachtet, die auch auf der rechten Seite schematisiert sind. NP = neurale Platte, pNC = vorwandernder Neuronistenkamm und mNC = migory neural erdenk. Skalenbalken = 250 m. (C) Höhere Vergrößerungsbilder einer Explantation aus genetisch gekennzeichneter Maus (Wnt1::cre; RosamTmG). Zellen ohne Cre-Treiber drücken Membrantomate (mT) in Rot aus. Die Expression von Cre unter der Kontrolle eines neuronalen Kamms-spezifischen Wnt1-Promotors führt zur Exzision der mT-Kassette und zur Expression der Membran GFP (mG) in Grün. Nuclei sind mit Hoescht (blau) befleckt. Skala bar = 200 m. (D–D'') Höhere Vergrößerungsbilder von Zugzellen, die Membran GFP (D) exzessiieren. (D') DNA ist mit Hoescht (blau) gekennzeichnet. (D'') Zusammenführung von D und D'. Maßstabsleiste = 20 m. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Explanten, die auf verschiedenen Substraten kultiviert werden.
(A–B) Phasenkontrastbilder von Explants, die auf kommerziellem ECM-Hydrogel kultiviert werden. (C–D) Explantationen, die auf 1 g/ml Fibronectin kultiviert werden. Neuralplatten (NP), Vorwander- (pNC) und migory (mNC) neurale Kammzellen können durch ihre unterschiedlichen Zellmorphologien unterschieden werden. Skalenbalken = 100 m. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Quantifizierung der kranialen neuralen Kammzellmigration und Zellformdynamik.
(A) Phasenkontrastrahmen aus der Zeitraffer-Bildgebung von Explant-Kulturen, die mit einzelnen neuronalen Kammzellspuren überlagert sind, mit dem ImageJ/Fiji Manual Cell Tracking-Plug-in. Zehn repräsentative mNC-Zellen wurden manuell über 18 h (217 Frames) und die XY- Koordinaten exportiert wurden. Daten werden als Überlagerungspunkt und Liniendiagramme dargestellt. Die Zellen wurden auf 1 g/ml Fibronectin plattiert. Skalenbalken = 200 m. (B) Repräsentatives Trajektodatendiagramm von 10 mNC-Zellen, das mit Migrationssoftware erzeugt wird. (C) Phasenkontrastrahmen aus der Zeitrafferanalyse von Explantkulturen. Gestrichelte Linien umreißen 8 repräsentative wandernde neurale Kammzellen, die für die Zellformdynamik analysiert wurden, wenn sie auf 1 g/ml Fibronectin plattiert werden. Skalenbalken = 200 m. (D) Schematische Darstellung der Berechnungen zur Quantifizierung der Zellfläche und der Zirkularität. Die Zellmorphologie des Schaltplans ist die der Zelle, die blau (C)hervorgehoben ist. Apx = Pixelbereich, Npx = Pixelzahl, A = Fläche, P = Umfang (1 Pixel = 1,60772 ' m2). (E–F) Quantifizierung der Zellfläche und Zellzirkularität misst im Laufe der Zeit. Die Daten stellen den Mittelwert von SEM dar. Jeder Punkt stellt eine Zelle (n = 60) dar, die aus 3 unabhängigen Experimenten entnommen und bei 0 h, 6 h, 12 h und 18 h analysiert wird (ns nicht signifikant, * p<0.05, **** p < 0.0001 one-way ANOVA, Tukeys Mehrfachvergleichstest). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Das Studium der neuralen Kammzellen von Säugetieren war eine Herausforderung für Wissenschaftler wegen der inutero Natur der Säugetierentwicklung. In-vivo-Studien sind schwer einzurichten, da der Embryo unter Bedingungen manipuliert werden muss, die das Leben in der Gebärmutter imitieren. In der Praxis ist es fast unmöglich, diese (E8+) Embryonen länger als 24 h reproduzierbar zu kultivieren, insbesondere für die Live-Bildgebung. Darüber hinaus treten Neuralkaminduktion und Migration gleichzeitig mit Neuralrohrverschluss und embryonalem Drehen in der Maus auf; Dies ist ein entscheidendes und belastendes morphogenetisches Ereignis, das häufig fehlschlägt, wenn Embryonen ex uterokultiviert werden. Somit ist die Erfolgsrate von Ex-utero-Ansätzen im Allgemeinen niedrig. Die Verwendung von verewigten NC-Zellen21 ist ein nützliches Werkzeug, um den Einsatz von Tieren zu reduzieren, und es kann eine bessere Quelle für neuronale Kammzellen für Langzeitanalysen, Transfektions- und Anreicherungsstudien bieten. Es besteht jedoch eindeutig die Notwendigkeit, primäre neurale Kammzellen zuverlässig zu kulturieren. Unsere Methode ist auf Maus-Knock-out oder bedingte genetische Modelle anwendbar. Eine vergleichbare Methode wie unsere wurde für andere neurale Kammpopulationenbeschrieben 20; Unsere Methode beschreibt jedoch gründlich die schrittweise Isolierung muriner Kranial-NC-Zellen. Wir beschreiben auch die Verwendung verschiedener Matrizen sowie das Migrationsanalyseverfahren im Detail.
Um konsistente Ergebnisse zu erzielen, stellten wir fest, dass der Inszenierung bei der Auswahl von Embryonen besondere Aufmerksamkeit gewidmet wurde. Es überrascht nicht, dass die Zahl der Sos mit verschiedenen Stadien der Cranial NC-Entwicklung korreliert. Daher ist das Wissen über die Embryoanatomie sehr wichtig, bevor experimentelle Daten erfasst werden. Dieser Ansatz kann dann an die Isolierung diskreter Populationen von neuralen Kammzellen angepasst werden, abhängig von der biologischen Frage und den Zielzellen.
Sobald die Embryonen ausgewählt und seziert sind, können mesodermale Zellen leicht unterschieden werden und sollten entfernt werden, um eine bessere Visualisierung zu ermöglichen und die Kontamination zu reduzieren. Bei längerfristigen Kulturen kann das neurale Plattengewebe bei 24 h Beschichtung entfernt werden, um eine Kontamination durch neuronales Gewebe zu verhindern. Eine weitere Verfeinerung könnte die Verwendung von fluoreszierender Linienkennzeichnung (z. B. mit einem Wnt1::cre oder Sox10::creERT-Treiber in Kombination mit fluoreszierenden Reportern24,25) sein, um neuronale Kammzellen von andere Gewebe, wie in Abbildung 2Cdargestellt.
Frühere Berichte haben das Potenzial der Beschichtung von Maus NC-Explantationskulturen auf verschiedenen Matrizen hervorgehoben, am häufigsten auf kommerziellen ECM-Hydrogelen, Fibronectin und Kollagen I20,21,26. In unseren Händen werden Maus-Kranial-NC-Explant-Kulturen erfolgreich auf allen drei Matrizen angebaut, in Denoriginalberichten (Daten nicht dargestellt). Der anfängliche verfeinerte Ansatz, den wir für unsere NC-Explantkulturen adaptierten, verwendete ein kommerzielles Hydrogel als Matrix der Wahl, das hauptsächlich aus Laminin und Kollagen bestand.21(Abbildung 3A–B). Die Zusammensetzung dieses Hydrogels ist jedoch nicht klar definiert, mit unbekanntem Wachstumsfaktor und Proteingehalt. Als solche haben wir seitdem unseren Ansatz zur Beschichtung von Maus-NC-Explantationskulturen auf Fibronectin (Abbildung 3C-D). Fibronectin ist gut definiert und stark exprimiert in den ECM- und Kellermembranen, entlang deren NC-Zellen migrierenin vivo28,29,30. Um eine Fibronectin-Matrix zu optimieren, die die Migration und Morphologie von neuralen Kammzellen am besten repliziert, wie sie mit dem Hydrogel beobachtet wurde, verglichen wir das am Hydrogel gezeigte NC-Zellverhalten mit einer Titration von 0,25–30 g/ml Fibronectin und definierte 1 idealen Eigenschaften bieten (Daten nicht dargestellt). Wir glauben, dass diese Vorarbeiten dazu beitragen können, einen Rahmen für den systematischen Vergleich von Matrizen, wie Fibronectin, mit den zuvor beschriebenen, nämlich Kollagen und Laminin, zu schaffen.32,33,34. Es wäre besonders interessant, Die Migrationskapazität von Maus-NC-Zellen auf Fibronectin mit Kollagen I zu vergleichen, da Kollagen-IA1 endogen von Maus-, Vogel- und menschlichen NC-Zellen abgesondert wird.28,30,31,32. Kollagen I ist daher bei der Berücksichtigung der Matrixwahl ebenso relevant wie Fibronectin. Es ist auch erwähnenswert, dass die Bioverfügbarkeit von Wachstumsfaktoren in den Medien durch verschiedene Matrixkomponenten verändert werden kann, insbesondere angesichts des hohen Serumgehalts unserer Medien. Um dies zu überwinden, arbeiten wir derzeit daran, serumfreie definierte Kulturbedingungen zu schaffen. Diese definierten Medien werden erfolgreich in neuralen Kamminduktionsprotokollen im pluripotenten Stammzellfeld eingesetzt, erfordern aber eine weitere Optimierung für unser NC-Explantenkultursystem.33,34. Unsere Arbeit kann auch als Ausgangspunkt für die Verfeinerung von Bedingungen für andere Arten von NC-Zellen wie Herz- und Rumpf-NC dienen und für nachfolgende Studien der NC-Differenzierung. Am wichtigsten ist, dass dieses Protokoll die Isolierung von Kranial-NC-Zellen für eine Vielzahl von Anwendungen ermöglicht. Wir stellen uns Studien zu gerichteter Migration, 3-D-Migration und Invasion vor. Auf diese Weise isolierte Zellen könnenin vitrofür eine Reihe von Analysen. Beispielsweise können Zellen leicht mit verschiedenen kleinen Molekülen behandelt werden, um bestimmte Proteine anzusprechen, sie können zu definierten Zeitpunkten behandelt werden, und Auswaschexperimente können entwickelt werden, um die Wiederherstellung des Zellverhaltens zu bestimmen (Abbildung 4). Längerfristige Kultur für Transfektion und Differenzierung assays ist möglich, sowie Durchgang von Zellen (Daten nicht gezeigt). Die Lebensfähigkeit, die Fähigkeit der Zellerneuerung und Multipotenz sollten jedoch nach dem Passieren validiert werden. Zellen, die auf Glasabdeckungen plattiert sind, können auch in immunfluoreszierenden Färbungsprotokollen nach Live-Bildgebung verwendet werden. Schließlich stellt dieser Ansatz ein enorm leistungsfähiges System zur Untersuchung der Migration von NC aus genetischen Mausmodellen dar.22,23,24,25.
Offenlegungen
Die Autoren haben keine Interessenkonflikte.
Danksagungen
Wir danken der King es College London Biological Services Unit, insbesondere Tiffany Jarvis und Lynsey Cashnella für ihre kontinuierliche Unterstützung. Wir danken Derek Stemple, Mamoru Ishii und Robert Maxson für die Beratung und Hilfe bei der Einrichtung dieses Protokolls. Wir danken Dheraj Taheem für die Hilfe bei der Gamma-Bestrahlung von STO-Zellen. Wir danken den Laboren von Liu und Krause, insbesondere Tommy Pallett, für die tolle Unterstützung. Diese Arbeit wurde durch Stipendien des BBSRC (BB/R015953/1 an KJL/MK), einer Studentin des MRC Doctoral Training Programme (LD), des Newland Pedley Fund (ALM) und kRIPIS II, Generalsekretariat für Forschung und Technologie (GSRT), Ministerium für Bildung und Religion, finanziert. Angelegenheiten, Griechenland und Fondation Santé (zu SGGM).
Materialien
| Name | Company | Catalog Number | Comments |
| bFGF | R&D systems | 233-FB | |
| b-mercaptoethanol | Gibco | 31350-010 | |
| Tissue culture flasks | Corning | 430639 | 25cm2 culture flasks |
| DMEM (4500 mg/L glucose) | Sigma | D5671 | |
| Dulbecco's phosphate-buffered saline (dPBS) | Sigma | D8537 | |
| Ethanol | Fisher Chemicals | E/0650DF/C17 | |
| Fetal Bovine Serum | Sigma | TFS AA 10-155 | |
| Fetal Bovine Serum (ES Cell FBS) | Gibco | 26140079/16141-079 | |
| Fibronectin bovine plasma | Sigma | F1141 | |
| Gelatin from bovine skin | Sigma | G9382 | |
| L-glutamine | Sigma | G7513 | |
| Glass-bottomed, multi-well 24-well tissue culture plate | Ibidi | 82406 | Glass-bottomed, No 1.5, 24-well tissue culture dish with black sides |
| Cell migration analysis tool | Ibidi | v2.0 | Manuals for this software can be found at: https://ibidi.com/manual-image-analysis/171-chemotaxis-and-migration-tool.html. |
| Circularity Plug-in | ImageJ | v1.29 or later | The circularity plug-in is an extended version of ImageJ/Fiji’s Measure command, designed by Rasband, W., (2000) (wsr@nih.gov). |
| LIF | ESGRO by Millipore | ESG1106 | |
| Manual Cell Tracking Plug-in | ImageJ | v1.34k or later | ref Cordelieres F. Institut Curie, Orsay (France) 2005 |
| Microscope Image Analysis Software | Universal Imaging Corporation | 6.3r7 | MetaMorph Software Series 6.3r7 |
| MEM non-essential aminoacids 100X | Gibco | 11140-050 | |
| Matrigel (ECM-based Hydrogel) | Corning | 356234 | |
| Microscope | Olympus | 1X81 | Olympus 1X81 inverted microscope |
| Microscope camera | Photometrics | 512B | Photometrics Cascadell 512B camera (4x UPlanFL N, 10x UPlanFL N, 20x UPlanFL N, 40x UPlanFL N objectives) |
| Microscope controller | Olympus | 1X2-UCB | Olympus 1X2-UCB microscope controller |
| Microscope temperature controller | Solent Scientific | RS232 | Maintained at 37°C |
| Penicillin/streptomycin | Sigma | A5955 | |
| Sodium Pyruvate | Sigma | S8636 | |
| Statistics software | Graphpad Prism | v7.0 | |
| STO feeder cells | ATCC | CRL-1503 | |
| Stereomicroscope | Nikon | SMZ | |
| XY microscope stage controller | Applied Scientific Instrumentation (ASI) | MS-4400 |
Referenzen
- Duband, J. L. Diversity in the molecular and cellular strategies of epithelium-to-mesenchyme transitions: Insights from the neural crest. Cell Adhesion & Migration. 4, 458-482 (2010).
- Mayor, R., Carmona-Fontaine, C. Keeping in touch with contact inhibition of locomotion. Trends in Cell Biology. 20, 319-328 (2010).
- Le Douarin, N., Kalcheim, C. . The neural crest. 2nd edn. , (1999).
- Trainor, P. A. . Neural Crest Cells : Evolution, Development, and Disease. , (2014).
- Jiang, R., Bush, J. O., Lidral, A. C. Development of the upper lip: morphogenetic and molecular mechanisms. Developmental dynamics : an official publication of the American Association of Anatomists. 235, 1152-1166 (2006).
- Ahola, J. A., et al. Increased incidence of Hirschsprung's disease in patients with hypoplastic left heart syndrome--a common neural crest-derived etiology?. Journal of Pediatric Surgery. 44, 1396-1400 (2009).
- Bajpai, R., et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 463, 958-962 (2010).
- Inoue, K., et al. Congenital hypomyelinating neuropathy, central dysmyelination, and Waardenburg-Hirschsprung disease: phenotypes linked by SOX10 mutation. Annals of Neurology. 52, 836-842 (2002).
- Kim, H., Kang, K., Ekram, M. B., Roh, T. Y., Kim, J. Aebp2 as an epigenetic regulator for neural crest cells. PloS one. 6, e25174 (2011).
- Barriga, E. H., Trainor, P. A., Bronner, M., Mayor, R. Animal models for studying neural crest development: is the mouse different?. Development. 142, 1555-1560 (2015).
- Bronner, M. E., LeDouarin, N. M. Development and evolution of the neural crest: an overview. Developmental Biology. 366, 2-9 (2012).
- De Calisto, J., Araya, C., Marchant, L., Riaz, C. F., Mayor, R. Essential role of non-canonical Wnt signalling in neural crest migration. Development. 132, 2587-2597 (2005).
- Carmona-Fontaine, C., et al. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 456, 957-961 (2008).
- Matthews, H. K., et al. Directional migration of neural crest cells in vivo is regulated by Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development. 135, 1771-1780 (2008).
- Banerjee, S., et al. A novel role for MuSK and non-canonical Wnt signaling during segmental neural crest cell migration. Development. 138, 3287-3296 (2011).
- Pryor, S. E., et al. Vangl-dependent planar cell polarity signalling is not required for neural crest migration in mammals. Development. 141, 3153-3158 (2014).
- Serbedzija, G. N., Bronner-Fraser, M., Fraser, S. E. Vital dye analysis of cranial neural crest cell migration in the mouse embryo. Development. 116, 297-307 (1992).
- Kawakami, M., Umeda, M., Nakagata, N., Takeo, T., Yamamura, K. Novel migrating mouse neural crest cell assay system utilizing P0-Cre/EGFP fluorescent time-lapse imaging. BMC Developmental Biology. 11, 68 (2011).
- Stemple, D. L., Anderson, D. J. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell. 71, 973-985 (1992).
- Etchevers, H. Primary culture of chick, mouse or human neural crest cells. Nature protocols. 6, 1568-1577 (2011).
- Ishii, M., et al. A stable cranial neural crest cell line from mouse. Stem Cells and Development. 21, 3069-3080 (2012).
- Gonzalez Malagon, S. G., Liu, K. J. ALK and GSK3: Shared Features of Neuroblastoma and Neural Crest Cells. Journal of Experimental Neuroscience. 12, 1179069518792499 (2018).
- Gonzalez Malagon, S. G., et al. Glycogen synthase kinase 3 controls migration of the neural crest lineage in mouse and Xenopus. Nature Communications. 9, 1126 (2018).
- Danielian, P. S., Muccino, D., Rowitch, D. H., Michael, S. K., McMahon, A. P. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Current Biology : CB. 8, 1323-1326 (1998).
- Laranjeira, C., et al. Glial cells in the mouse enteric nervous system can undergo neurogenesis in response to injury. Journal of Clinical Investigation. 121, 3412-3424 (2011).
- Pfaltzgraff, E. R., Mundell, N. A., Labosky, P. A. Isolation and culture of neural crest cells from embryonic murine neural tube. Journal Of Visualized Experiments : JoVE. , e4134 (2012).
- Sternberg, J., Kimber, S. J. The relationship between emerging neural crest cells and basement membranes in the trunk of the mouse embryo: a TEM and immunocytochemical study. Journal of Embryology and Experimental Morphology. 98, 251-268 (1986).
- Sternberg, J., Kimber, S. J. Distribution of fibronectin, laminin and entactin in the environment of migrating neural crest cells in early mouse embryos. Journal of Embryology and Experimental Morphology. 91, 267-282 (1986).
- George, E. L., Georges-Labouesse, E. N., Patel-King, R. S., Rayburn, H., Hynes, R. O. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 119, 1079-1091 (1993).
- Henderson, D. J., Copp, A. J. Role of the extracellular matrix in neural crest cell migration. Journal of Anatomy. 191 (Pt 4), 507-515 (1997).
- Thomas, S., et al. Human neural crest cells display molecular and phenotypic hallmarks of stem cells. Human Molecular Genetics. 17 (21), 3411-3425 (2008).
- Greenberg, J. H., Seppa, S., Seppa, H., Hewitt, T. Role of collagen and fibronectin in neural crest cell adhesion and migration. Developmental Biology. 87 (2), 259-266 (1981).
- Leung, A. W., et al. WNT/β-catenin signaling mediates human neural crest induction via a pre-neural border intermediate. Development. 143 (3), 398-410 (2016).
- Zhu, Q., Lu, Q., Gao, R., Cao, T. Prospect of Human Pluripotent Stem Cell-Derived Neural Crest Stem Cells in Clinical Application. Stem Cells International. 2016, 7695836 (2016).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten