
Method Article
Überwachung der Protein-RNA-Interaktionsdynamik in vivo mit hoher zeitlicher Auflösung unter Verwendung von χCRAC
In diesem Artikel
Erratum Notice
Zusammenfassung
Die kinetische Vernetzung und Analyse von cDNA ist eine Methode, die es ermöglicht, die Dynamik von Protein-RNA-Interaktionen in lebenden Zellen mit hoher zeitlicher Auflösung zu untersuchen. Hier wird das Protokoll detailliert beschrieben, einschließlich des Wachstums von Hefezellen, der UV-Vernetzung, der Ernte, der Proteinreinigung und der Vorbereitungsschritte der Next-Generation-Sequencing-Bibliothek.
Zusammenfassung
Die Interaktion zwischen RNA-bindenden Proteinen (RBPs) und ihren RNA-Substraten weist Fluidität und Komplexität auf. Innerhalb ihrer Lebensdauer kann eine einzelne RNA von vielen verschiedenen RBPs gebunden werden, die ihre Produktion, Stabilität, Aktivität und ihren Abbau regulieren. Daher wurde viel getan, um die Dynamik zu verstehen, die zwischen diesen beiden Arten von Molekülen besteht. Ein besonders wichtiger Durchbruch kam mit dem Aufkommen der "c ross-linking and immunoprecipitation" (CLIP). Diese Technik ermöglichte eine stringente Untersuchung, welche RNAs an ein bestimmtes RBP gebunden sind. Kurz gesagt, das zu untersuchende Protein wird in vivo mit seinen RNA-Substraten UV-vernetzt, unter sehr strengen Bedingungen gereinigt und dann werden die kovalent mit dem Protein vernetzten RNAs in cDNA-Bibliotheken umgewandelt und sequenziert. Seit seiner Konzeption wurden viele abgeleitete Techniken entwickelt, um CLIP für bestimmte Studienbereiche zugänglich zu machen. Die Vernetzung mit ultraviolettem Licht ist jedoch notorisch ineffizient. Dies führt zu verlängerten Expositionszeiten, die eine zeitliche Untersuchung von RBP-RNA-Interaktionen unmöglich machen. Um dieses Problem zu lösen, haben wir kürzlich stark verbesserte UV-Bestrahlungs- und Zellerntegeräte entwickelt und gebaut. Mit diesen neuen Werkzeugen haben wir ein Protokoll für zeitaufgelöste Analysen von RBP-RNA-Interaktionen in lebenden Zellen mit hoher zeitlicher Auflösung entwickelt: Kinetic CRoss-linking und Analysis of cDNAs (χCRAC). Wir haben diese Technik kürzlich verwendet, um die Rolle von Hefe-RBPs bei der Anpassung an Nährstoffstress zu untersuchen. Dieses Manuskript gibt einen detaillierten Überblick über die χCRAC-Methode und präsentiert die jüngsten Ergebnisse, die mit dem Nrd1 RBP erzielt wurden.
Einleitung
RNAs sind oft auf RBPs angewiesen, um ihre Funktion auszuüben, was zu einem großen Interesse am Verständnis der Dynamik zwischen diesen Molekülen geführt hat. Viele RBPs wurden in einer Vielzahl von Organismen identifiziert. Es war jedoch schon immer bekanntermaßen schwierig, RBP-RNA-Interaktionen in vivo zu untersuchen. Ein großer Durchbruch bei der Untersuchung solcher Wechselwirkungen kam mit dem Aufkommen von CLIP1. Diese Methode nutzt ultraviolette (UV, 254 nm) Bestrahlung, um kovalente Bindungen zwischen RBPs und ihren direkt gebundenen RNAs zu induzieren (d. h. Quervernetzung ohne Abstand). Anschließend wird das zu untersuchende RBP unter strengen Bedingungen immungereinigt, um sicherzustellen, dass nur RNAs identifiziert werden, die kovalent mit den Proteinen vernetzt sind. Gebundene RNAs werden dann teilweise mit RNasen verdaut und anschließend zur Sequenzierung in cDNA-Bibliotheken umgewandelt. Die hohe Aufreinigungsstringenz ist wichtig, da sie die Spezifität der Protein- und RNA-Rückgewinnung erheblich erhöht, was auch durch die SDS-PAGE-Aufreinigung des vernetzten Ribonukleoprotein-Komplexes (RNP) weiter verbessert wird. CLIP und verwandte Methoden liefern auch Einblicke in die Nukleotidauflösung der Proteinbindungsstelle, da während der Herstellung der Sequenzierungsbibliothek Aminosäuren, die mit der RNA vernetzt sind, häufig die reverse Transkriptase beenden oder das Enzym veranlassen, Mutationen an dieser Stelle einzuführen 1,2,3.
Seit seiner Einführung hat das ursprüngliche CLIP-Protokoll eine bemerkenswerte Vielfalt an abgeleiteten Methoden hervorgebracht. Ein besonders wichtiger Durchbruch war die Entwicklung von HITS-CLIP (oder CLIP-seq), das die Hochdurchsatzsequenzierung mit dem CLIP-Ansatz3 verbindet. Dies wurde seither von allen CLIP-basierten Methoden übernommen. iCLIP führte Verbesserungen in den RNase-vermittelten Trimm- und Adaptorligationstechniken ein, die eine genauere Kartierung der RBP-Bindungsstellen ermöglichen4. PAR-CLIP kombinierte die 4thio-uridin/Uracil-Markierung mit der Vernetzung bei 365 nm und ermöglichte so die Kartierung von Vernetzungsstellen durch die Analyse von T-C-Substitutionen5. CRAC, Urea-iCLIP, dCLIP und uvCLAP führten Denaturierungsbedingungen und duale Affinitätsreinigungsschritte ein, die die Hintergrundbindung an das Affinitätsharz weiter reduzieren und die Spezifität des Proteineinfangs weiter erhöhen 2,6,7,8,9. Darüber hinaus führten CRAC, uvCLAP und dCLIP die Markierung des interessierenden RBP mit einem Affinitäts-Tag ein, wodurch die Notwendigkeit der Generierung spezifischer Antikörper überwunden wurde.
Es wurden auch mehrere Optimierungen vorgenommen, um die CLIP-Methodik zu beschleunigen. Das ursprüngliche CLIP-Protokoll nutzte die radioaktive Markierung der gefangenen RNAs, um die RBP-RNA-Komplexe nach SDS-PAGE sichtbar zu machen. Der Einsatz von Radioaktivität kann jedoch für Laboratorien, die nicht für solche Arbeiten eingerichtet sind, problematisch sein. irCLIP enthält einen Fluorophor-gekoppelten Adapter, der die Visualisierung durch Infrarot-Bildgebung10 erleichtert, und sCLIP nutzt die Biotinylierung von eingefangenen RNAs, um sie durch Streptavidin-konjugiertes HRP11 zu visualisieren. Darüber hinaus verzichtet eCLIP komplett auf die RNA-Markierung; Stattdessen wird das Protein allein aufgrund seiner bekannten Größe12 herausgeschnitten. Die Streptavidin-basierte Aufreinigung wurde auch verwendet, um den Prozess der Bibliotheksvorbereitung in FAST-iCLIP zu beschleunigen, bei dem ein biotinylierter 3'-Adapter auf die RNAs ligiert und verwendet wird, um die Aufreinigung nach reverser Transkription und Zirkularisierung zu ermöglichen13. Zusätzliche Erweiterungen des iCLIP-Protokolls erhöhten auch die Komplexität der Bibliothekenerheblich 4.
Schließlich wurde CLIP modifiziert, um das Einfangen von RBPs aus verschiedenen zellulären Subkompartimenten zu ermöglichen 14,15, um neu transkribierte RNAs durch gepulste Induktion der photoaktivierbaren Ribonukleoside 5,16,17 sichtbar zu machen, um methylierte RNAseinzufangen 18,19,20, um RNA-RNA-Interaktionen 21,22 zu untersuchen und um 3'-Enden zu kartieren 23,24.
Trotz des großen Beitrags von CLIP-basierten Techniken zum Verständnis der Wechselwirkungen zwischen RBPs und RNAs wurde dies durch die Ineffizienz der UV-Vernetzung eingeschränkt. Während in einer Monoschicht gezüchtete Kulturzellen in der Regel relativ einfach zu vernetzen sind, ist dies in Geweben oder Zellen in Lösung deutlich schwieriger. Gewebe kann mehrere Runden UV-Bestrahlung erfordern, um in die erforderlichen Zellschichten einzudringen, während mikrobielle Zellen oft in reichhaltigen Medien gezüchtet werden, die aromatische, UV-absorbierende Verbindungen enthalten25. Tatsächlich wurden UV-Bestrahlungszeiten von bis zu 30 Minuten verwendet, um eine ausreichende Vernetzung zwischen RBPs und ihren gebundenen RNAs für solche Proben zu erzeugen26,27,28. Diese ausgedehnte UV-Exposition induziert Stressreaktionen innerhalb der Zelle, wie z. B. UV-induzierte DNA-Schäden, die in einigen Anwendungen die endgültigen Daten kontaminieren können.
Die Mehrzahl der CLIP-Studien konzentrierte sich auf die Erstellung einzelner "Momentaufnahmen" spezifischer Protein-RNA-Interaktionen in einer Zelle. Protein-RNA-Interaktionen sind jedoch von Natur aus dynamisch, insbesondere wenn Zellen Veränderungen in ihrer Umgebung unterliegen. Dies kann eine plötzliche Verringerung der Verfügbarkeit von essentiellen Nährstoffen oder schnelle Temperaturänderungen umfassen. Um die Rolle eines RBP während Stress wirklich zu verstehen, ist es daher am besten, zeitaufgelöste Analysen durchzuführen, da sie das gesamte Spektrum der RBP-Ziele während des Stresses erfassen und bestimmen können, in welchem Stadium der Stressantwort das gewählte RBP aktiv ist. Insbesondere Studien an Hefen zeigten, dass die ersten Minuten der Anpassung absolut überlebenswichtig sind und die RNA-Halbwertszeiten in Bakterien von Minuten bis Sekunden variieren können 29,30,31,32,33. Daher sollten solche zeitaufgelösten Analysen idealerweise mit hoher zeitlicher Auflösung durchgeführt werden. Die langen Vernetzungszeiten machen es jedoch besonders schwierig, adaptive Reaktionen im Frühstadium zu untersuchen.
Um diese Probleme zu lösen, haben wir kürzlich eine verbesserte Methode entwickelt, die in der Lage ist, Zellen auf winzigen Zeitskalen zu vernetzen und zu ernten. Unsere χCRAC-Methode ermöglicht die quantitative Messung dynamischer Änderungen in RBP-RNA-Wechselwirkungen mit bisher unerreichter Auflösung. Entscheidend für dieses Verfahren war die Entwicklung einer neuartigen UV-Bestrahlungsvorrichtung32 , die die erforderliche Vernetzungszeit in Hefen und Bakterien in Lösung um das 10-fache reduziert, wodurch RBP-RNA-Wechselwirkungen sofort effektiv eingefroren werden. Um die Zellen nach UV-Bestrahlung schnell zu ernten, haben wir außerdem ein Vakuumfiltrationsgerät entwickelt, das exponentiell wachsende Hefe in einer 0,5-Liter-Kultur in etwa 30 s32 ernten kann. Diese technologischen Innovationen ermöglichen die Untersuchung der RBP-RNA-Dynamik mit einer Auflösung im Minutenbereich. Darüber hinaus haben wir auch mehrere Optimierungen am ursprünglichen CRAC-Protokoll2 vorgenommen, um seine Praktikabilität zu erhöhen.
Mit χCRAC haben wir kürzlich das Targetom eines Hefekern-RBP, Nab3, als Reaktion auf Glukoseentzug untersucht. In Saccharomyces cerevisiae kann Nab3 einen Komplex mit Nrd1, einem RBP und der RNA-Helikase Sen1 bilden, um den NNS-Komplex zu bilden. Die Bindung von NNS an die RNA-Polymerase und das entstehende Transkript kann eine transkriptionelle Terminierung auslösen34. Dieser Komplex ist hauptsächlich an der Entfernung kryptischer, nicht-kodierender RNA-Transkripte beteiligt, reguliert aber auch die Expression proteinkodierender Gene. Die Studie zeigte ein differentielles Targeting von Nab3 auf nicht-kodierende und kodierende Transkripte nach nur einer Minute Stress32. Wir konnten zeigen, dass die co-transkriptionelle Terminierung durch Nab3 zu einer sehr transienten, pulsartigen Expression von Retrotransposon-Genen führt, die mit herkömmlichen CLIP-basierten Ansätzen nur schwer zu detektieren gewesen wäre. Darüber hinaus erhöhten die kurzen UV-Bestrahlungszeiten in unserem UV-Vernetzer auch die Rückgewinnung kurzlebiger nicht-kodierender RNAssignifikant 32. χCRAC wird wahrscheinlich ein entscheidendes Instrument sein, um nicht nur zu verdeutlichen, wie RBPs die Reaktion auf Stress auf unmittelbaren Zeitskalen gestalten, sondern auch ihre sich ändernden Rollen während des gesamten Lebenszyklus einer Reaktion. Dieses Manuskript bietet einen detaillierten Überblick über alle Schritte des χCRAC-Protokolls. Zur Veranschaulichung wurde die Methode verwendet, um das Hefeprotein Nrd1, das an der transkriptionellen Terminierung und dem RNA-Zerfall beteiligt ist35,36, und sein RNA-Targetom als Reaktion auf Glukoseentzug über eine Vielzahl von Zeitpunkten hinweg zu untersuchen. Schließlich zeigen wir auch, dass unsere UV-Bestrahlungseinheit RBPs schnell mit RNA in HeLa-Zellen vernetzen kann, wodurch es möglich ist, auch hochauflösende zeitaufgelöste Analysen in adhärenten Zellen durchzuführen.
Protokoll
| TN150 |
| 50 mM Tris pH 7,8 |
| 150 mM NaCl |
| 0,1 % NP-40 |
| 1X Proteasehemmer |
| TN1000 |
| 50 mM Tris pH 7,8 |
| 1M NaCl |
| 0,1 % NP-40 |
| NP-PNK |
| 50 mM Tris-HCl pH 7,8 |
| 10 mM MgCl2 |
| 0,1 % NP-40 |
| 5 mM Beta-Mercaptoethanol |
| 5 x PNK |
| 250 mM Tris-HCl pH 7,8 |
| 50 mM MgCl2 |
| 50 mM Beta-Mercaptoethanol |
| WB I |
| 50 mM Tris-HCl pH 7,8 |
| 300 mM NaCl |
| 10 mM Imidazol |
| 6M Guanidin-HCl |
| 0,1 % NP-40 |
| 5 mM Beta-Mercaptoethanol |
| WB II |
| 50 mM Tris-HCl pH 7,8 |
| 50 mM NaCl |
| 10 mM Imidazol |
| 0,1 % NP-40 |
| 5 mM Beta-Mercaptoethanol |
| Elutionspuffer |
| 50 mM Tris pH 7,8 |
| 50 mM NaCl |
| 250 mM Imidazol |
| 0,1 % NP-40 |
| 5 mM Beta-Mercaptoethanol |
| Protease-K-Puffer |
| 50 mM Tris |
| 0,1 % NP-40 |
| 5 mM β-Mercaptoethanol |
| 1% Sicherheitsdatenblatt |
| 5 mM EDTA |
| 50 mM NaCl2 |
| Säugetier-Lysepuffer |
| 50 mM Tris-HCl pH 8 |
| 100 mM NaCl |
| 0,5 % v/v Triton X-100 |
| 0,25 % w/v Na-Desoxycholat |
| 0,1 % w/v SDS |
| 5 mM EDTA |
| 1 mM DVB-T (frisch hinzugefügt) |
| 1X Proteasehemmer |
Tabelle 1: Die für χCRAC erforderlichen Puffer und ihre Zusammensetzungen.
1. UV-Vernetzung und Lysatherstellung
- Mikroorganismen in Lösung
- 3,5 l des gewünschten Mediums werden mit Hefe aus einer Übernachtkultur auf eine Start-OD600 von 0,05 geimpft. Anbau bei 30 °C mit kontinuierlichem Schütteln bei 180 U/min.
- Bereiten Sie während des Wachstums andere notwendige Materialien vor.
- Bereiten Sie einen Behälter mit flüssigem Stickstoff vor.
- 3 l Stressmedium zubereiten und im Wasserbad auf 30 °C erwärmen.
- Richten Sie die Filtervorrichtung ein, schalten Sie den Vernetzer ein (Abbildung 2A) und beschriften Sie konische 50-ml-Röhrchen, eines für jeden Zeitpunkt.
- Sobald die Zellen den gewünschten OD 600 erreicht haben, gießen Sie500 ml Zellen direkt in den Vernetzer und bestrahlen Sie sie mit 250 mJ 254 nm UV. Siehe Abbildung 2A und Abbildung 3A für Details zur Verwendung des Vernetzers.
HINWEIS: Die UV-Bestrahlungsenergie muss für jedes Protein von Interesse sorgfältig optimiert werden. Weitere Informationen finden Sie in der Diskussion. - Filtern Sie die Zellen nach der Vernetzung mit einem der Vakuumfiltrationsgeräte (Abbildung 2B,C). Rollen Sie die Membran mit den filtrierten Zellen auf, legen Sie sie in das konische 50-ml-Röhrchen t = 0 (Zeit Null) und frieren Sie sie in flüssigem Stickstoff ein.
- Filtern Sie die restlichen Zellen über sechs verschiedene Filter. Resuspendieren Sie die gesammelten Zellen in den 3 l des zuvor erwärmten stressinduzierenden Mediums, indem Sie die Membranen in das Medium fallen lassen und 50 s lang kräftig mit einem Stripette mischen. Bereiten Sie sich nach den 50 s auf die Entnahme der t = 1 Probe vor.
- Nach 1 min werden 500 ml Zellen vernetzt und durch Filtration wie in den Schritten 1.1.3–1.1.4 geerntet. Wiederholen Sie den Vorgang nach 2, 4, 8, 14 und 20 Minuten oder je nach Bedarf zu verschiedenen Zeitpunkten.
- Lagern Sie die konischen Röhrchen mit den Zellen bei -80 °C. Phosphatgepufferte Kochsalzlösung (PBS) über Nacht auf 4 °C einstellen.
- Am nächsten Tag nehmen Sie jedes konische Röhrchen, das eine vernetzte Probe enthält, und resuspendieren Sie die Zellen in 25 ml kaltem PBS durch kräftiges Schütteln.
- Übertragen Sie die Zellsuspensionen in neue konische Röhrchen und schleudern Sie sie bei 4.600 x g, 5 min bei 4 °C.
- Gießen Sie das PBS ab, drehen Sie es schnell erneut, um Rest-PBS aufzufangen, und dekantieren Sie dann die restliche Flüssigkeit mit einer Pipette.
- Berechnen Sie das Gewicht des Pellets im Röhrchen, indem Sie es mit einem leeren Röhrchen vergleichen.
- Fügen Sie zwei Pelletvolumina eiskaltes TN150, 60 μl DNase 1 und 10 μl RNase-Inhibitor hinzu. 30 Minuten auf Eis inkubieren.
- Fügen Sie beispielsweise für 400 mg Zellen 800 μl eiskaltes TN150 hinzu.
- Die Zugabe der DNase ist für die meisten löslichen Proteine nicht essentiell, aber sehr wichtig bei der Untersuchung von Chromatin-gebundenen Proteinen wie der RNA-Polymerase. Darüber hinaus reduziert es die Viskosität von bakteriellen Lysaten. Es ist sehr wichtig, genau zwei Pelletvolumina des Lysepuffers zu verwenden, da sonst die Lyseeffizienz abnehmen kann.
- Fügen Sie der Zellsuspension drei Pelletvolumina (in ml) Zirkonoxidkügelchen hinzu. Verwenden Sie für Hefe Perlen mit einem Durchmesser von 0,5 mm und für Bakterien 0,1 mm.
- Messen Sie beispielsweise für 400 mg Zellen 1,2 ml Zirkonoxidkügelchen in einem 1,5-ml-Röhrchen ab und geben Sie sie zu den in Lysepuffer resuspendierten Zellen.
- Wirbeln Sie die Zellsuspensionen 1 Minute lang und legen Sie sie dann 1 Minute lang auf Eis. Wiederholen Sie den Vorgang insgesamt 5x.
- Fügen Sie zwei Pelletvolumina TN150-Puffer hinzu und wirbeln Sie kräftig zum Mischen.
- Zentrifugieren Sie die Suspension im konischen Röhrchen bei 4.600 g für 20 min bei 4 °C in einer Tischzentrifuge.
- Entnehmen Sie nach dem Zentrifugieren eine 50-μl-Probe des Überstands für zukünftige Western-Blot-Analysen, um die Proteinexpression der gesamten Zelle zu untersuchen.
- Die Überstände werden in 1,5-ml-Röhrchen überführt und das Lysat 20 Minuten lang bei 20.000 x g bei 4 °C in einer Mikrofuge geschleudert.
- Wenn Sie 5-ml-Röhrchen verwenden, zentrifugieren Sie alternativ 20 Minuten lang bei 13.000 x g .
- Entnehmen Sie nach der Zentrifugation eine 50-μl-Probe des Überstands für zukünftige Western-Blot-Analysen, um die lösliche Expression des Proteins zu untersuchen.
- Fahren Sie mit der RBP-Erfassung fort (Abschnitt 2).
- Kultivierte adhärente Zellen
- Säen Sie 24 Stunden vor der UV-Vernetzung genügend adhärente Zellen in eine Petrischale, damit sie am nächsten Tag eine Konfluenz von 80 % erreichen können. Wachsen Sie über Nacht im gewünschten Medium in einem Zellkultur-Inkubator bei 37 °C, 5% CO2.
HINWEIS: Bei der Verwendung von Quarz-Petrischalen ist es von Vorteil, die Zelladhäsion durch die Behandlung der Kulturware mit Poly-D-Lysin (70.000–140.000 Gew.-Gewicht) und fetalem Kälberserum (FCS) 2,5 h vor der Aussaat zu fördern. Fügen Sie so viel Poly-D-Lysin hinzu, dass die gesamte Wachstumsfläche bedeckt ist, und inkubieren Sie es 5 Minuten lang bei Raumtemperatur (RT). Als nächstes sollte die Quarz-Petrischale gründlich mit Wasser gespült und im Zellkultur-Inkubator für 2 h oder bis sie vollständig trocken ist, getrocknet werden. Geben Sie anschließend so viel FCS hinzu, dass die Wachstumsfläche vollständig bedeckt ist, und legen Sie es für mindestens 30 Minuten in den Inkubator. Das FCS sollte vor der Aussaat der Zellen vollständig entfernt werden. - Sobald die Zellen eine Konfluenz von 80 % erreicht haben, entfernen Sie das Medium und waschen Sie es mit 15 ml eiskaltem PBS. Entfernen Sie anschließend die restliche Flüssigkeit vollständig und fahren Sie sofort mit dem nächsten Schritt fort.
- Übertragen Sie die Petrischale in das Tablett für adhärente Zellen (Abbildung 3B) und bestrahlen Sie sie mit 300 mJ 254 nm UV. Siehe Abbildung 2A und Abbildung 3B für Details zur Verwendung des Vernetzers.
HINWEIS: Die UV-Bestrahlungsenergie muss für jedes Protein von Interesse sorgfältig optimiert werden. Weitere Informationen finden Sie in der Diskussion. - Stellen Sie die Petrischale unmittelbar nach der Vernetzung auf Eis und fügen Sie 10 ml eiskaltes PBS hinzu. Sammeln Sie die Zellen durch Schaben und übertragen Sie sie in ein konisches 15-ml-Röhrchen. Pellet durch Zentrifugation bei 300 x g für 5 min bei 4 °C.
- Entfernen Sie das PBS, resuspendieren Sie das Zellpellet in 1 ml eiskaltem PBS und geben Sie es in ein 1,5-ml-Mikrozentrifugenröhrchen. Pelletzellen nochmals durch Zentrifugation für 5 min bei 300 x g bei 4 °C.
- Entfernen Sie das PBS und frieren Sie die Zellpellets auf Trockeneis ein. Lagern Sie die Zellpellets bis zur Verwendung bei -80 °C.
- Wiederholen Sie die Schritte 1.2.3 bis 1.2.6 für jeden Zeitpunkt.
- Die Zellpellets werden in 1 ml Lysepuffer resuspendiert und in ein konisches 15-ml-Röhrchen überführt. Fügen Sie anschließend 1 ml Lysepuffer hinzu, so dass insgesamt 2 ml vorhanden sind.
- Fügen Sie 5 μl Säugetier-RNase-Inhibitor hinzu.
- Beschallen Sie 5x für 10 s auf Eis bei 10 Ampere. Warten Sie 30 Sekunden zwischen den Beschallungsrunden.
- Berechnen Sie die Proteinkonzentration jeder Probe und normalisieren Sie sie auf die niedrigste Konzentration.
- Übertragen Sie 1,98 ml Lysat in ein 2-ml-Röhrchen.
- 10 μl DNase I zugeben und bei 37 °C 5 min unter Schütteln mit 1.200 U/min inkubieren.
- Zentrifugieren Sie das Lysat bei 16.000 x g für 20 min bei 4 °C.
- Entnehmen Sie nach der Zentrifugation eine 50-μl-Probe des Überstands für zukünftige Western-Blot-Analysen, um die lösliche Expression des Proteins zu untersuchen.
- Fahren Sie mit der RBP-Erfassung fort (Abschnitt 2).
- Säen Sie 24 Stunden vor der UV-Vernetzung genügend adhärente Zellen in eine Petrischale, damit sie am nächsten Tag eine Konfluenz von 80 % erreichen können. Wachsen Sie über Nacht im gewünschten Medium in einem Zellkultur-Inkubator bei 37 °C, 5% CO2.
2. RBP-Erfassung
- Waschen Sie die magnetischen Anti-FLAG-Beads (75 μl Aufschlämmung pro Probe) oder IgG-Agarose (500 μl Aufschlämmung pro Probe) 3x mit 5 ml TN150. Resuspendieren Sie in einem Endvolumen von 700 μl TN150 und geben Sie 100 μl gewaschene Kügelchen in sieben konische 15-ml-Röhrchen.
- Bis zur Verwendung auf Eis lagern.
- Sobald die Lysate geklärt sind, geben Sie den Überstand in das Röhrchen mit den Anti-FLAG/IgG-Kügelchen.
- Nuss bei 4 °C für 2 h.
HINWEIS: Einige Protokolle beschreiben Inkubationen über Nacht mit den Beads, dies wird jedoch nicht empfohlen, da lange Inkubationszeiten die Rückgewinnung von vernetzten RNAs drastisch reduzieren können.
3. Waschen der Perlen und TEV-Spaltung der Tags
- Ernten Sie die Perlen und entfernen Sie das Lysat.
- Nehmen Sie eine 50-μl-Probe des Überstands für zukünftige Western-Blot-Analysen, um das nicht eingefangene Protein zu untersuchen.
- Resuspendieren Sie die Kügelchen in eiskaltem TN1000 und füllen Sie sie in ein 1,5-ml-Röhrchen um. 10 min, 4 °C, mit Nutation waschen. Wiederholen Sie den Vorgang für insgesamt drei Wäschen.
- Wenn Sie IgG-Agarose-Kügelchen verwenden, waschen Sie sie mit 5 ml TN1000. Wenn Sie Anti-FLAG-Perlen verwenden, verwenden Sie 2 ml.
- Als nächstes waschen Sie die Perlen 3x mit TN150, mit dem gleichen Volumen wie oben.
- Nach dem dritten Waschgang resuspendieren Sie die Kügelchen in 600 μl TN150.
- Geben Sie 30 U hausgemachte GST-TEV-Protease in die Wulstsuspension und drehen Sie sie 2 h lang bei RT.
HINWEIS: Die rekombinante GST-TEV-Protease ist jetzt auch kommerziell erhältlich, wurde aber nicht mit diesem Protokoll getestet.- Bereiten Sie sich während des Aufschlusses auf die nächsten Schritte vor, indem Sie für jede Probe Säulen mit drei 1,5-ml-Röhrchen aufstellen (d. h. für sieben Proben drei Reihen mit je sieben Säulen).
- In die letzte Reihe der Röhrchen werden 0,4 g Guanidiahydrochlorid, 27 μl 5M Natriumchlorid und 3 μl 2,5 M Imidazol (pH = 8) gegeben. Beachten Sie, dass der pH-Wert des Imidazols 8 betragen muss. Dies ist entscheidend für die Aufrechterhaltung der RNA-Integrität.
- Waschen Sie zusätzlich die erforderliche Menge an Nickelperlen in WB I 3x. Verwenden Sie 100 μl Aufschlämmung pro Probe. Nach der letzten Wäsche resuspendieren Sie die Perlen im gleichen Originalvolumen von WB I und lagern Sie sie auf Eis.
- Sobald der TEV-Aufschluss abgeschlossen ist, sammeln Sie den Überstand mit einem magnetischen Gestell für Anti-FLAG-Beads oder Zentrifugation für IgG-Beads und geben Sie ihn in die erste Reihe der zuvor aufgestellten Röhrchen.
- Nehmen Sie eine 50-μl-Probe des TEV-Eluats für die Western-Blot-Analyse.
- Stellen Sie einen Thermoblock-Inkubator auf 37 °C ein. In die zweite Reihe der Röhrchen geben Sie 1 μl RNase-Cocktail (1:50 Verdünnung).
- Nehmen Sie 550 μl TEV-Eluat aus der ersten Röhrchenreihe und geben Sie es in die zweite Reihe (mit dem RNase-Cocktail). Pipettieren Sie kräftig, um das Mischen zu gewährleisten.
- Nachdem Sie dies für die erste Probe abgeschlossen haben, legen Sie das Röhrchen sofort in den Thermoblock und starten Sie einen Timer. Fahren Sie mit den nachfolgenden Beispielen fort, sodass jedes gestaffelt ist.
- Genau 5 Minuten inkubieren. Wenn Sie fertig sind, nehmen Sie die erste Probe aus dem Thermoblock und übertragen Sie die Lösung in die dritte Reihe von Röhrchen (mit dem Guanidiumhydrochloridpulver).
HINWEIS: Eine 5-minütige Inkubation mit einer 1:50-Verdünnung des RNase-Cocktails ist normalerweise für die meisten Proteine geeignet, aber dieser Schritt muss sorgfältig mit unterschiedlichen Inkubationszeiten oder -konzentrationen für jedes Protein optimiert werden, um sicherzustellen, dass die vernetzten RNAs die richtige Größe haben (30–100 nt). - Sofort ein paar Sekunden lang bei voller Geschwindigkeit wirbeln, um das Guanidiumpulver aufzulösen, und dann zur nächsten Probe übergehen.
- Nachdem alle Proben in das Guanidiumpulver überführt wurden, wirbeln Sie erneut, um sicherzustellen, dass das gesamte Pulver vollständig aufgelöst ist.
- Fügen Sie 100 μl gewaschene Nickelkügelchen hinzu und lassen Sie sie über Nacht bei 4 °C drehen. Diese Inkubationszeit kann auf 2 h verkürzt werden.
4. Behandlung mit alkalischer Phosphatase auf dem Bead
- Stellen Sie einen Thermoblock auf 37 °C ein.
- Platzieren Sie eine Aufreinigungs-Spin-Säule in einem 2-ml-Röhrchen, eine für jede Probe. Übertragen Sie die Nickelperlen auf die Säulen und lassen Sie den Überstand abfließen. Stellen Sie anschließend sicher, dass alle Nickelkügelchen aus dem 1,5-ml-Röhrchen entfernt wurden, indem Sie sie mit WB I spülen und auf die Säule auftragen.
- Richten Sie 2 ml-Röhrchen ein, sechs pro Probe (eines zum Auffangen jeder Wäsche). Halten Sie die Außenseite der Säulen trocken, um den Durchfluss aufrechtzuerhalten. Waschen Sie die Perlen 3x mit 500 μL WB I und dann 3x mit 500 μL NP-PNK.
- Schließen Sie den Deckel der Schleudersäule und schleudern Sie kurz die Perlen, um überschüssigen Puffer zu entfernen.
- Setzen Sie den Stopfen auf die Säule, füllen Sie die Säulen in 1,5-ml-Röhrchen und fügen Sie 60 μl des in Tabelle 2 gezeigten Reaktionsgemisches hinzu.
| Bestandteil | 1x | 7,5-fach |
| 5 x PNK-Puffer | 12 | 90 |
| Alkalische Phosphatase | 4 | 30 |
| RNase-Hemmer | 2 | 15 |
| H2O | 42 | 315 |
| Endgültiges Volumen | 60 μL | 450 μL |
Tabelle 2: Alkalisches Phosphatase-Reaktionsgemisch.
- Die Kügelchen 1 h bei 37 °C inkubieren.
- Waschen Sie die Kügelchen 1x mit 500 μL WB I, um die alkalische Phosphatase zu inaktivieren, und anschließend 3x mit 500 μL NP-PNK-Puffer. Achten Sie darauf, das Innere der Säule gründlich mit dem NP-PNK-Puffer zu spülen, um alle Spuren von Guanidium zu entfernen.
5. On-Bead-Ligation des App-PE-Linkers an das 3'-Ende der RNA
- Schleudern Sie den verbleibenden Puffer aus und geben Sie 60 μl des in Tabelle 3 angegebenen Gemisches (siehe Tabelle 4 für die App-PE-Sequenz) in die Säulen. Die Reaktion wird 6 h bei 25 °C inkubiert.
| Bestandteil | 1x | 7,5-fach |
| 5 x PNK-Puffer | 12 | 90 |
| App-PE-Adapter (100 μM) | 0.6 | 4.5 |
| T4 RNA Ligase 2 verkürzt K227Q | 3 | 22.5 |
| RNase-Hemmer | 1.5 | 11.25 |
| 50% PEG 8000 | 12 | 90 |
| H2O | 30.9 | 231.75 |
| Endgültiges Volumen | 60 μL | 450 μL |
Tabelle 3: App-PE-Linker-Ligations-Reaktionsgemisch.
| Name des Oligonukleotids | Sequenz (5'-3') | |||
| L5Aa | invddT-ACACrGrArCrGrCrUrCrUrCrCrCrGrArUrCrUrNrNrUrArArGrCrN-OH | |||
| L5Ab | invddT-ACACrGrArCrGrCrUrCrUrCrCrCrGrArUrCrUrNrNrNrArUrArArGrCrN-OH | |||
| L5Ac | invddT-ACACrGrArCrCrCrUrCrUrCrCrGrArUrCrUrNrNrNrGrCrGrCrArCrCrN-OH | |||
| L5Ad | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrCrGrGrCrUrArGrCrN-OH | |||
| L5Ba | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrArArGrCrN-OH | |||
| L5Bb | invddT-ACACrGrArCrCrCrUrCrUrCrCrGrArUrCrUrNrNrNrGrUrGrCrN-OH | |||
| L5BC | invddT-ACACrGrArCrGrCrUrCrUrCrCrCrGrArUrCrUrNrNrNrCrArCrUrArGrCrN-OH | |||
| L5Bd | invddT-ACACrGrArCrCrCrUrCrUrCrCrGrArUrCrUrNrNrUrCrUrCrUrArGrCrN-OH | |||
| L5Ca | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrNrUrArGrCrN-OH | |||
| L5Cb | invddT-ACACrGrArCrGrCrUrCrUrCrCrCrGrArUrCrUrNrNrNrGrCrN-OH | |||
| L5Cc | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrArCrUrCrArGrCrN-OH | |||
| L5Cd | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrArCrUrUrArGrCrN-OH | |||
| L5Da | invddT-ACACrGrArCrGrCrUrCrUrCrCrCrGrArUrCrNrNrNrCrGrGrUrUrN-OH | |||
| L5Db | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrGrCrArN-OH | |||
| L5Dc | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrUrArGrCrN-OH | |||
| L5Dd | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrArCrArCrCrGrN-OH | |||
| L5Ea | invddT-ACACrGrArCrGrCrUrCrUrCrCrCrGrArUrCrUrNrNrNrCrArCrUrGrUrN-OH | |||
| L5Eb | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrGrUrCrArArN-OH | |||
| L5Ec | invddT-ACACrGrArCrGrCrUrCrUrCrCrCrGrArUrCrUrNrNrUrGrUrCrArCrN-OH | |||
| L5Ed | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrArCrArArGrGrN-OH | |||
| App_PE | App-NAGATCGGAAGAGCACACGTCTG-ddC | |||
Tabelle 4: Die Sequenzen der DNA- und RNA-Adapter, die für die Ligation an den 5'- und 3'-Enden der eingefangenen RNAs erforderlich sind. Diese wurden mittels RNase-freier HPLC aufgereinigt.
- Waschen Sie die Perlen 1x mit 500 μL WB I und 3x mit 500 μL NP-PNK-Puffer. Legen Sie die Säule in ein neues Rohr und drehen Sie den restlichen Puffer aus.
6. On-Bead-Phosphorylierung der 5'-Enden der RNA
- 80 μl des in Tabelle 5 angegebenen Gemisches in die Spalten geben. Inkubieren Sie die Reaktion für 40 min bei 37 °C.
HINWEIS: Die Proben werden nun hochradioaktiv sein. Daher sollten alle nachfolgenden Arbeiten hinter einem Schutzschirm durchgeführt werden und Abfälle sollten gemäß den örtlichen Gesundheits- und Sicherheitsvorschriften entsorgt werden.
| Bestandteil | 1x | 7,5-fach |
| 5 x PNK-Puffer | 16 | 120 |
| 32P-ɣATP (10 μCi/μL) | 3 | 22.5 |
| T4 PNK | 3 | 22.5 |
| H2O | 58 | 435 |
| Endgültiges Volumen | 80 μL | 600 μL |
Tabelle 5: Phosphorylierungsreaktionsgemisch.
- Fügen Sie 1 μL 100 mM ATP hinzu und lassen Sie die Reaktion weitere 20 Minuten ablaufen. Dadurch wird sichergestellt, dass fast alle 5'-Enden Phosphate enthalten, um die Ligation des 5'-Linkers zu erleichtern.
- Stellen Sie 2 ml-Röhrchen auf, fünf pro Probe.
- Waschen Sie die Perlen 1x mit 500 μL WB I und 3x mit 500 μL NP-PNK-Puffer. Beachten Sie, dass diese Elution sehr radioaktiv sind und daher ordnungsgemäß entsorgt werden sollten.
- Bewegen Sie die Säule in die letzte Röhre und drehen Sie den verbleibenden Puffer aus.
7. On-Bead-Ligatur des 5'-Linkers
HINWEIS: Die 5'-Linker enthalten einen RNA-Barcode, der zur Identifizierung jeder Probe nach der Sequenzierung verwendet wird. Daher ist es absolut wichtig zu beachten, welcher Linker für welche Probe verwendet wird.
- 78 μl der in Tabelle 6 beschriebenen Mischung in die Spalten geben. Geben Sie 2 μl 5'-Adapter (100 μM; siehe Tabelle 4) in jedes Röhrchen und inkubieren Sie es über Nacht bei 18 °C.
| Bestandteil | 1x | 7,5-fach |
| 5 x PNK-Puffer | 16 | 120 |
| ATP (10 mM) | 8 | 60 |
| RNase-Hemmer | 2 | 15 |
| T4-RNA-Ligase | 4 | 30 |
| H2O | 48 | 360 |
| Endgültiges Volumen | 78 μL | 585 μL |
Tabelle 6: 5'-Linker-Ligations-Reaktionsgemisch.
- Waschen Sie die Beads am nächsten Tag 1x mit 500 μL WB I und 3x mit 500 μL WB II und überführen Sie die Säulen in ein neues 2 mL Röhrchen.
8. Elution, SDS-PAGE und RNA-Extraktion
- Stellen Sie die Zentrifuge auf 4 °C ein. Bereiten Sie zwei Reihen von 1,5-ml-Röhrchen pro Probe für die Elution vor.
- Schleudern Sie das Hohlraumvolumen der Säulen mit Nickelperlen mit einer schnellen Drehung aus. Legen Sie die Säulen in die erste Reihe der Elutionsröhrchen und fügen Sie 200 μl Elutionspuffer hinzu. Warten Sie 2 Minuten und zwingen Sie dann den Puffer mit einer schnellen Drehung durch die Spalte.
- Verschieben Sie die Spalten in die zweite Reihe der Röhren und wiederholen Sie Schritt 8.2. Jede Probe enthält nun insgesamt 400 μl Eluat, aufgeteilt auf zwei 1,5-ml-Röhrchen.
- Nehmen Sie alle Eluate und füllen Sie sie zusammen in ein 5-ml-Röhrchen. Fügen Sie 2 μl von 20 mg/ml Glykogen hinzu. Wenn also sieben Proben verwendet werden, befinden sich nun 2,8 ml gepooltes Eluat im 5-ml-Röhrchen.
- Geben Sie 100 μl Trichloressigsäure (TCA) pro Probe [z. B. 700 μl TCA für 7 Proben (2,8 ml gepooltes Eluat)] in das 5-ml-Röhrchen und wirbeln Sie es 30 s lang durch.
- 20 Minuten auf Eis inkubieren.
- 30 min bei 17.000 x g, 4 °C, in einer Tischzentrifuge zentrifugieren.
- Entfernen Sie den Überstand vorsichtig aus dem konischen Röhrchen und überprüfen Sie die Pipette mit einem Geigerzähler, um sicherzustellen, dass das Pellet nicht versehentlich entfernt wurde. Wenn dies der Fall ist, setzen Sie den Überstand wieder in das Röhrchen ein und zentrifugieren Sie ihn weitere 10 Minuten lang.
HINWEIS: Der Überstand kann noch hochradioaktiv sein. Stellen Sie sicher, dass Sie eine ordnungsgemäße Abschirmung verwenden. - Resuspendieren Sie das Pellet vollständig in 2 ml eiskaltem Aceton.
- 15 min bei 17.000 x g, 4 °C zentrifugieren.
- Entfernen Sie so viel Aceton wie möglich mit einer P1000-Pipette. Drehen Sie anschließend kurz das Röhrchen, um kleine Acetontröpfchen zu sammeln, und entfernen Sie es dann mit einer P10-Pipette. 2 Minuten in einem Abzug trocknen.
HINWEIS: Der Acetonüberstand kann immer noch radioaktiv sein. Stellen Sie sicher, dass Sie eine ordnungsgemäße Abschirmung verwenden. - Resuspendieren Sie die Probe in 30 μL 1x Proteinladepuffer. Um sicherzustellen, dass das Pellet ordnungsgemäß resuspendiert wird, überprüfen Sie, ob der überwiegende Teil der Radioaktivität jetzt im Ladepuffer vorhanden ist und nicht im 1,5-ml-Röhrchen verbleibt, indem Sie die Lösung in einer P200-Pipette entfernen und die im 1,5-ml-Röhrchen verbleibende Aktivität mit einem Geigerzähler messen.
- Die Probe wird 10 min bei 65 °C erhitzt. Laden Sie ein 1 mm, 4–12 % vorgefertigtes Bis-Tris-Gel auf und lassen Sie es 1,5 h bei 125 V im MOPS-Puffer laufen.
- Nachdem das Gel zu Ende gelaufen ist, öffnen Sie die Gelkassette. Das Gel sollte auf der Bodenplatte zurückgehalten werden. Entsorgen Sie das Oberteil.
- Wickeln Sie das Gel in Frischhaltefolie ein und befestigen Sie es dann mit Klebeband an der Innenseite einer lichtdichten Kassette. Stellen Sie sicher, dass die Kassette über einen Verstärkungsbildschirm verfügt, um das Signal zu verbessern.
- Belichten Sie das Gel mit einem autoradiographischen Film und lagern Sie die Kassette während der Belichtung bei -80 °C. Die Belichtungszeit variiert zwischen Proteinen mit unterschiedlichen Vernetzungseffizienzen.
- Beim Einlegen des Films muss es eine Möglichkeit geben, ihn auf die Kassette neu auszurichten, um das gewünschte Band im nächsten Schritt auszuschneiden. Um dies zu gewährleisten, verwenden Sie ein fluoreszierendes Lineal und achten Sie auch darauf, dass sich das Gel an einer Ecke der Kassette befindet, die dann von der Folie bedeckt wird, die ebenfalls in der äußersten Ecke platziert ist.
HINWEIS: Als Faustregel gilt, dass Eluate im Ladepuffer, die einen Messwert von mindestens ~250 cps liefern, wenn sie auf einem Geigerzähler angezeigt werden, ein ausreichendes Signal für eine Belichtung von 3 h liefern. Andernfalls wird eine Belichtung über Nacht durchgeführt.
- Beim Einlegen des Films muss es eine Möglichkeit geben, ihn auf die Kassette neu auszurichten, um das gewünschte Band im nächsten Schritt auszuschneiden. Um dies zu gewährleisten, verwenden Sie ein fluoreszierendes Lineal und achten Sie auch darauf, dass sich das Gel an einer Ecke der Kassette befindet, die dann von der Folie bedeckt wird, die ebenfalls in der äußersten Ecke platziert ist.
- Entwickle den Film. Schneiden Sie die Frischhaltefolie ab, die das Gel bedeckt, aber bewegen Sie das Gel nicht. Andernfalls wird das Bild vom Gel versetzt.
HINWEIS: Das Gel wird wahrscheinlich hochradioaktiv sein. Achten Sie darauf, dass Sie beim Ausschneiden der Gelscheibe eine ordnungsgemäße Abschirmung verwenden. - Legen Sie die Folie über das Gel und schneiden Sie das gewünschte Band heraus. Geben Sie die Gelscheibe in ein 2-ml-Röhrchen.
- Zerdrücken Sie die Gelscheibe mit einer P1000-Pipettenspitze und fügen Sie 600 μl Proteinase-K-Puffer plus 200 μg Proteinase K hinzu (dieses Protokoll verwendet 10 μl einer 20 mg/ml-Proteinase-K-Lösung). 2 h bei 55 °C unter kräftigem Schütteln inkubieren.
- Schneiden Sie anschließend das Ende einer P1000-Spitze mit einem sauberen Skalpell ab und übertragen Sie den Überstand und die Gelstücke in eine Spin-Säule, die sich in einem 2-ml-Röhrchen befindet.
- Drehen Sie die Säule 1 Minute lang bei 17.000 x g bei RT. Sammeln Sie den Durchfluss, der die radioaktiven, isolierten RNAs enthält.
- Führen Sie eine Phenol:Chloroform-Extraktion durch.
- Fügen Sie 50 μl 3 M Natriumacetat, pH = 5,2, und 500 μl Phenol:Chloroform hinzu und wirbeln Sie gut vortex. 5 Minuten bei 17.000 x g schleudern. Entfernen Sie die wässrige oberste Schicht und geben Sie sie in ein neues 1,5-ml-Röhrchen.
- Fügen Sie 500 μl Chloroform hinzu und wirbeln Sie 10–15 s lang kräftig vortex. 5 min bei 17.000 x g bei RT schleudern. Entfernen Sie die wässrige Schicht und geben Sie sie in ein neues 1,5-ml-Röhrchen.
- Fügen Sie 1 μl 20 mg/ml Glykogen und 1 ml eiskaltes, 96%iges Ethanol hinzu. 30 min bei -80 °C oder über Nacht bei -20 °C ausfallen.
- 30 min bei 4 °C, 17.000 x g zentrifugieren. Entfernen Sie den Überstand, fügen Sie 500 μl 70%iges Ethanol hinzu und zentrifugieren Sie 5 Minuten lang, 4 °C bei 17.000 x g. Entfernen Sie das gesamte Ethanol, drehen Sie schnell, um Rückstände zu sammeln, und entfernen Sie überschüssiges Ethanol mit einer P10-Pipette.
- Trocknen Sie das Pellet ~3 Minuten lang in einem Abzug. Resuspendieren Sie in 20 μl DEPC-behandeltem Wasser.
- Lagern Sie die RNA bei -80 °C über Nacht oder fahren Sie sofort mit dem Schritt der reversen Transkription fort.
9. Reverse Transkription
- Zu den 20 μl RNA werden 2 μl 10 μM RT-Oligo (PE_reverse; siehe Tabelle 7) und 4 μl 5 mM dNTPs hinzugefügt.
| Name des Oligonukleotids | Sequenz (5'-3') | |||
| P5 vorwärts | AATGATACGGCGACCACCGAGATCTACCTCTTTCCCTACACGACGACGCTCTTCCGATCT | |||
| BC1 | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC3 | CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC4 | CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC5 | CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC7 | CAAGCAGAAGACGGCATACGAGATCAGATCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC8 | CAAGCAGAAGACGGCATACGAGATTAGCTTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC9 | CAAGCAGAAGACGGCATACGAGATGATCAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC10 | CAAGCAGAAGACGGCATACGAGATATCACGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| PE_reverse | CAGACGTGTGCTCTTCCGATCT | |||
Tabelle 7: Die PCR-Primer (einschließlich der Barcode-Sequenzen) und der Primer für die reverse Transkription.
- In einen vorgeheizten Thermoblock bei 85 °C für 3 min geben, dann 5 min auf Eis kalt stellen. Sammeln Sie den Inhalt des Röhrchens durch kurzes Zentrifugieren und fügen Sie dann 8 μl 5x Reverse-Transkriptase-Puffer, 2 μl 100 mM DTT und 2 μl RNase-Inhibitor hinzu.
- Inkubieren Sie die Mischung bei 50 °C für 3 min und fügen Sie dann 2 μl reverse Transkriptase hinzu und inkubieren Sie sie 1 h lang bei 50 °C.
- Inaktivieren Sie die reverse Transkriptase durch Inkubation bei 65 °C für 15 min.
- Die Röhrchen in einen bei 37 °C vorgeheizten Thermoblock geben und 3 min akklimatisieren lassen.
- 2 μl RNase H zugeben und 30 min bei 37 °C inkubieren.
- Isolieren Sie die cDNA mit Hilfe von SPRI-Kügelchen.
- Fügen Sie zwei Volumina mit 84 μl Perlen hinzu. 15 Minuten inkubieren. Legen Sie die Perlen auf ein Magnetgestell und lassen Sie sie 1 Minute lang ernten.
- Entfernen und entsorgen Sie den Überstand und fügen Sie 200 μl 70%iges Ethanol hinzu. Entfernen Sie die Perlen nicht vom Magnetgestell. Inkubieren Sie die Kügelchen mit dem Ethanol für 30 s.
- Entfernen Sie das Ethanol und wiederholen Sie den Waschschritt. Entfernen Sie das restliche Ethanol mit einer P10-Spitze.
- Legen Sie die Perlen für 2 Minuten in einen Abzug, um sie zu trocknen. Nehmen Sie die Perlen aus dem Rack, suspendieren Sie sie in 12 μl Wasser und legen Sie die Perlen dann wieder auf das Rack. Entfernen Sie 11 μl des Überstands.
- Frieren Sie die cDNA bei -20 °C ein oder fahren Sie sofort mit dem PCR-Schritt fort.
10. qPCR-Reaktion
- Vor der abschließenden PCR zur Amplifikation der cDNAs wird eine quantitative Polymerase-Kettenreaktion (qPCR) durchgeführt, um die optimale Anzahl von Zyklen für die Amplifikation der cDNAs zu ermitteln und eine Überamplifikation der Bibliothek zu verhindern.
- Richten Sie eine qPCR-Reaktion auf Eis gemäß Tabelle 8 ein. Siehe Tabelle 7 für alle Grundierungen.
| Bestandteil | 1x |
| 2x qPCR-Reaktions-Mastermix | 5 |
| 0,1 μM P5-Primer (vorwärts) | 0.8 |
| 0,1 μM BC Primer (umgekehrt) | 0.8 |
| cDNA (oder Wasser als Negativkontrolle) | 1 |
| H2O | 2.4 |
| Endgültiges Volumen | 10 μL |
Tabelle 8: qPCR-Reaktionsgemisch.
- Für eine korrekte Quantifizierung der für die Amplifikation erforderlichen Zyklen werden drei technische Replikate für die cDNA und drei negative Kontrollen (d. h. Wasserkontrollen) verwendet.
- Versiegeln Sie die Platten mit optisch transparenter Folie und führen Sie die qPCR gemäß den Anweisungen des Kit-Herstellers durch.
- Analysieren Sie die Proben durch eine absolute Quantifizierungsmethode, um die Anzahl der Zyklen (n) zu ermitteln, bei denen das Knie des exponentiellen Wachstums erreicht wird (siehe Abbildung 4C für ein Beispiel). Diese Anzahl von Zyklen wird dann für die endgültige Amplifikation des Rests der cDNA verwendet.
11. PCR-Reaktion und Gelextraktion
- Richten Sie die PCR-Reaktion auf Eis gemäß Tabelle 9 ein. Siehe Tabelle 7 für alle Grundierungen.
HINWEIS: Es werden nur 5 μl der cDNA-Bibliothek verwendet.
| Bestandteil | 1x |
| 10 x Korrekturlese-Polymerase-Puffer | 5 |
| 10 μM P5 Primer (vorwärts) | 1 |
| 10 μM BC Primer (umgekehrt) | 1 |
| 5 mM dNTPs | 2.5 |
| Korrekturlesen des Polymerase-Enzyms | 1 |
| cDNA | 5 |
| H2O | 34.5 |
| Endgültiges Volumen | 50 μL |
Tabelle 9: PCR-Reaktionsgemisch.
- Führen Sie die PCR wie folgt durch: 95 °C für 2 min; n Zyklen von 98 °C für 20 s, 52 °C für 30 s und 72 °C für 1 min; und 72 °C für 5 min. Die Anzahl (n) der Zyklen für die Amplifikation der χCRAC-Bibliothek wird durch die in Abschnitt 10 beschriebene qPCR bestimmt.
- 1 μl Exonuklease I zugeben und bei 37 °C 60 min inkubieren.
- Bereinigen Sie die amplifizierte cDNA mit SPRI-Beads wie oben beschrieben unter Verwendung von zwei Beads-Volumina (d. h. 100 μL). Elute in 11 μL.
- Fügen Sie 3 μl 6x Beladungsfarbstoff hinzu und lassen Sie ein vorgefertigtes 6%iges FSME-Gel bei 100 V für 1 h in 1x FSME-Puffer laufen. Verwenden Sie eine Leiter, die für die Quantifizierung kurzer DNA-Fragmente geeignet ist.
- Wenn Sie fertig sind, nehmen Sie das Gel aus der Kassette und geben Sie es in einen geeigneten, flüssigkeitsdichten Behälter mit ausreichend 1x TBE, um das Gel zu bedecken (z. B. ~50 ml). Fügen Sie eine angemessene Menge eines sicheren SYBR-Farbstoffs hinzu (z. B. verwenden Sie für 50 ml 5 μl eines 10.000-fachen Farbstoffs)
- Lassen Sie das Gel durch sanftes Mischen 15 min bei RT einwirken. Lassen Sie das SYBR-haltige 1x TBE abtropfen und ersetzen Sie es durch sauberes 1x TBE. Waschen Sie das Gel 10 Minuten lang unter leichtem Schütteln bei RT.
- Das 1x TBE abtropfen lassen und das Gel in eine Klarsichtmappe geben. Schneiden Sie den Ordner auf eine geeignete Größe zu.
- Bilde das Gel mit einem geeigneten Mittel wie einem Phosphorimager ab. Exzisieren Sie DNA-Fragmente zwischen ~175 bp und ~400 bp. Geben Sie die Gelscheibe in eine 1,5-ml-Tube.
- Die Gelscheibe mit einer P1000-Spitze gründlich zerdrücken und 400 μLH2Ozugeben. Bei 37 °C unter Schütteln 1 h in einem Thermoblock inkubieren.
- Die Probe 10 min auf Trockeneis einfrieren, dann bei 37 °C unter Schütteln für 1 h wieder in den Thermoblock legen.
- Erstellen Sie eine Filtereinheit, indem Sie eine Filtersäule nehmen und zwei Glasmikrofaserfilter einsetzen. Legen Sie das Gerät in ein 1,5-ml-Röhrchen.
- Schneiden Sie das Ende einer P1000-Spitze mit einem sauberen Skalpell ab, nehmen Sie die zerkleinerte TBE-Gelsuspension auf und geben Sie sie dann in die in Schritt 11.12 erstellte Filtereinheit. 30 Sekunden lang mit 17.000 x g drehen.
- Fügen Sie dem Überstand 1 μl Glykogen zusammen mit 40 μl Natriumacetat (pH = 5,2) und 1 ml 96%igem Ethanol hinzu. Bei -80 °C für 30 min inkubieren.
- 30 min bei 17.000 x g, 4 °C zentrifugieren. Entsorgen Sie den Überstand und waschen Sie ihn mit 500 μl 70%igem Ethanol.
- 5 min schleudern, das Ethanol vollständig entfernen und das Pellet dann 3 min in einem Abzug trocknen.
- Resuspendieren Sie in 10 μLH2Ound messen Sie die DNA-Konzentration.
Ergebnisse
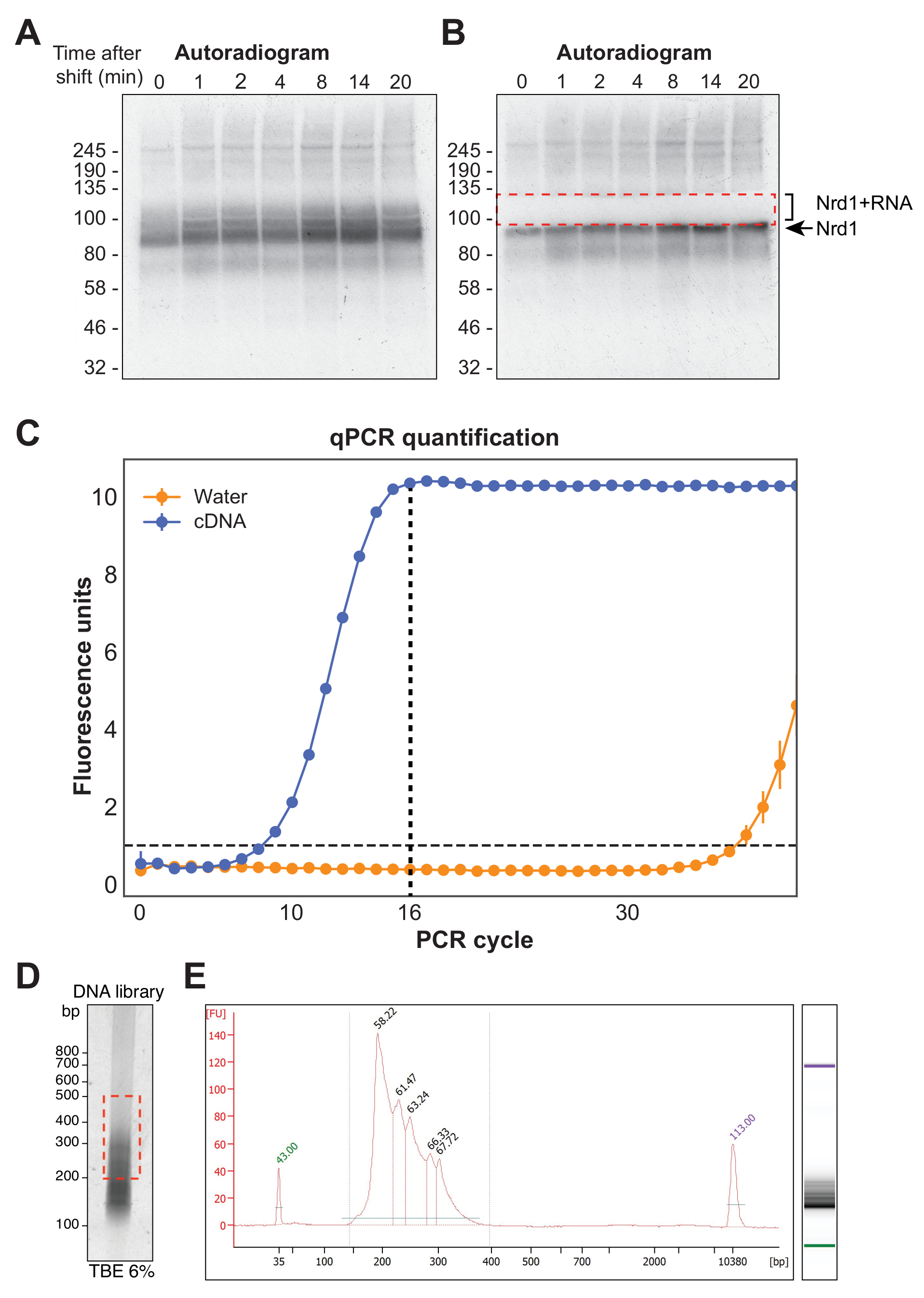
Um die Wirksamkeit der χCRAC-Methode zu demonstrieren, wurde ein Zeitverlaufsexperiment mit Hefestämmen durchgeführt, die ein HTP-markiertes Nrd1-Protein exprimieren. Eine detaillierte schematische Darstellung, die die Funktionsweise des Verfahrens beschreibt, ist in Abbildung 1 dargestellt. Wie Nab3 ist auch Nrd1 am Zerfall der nukleären RNA einer Vielzahl von RNA-Transkripten beteiligt37. Frühere Arbeiten aus dem Corden-Labor deuteten darauf hin, dass sich die Bindung von Nrd1 an seine RNA-Ziele signifikant ändert, wenn Zellen einem Glukosemangel ausgesetzt sind28,38. Daher wurden Zellen, die exponentiell in glukosehaltigem Medium (SD-TRP) wuchsen, über einen bestimmten Zeitverlauf in dasselbe Medium ohne Glukose (S-TRP) verschoben, um dynamische Veränderungen in den Nrd1-RNA-Interaktionen zu beobachten. Die Proben wurden in der Vari-X-Linkerkammer (Abbildung 3A) vor der Schicht und dann nach 1, 2, 4, 8, 14 und 20 min entnommen und vernetzt. Das für das Zellwachstum verwendete Medium enthielt absichtlich einen Mangel an Tryptophan, um die UV-Absorption durch diese aromatische Aminosäure zu reduzieren. Beachten Sie, dass es am besten ist, synthetisches Medium zu verwenden, das filtersterilisiert ist, da das Autoklavieren des Mediums zu einer Karamellisierung des Zuckers führen kann. Dies verringert dann die Vernetzungseffizienz.
Abbildung 4A zeigt eine repräsentative Autoröntgenaufnahme aus einem χCRAC-Experiment. Beachten Sie, dass in diesem Beispiel die Stichproben nicht zusammengefasst wurden. Stattdessen wurde jeder einzeln auf dem Gel ausgeführt. Dies wird für erste experimentelle Tests empfohlen, um zu zeigen, dass das Protein zu allen getesteten Zeitpunkten effektiv mit RNA vernetzt. Ein besonders intensives Signal wurde beim erwarteten Molekulargewicht des RBP beobachtet, das das Protein repräsentiert, das an sehr kurze, radioaktiv markierte RNAs gebunden ist, die nicht für eine Sequenzierung geeignet sind. Dazu wurde das Schmiersignal oberhalb dieser Bande, das mit längeren RNA-Fragmenten vernetzt ist, isoliert. Das Fragment wurde knapp oberhalb der Proteinbande plus etwa 30 kDa geschnitten. Abbildung 4B zeigt ein Autoradiogramm nach der Exzision, bei dem das Protein mit kurzen RNAs vernetzt ist, die im Gel verbleiben, und das zuvor abstrichige Signal nun herausgeschnitten wird.
Nach der reversen Transkription muss die cDNA-Bibliothek mittels PCR amplifiziert werden. Eine Überamplifikation der Bibliothek muss jedoch vermieden werden, da dies zu einer Verzerrung gegenüber Sequenzen führen kann, die bevorzugt von der Polymerase amplifiziert werden, und PCR-Artefakte erzeugen kann. Überverstärkte Bibliotheken enthalten auch eine große Anzahl doppelter Sequenzen, die Lesevorgänge auf dem Sequenzer verschwenden. Um die ideale Anzahl von PCR-Zyklen für die Amplifikation der finalen Bibliothek zu berechnen, wurde ein aliquoter Teil der cDNA mittels qPCR mit den P5- und BC-Oligonukleotiden amplifiziert. Der erste Zyklus, bei dem die Bibliothek den Höhepunkt der Fluoreszenz erreichte, wurde als PCR-Zykluszählung gewählt. Abbildung 4C zeigt ein Beispiel für eine qPCR aus einer typischen cDNA-Bibliothek, die eine maximale Zykluszahl von 16 ergab. Dieser Wert wurde dann für die abschließende χCRAC-PCR verwendet. Um die sequenzierten Daten zu verarbeiten, verwendeten wir eine zuvor in unserem Labor entwickelte Software (pyCRAC) und die entsprechende Pipeline zur Analyse der kinetischen CRAC-Daten (Nues et al., 2017; https://git.ecdf.ed.ac.uk/sgrannem/pycrac, https://bitbucket.org/sgrann/kinetic_crac_pipeline/src/default/). Diese Open-Source-Softwarewerkzeuge ermöglichen das Demultiplexen und Trimmen der Daten, das Entfernen von PCR-Duplikaten, die Identifizierung statistisch signifikanter Peaks, das Einlesen von Clustern in zusammenhängende Sequenzen und die Identifizierung von Bindungsmotiven39. Weitere Informationen zur Funktionsweise dieser Tools finden Sie auf den jeweiligen Webseiten.
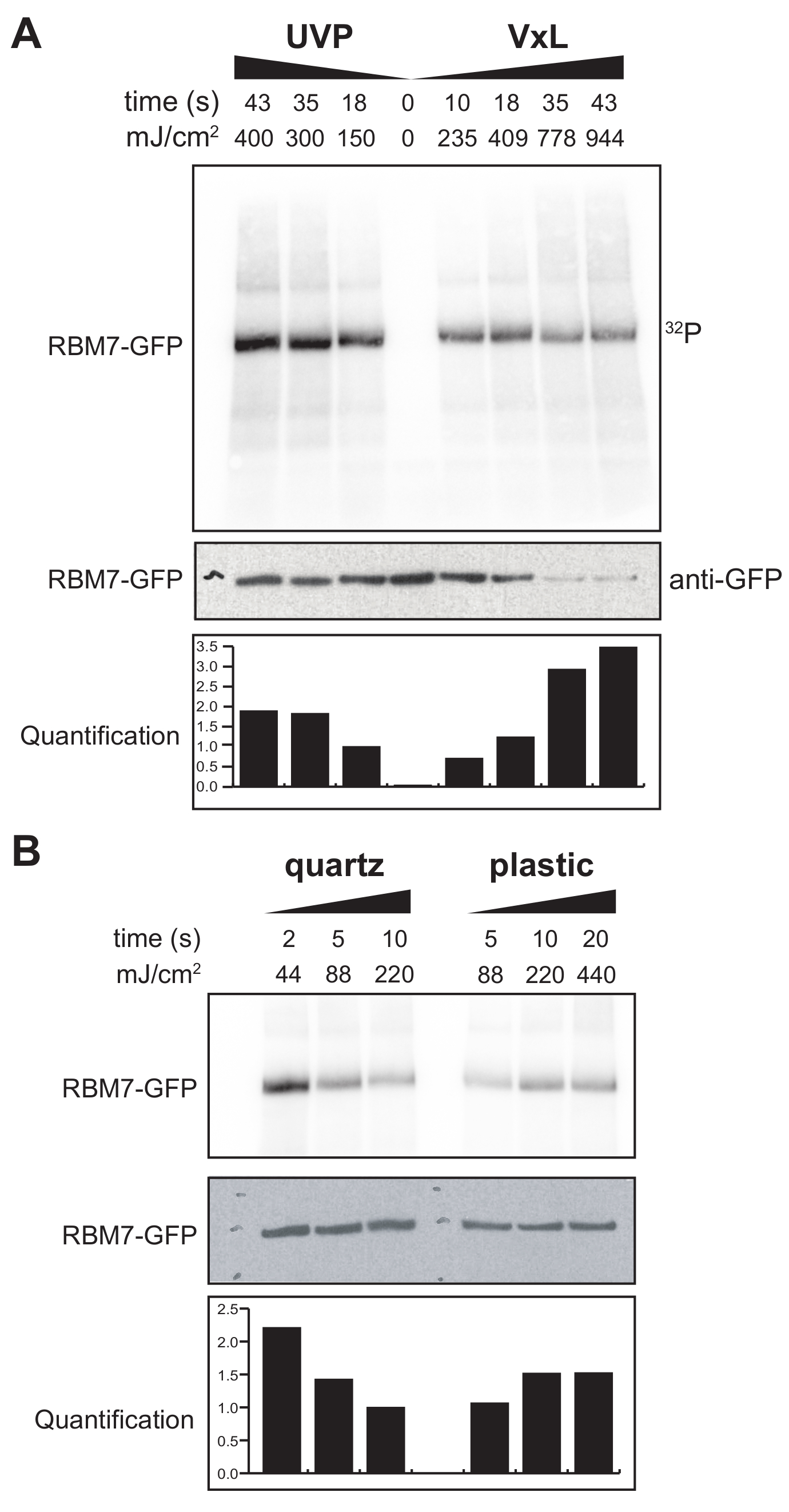
Wir haben auch mit der Entwicklung eines χCRAC-Protokolls für Säugetierzellen begonnen. Die Mehrzahl der Säugetierzelllinien wird als Monoschicht gezüchtet und die Schale in unserem Vernetzer mit dem UV-durchlässigen Beutel ist für Experimente mit adhärenten Zellen nicht geeignet. Um dieses Problem zu lösen, haben wir eine Stufe entwickelt, bei der Benutzer 1–2 Petrischalen (150 mm Durchmesser und 25 mm Tiefe) mit adhärenten Zellen UV-bestrahlen können (Abbildung 3B). In einem ersten Test wurde die Effizienz des Vernetzers für Säugetierzellen durch Vernetzung und Einfangen von stabil markiertem GFP-RBM7 unter Verwendung von Anti-GFP-Antikörpern und einer traditionellen CLIP-basierten Aufreinigung gemessen. Wie in Abbildung 5A gezeigt, war der Vernetzer in der Lage, Protein-RNA-Komplexe aus Säugetierzellen, die als Monoschicht gezüchtet wurden, unter Verwendung von 254 nm UV-Bestrahlung mit Wirkungsgraden zu gewinnen, die mit einem weit verbreiteten UV-Bestrahlungsgerät vergleichbar sind. Standard-Zellkulturkunststoffe, die normalerweise für UV-Vernetzungsexperimente verwendet werden, sind jedoch bis 254 nm UV undurchdringlich. Daher würden die Zellen in unserem Vernetzer nur von der oberen Bank der UV-Lampen bestrahlt. Um dies zu überwinden, haben wir eine UV-durchlässige Quarz-Petrischale für Zellwachstum und Vernetzung entwickelt. Die Verwendung der Quarzkulturen zeigte eine robuste Rückgewinnung von Protein-RNA-Komplexen mit nur 2 s UV-Bestrahlung (Abbildung 5B). In Kombination mit RBP-Capture-Methoden für Säugetierzellen wie der CLIP-Technologie sind diese kurzen Vernetzungszeiten mit Zeitverläufen möglich, um raumzeitliche RNA-Bindungsprofile von RBPs als Reaktion auf genotoxischen Stress oder schnelle Verarmung von Proteinfaktoren oder parallel zur Transkriptions- oder Zellzyklussynchronisation zu erhalten.
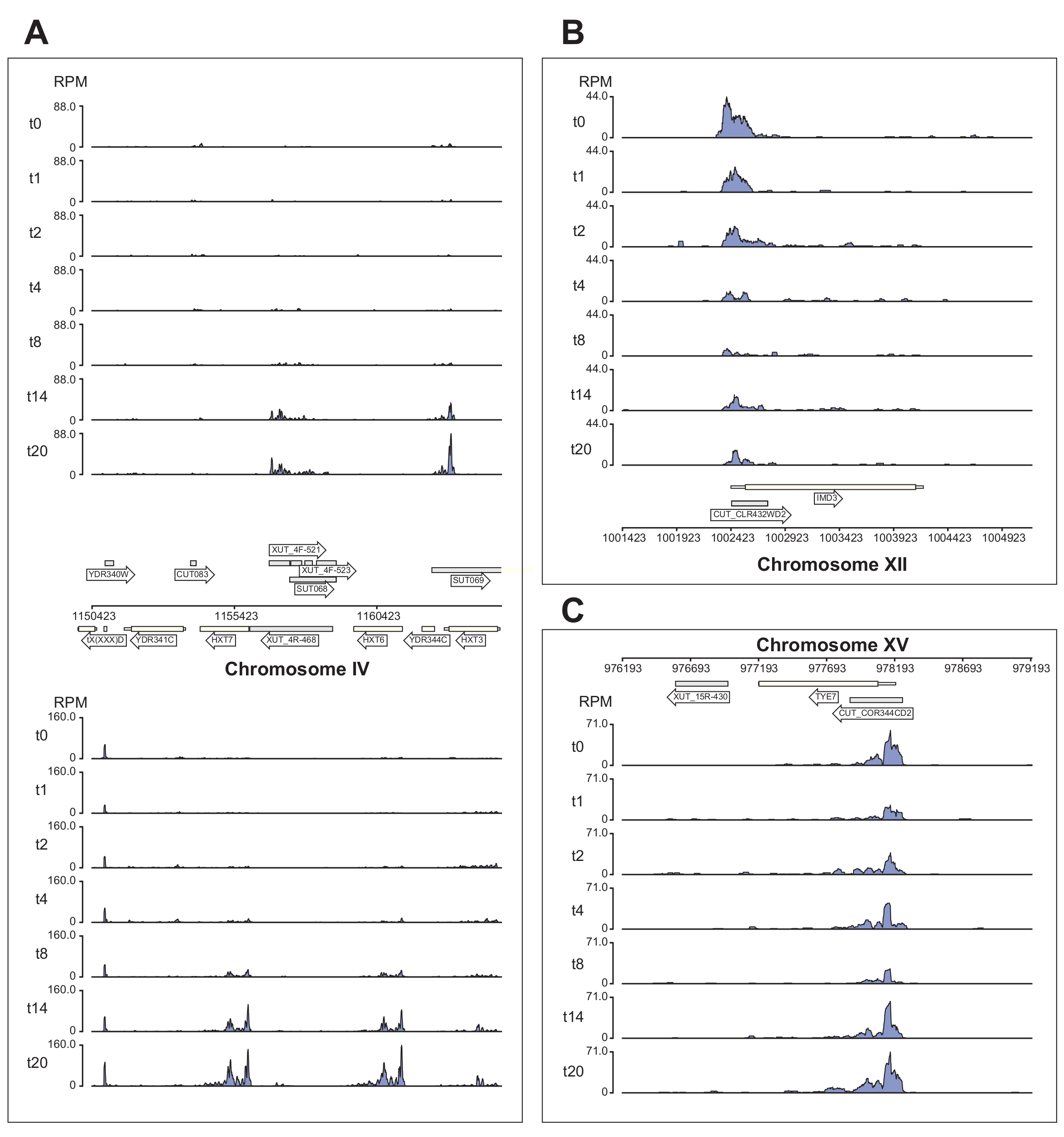
Abbildung 6 zeigt mehrere Beispiele für die Nrd1-Daten, die von der χCRAC-Pipeline verarbeitet werden. Diese Abbildung wurde unter Verwendung der von der Pipeline generierten Bedgraph-Dateien und des Python-GenomeBrowser-Pakets (https://pypi.org/project/GenomeBrowser/1.6.3/) erstellt, das wir entworfen haben, um die Erstellung von Genombrowserbildern der Daten in Publikationsqualität zu vereinfachen. Die grauen Rechtecke stellen genomische Regionen dar, die nicht-kodierende RNAs exprimieren, wie z. B. das kryptische instabile Transkript (CUTs), stabile uncharakterisierte Transkripte (SUTs)40 und Xrn1-sensitive instabile Transkripte (XUTs)41. Die Daten in Abbildung 6 zeigen, dass Nrd1 an viele dieser nicht-kodierenden RNA-Transkripte bindet, was mit der Idee übereinstimmt, dass dieses Protein am Abbau dieser Klasse von Transkripten beteiligt ist42. Abbildung 6A zeigt eine ~15 kb große Region auf Chromosom IV. Hier kam es zu einem signifikanten Anstieg der Bindung von Nrd1 an Transkripte, die für die hochaffinen Glukosetransporter HXT6 und HXT7 kodieren, die beide während des Glukosemangels hochreguliert werden. Es ist wahrscheinlich, dass die Beendigung der Transkription durch den NNS-Komplex die Induktionskinetik dieser Gene während des Glukosemangels beeinflussen kann. Abbildung 6B zeigt ein Beispiel für die Quervernetzung von Nrd1 zum Imd3-Transkript, von dem bekannt ist, dass es durch Nab3reguliert wird 43. In diesem Fall zeigten die Daten eine signifikante Verringerung der Bindung bei Glukosemangel. Frühere Arbeiten zeigten eine verminderte Bindung von Nab3 an das Tye7-Transkript während des Glukosemangels44. In Übereinstimmung mit dieser Beobachtung deuten die χCRAC-Daten darauf hin, dass die Bindung von Nrd1 während des Glukosemangels abnahm und die Quervernetzung von Nrd1 zu Tye7 nach 8 Minuten Stress am niedrigsten war (Abbildung 4C). Es scheint jedoch, dass dieser Effekt nur vorübergehend war, denn nach 14 Minuten Glukosemangel ging die Nrd1-Bindung wieder auf das Ausgangsniveau zurück.

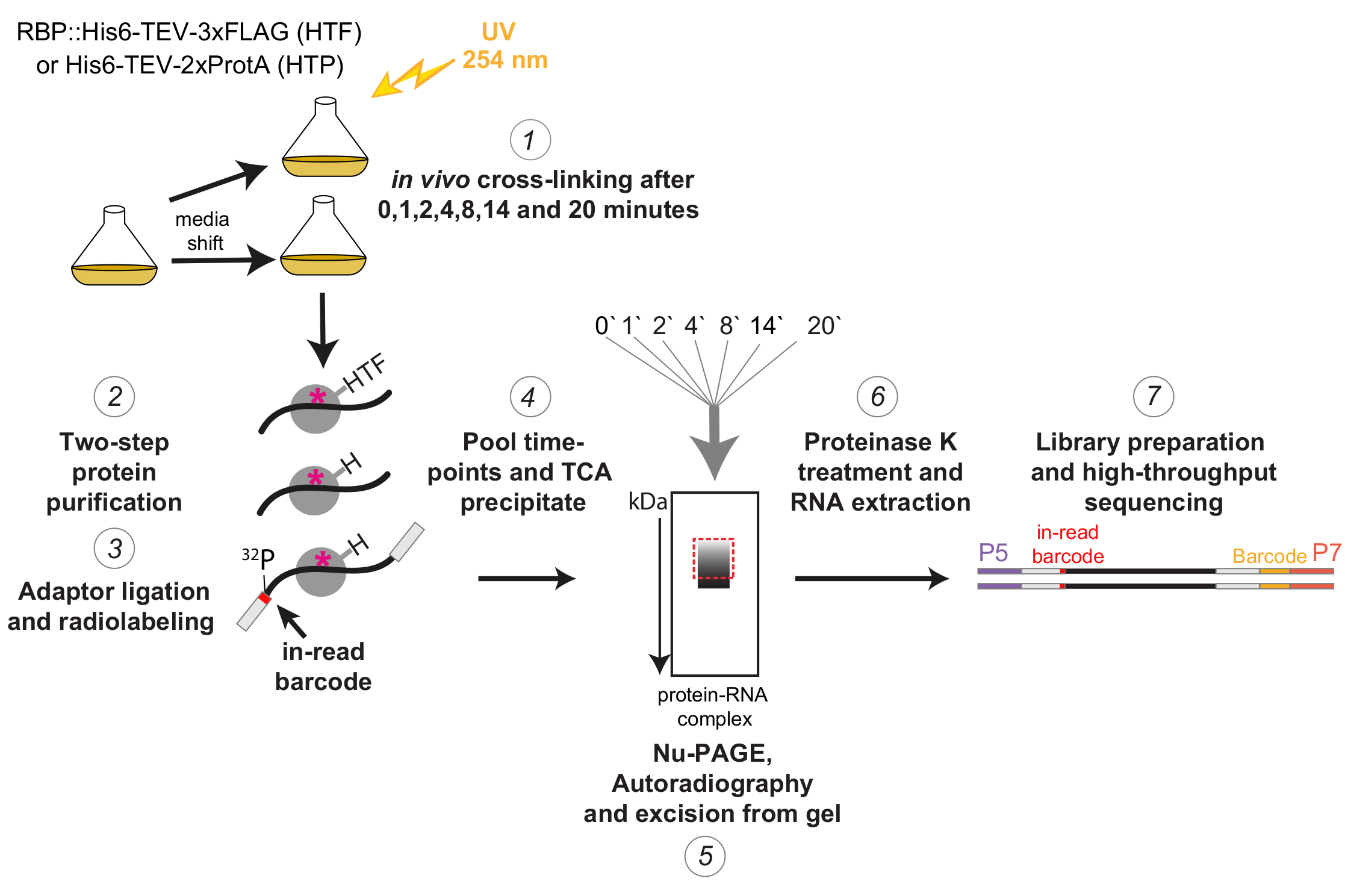
Abbildung 1: Schematische Darstellung des χCRAC-Protokolls. Getaggte Sorten wurden bis zur gewünschten Dichte angebaut. RBP steht für ein RNA-bindendes Protein. Anschließend wurde eine Referenzprobe entnommen und mit 254 nm UV-Licht vernetzt. Die verbleibenden Zellen wurden durch Filtration geerntet und dann schnell in das stressauslösende Medium überführt. Für das hier beschriebene χCRAC-Experiment wurden 1, 2, 4, 8, 14 und 20 min nach der Verschiebung Proben entnommen und vernetzt (1). Das zu untersuchende RBP wurde dann mit einer hochstringenten zweistufigen Affinitätsreinigung gereinigt (2). Als nächstes wurden die eingefangenen vernetzten RNAs teilweise mit RNasen verdaut, am 5'-Ende radioaktiv markiert und mit Adaptern ligiert (3). Die 5'-Adapter enthielten eindeutige "In-Read"-Barcode-Sequenzen, so dass die einzelnen Proben nach der Sequenzierung bioinformatisch getrennt werden konnten. Die RBP-RNA-Komplexe wurden dann eluiert, gepoolt und zusammen ausgefällt (4), mittels SDS-PAGE aufgelöst und mittels Autoradiographie sichtbar gemacht (5). Anschließend wurde ein einzelner Gelschnitt mit dem radioaktiven Signal direkt über dem Hauptband, der im Autoradiographiebild mit einem gestrichelten roten Kästchen dargestellt ist, aus dem Gel herausgeschnitten (5). Die Gelschnitte wurden mit der Protease K behandelt und die RNA anschließend extrahiert (6), in cDNAs umgewandelt und mittels PCR amplifiziert (7). Im PCR-Schritt wurden zusätzliche Barcodes (gelber Block, der durch das P7-Oligo eingeführt wurde) eingeführt, so dass viele Bibliotheken in einer einzigen Spur gemultiplext werden konnten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Vernetzung und Vakuumfiltration. (A) Der Vernetzer. Die Zellsuspension wird in einen Trichter gegossen, der sich oben rechts in der Maschine befindet (siehe auch Abbildung 3A für eine Nahaufnahme) und in einem UV-transparenten Beutel gehalten, der sich in der mittleren Schale befindet. Dieser Beutel wird von zwei Verschlüssen flankiert, die geschlossen bleiben, bis der Benutzer die Maschine anweist, den Bestrahlungsschritt zu starten. Die Zellen werden sowohl oben als auch unten von den Trays mit UV-Licht bestrahlt. Das Gerät wird mit 254- und 365-nm-UV-Lampen geliefert, wobei letztere für PAR-CLIP-Experimente geeignet sind. Die Maschine wird über ein Touchscreen-Panel oben rechts bedient, mit dem die UV-Dosierung oder die Belichtungszeit gesteuert werden können. (B) Nach der Vernetzung werden die Zellen von der linken Seite der Maschine entleert. Die Zellsuspensionen werden durch Vakuum zurückgewonnen und in einen Glaskolben entleert, wo sie anschließend zur Ernte in eine Vakuumfiltrationsvorrichtung gegossen werden können. (C) Vakuum-Filtrationsgeräte. Diese werden über einen Clip geöffnet und geschlossen und ein Filter dazwischen gesteckt. Vier Filtrationsgeräte wurden parallel für sehr kurze Zeitreihen eingesetzt, um keine Zeit durch Filterwechsel zu verlieren. (D) Nach der Filtration wurde der Medienüberstand zur anschließenden Entsorgung in Kolben entleert. Unterhalb der Vakuumfiltrationsgeräte wurden Ventile installiert, um das Vakuum im System aufrechtzuerhalten, wenn der Filter entfernt wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: Vernetzung von suspendierten vs. adhärenten Zellen. (A) Der Vernetzer mit der Vari-X-Linkerkammer für Suspensionszellen. Die Zellkultur wird in den Probeneinlass (Trichter) gegossen, der sich oben rechts im Tray befindet. (B) Tablett, das Petrischalen aus Kunststoff oder Quarz aufnehmen kann, um adhärente Zellen oder kleine Volumina von Suspensionszellen zu vernetzen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Vorbereitung der Bibliothek. (A) Beispiel eines Autoradiogramms aus einem Nrd1-HTP χCRAC-Experiment. Das starke, konzentrierte Signal repräsentiert das Protein, das zu sehr kurzen RNAs vernetzt ist, während der obige Abstrich das Protein darstellt, das mit RNAs vernetzt ist, die für die Sequenzierung ausreichend lang sind. (B) Der Abstrich wurde wie in einem Autoradiogramm nach der Gelexzision gezeigt. (C) Eine repräsentative qPCR aus einer χCRAC cDNA-Bibliothek. In diesem Beispiel wurde die maximale Amplifikation der cDNA nach 16 Zyklen erreicht. Somit wurden 16 Zyklen für die finale Verstärkung verwendet. Der Fehlerbalken stellt die Standardabweichung von drei technischen qPCR-Replikaten dar. (D) Beispiel für ein Phosphorbild aus einer cDNA-Bibliothek auf einem 6%igen FSME-Gel. (E) cDNA-Längen- und Qualitätsanalyse aus einer chipbasierten Kapillarelektrophorese. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 5: Hoch-RNase-Test-iCLIP-Experiment zum Testen der Vernetzung in Säugetierzellen. Gezeigt werden Autoradiogramme aus GFP-RBM7 iCLIP-Experimenten, die die Effizienz der RNP-Rückgewinnung über verschiedene Vernetzungsenergien hinweg testeten. Immunpräzipitationen wurden mit Anti-GFP-Antikörpern durchgeführt, die an magnetische Beads auf vernetzten Zellen gekoppelt waren, die GFP-RBM7 stabil exprimierten. Immunpräzipitate wurden mit hohen Konzentrationen von RNase I inkubiert, um assoziierte RNAs auf kurze, gleichmäßige Längen zu trimmen. RNPs wurden durch 32P-Markierung und SDS-PAGE sichtbar gemacht und wandern als definierte Bande, nahe der Migration des unvernetzten Proteins. Die Quantifizierung zeigt die Ergebnisse densitometrischer Analysen von radioaktiv markierten RBM7-RNA-Signalen, die auf das Anti-GFP-Western-Blot-Signal normalisiert wurden. (A) Vernetzungszeitverlauf des üblicherweise verwendeten UVP-Vernetzers im Vergleich zu unserem Vernetzer (Vari-X-Vernetzer; (B) Vernetzungszeitverlauf unseres Vernetzers auf Quarz- (links) und Kunststoffkulturwaren. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 6: Beispielhafte Genom-Browser-Diagramme, die die Leistungsfähigkeit von χCRAC zeigen, die differentielle, zeitliche Bindung von Nrd1 an seine Ziele zu zeigen. Jedes Feld zeigt Diagramme für einzelne Genomregionen. Die Pfeile zeigen an, auf welchem Strang die Gene kodiert sind (linker Zeigepfeil = Minusstrang; rechter Zeigepfeil = Plus-Strang). Die Zeitpunkte (min) werden durch t0, t1, t2 usw. auf den y-Achsen jedes Teildiagramms angegeben. Römische Ziffern, die die Chromosomen und die Koordinaten angeben, werden angezeigt. (A) Bei Glukoseentzug bindet Nrd1 zwei hochaffine Glukosetransporter, HXT6 und HXT7, die beide in diesem Zustand hochreguliert sind. (B) Es wurde beobachtet, dass Nrd1 an Imd3, ein bereits validiertes Ziel von Nab344, bindet, wobei die Intensität nach Glukosemangel abnimmt. (C) Die Nrd1-Bindung von Tye7 weist eine dynamische und transiente Natur auf, die nach Glukosemangel nach 8 min Stress auf ein Minimum abnimmt. Nach 14 min kehrt die Bindung jedoch wieder auf das Basalniveau zurück. Die Lesevorgänge wurden auf "Lesevorgänge pro Million" (RPM; y-Achse) normalisiert. Graue Kästchen kennzeichnen Regionen, die nicht-kodierende RNAs kodieren. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Diskussion
Die χCRAC-Methode hat in Kombination mit den neuen Vernetzungs- und Zellerntegeräten ein großes Potenzial, da sie auf ein breites Spektrum von Modellorganismen anwendbar ist und daher für das RNA-Feld von allgemeinem Interesse sein sollte. Es gibt viele Bereiche, in denen χCRAC eingesetzt werden kann. Mit der Methode ließe sich beispielsweise die hierarchische Anordnung von Proteinen zu großen makromolekularen Komplexen wie dem Spleißosom und dem Ribosom messen, bei der es häufig zu dynamischen Wechselwirkungen zwischen Proteinen und RNA-Molekülen kommt. Wir verwenden es jetzt auch routinemäßig, um Wechselwirkungen zwischen RNA-Zerfallsfaktoren und ihren Substraten zu überwachen, wenn Zellen verschiedenen Arten von Stress ausgesetzt sind. Auf diese Weise können wir bestimmen, in welchem Stadium der adaptiven Reaktion diese Faktoren am aktivsten sind, an welche Substrate sie binden und wie dynamisch diese Wechselwirkungen sind. Solche Daten sollen es den Forschern ermöglichen, den relativen Beitrag jedes Faktors zur Anpassung an Umweltveränderungen zu bestimmen.
χCRAC verwendet duale Affinitätsreinigungs-Tags (HTF oder HTP), um das Protein unter hochstringenten und denaturierenden Bedingungen zu reinigen. Dadurch wird sichergestellt, dass die kogereinigte RNA stark für RNAs angereichert ist, die kovalent mit dem interessierenden Protein vernetzt waren. Die Verwendung von Affinitätstags hat jedoch Nachteile. Zum Beispiel könnte der Tag die Funktion des Proteins stören, was zu einem verzerrten Auslesen seines RNA-bindenden Interaktoms führen könnte. Darüber hinaus ist es für einige Modellorganismen möglicherweise nicht immer möglich, Tags zu verwenden, da die genetischen Werkzeuge zur Integration von DNA-Fragmenten in das Genom oder zur Transformation von Expressionsplasmiden noch nicht verfügbar sind. Es ist jedoch einfach, einige Teile des χCRAC-Protokolls zu ändern, um es mit CLIP-basierten Protokollen kompatibel zu machen, die auf Antikörper zur Aufreinigung des RBP angewiesen sind. Tatsächlich hat diese Studie gezeigt, dass es möglich ist, iCLIP-basierte Reinigungen mit unserem Vernetzer zu kombinieren. Wir sind gerade dabei, CLIP-Protokolle zu entwickeln, um die zeitliche Assoziation von humanen RNA-bindenden Proteinen mit entstehenden RNA-Transkripten zu untersuchen.
Bei der Durchführung von χCRAC an einem neuen Protein muss die UV-Belichtung optimiert werden, um eine maximale Vernetzung zu induzieren. Dies ist wichtig, da hohe UV-Strahlen die Rückgewinnung von RNA während des Aufreinigungsschritts verringern können. Zellen, die das rekombinante RBP exprimierten, wurden verschiedenen UV-Dosen ausgesetzt: 100 mJ/cm2, 250 mJ/cm2, 500 mJ/cm2 und 1 J/cm2. Anschließend wurden die RNPs eingefangen und die RNAs fragmentiert und radioaktiv markiert. Anschließend wurden die RNPs mittels SDS-PAGE aufgelöst und ein Autoradiogramm erstellt, um daraus abzuleiten, welche Belichtung das intensivste Signal lieferte (d.h. die maximale Vernetzung).
Sobald die Versuchsbedingungen optimiert sind, werden bei der Durchführung von χCRAC mehrere Kontrollexperimente empfohlen. Zunächst kann eine UV-bestrahlte, nicht markierte Probe verwendet werden, um die Hintergrundbindung an die Reinigungsperlen zu überwachen. Zweitens ermöglicht eine zweite Zeitreihe, in der die Zellen in das ursprüngliche Medium zurückverschoben werden, bei der Anwendung von χCRAC während eines Shift-Experiments untersucht, ob die Filtration der Zellen selbst Veränderungen des RNA-Niveaus oder Protein-RNA-Interaktionen induziert.
Wie in der Einleitung erwähnt, schlagen zahlreiche kürzlich veröffentlichte Arbeiten eine Reihe von Optimierungen des CLIP-Protokolls vor. Dies schließt die Verwendung von fluoreszenzmarkierten Adaptern zur Detektion des Protein-RNA-Komplexes durch Infrarot-Abtastung10 sowie Optimierungen an verschiedenen Nukleinsäurereinigungs- und Größenauswahlschritten ein, die die Komplexität der resultierenden Bibliotheken 12,45 erhöhen. Wir implementieren derzeit einige dieser Verbesserungen, um das χCRAC-Protokoll weiter zu verfeinern. Das hier vorgestellte Protokoll enthält bereits eine Reihe von Verbesserungen gegenüber den ursprünglichen CRAC- und χCRAC-Protokollen, die die Komplexität der Daten erhöhen. Zuvor wurden beispielsweise die vernetzten, radioaktiven Protein-RNA-Komplexe auf SDS-PAGE-Gelen nach der Auflösung auf eine Nitrozellulosemembran übertragen und die vernetzte RNA aus dem Blot isoliert. Der Transfer des RNP und die anschließende RNA-Extraktion können jedoch sehr ineffizient sein, insbesondere wenn es sich um große RBPs wie RNA-Polymerase-Untereinheiten handelt. Dies kann zu einer deutlichen Verringerung der Rückgewinnung der vernetzten RNA führen. Im aktuellen Protokoll wird die vernetzte RNA direkt aus SDS-PAGE-Gelschnitten extrahiert, wie in Abbildung 1 dargestellt. Dies erhöhte die Rückgewinnung von vernetzten RNAs. Zusätzlich wurde das Produkt nach PCR-Amplifikation der cDNAs ursprünglich auf 3%-Agarosegele mit niedriger Schmelztemperatur aufgelöst und anschließend 175–300 bp PCR-Produkte aus dem Gel extrahiert. Diese Gele können jedoch leicht überladen werden, was zu einer sehr schlechten Trennung der DNA führt. Der Ersatz von Agarosegelen durch vorgefertigte FSME-Gele führte zu einer gleichmäßigeren Größentrennung und einer besseren Rückgewinnung von PCR-Produkten.
Offenlegungen
A. Langford und W. Worboys sind mit UVO3, einem Handelsunternehmen, verbunden. Sie spielten keine Rolle beim Studiendesign, bei der Datenerhebung und -interpretation oder bei der Entscheidung, die Arbeit zur Veröffentlichung einzureichen.
Danksagungen
Diese Arbeit wurde unterstützt durch Zuschüsse des Wellcome Trust (091549 an S.G und 109093/Z/15/A an S.M.), des Wellcome Trust Centre for Cell Biology Core Grant (092076) und des Medical Research Council Non-Clinical Senior Research Fellowship (MR/R008205/1 an S.G.), der European Molecular Biology Organization im Rahmen eines langfristigen Postdoc-Stipendiums (ALTF 1070-2017 an R.A.C.), und dem Independent Research Fund Denmark (T.H.J).
Materialien
| Name | Company | Catalog Number | Comments |
| 1,4-dithioreitol | Merck | 10708984001 | Buffer component in mammalian cell lysis |
| 1.5 mL tubes | Eppendorf | 0030 120.086 | General reaction tube |
| 2 mL tubes | Eppendorf | 0030 123.344 | For holding columns and collection of waste |
| 32P-yATP | Perkin Elmer | NEG502Z-250 | For radiolabelling the 5' end of the RNA |
| 4-12% Bis-Tris gel | Invitrogen | NP0321BOX | SDS-PAGE gel |
| 4X loading buffer | Novex | NP0008 | Protein loading dye concentrate |
| 50 bp ladder | New England Biolabs | N3236 | Reference ladder for excising region of interest from the amplified cDNA library |
| 50% PEG | NEB | B100045 | For the L5 linker ligation |
| 6% TBE gel | Invitrogen | EC6265BOX | For separation and purification of the cDNA library |
| Acetone | ACROS Organics | 423245000 | Washing of TCA-precipitated proteins |
| anti-FLAG beads | Sigma Aldrich | M8823-1ML | For purifcation of FLAG-tagged RBPs |
| ATP (100 mM) | Thermo Fisher Scientific | R0441 | For ligation of the L5 linker onto the 5' end of captured RNAs |
| Beta-mercaptoethanol | Sigma Aldrich | M3148-100ML | Buffer component |
| Biomax MS intensifying screen | Sigma Aldrich | Z363162-1EA | For intensifying the autoradiogram signal |
| Chloroform | Thermo Fisher Scientific | 1010219 | For phenol-chloroform extraction following RNA purification |
| cOmplete EDTA-free protease inhibitor cocktail | Roche | 11873580001 | For inhibition of cellular proteases after lysis |
| Complete supplement mixture -TRP | Formedium | DCS0149 | For preparation of synthetic defined medium |
| Costar Spin-X 0.22 µm filters | Sigma Aldrich | CLS8160 | For isolating the excised cDNAs following gel extraction |
| DNase RQ1 | Promega | M6101 | For DNA digest following cell lysis |
| dNTPs (10 mM) | Sigma Aldrich | 4638956001 | For reverse transcription and PCR |
| Ethanol | Thermo Fisher Scientific | 10041814 | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Ethylenediaminetetraacetic acid | Invitrogen | AM9261 | For protease K buffer |
| Exonuclease I | New England Biolabs | M0293 | For degradation of primers following PCR |
| Glass microfiber filters | Whatman | 1823-010 | For isolating the excised cDNAs following gel extraction |
| Glucose | Formedium | GLU03 | For preparation of glucose-containing, synthetic defined medium |
| Glycogen (20 mg/mL) | Roche | 10901393001 | Precipitation of proteins, RNA and DNA |
| GST-TEV | Homemade | Construct and purification protocol is available upon request | |
| Guanidium hydrochloroide | Thermo Fisher Scientific | 10071503 | Required for pulldown denaturing conditions and washing buffer |
| IgG beads | GE Healthcare | 17-0969-01 | For purification of protein A-tagged RBPs |
| Imidazole | Sigma Aldrich | I2399-100G | For elution of captured proteins from Nickel beads |
| Isoamyl alcohol | Thermo Fisher Scientific | A393-500 | For phenol-chloroform extraction following RNA purification |
| Luna Universal One-Step RT-qPCR | NEB | E3005S | For qPCR of the cDNA in order to calculate required number of PCR cycles |
| Magnesium chloride | Fluka Analytical | 63020-1L | For PNK buffer |
| Membrane filters | Millipore | AAWP09000 for yeast or HAWP09000 for bacteria | For vacuum filtration of cells |
| Micro bio-spin columns | Biorad | 732-6204 | For collecting eluate after gel extraction |
| Ni-NTA beads | Qiagen | 30210 | For secondary protein capture |
| NP-40 | Sigma Aldrich | I8896-100ML | Buffer component |
| Pfu polymerase | Promega | M7741 | For amplification of the cDNA library |
| Phenol | Sigma Aldrich | P4682-400ML | For phenol-chloroform extraction following RNA purification |
| Pierce spin columns | Thermo Fisher Scientific | 69725 | For on-column enzymatic reactions |
| Protease K | Roche | 3115887001 | For degradation of the RBP following gel extraction |
| Quartz Petri dish | UVO3 | N/A | For cross-linking of adherent cells. Available from https://www.vari-x-link.com for 400 GBP |
| Radiography films | Amersham | 28906843 | For autoradiography visualisation |
| RNAClean XP beads | Beckmann | A63987 | SPRI beads for clean up of RNAs and cDNAs |
| RNase H | New England Biolabs | M0297 | For degradation of RNAs following reverse transcription |
| RNase-It | Agilent | 400720 | For RNA digestion |
| rRNasin | Promega | N2511 | For inhibition of any contaminating RNases during enzymatic reaction |
| Sodium acetate | Sigma Aldrich | S2889-1KG | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Sodium chloride | Thermo Fisher Scientific | 7647-14-5 | Buffer component |
| Sodium deoxycholate | Sigma Aldrich | D6750-100G | Buffer component in mammalian cell lysis |
| Sodium dodecylsulfate | Sigma Aldrich | L3771-1KG | For protease K buffer |
| SUPERase-In | Invitrogen | AM2694 | For inhibition of cellular RNases after lysis |
| SuperScript IV | Thermo Fisher Scientific | 18090010 | For reverse transcription |
| T4 PNK | New England Biolabs | M0201 | For radiolabelling the 5' end of the RNA |
| T4 RNA ligase 1 | New England Biolabs | M0204 | For ligation of the L5 adaptor onto the RNA 5' end |
| T4 RNase ligase 2, truncated K222Q | NEB | M0351S | For ligation of the App_PE linker onto the 3' end of captured RNAs |
| TBE buffer (10X) | Invitrogen | 15581-028 | For running TBE gels |
| TEV protease | Homemade | For eluting captured proteins following FLAG capture | |
| Thermosensitive alkaline phosphatase | Promega | M9910 | For 5' and 3' dephosphorylation of RNAs |
| Trichloroacetic acid (100%) | Sigma Aldrich | T0699-100ML | For precipitation of RBP-RNA complexes |
| Tris hydrochloride | Invitrogen | 15504-020 | Buffer component |
| Triton X-100 | Sigma Aldrich | T8787-100ML | Buffer component in mammalian cell lysis |
| Vari Filter | UVO3 | N/A | Device for vacuum harvesting cells. Available from https://www.vari-x-link.com for 100 GBP |
| Vari-X-Linker | UVO3 | N/A | Cross-linker for cross-linking cells. Available from https://www.vari-x-link.com for 16,000 GBP |
| Yeast nitrogen base | Formedium | CYN0410 | For preparation of synthetic defined medium |
| Zirconia beads | Thistle | 11079105Z for yeast or 11079101Z for bacteria | For cell lysis via bead beating |
Referenzen
- Ule, J., et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 302 (5648), 1212-1215 (2003).
- Granneman, S., Kudla, G., Petfalski, E., Tollervey, D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proceedings of the National Academy of Sciences. 106 (24), 9613-9618 (2009).
- Licatalosi, D. D., et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 456 (7221), 464-469 (2008).
- König, J., et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nature Structural & Molecular Biology. 17 (7), 909-915 (2010).
- Hafner, M., et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell. 141 (1), 129-141 (2010).
- Aktaş, T., et al. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature. 544 (7648), 115-119 (2017).
- Huppertz, I., et al. iCLIP: Protein–RNA interactions at nucleotide resolution. Methods. 65 (3), 274-287 (2014).
- Li, X., et al. Comprehensive in vivo RNA-binding site analyses reveal a role of Prp8 in spliceosomal assembly. Nucleic Acids Research. 41 (6), 3805-3818 (2013).
- Rosenberg, M., et al. Denaturing CLIP, dCLIP, Pipeline Identifies Discrete RNA Footprints on Chromatin-Associated Proteins and Reveals that CBX7 Targets 3′ UTRs to Regulate mRNA Expression. Cell Systems. 5 (4), 368-385 (2017).
- Zarnegar, B. J., et al. irCLIP platform for efficient characterization of protein–RNA interactions. Nature Methods. 13 (6), 489-492 (2016).
- Kargapolova, Y., Levin, M., Lackner, K., Danckwardt, S. sCLIP—an integrated platform to study RNA–protein interactomes in biomedical research: identification of CSTF2tau in alternative processing of small nuclear RNAs. Nucleic Acids Research. 45 (10), 6074-6086 (2017).
- Van Nostrand, E. L., et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nature Methods. 13 (6), 508-514 (2016).
- Flynn, R. A., et al. Dissecting noncoding and pathogen RNA–protein interactomes. RNA. 21 (1), 135-143 (2015).
- Brugiolo, M., Botti, V., Liu, N., Müller-McNicoll, M., Neugebauer, K. M. Fractionation iCLIP detects persistent SR protein binding to conserved, retained introns in chromatin, nucleoplasm and cytoplasm. Nucleic Acids Research. 45 (18), 10452-10465 (2017).
- Sanford, J. R., et al. Identification of Nuclear and Cytoplasmic mRNA Targets for the Shuttling Protein SF2/ASF. PLOS ONE. 3 (10), e3369(2008).
- Garzia, A., Meyer, C., Morozov, P., Sajek, M., Tuschl, T. Optimization of PAR-CLIP for transcriptome-wide identification of binding sites of RNA-binding proteins. Methods. 118-119, 24-40 (2017).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Research. 22 (10), 2031-2042 (2012).
- Chen, K., et al. High-Resolution N6-Methyladenosine (m6A) Map Using Photo-Crosslinking-Assisted m6A Sequencing. Angewandte Chemie International Edition. 54 (5), 1587-1590 (2015).
- Ke, S., et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes & Development. 29 (19), 2037-2053 (2015).
- Linder, B., et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature Methods. 12 (8), 767-772 (2015).
- Kudla, G., Granneman, S., Hahn, D., Beggs, J. D., Tollervey, D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proceedings of the National Academy of Sciences. 108 (24), 10010-10015 (2011).
- Sugimoto, Y., et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature. 519 (7544), 491-494 (2015).
- Hwang, H. W., et al. cTag-PAPERCLIP Reveals Alternative Polyadenylation Promotes Cell-Type Specific Protein Diversity and Shifts Araf Isoforms with Microglia Activation. Neuron. 95 (6), 1334-1349 (2017).
- Hwang, H. W., et al. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Reports. 15 (2), 423-435 (2016).
- Lee, F. C. Y., Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Molecular Cell. 69 (3), 354-369 (2018).
- Beckmann, B. M. RNA interactome capture in yeast. Methods. 118-119, 82-92 (2017).
- Granneman, S., Petfalski, E., Tollervey, D. A cluster of ribosome synthesis factors regulate pre-rRNA folding and 5.8S rRNA maturation by the Rat1 exonuclease. The EMBO Journal. 30 (19), 4006-4019 (2011).
- Schaughency, P., Merran, J., Corden, J. L. Genome-Wide Mapping of Yeast RNA Polymerase II Termination. PLOS Genetics. 10 (10), e1004632(2014).
- Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S., Cohen, S. N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proceedings of the National Academy of Sciences. 99 (15), 9697-9702 (2002).
- Kresnowati, M. T. A. P., et al. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Molecular Systems Biology. 2, 49(2006).
- Marguerat, S., Lawler, K., Brazma, A., Bähler, J. Contributions of transcription and mRNA decay to gene expression dynamics of fission yeast in response to oxidative stress. RNA Biology. 11 (6), 702-714 (2014).
- van Nues, R., et al. Kinetic CRAC uncovers a role for Nab3 in determining gene expression profiles during stress. Nature Communications. 8 (1), 12(2017).
- Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M., Rosenow, C. Global RNA Half-Life Analysis in Escherichia coli Reveals Positional Patterns of Transcript Degradation. Genome Research. 13 (2), 216-223 (2003).
- Tudek, A., Candelli, T., Libri, D. Non-coding transcription by RNA polymerase II in yeast: Hasard or nécessité? Biochimie. 117, 28-36 (2015).
- Lingaraju, M., et al. The MTR4 helicase recruits nuclear adaptors of the human RNA exosome using distinct arch-interacting motifs. Nature Communications. 10 (1), 1-11 (2019).
- Lubas, M., et al. Interaction Profiling Identifies the Human Nuclear Exosome Targeting Complex. Molecular Cell. 43 (4), 624-637 (2011).
- Conrad, N. K., et al. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics. 154 (2), 557-571 (2000).
- Darby, M. M., Serebreni, L., Pan, X., Boeke, J. D., Corden, J. L. The Saccharomyces cerevisiae Nrd1-Nab3 Transcription Termination Pathway Acts in Opposition to Ras Signaling and Mediates Response to Nutrient Depletion. Molecular and Cellular Biology. 32 (10), 1762-1775 (2012).
- Webb, S., Hector, R. D., Kudla, G., Granneman, S. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biology. 15 (1), R8(2014).
- Jensen, T. H., Jacquier, A., Libri, D. Dealing with Pervasive Transcription. Molecular Cell. 52 (4), 473-484 (2013).
- van Dijk, E. L., et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 475 (7354), 114-117 (2011).
- Thiebaut, M., et al. Futile Cycle of Transcription Initiation and Termination Modulates the Response to Nucleotide Shortage in S. cerevisiae. Molecular Cell. 31 (5), 671-682 (2008).
- Merran, J., Corden, J. L. Yeast RNA-Binding Protein Nab3 Regulates Genes Involved in Nitrogen Metabolism. Molecular and Cellular Biology. 37 (18), e00154-e00117 (2017).
- Bresson, S., Tuck, A., Staneva, D., Tollervey, D. Nuclear RNA Decay Pathways Aid Rapid Remodeling of Gene Expression in Yeast. Molecular Cell. 65 (5), 787-800 (2017).
- Buchbender, A., et al. Improved library preparation with the new iCLIP2 protocol. Methods. , (2019).
Erratum
Formal Correction: Erratum: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC
Posted by JoVE Editors on 2/17/2023. Citeable Link.
An erratum was issued for: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC. The Authors section was updated from:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Synthetic and Systems Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
to:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Mehak Chauhan1
Niki Christopoulou1
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Engineering Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten