
Method Article
Monitoraggio delle dinamiche di interazione proteina-RNA in vivo ad alta risoluzione temporale utilizzando χCRAC
In questo articolo
Erratum Notice
Riepilogo
Il cross-linking cinetico e l'analisi del cDNA è un metodo che consente di studiare la dinamica delle interazioni proteina-RNA nelle cellule viventi ad alta risoluzione temporale. Qui il protocollo è descritto in dettaglio, compresa la crescita delle cellule di lievito, la reticolazione UV, la raccolta, la purificazione delle proteine e le fasi di preparazione della libreria di sequenziamento di prossima generazione.
Abstract
L'interazione tra le proteine leganti l'RNA (RBP) e i loro substrati di RNA mostra fluidità e complessità. Nel corso della sua vita, un singolo RNA può essere legato da molti RBP diversi che ne regoleranno la produzione, la stabilità, l'attività e la degradazione. Come tale, molto è stato fatto per comprendere le dinamiche che esistono tra questi due tipi di molecole. Un passo avanti particolarmente importante è arrivato con l'emergere della "cross-l inchiostrazione e la ricetta immunop" (CLIP). Questa tecnica ha permesso indagini rigorose su quali RNA sono legati da un particolare RBP. In breve, la proteina di interesse è UV cross-linked ai suoi substrati di RNA in vivo, purificata in condizioni altamente rigorose, e quindi gli RNA covalentemente reticolati alla proteina vengono convertiti in librerie di cDNA e sequenziati. Fin dalla sua concezione, sono state sviluppate molte tecniche derivate al fine di rendere CLIP suscettibile a particolari campi di studio. Tuttavia, la reticolazione mediante luce ultravioletta è notoriamente inefficiente. Ciò si traduce in tempi di esposizione prolungati che rendono impossibile lo studio temporale delle interazioni RBP-RNA. Per ovviare a questo problema, abbiamo recentemente progettato e costruito dispositivi di irradiazione UV e di raccolta cellulare molto migliorati. Utilizzando questi nuovi strumenti, abbiamo sviluppato un protocollo per analisi risolte nel tempo delle interazioni RBP-RNA in cellule viventi ad alta risoluzione temporale: Kinetic CRoss-linking e Analysis of cDNAs (χCRAC). Recentemente abbiamo usato questa tecnica per studiare il ruolo degli RBP del lievito nell'adattamento allo stress dei nutrienti. Questo manoscritto fornisce una panoramica dettagliata del metodo χCRAC e presenta i recenti risultati ottenuti con il RBP Nrd1.
Introduzione
Gli RNA spesso si affidano agli RBP per esercitare la loro funzione, il che ha portato a un grande interesse nella comprensione delle dinamiche tra queste molecole. Molti RBP sono stati identificati in un'ampia varietà di organismi. Tuttavia, è sempre stato notoriamente difficile studiare le interazioni RBP-RNA in vivo. Un importante passo avanti nello studio di tali interazioni è arrivato con l'emergere di CLIP1. Questo metodo utilizza l'irradiazione ultravioletta (UV, 254 nm) per indurre legami covalenti tra RBP e i loro RNA direttamente legati (cioè cross-linking a distanza zero). Successivamente, il RBP di interesse viene immunopurificato in condizioni rigorose per garantire che vengano identificati solo gli RNA covalentemente reticolati alle proteine. Gli RNA legati vengono quindi parzialmente digeriti con RNasi e successivamente convertiti in librerie di cDNA per il sequenziamento. L'elevato rigore di purificazione è importante in quanto aumenta notevolmente la specificità del recupero di proteine e RNA, che è anche ulteriormente migliorata attraverso la purificazione SDS-PAGE del complesso ribonucleoproteina reticolato (RNP). CLIP e metodi correlati forniscono anche informazioni sulla risoluzione nucleotidica nel sito di legame della proteina, perché durante la preparazione della libreria di sequenziamento, gli amminoacidi che si reticolano all'RNA spesso terminano la trascrittasi inversa o causano l'introduzione di mutazioni dell'enzima in questo sito 1,2,3.
Fin dalla sua introduzione, il protocollo CLIP originale ha prodotto una notevole varietà di metodologie derivate. Un passo avanti particolarmente importante è arrivato con lo sviluppo di HITS-CLIP (o CLIP-seq), che fonde il sequenziamento ad alto throughput con l'approccio CLIP3. Da allora questo è stato adottato da tutte le metodologie basate su CLIP. iCLIP ha introdotto miglioramenti nelle tecniche di trimming e legatura dell'adattatore mediate dalla RNasi che facilitano una mappatura più accurata dei siti di legame RBP4. PAR-CLIP ha combinato la marcatura 4tio-uridina/uracile con la reticolazione a 365 nm, rendendo possibile la mappatura dei siti di reticolazione analizzando le sostituzioni T-C5. CRAC, urea-iCLIP, dCLIP e uvCLAP hanno introdotto condizioni di denaturazione e fasi di purificazione a doppia affinità che riducono ulteriormente il legame di fondo alla resina di affinità e aumentano ulteriormente la specificità della cattura proteica 2,6,7,8,9. Inoltre, CRAC, uvCLAP e dCLIP hanno introdotto il tagging dell'RBP di interesse con un tag di affinità, superando così la necessità di generare anticorpi specifici.
Sono state inoltre apportate diverse ottimizzazioni per accelerare la metodologia CLIP. Il protocollo CLIP originale utilizzava la radiomarcatura degli RNA catturati per visualizzare i complessi RBP-RNA dopo SDS-PAGE. Tuttavia, l'uso della radioattività può essere problematico per i laboratori non istituiti per tale lavoro. irCLIP incorpora un adattatore accoppiato a fluorofori che facilita la visualizzazione attraverso l'imaging a infrarossi10 e sCLIP utilizza la biotinilazione degli RNA catturati per visualizzarli attraverso HRP11 coniugato con streptavidina. Inoltre, eCLIP rinuncia completamente alla marcatura dell'RNA; Invece la proteina viene asportata esclusivamente in base alla sua dimensione nota12. La purificazione a base di streptavidina è stata utilizzata anche per accelerare il processo di preparazione della libreria in FAST-iCLIP, dove un adattatore 3' biotinilato viene legato agli RNA e utilizzato per consentire la purificazione dopo la trascrizione inversa e la circolarizzazione13. Ulteriori miglioramenti al protocollo iCLIP hanno anche aumentato notevolmente la complessità delle librerie4.
Infine, CLIP è stato modificato per consentire la cattura di RBP da diversi sottocompartimenti cellulari 14,15, per visualizzare RNA appena trascritti utilizzando l'induzione pulsata di ribonucleosidi fotoattivabili 5,16,17, per catturare RNA metilati18,19,20, per esaminare le interazioni RNA-RNA 21,22 e per mappare 3' estremità 23,24.
Nonostante i grandi contributi delle tecniche basate su CLIP nell'aiutare la nostra comprensione delle interazioni tra RBP e RNA, è stato limitato dall'inefficienza del cross-linking UV. Sebbene le cellule di coltura coltivate in un monostrato siano generalmente relativamente facili da reticolare, questo è significativamente più impegnativo nei tessuti o nelle cellule in soluzione. I tessuti possono richiedere più cicli di esposizione ai raggi UV per penetrare negli strati cellulari richiesti, mentre le cellule microbiche sono spesso coltivate in terreni ricchi che contengono composti aromatici che assorbono i raggi UV25. Infatti, sono stati utilizzati tempi di irradiazione UV fino a 30 minuti per generare una sufficiente reticolazione tra RBP e i loro RNA legati per tali campioni26,27,28. Questa esposizione UV prolungata induce risposte allo stress all'interno della cellula, come danni al DNA indotti dai raggi UV, che possono contaminare i dati finali in alcune applicazioni.
La maggior parte degli studi CLIP si è concentrata sulla generazione di singole "istantanee" di specifiche interazioni proteina-RNA in una cellula. Tuttavia, le interazioni proteina-RNA sono intrinsecamente dinamiche, in particolare quando le cellule sono soggette a cambiamenti nel loro ambiente. Ciò può includere un'improvvisa riduzione della disponibilità di nutrienti essenziali o rapidi cambiamenti di temperatura. Pertanto, per comprendere veramente il ruolo di un RBP durante lo stress, è meglio eseguire analisi risolte nel tempo perché possono catturare l'intero spettro di obiettivi RBP durante lo stress e determinare in quale fase della risposta allo stress è attivo l'RBP scelto. In particolare, studi sul lievito hanno dimostrato che i primi minuti di adattamento sono assolutamente cruciali per la sopravvivenza e le emivite dell'RNA nei batteri possono variare da minuti a secondi 29,30,31,32,33. Pertanto, tali analisi risolte nel tempo dovrebbero idealmente essere eseguite ad alta risoluzione temporale. Tuttavia, i lunghi tempi di cross-linking rendono particolarmente impegnativo lo studio delle risposte adattative in fase iniziale.
Al fine di superare questi problemi, abbiamo recentemente sviluppato un metodo migliorato che è in grado di reticolare e raccogliere cellule su scale temporali di un minuto. Il nostro metodo χCRAC consente la misurazione quantitativa dei cambiamenti dinamici nelle interazioni RBP-RNA a risoluzione precedentemente non testimoniata. Cruciale per questo metodo è stato lo sviluppo di un nuovo dispositivo di irradiazione UV32 che riduce il tempo di reticolazione richiesto nel lievito e nei batteri in soluzione di circa 10 volte, congelando efficacemente istantaneamente le interazioni RBP-RNA. Inoltre, al fine di raccogliere rapidamente le cellule dopo l'irradiazione UV, abbiamo sviluppato un dispositivo di filtrazione sottovuoto in grado di raccogliere lieviti in crescita esponenziale in una coltura da 0,5 L in circa 30 s32. Queste innovazioni tecnologiche permettono lo studio della dinamica RBP-RNA a risoluzione su scala minuta. Inoltre, abbiamo anche introdotto diverse ottimizzazionial protocollo CRAC 2 originale al fine di aumentarne la praticità.
Utilizzando χCRAC, abbiamo recentemente studiato il targetoma di un RBP nucleare di lievito, Nab3, in risposta alla privazione di glucosio. In Saccharomyces cerevisiae, Nab3 può formare un complesso con Nrd1, un RBP e l'RNA elicasi Sen1 per formare il complesso NNS. Il legame di NNS alla RNA polimerasi e al trascritto nascente può innescare la terminazione trascrizionale34. Questo complesso è principalmente coinvolto nella rimozione di trascritti criptici di RNA non codificanti, ma ha anche dimostrato di regolare l'espressione di geni codificanti proteine. Lo studio ha mostrato un targeting differenziale di Nab3 verso trascrizioni non codificanti e codificanti dopo solo un minuto di stress32. Abbiamo dimostrato che la terminazione co-trascrizionale da parte di Nab3 si traduce in un'espressione molto transitoria, simile a un polso, di geni retrotrasposoni, che sarebbe stata difficile da rilevare utilizzando approcci tradizionali basati su CLIP. Inoltre, i brevi tempi di irradiazione UV nel nostro reticolante UV hanno anche aumentato significativamente il recupero di RNA non codificanti di breve durata32. χCRAC sarà probabilmente uno strumento cruciale per chiarire non solo come i RBP modellano la risposta allo stress su scale temporali immediate, ma anche i loro ruoli mutevoli durante l'intero ciclo di vita di una risposta. Questo manoscritto fornisce una panoramica dettagliata di tutte le fasi del protocollo χCRAC. A scopo illustrativo, il metodo è stato utilizzato per studiare la proteina Nrd1 del lievito, che è coinvolta nella terminazione trascrizionale e nel decadimento dell'RNA35,36, e il suo bersaglioma di RNA in risposta alla deprivazione di glucosio in una moltitudine di punti temporali. Infine, dimostriamo anche che la nostra unità di irradiazione UV può rapidamente reticolare RBP con RNA in cellule HeLa, rendendo possibile eseguire anche analisi ad alta risoluzione risolte nel tempo in cellule aderenti.
Protocollo
| TN150 |
| 50 mM Tris pH 7,8 |
| 150 mM NaCl |
| 0,1% NP-40 |
| 1X inibitore della proteasi |
| TN1000 |
| 50 mM Tris pH 7,8 |
| 1M NaCl |
| 0,1% NP-40 |
| NP-PNK |
| 50 mM Tris-HCl pH 7,8 |
| 10 mM MgCl2 |
| 0,1% NP-40 |
| 5 mM di beta-mercaptoetanolo |
| 5 x PNK |
| 250 mM Tris-HCl pH 7,8 |
| 50 mM MgCl2 |
| 50 mM di beta-mercaptoetanolo |
| WB I |
| 50 mM Tris-HCl pH 7,8 |
| 300 mM NaCl |
| 10 mM di imidazolo |
| 6M guanidina-HCl |
| 0,1% NP-40 |
| 5 mM di beta-mercaptoetanolo |
| Terza Guerra Mondiale II |
| 50 mM Tris-HCl pH 7,8 |
| 50 mM NaCl |
| 10 mM di imidazolo |
| 0,1% NP-40 |
| 5 mM di beta-mercaptoetanolo |
| Tampone di eluizione |
| 50 mM Tris pH 7,8 |
| 50 mM NaCl |
| 250 mM di imidazolo |
| 0,1% NP-40 |
| 5 mM di beta-mercaptoetanolo |
| Tampone Proteasi K |
| 50 mM Tris |
| 0,1% NP-40 |
| 5 mM β-mercaptoetanolo |
| 1% SDS |
| 5 mM EDTA |
| 50 mM NaCl2 |
| Tampone di lisi dei mammiferi |
| 50 mM Tris-HCl pH 8 |
| 100 mM NaCl |
| 0,5% v/v Triton X-100 |
| 0,25% p/v Na-desossicolato |
| 0,1% w/v SDS |
| 5 mM EDTA |
| 1 mM DTT (aggiunto fresco) |
| 1X inibitore della proteasi |
Tabella 1: Riserve necessarie per χCRAC e loro composizione.
1. Produzione di reticolazione UV e lisato
- Microrganismi in soluzione
- Inoculare 3,5 L del mezzo desiderato con lievito da una coltura notturna a un OD600 iniziale di 0,05. Crescere a 30 °C con agitazione continua a 180 giri/min.
- Durante la crescita, preparare altri materiali necessari.
- Preparare un contenitore di azoto liquido.
- Preparare 3 L di mezzo che induce stress e caldo a 30 °C a bagnomaria.
- Impostare l'apparato filtrante, accendere il reticolante (Figura 2A) ed etichettare tubi conici da 50 ml, uno per ogni punto temporale.
- Una volta che le cellule raggiungono l'OD 600 desiderato, versare500 ml di cellule direttamente nel reticolante e irradiare UV con 250 mJ di 254 nm UV. Vedere la Figura 2A e la Figura 3A per informazioni dettagliate sull'uso del crosslinker.
NOTA: L'energia di irradiazione UV deve essere attentamente ottimizzata per ogni proteina di interesse. Vedi la discussione per ulteriori dettagli. - Dopo la reticolazione, filtrare le celle utilizzando uno dei dispositivi di filtrazione sotto vuoto (Figura 2B,C). Arrotolare la membrana con le celle filtrate, posizionare nel tubo conico t = 0 (tempo zero) 50 mL e congelare in azoto liquido.
- Filtrare le celle rimanenti su sei filtri diversi. Risospendere le cellule raccolte nei 3 L del mezzo che induce stress precedentemente riscaldato facendo cadere le membrane nel mezzo e mescolando vigorosamente con una striscia per 50 s. Dopo i 50 s, prepararsi per prendere il campione t = 1.
- Dopo 1 minuto, reticolare 500 ml di cellule e raccogliere mediante filtrazione come nei passaggi 1.1.3-1.1.4. Ripetere l'operazione dopo 2, 4, 8, 14 e 20 minuti o punti temporali diversi in base alle esigenze.
- Conservare i tubi conici contenenti le celle a -80 °C. Impostare tampone fosfato salino (PBS) a 4 °C durante la notte.
- Il giorno successivo, prelevare ogni tubo conico contenente un campione reticolato e risospendere le cellule in 25 ml di PBS freddo agitando vigorosamente.
- Trasferire le sospensioni cellulari in nuovi tubi conici e ruotare a 4.600 x g, 5 min a 4 °C.
- Versare il PBS, girare rapidamente di nuovo per raccogliere il PBS residuo e quindi decantare il liquido rimanente con una pipetta.
- Calcola il peso del pellet nel tubo confrontandolo con un tubo vuoto.
- Aggiungere due volumi di pellet di TN150 ghiacciato, 60 μL di DNasi 1 e 10 μL di inibitore della RNasi. Incubare su ghiaccio per 30 min.
- Ad esempio, per 400 mg di cellule, aggiungere 800 μL di TN150 ghiacciato.
- L'aggiunta della DNasi non è essenziale per la maggior parte delle proteine solubili, ma è molto importante quando si studiano proteine legate alla cromatina come la RNA polimerasi. Inoltre, riduce la viscosità dei lisati batterici. È molto importante utilizzare esattamente due volumi di pellet del tampone di lisi, altrimenti l'efficienza della lisi può diminuire.
- Aggiungere tre volumi di pellet (in ml) di perle di zirconia alla sospensione cellulare. Per il lievito, utilizzare perline di 0,5 mm di diametro e per i batteri utilizzare 0,1 mm.
- Ad esempio, per 400 mg di cellule, misurare 1,2 ml di perle di zirconia in un tubo da 1,5 ml e aggiungerle alle cellule risospese nel tampone di lisi.
- Vortice le sospensioni cellulari per 1 minuto, quindi posizionare sul ghiaccio per 1 minuto. Ripetere l'operazione per un totale di 5x.
- Aggiungere due volumi di pellet di tampone TN150 e vortice vigorosamente per miscelare.
- Centrifugare la sospensione nel tubo conico a 4.600 g per 20 minuti a 4 °C in una centrifuga da banco.
- Dopo la centrifugazione, prelevare un campione di 50 μL del surnatante per la futura analisi Western blot per esaminare l'intera espressione proteica cellulare.
- Trasferire i surnatanti in provette da 1,5 mL e centrifugare il lisato per 20 minuti a 20.000 x g a 4 °C, in un microfugo.
- In alternativa, se si utilizzano provette da 5 ml, centrifugare a 13.000 x g per 20 minuti.
- Dopo la centrifugazione, prelevare un campione di 50 μL del surnatante per la futura analisi Western blot per esaminare l'espressione solubile della proteina.
- Procedere all'acquisizione RBP (sezione 2).
- Cellule aderenti in coltura
- Seminare abbastanza cellule aderenti in una capsula di Petri 24 ore prima della reticolazione UV in modo che possano raggiungere l'80% di confluenza il giorno successivo. Crescere durante la notte nel terreno desiderato in un incubatore di coltura cellulare a 37 °C, 5% CO2.
NOTA: Se si utilizzano piastre di Petri al quarzo, è utile promuovere l'adesione cellulare attraverso il trattamento della coltura con poli-D-lisina (70.000-140.000 wt) e siero fetale di vitello (FCS) 2,5 ore prima della semina. Aggiungere abbastanza poli-D-lisina per coprire l'intera superficie di crescita e incubare a temperatura ambiente (RT) per 5 minuti. Successivamente, la capsula di Petri al quarzo deve essere risciacquata accuratamente con acqua e asciugata nell'incubatore di coltura cellulare per 2 ore o fino a completa asciugatura. Successivamente, aggiungere abbastanza FCS per coprire completamente la superficie di crescita e posizionarlo nell'incubatore per almeno 30 minuti. L'FCS deve essere completamente rimosso prima di seminare le cellule. - Una volta che le cellule hanno raggiunto l'80% di confluenza, rimuovere il mezzo e lavare con 15 ml di PBS ghiacciato. Quindi, rimuovere completamente tutto il liquido rimanente e procedere immediatamente al passaggio successivo.
- Trasferire la capsula di Petri nel vassoio per le celle aderenti (Figura 3B) e irradiare UV con 300 mJ di UV a 254 nm. Vedere la Figura 2A e la Figura 3B per i dettagli sull'uso del reticolante.
NOTA: L'energia di irradiazione UV deve essere attentamente ottimizzata per ogni proteina di interesse. Vedi la discussione per ulteriori dettagli. - Immediatamente dopo la reticolazione, posizionare la capsula di Petri sul ghiaccio e aggiungere 10 ml di PBS ghiacciato. Raccogliere le cellule raschiando e trasferirle in un tubo conico da 15 ml. Pellet mediante centrifugazione a 300 x g per 5 minuti a 4 °C.
- Rimuovere il PBS e risospendere il pellet cellulare in 1 ml di PBS ghiacciato e trasferirlo in una provetta da microcentrifuga da 1,5 ml. Celle a pellet nuovamente mediante centrifugazione per 5 minuti a 300 x g a 4 °C.
- Rimuovere il PBS e congelare a scatto i pellet cellulari sul ghiaccio secco. Conservare il pellet cellulare a -80 °C fino al momento del bisogno.
- Ripetere i passaggi 1.2.3–1.2.6 per ogni timepoint.
- Risospendere i pellet cellulari in 1 mL di tampone di lisi e trasferirli in un tubo conico da 15 ml. Successivamente, aggiungere 1 mL di tampone di lisi per un totale di 2 ml.
- Aggiungere 5 μL di inibitore della RNasi dei mammiferi.
- Sonicate 5x per 10 s su ghiaccio a 10 ampere. Attendere 30 secondi tra i round di sonicazione.
- Calcolare la concentrazione proteica di ciascun campione e normalizzare alla concentrazione più bassa.
- Trasferire 1,98 mL di lisato in una provetta da 2 mL.
- Aggiungere 10 μL di DNasi I e incubare a 37 °C per 5 minuti agitando a 1.200 giri/min.
- Centrifugare il lisato a 16.000 x g per 20 minuti a 4 °C.
- Dopo la centrifugazione, prelevare un campione di 50 μL del surnatante per la futura analisi Western blot per esaminare l'espressione solubile della proteina.
- Procedere all'acquisizione RBP (sezione 2).
- Seminare abbastanza cellule aderenti in una capsula di Petri 24 ore prima della reticolazione UV in modo che possano raggiungere l'80% di confluenza il giorno successivo. Crescere durante la notte nel terreno desiderato in un incubatore di coltura cellulare a 37 °C, 5% CO2.
2. Acquisizione RBP
- Lavare le perle magnetiche anti-FLAG (75 μL di liquame per campione) o IgG agarosio (500 μL di liquame per campione) 3x con 5 mL di TN150. Risospendere in un volume finale di 700 μL di TN150 e aggiungere 100 μL di perline lavate a sette tubi conici da 15 ml.
- Conservare in ghiaccio fino a esaurimento.
- Una volta chiarificati i lisati, aggiungere il surnatante al tubo contenente le sfere anti-FLAG/IgG.
- Nutate a 4 °C per 2 h.
NOTA: Alcuni protocolli descrivono incubazioni notturne con le perle, ma questo non è raccomandato, perché lunghi tempi di incubazione possono ridurre drasticamente il recupero di RNA reticolati.
3. Lavaggio delle perline e scollatura TEV delle etichette
- Raccogliere le perline e rimuovere il lisato.
- Prelevare un campione di 50 μL del surnatante per la futura analisi Western blot per esaminare la proteina non catturata.
- Risospendere le perle in TN1000 ghiacciato e trasferirle in un tubo da 1,5 ml. Lavare per 10 min, 4 °C, con nutazione. Ripetere l'operazione per un totale di tre lavaggi.
- Se si utilizzano perle di agarosio IgG, lavare con 5 ml di TN1000. Se si utilizzano perline anti-FLAG, utilizzare 2 ml.
- Quindi, lavare le perline 3x con TN150, con lo stesso volume di cui sopra.
- Dopo il terzo lavaggio, risospendere le perle in 600 μL di TN150.
- Aggiungere 30 U di proteasi GST-TEV fatta in casa alla sospensione del tallone e ruotare per 2 ore a RT.
NOTA: La proteasi GST-TEV ricombinante è ora disponibile in commercio, ma non è stata testata con questo protocollo.- Durante la digestione, prepararsi per i passaggi successivi impostando colonne di tre tubi da 1,5 ml per ciascun campione (cioè per sette campioni, avere tre file di sette colonne).
- All'ultima fila di provette, aggiungere 0,4 g di guanidio cloridrato, 27 μL di cloruro di sodio 5M e 3 μL di imidazolo 2,5 M (pH = 8). Si noti che il pH dell'imidazolo deve essere 8. Questo è fondamentale per mantenere l'integrità dell'RNA.
- Inoltre, lavare il volume richiesto di perle di nichel in WB I 3x. Utilizzare 100 μL di liquame per campione. Dopo il lavaggio finale, risospendere le perle nello stesso volume originale di WB I e conservare su ghiaccio.
- Una volta completata la digestione TEV, raccogliere il surnatante utilizzando una cremagliera magnetica per le perle anti-FLAG o la centrifugazione per le perle IgG, e trasferirlo nella prima fila dei tubi precedentemente allestiti.
- Prelevare un campione di 50 μL dell'eluato TEV per l'analisi Western blot.
- Impostare un'incubatrice termoblocco a 37 °C. Alla seconda fila di provette, aggiungere 1 μL di cocktail RNasi (diluizione 1:50).
- Prelevare 550 μL di TEV eluato dalla prima fila di provette e aggiungerlo alla seconda fila (contenente il cocktail RNasi). Pipettare energicamente per garantire la miscelazione.
- Dopo aver completato questo per il primo campione, posizionare immediatamente il tubo nel blocco termico e avviare un timer. Passare ai campioni successivi, in modo che ciascuno sia sfalsato.
- Incubare per esattamente 5 minuti. Una volta completato, rimuovere il primo campione dal blocco termico e trasferire la soluzione nella terza fila di provette (contenente la polvere di guanidio cloridrato).
NOTA: Un'incubazione di 5 minuti con una diluizione 1:50 del cocktail RNasi è solitamente adatta per la maggior parte delle proteine, ma questo passaggio dovrà essere attentamente ottimizzato con tempi di incubazione o concentrazioni diversi per ciascuna proteina per assicurarsi che gli RNA reticolati siano della dimensione corretta (30-100 nt). - Immediatamente vortice per un paio di secondi a tutta velocità per sciogliere la polvere di guanidio e quindi passare al campione successivo.
- Dopo che tutti i campioni sono stati trasferiti nella polvere di guanidio, vortice di nuovo per assicurarsi che tutta la polvere sia completamente disciolta.
- Aggiungere 100 μL di perle di nichel lavate e ruotare per una notte a 4 °C. Questa incubazione può essere ridotta a 2 ore.
4. Trattamento con fosfatasi alcalina on-bead
- Impostare un blocco termico a 37 °C.
- Posizionare una colonna di centrifuga di purificazione in un tubo da 2 ml, uno per ogni campione. Trasferire le sfere di nichel sulle colonne e lasciare che il surnatante defluisca. Successivamente, assicurarsi che tutte le sfere di nichel siano state rimosse dal tubo da 1,5 mL risciacquando con WB I e applicando sulla colonna.
- Impostare 2 mL di provette, sei per campione (una per raccogliere ogni lavaggio). Mantenere l'esterno delle colonne asciutto per mantenere il flusso. Lavare le perline 3x con 500 μL di WB I e poi 3x con 500 μL di NP-PNK.
- Chiudere il coperchio della colonna di rotazione e ruotare brevemente le perline per rimuovere il buffer in eccesso.
- Mettere il tappo sulla colonna, mettere le colonne in tubi da 1,5 mL e aggiungere 60 μL della miscela di reazione vista nella Tabella 2.
| Componente | 1x | 7,5x |
| 5 x buffer PNK | 12 | 90 |
| Fosfatasi alcalina | 4 | 30 |
| Inibitore della RNasi | 2 | 15 |
| H2O | 42 | 315 |
| Volume finale | 60 μL | 450 μL |
Tabella 2: Miscela di reazione della fosfatasi alcalina.
- Incubare le perle per 1 h a 37 °C.
- Lavare le perle 1x con 500 μL di WB I per inattivare la fosfatasi alcalina e poi 3x con 500 μL di tampone NP-PNK. Assicurarsi di sciacquare accuratamente l'interno della colonna con il tampone NP-PNK per rimuovere eventuali tracce di guanidio.
5. Legatura on-bead del linker App-PE all'estremità 3' dell'RNA
- Estrarre il buffer rimanente e aggiungere 60 μL della miscela specificata nella Tabella 3 (vedere Tabella 4 per la sequenza App-PE) alle colonne. Incubare la reazione per 6 ore a 25 °C.
| Componente | 1x | 7,5x |
| 5 x buffer PNK | 12 | 90 |
| Adattatore App-PE (100 μM) | 0.6 | 4.5 |
| T4 RNA ligasi 2 troncata K227Q | 3 | 22.5 |
| Inibitore della RNasi | 1.5 | 11.25 |
| 50% PEG 8000 | 12 | 90 |
| H2O | 30.9 | 231.75 |
| Volume finale | 60 μL | 450 μL |
Tabella 3: Miscela di reazione di legatura del linker App-PE.
| Nome oligonucleotide | Sequenza (5'-3') | |||
| L5Aa | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrUrArArGrCrN-OH | |||
| L5Ab | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrRArUrArGrCrN-OH | |||
| L5Ac | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrGrCrGrCrArGrCrN-OH | |||
| L5Ad | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrCrGrCrUrUrArGrCrN-OH | |||
| L5Ba | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrArGrArGrCrN-OH | |||
| L5BB | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrGrUrGrArGrCrN-OH | |||
| L5Bc | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrCrArCrUrArGrCrN-OH | |||
| L5Bd | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrUrCrUrCrUrArGrCrN-OH | |||
| L5Ca | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrCrUrArGrCrN-OH | |||
| L5Cb | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrUrGrGrArGrCrN-OH | |||
| L5CC | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrArCrUrCrArGrCrN-OH | |||
| L5CD | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrGrArCrUrArGrCrN-OH | |||
| L5Da | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrCrGrUrGrArUrN-OH | |||
| L5Db | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrGrCrArCrUrArN-OH | |||
| L5CC | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrUrArGrUrGrCrN-OH | |||
| L5Dd | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrArUrCrArCrGrN-OH | |||
| L5Ea | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrCrArCrUrGrUrN-OH | |||
| L5Eb | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrGrUrGrArCrArN-OH | |||
| L5Ec | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrUrGrUrCrArCrN-OH | |||
| L5Ed | invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrArCrArGrUrGrN-OH | |||
| App_PE | App-NAGATCGGAAGAGCACACGTCTG-ddC | |||
Tabella 4: Le sequenze degli adattatori di DNA e RNA necessari per la legatura sulle estremità 5' e 3' degli RNA catturati. Questi sono stati purificati attraverso HPLC privo di RNasi.
- Lavare le perline 1x con 500 μL di WB I e 3x con 500 μL di tampone NP-PNK. Inserire la colonna in un nuovo tubo e ruotare il buffer rimanente.
6. Fosforilazione on-bead delle estremità 5' dell'RNA
- Aggiungere 80 μL della miscela specificata nella tabella 5 alle colonne. Incubare la reazione per 40 minuti a 37 °C.
NOTA: I campioni saranno ora altamente radioattivi. Pertanto, tutti i lavori successivi dovrebbero essere eseguiti dietro uno schermo protettivo e i rifiuti dovrebbero essere smaltiti secondo le norme locali in materia di salute e sicurezza.
| Componente | 1x | 7,5x |
| 5 x buffer PNK | 16 | 120 |
| 32P-ɣATP (10 μCi/μL) | 3 | 22.5 |
| T4 PNK | 3 | 22.5 |
| H2O | 58 | 435 |
| Volume finale | 80 μL | 600 μL |
Tabella 5: Miscela di reazione di fosforilazione.
- Aggiungere 1 μL di 100 mM ATP e lasciare che la reazione proceda per altri 20 minuti. Questo farà in modo che quasi tutte le estremità 5' abbiano fosfati per facilitare la legatura del linker 5'.
- Impostare 2 provette da ml, cinque per campione.
- Lavare le perline 1x con 500 μL di WB I e 3x con 500 μL di tampone NP-PNK. Si noti che queste eluizioni saranno molto radioattive e quindi dovrebbero essere smaltite in modo appropriato.
- Spostate la colonna sul tubo finale ed estraete il buffer rimanente.
7. Legatura on-bead del linker 5'
NOTA: I linker 5' contengono un codice a barre RNA che viene utilizzato per l'identificazione di ciascun campione dopo il sequenziamento. Pertanto, è assolutamente fondamentale notare quale linker viene utilizzato per quale campione.
- Aggiungere alle colonne 78 μL della miscela descritta nella tabella 6 . Aggiungere 2 μL di adattatore da 5' (100 μM; vedere Tabella 4) a ciascuna provetta e incubare per una notte a 18 °C.
| Componente | 1x | 7,5x |
| 5 x buffer PNK | 16 | 120 |
| ATP (10 mM) | 8 | 60 |
| Inibitore della RNasi | 2 | 15 |
| T4 RNA ligasi | 4 | 30 |
| H2O | 48 | 360 |
| Volume finale | 78 μL | 585 μL |
Tabella 6: Miscela di reazione di legatura del linker 5'.
- Il giorno seguente, lavare i grani 1x con 500 μL di WB I e 3x con 500 μL di WB II e trasferire le colonne in un nuovo tubo da 2 ml.
8. Eluizione, SDS-PAGE ed estrazione dell'RNA
- Impostare la centrifuga a 4 °C. Preparare due file di provette da 1,5 mL per campione per l'eluizione.
- Ruotare il volume vuoto delle colonne con perline di nichel con una rotazione rapida. Posizionare le colonne nella prima fila di tubi di eluizione e aggiungere 200 μL di tampone di eluizione. Attendere 2 minuti, quindi forzare il buffer attraverso la colonna con un giro rapido.
- Spostare le colonne sulla seconda riga di tubi e ripetere il passaggio 8.2. Ogni campione avrà ora 400 μL di eluato in totale, suddivisi in due provette da 1,5 ml.
- Prendere tutti gli eluati e trasferirli insieme in un tubo da 5 ml. Aggiungere 2 μL di 20 mg/ml di glicogeno. Pertanto, se si utilizzano sette campioni, ora ci saranno 2,8 ml di eluato aggregato nel tubo da 5 ml.
- Aggiungere 100 μL di acido tricloroacetico (TCA) per campione [ad esempio 700 μL di TCA per 7 campioni (2,8 mL di eluato aggregato)] al tubo da 5 mL e vortice bene per 30 s.
- Incubare su ghiaccio per 20 min.
- Centrifugare per 30 minuti a 17.000 x g, 4 °C, in centrifuga da banco.
- Rimuovere con cautela il surnatante dal tubo conico, controllando la pipetta con un contatore Geiger per assicurarsi che il pellet non sia stato rimosso accidentalmente. In caso affermativo, riportare il surnatante nel tubo e centrifugare per altri 10 minuti.
NOTA: il surnatante potrebbe essere ancora altamente radioattivo. Assicurati di utilizzare una schermatura adeguata. - Risospendere completamente il pellet in 2 ml di acetone ghiacciato.
- Centrifugare per 15 minuti a 17.000 x g, 4 °C.
- Rimuovere il più possibile l'acetone con una pipetta P1000. Successivamente, ruotare brevemente il tubo per raccogliere piccole goccioline di acetone, quindi rimuovere con una pipetta P10. Asciugare per 2 minuti in una cappa aspirante.
NOTA: L'acetone surnatante può ancora essere radioattivo. Assicurati di utilizzare una schermatura adeguata. - Risospendere il campione in 30 μL di tampone di carico proteico 1x. Per garantire che il pellet sia correttamente sospeso, controllare che la stragrande maggioranza della radioattività sia ora presente nel tampone di carico e non lasciata nel tubo da 1,5 mL rimuovendo la soluzione in una pipetta P200 e misurando l'attività rimasta nel tubo da 1,5 mL utilizzando un contatore Geiger.
- Riscaldare il campione per 10 minuti a 65 °C. Caricare su un gel Bis-Tris prefabbricato da 1 mm, 4-12% e correre per 1,5 ore a 125 V in tampone MOPS.
- Dopo che il gel ha terminato di funzionare, aprire la cassetta del gel. Il gel deve essere trattenuto sulla piastra inferiore. Smaltire la parte superiore.
- Avvolgere il gel nella pellicola trasparente e quindi fissarlo con del nastro adesivo all'interno di una cassetta a tenuta di luce. Assicurarsi che la cassetta abbia uno schermo di amplificazione per migliorare il segnale.
- Esporre una pellicola autoradiografica al gel e conservare la cassetta a -80 °C durante l'esposizione. Il tempo di esposizione varierà tra proteine con diverse efficienze di reticolazione.
- Quando si posiziona la pellicola, ci deve essere un modo per riallinearlo alla cassetta per tagliare la banda di interesse nella fase successiva. Per garantire ciò, utilizzare un righello fluorescente e assicurarsi anche che il gel si trovi in un angolo della cassetta, che viene quindi coperto dal film posizionato anche nell'angolo più alto.
NOTA: Come regola generale, gli eluati nel buffer di caricamento che danno una lettura di almeno ~ 250 cps quando visualizzati su un contatore Geiger danno un segnale sufficiente per un'esposizione di 3 ore. In caso contrario, viene eseguita l'esposizione notturna.
- Quando si posiziona la pellicola, ci deve essere un modo per riallinearlo alla cassetta per tagliare la banda di interesse nella fase successiva. Per garantire ciò, utilizzare un righello fluorescente e assicurarsi anche che il gel si trovi in un angolo della cassetta, che viene quindi coperto dal film posizionato anche nell'angolo più alto.
- Sviluppa il film. Tagliare via la pellicola trasparente che copre il gel ma non spostare il gel. In caso contrario, l'immagine verrà spostata dal gel.
NOTA: Il gel sarà probabilmente altamente radioattivo. Assicurati di utilizzare una schermatura adeguata quando tagli la fetta di gel. - Posizionare il film sul gel ed asportare la fascia di interesse. Mettere la fetta di gel in un tubo da 2 ml.
- Schiacciare la fetta di gel con una punta di pipetta P1000 e aggiungere 600 μL di tampone proteinasi K più 200 μg di proteinasi K (questo protocollo utilizza 10 μL di una soluzione di proteinasi K da 20 mg/ml). Incubare per 2 ore a 55 °C agitando energicamente.
- Successivamente, tagliare l'estremità di una punta P1000 con un bisturi pulito e trasferire i pezzi di surnatante e gel in una colonna di rotazione posta in un tubo da 2 ml.
- Ruotare la colonna per 1 minuto a 17.000 x g a RT. Raccogliere il flusso attraverso, che contiene gli RNA isolati radioattivi.
- Eseguire un'estrazione fenolo:cloroformio.
- Aggiungere 50 μL di acetato di sodio 3 M, pH = 5,2, e 500 μL di fenolo:cloroformio e vortice. Centrifugare per 5 minuti a 17.000 x g. Rimuovere lo strato superiore acquoso e metterlo in un nuovo tubo da 1,5 ml.
- Aggiungere 500 μL di cloroformio e vortice vigorosamente per 10-15 s. Centrifugare per 5 minuti a 17.000 x g a RT. Rimuovere lo strato acquoso e metterlo in un nuovo tubo da 1,5 ml.
- Aggiungere 1 μL di 20 mg/mL di glicogeno e 1 mL di etanolo ghiacciato al 96%. Precipitare per 30 min a -80 °C o per una notte a -20 °C.
- Centrifugare per 30 min a 4 °C, 17.000 x g. Rimuovere il surnatante, aggiungere 500 μL di etanolo al 70% e centrifugare per 5 minuti, 4 °C a 17.000 x g. Rimuovere tutto l'etanolo, eseguire una rapida rotazione per raccogliere i residui e rimuovere l'eccesso con una pipetta P10.
- Asciugare il pellet per ~ 3 minuti in una cappa aspirante. Risospendere in 20 μL di acqua trattata con DEPC.
- Conservare l'RNA a -80 °C durante la notte o procedere immediatamente alla fase di trascrizione inversa.
9. Trascrizione inversa
- Aggiungere 2 μL di 10 μM RT oligo (PE_reverse; vedere Tabella 7) e 4 μL di 5 mM dNTPs ai 20 μL di RNA.
| Nome oligonucleotide | Sequenza (5'-3') | |||
| P5 avanti | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | |||
| BC1 | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC3 | CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC4 | CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC5 | CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC7 | CAAGCAGAAGACGGCATACGAGATCAGATCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC8 | CAAGCAGAAGACGGCATACGAGATTAGCTTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC9 | CAAGCAGAAGACGGCATACGAGATGATCAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC10 | CAAGCAGAAGACGGCATACGAGATATCACGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| PE_reverse | CAGACGTGTGCTCTTCCGATCT | |||
Tabella 7: I primer PCR (comprese le sequenze di codici a barre) e il primer di trascrizione inversa.
- Trasferire in un blocco termico preriscaldato a 85 °C per 3 minuti, quindi raffreddare a scatto sul ghiaccio per 5 minuti. Raccogliere il contenuto della provetta mediante breve centrifugazione e quindi aggiungere 8 μL di 5x tampone trascrittasi inversa, 2 μL di 100 mM DTT e 2 μL di inibitore della RNasi.
- Incubare la miscela a 50 °C per 3 minuti, quindi aggiungere 2 μL di trascrittasi inversa e incubare per 1 ora a 50 °C.
- Inattivare la trascrittasi inversa mediante incubazione a 65 °C per 15 minuti.
- Trasferire i tubi in un blocco termico preriscaldato a 37 °C e lasciare per 3 minuti per acclimatarsi.
- Aggiungere 2 μL di RNasi H e incubare per 30 minuti a 37 °C.
- Isolare il cDNA usando le perline SPRI.
- Aggiungere due volumi da 84 μL di perline. Incubare per 15 min. Metti le perline su una griglia magnetica e lascia per 1 minuto per raccogliere le perline.
- Rimuovere e smaltire il surnatante e aggiungere 200 μL di etanolo al 70%. Non rimuovere le perle dal rack magnetico. Incubare le perle con l'etanolo per 30 s.
- Rimuovere l'etanolo e ripetere la fase di lavaggio. Rimuovere tutto l'etanolo residuo utilizzando una punta P10.
- Mettere le perline in una cappa aspirante per 2 minuti per asciugarle. Rimuovere le perline dal rack, risospenderle in 12 μL di acqua, quindi rimettere le perline sul rack. Rimuovere 11 μL di surnatante.
- Congelare il cDNA a -20 °C o procedere immediatamente alla fase PCR.
10. Reazione qPCR
- Prima della PCR finale per l'amplificazione dei cDNA, viene eseguita una reazione a catena della polimerasi quantitativa (qPCR) per identificare il numero ottimale di cicli per amplificare i cDNA per prevenire la sovraamplificazione della libreria.
- Impostare una reazione qPCR sul ghiaccio secondo la Tabella 8. Vedere la tabella 7 per tutti i primer.
| Componente | 1x |
| 2x mastermix di reazione qPCR | 5 |
| Primer P5 da 0,1 μM (avanti) | 0.8 |
| Primer da 0,1 μM BC (inverso) | 0.8 |
| cDNA (o acqua come controllo negativo) | 1 |
| H2O | 2.4 |
| Volume finale | 10 μL |
Tabella 8: miscela di reazione qPCR.
- Per una corretta quantificazione dei cicli necessari per l'amplificazione, utilizzare tre repliche tecniche per il cDNA e tre controlli negativi (cioè acqua).
- Sigillare le piastre con pellicola otticamente trasparente ed eseguire la qPCR secondo le istruzioni del produttore del kit.
- Analizzare i campioni attraverso un metodo di quantificazione assoluta per identificare il numero di cicli (n) in cui viene raggiunto il ginocchio di crescita esponenziale (vedi Figura 4C per un esempio). Questo numero di cicli viene quindi utilizzato per l'amplificazione finale del resto del cDNA.
11. Reazione PCR ed estrazione del gel
- Impostare la reazione PCR sul ghiaccio secondo la Tabella 9. Vedere la tabella 7 per tutti i primer.
NOTA: vengono utilizzati solo 5 μL della libreria di cDNA.
| Componente | 1x |
| 10 x tampone polimerasi a correzione di bozze | 5 |
| Primer P5 da 10 μM (avanti) | 1 |
| Primer BC 10 μM (retro) | 1 |
| 5 mM dNTP | 2.5 |
| Enzima polimerasi di lettura della prova | 1 |
| cDNA | 5 |
| H2O | 34.5 |
| Volume finale | 50 μL |
Tabella 9: Miscela di reazione PCR.
- Eseguire la PCR come segue: 95 °C per 2 minuti; n cicli di 98 °C per 20 s, 52 °C per 30 s e 72 °C per 1 min; e 72 °C per 5 min. Il numero (n) di cicli per l'amplificazione della libreria χCRAC è determinato dalla qPCR descritta nella sezione 10.
- Aggiungere 1 μL di esonucleasi I e incubare a 37 °C per 60 minuti.
- Pulire il cDNA amplificato usando perline SPRI come descritto sopra usando due volumi di perline (cioè 100 μL). Eluire in 11 μL.
- Aggiungere 3 μL di colorante caricante 6x ed eseguire su un gel prefabbricato TBE al 6% a 100 V per 1 ora in 1x tampone TBE. Utilizzare una scala appropriata per la quantificazione di brevi frammenti di DNA.
- Una volta terminato, rimuovere il gel dalla cassetta e metterlo in un contenitore adatto a tenuta di liquidi con abbastanza 1x TBE per coprire il gel (ad esempio ~ 50 ml). Aggiungere una quantità appropriata di colorante sicuro SYBR (ad es. per 50 ml, utilizzare 5 μL di un colorante 10.000x)
- Lasciare macchiare il gel mescolando delicatamente per 15 minuti a RT. Scolare il TBE 1x contenente SYBR e sostituirlo con 1x TBE pulito. Lavare il gel per 10 minuti agitando delicatamente a RT.
- Scolare il TBE 1x e posizionare il gel in una cartella trasparente. Tagliare la cartella a una dimensione appropriata.
- Immagine del gel attraverso un mezzo appropriato come un phosphorimager. Frammenti di DNA soggetti ad accisa tra ~175 bp e ~400 bp. Mettere la fetta di gel in un tubo da 1,5 ml.
- Schiacciare accuratamente la fetta di gel con una punta P1000 e aggiungere 400 μL di H2O. Incubare a 37 °C agitando per 1 ora in un blocco termico.
- Congelare il campione su ghiaccio secco per 10 minuti, quindi riporre nel blocco termico a 37 °C agitando per 1 ora.
- Creare un'unità filtrante prendendo una colonna filtrante e inserendo due filtri in microfibra di vetro all'interno. Posizionare l'unità in un tubo da 1,5 ml.
- Tagliare l'estremità di una punta P1000 con un bisturi pulito e assorbire la sospensione di gel TBE distrutta, quindi erogare nell'unità filtrante creata nel passaggio 11.12. Girare a 17.000 x g per 30 s.
- Aggiungere 1 μL di glicogeno al surnatante, insieme a 40 μL di acetato di sodio, pH = 5,2 e 1 mL di etanolo al 96%. Incubare a -80 °C per 30 min.
- Centrifugare per 30 minuti a 17.000 x g, 4 °C. Scartare il surnatante e lavare con 500 μL di etanolo al 70%.
- Girare per 5 minuti, rimuovere completamente l'etanolo e quindi asciugare il pellet in una cappa aspirante per 3 minuti.
- Risospendere in 10 μL di H2O e misurare la concentrazione di DNA.
Risultati
Per dimostrare l'efficacia del metodo χCRAC, è stato eseguito un esperimento a tempo con ceppi di lievito che esprimono una proteina Nrd1 marcata HTP. Una rappresentazione schematica dettagliata che descrive il funzionamento del metodo è fornita nella Figura 1. Come Nab3, Nrd1 è coinvolto nel decadimento dell'RNA nucleare di una varietà di trascritti di RNA37. Precedenti lavori del laboratorio di Corden hanno suggerito che il legame di Nrd1 ai suoi bersagli RNA cambia significativamente quando le cellule sono sottoposte a fame di glucosio28,38. Pertanto, le cellule che crescono esponenzialmente in un mezzo contenente glucosio (SD-TRP) sono state spostate sullo stesso mezzo senza glucosio (S-TRP) nel corso di un corso di tempo per monitorare i cambiamenti dinamici nelle interazioni Nrd1-RNA. I campioni sono stati prelevati e reticolati nella camera Vari-X-linker (Figura 3A) prima dello spostamento e poi dopo 1, 2, 4, 8, 14 e 20 minuti. Il mezzo utilizzato per la crescita cellulare era deliberatamente carente di triptofano per ridurre l'assorbimento UV da parte di questo amminoacido aromatico. Si noti che è meglio utilizzare un mezzo sintetico sterilizzato con filtro perché l'autoclave del mezzo può portare alla caramellizzazione degli zuccheri. Ciò riduce quindi l'efficienza di reticolazione.
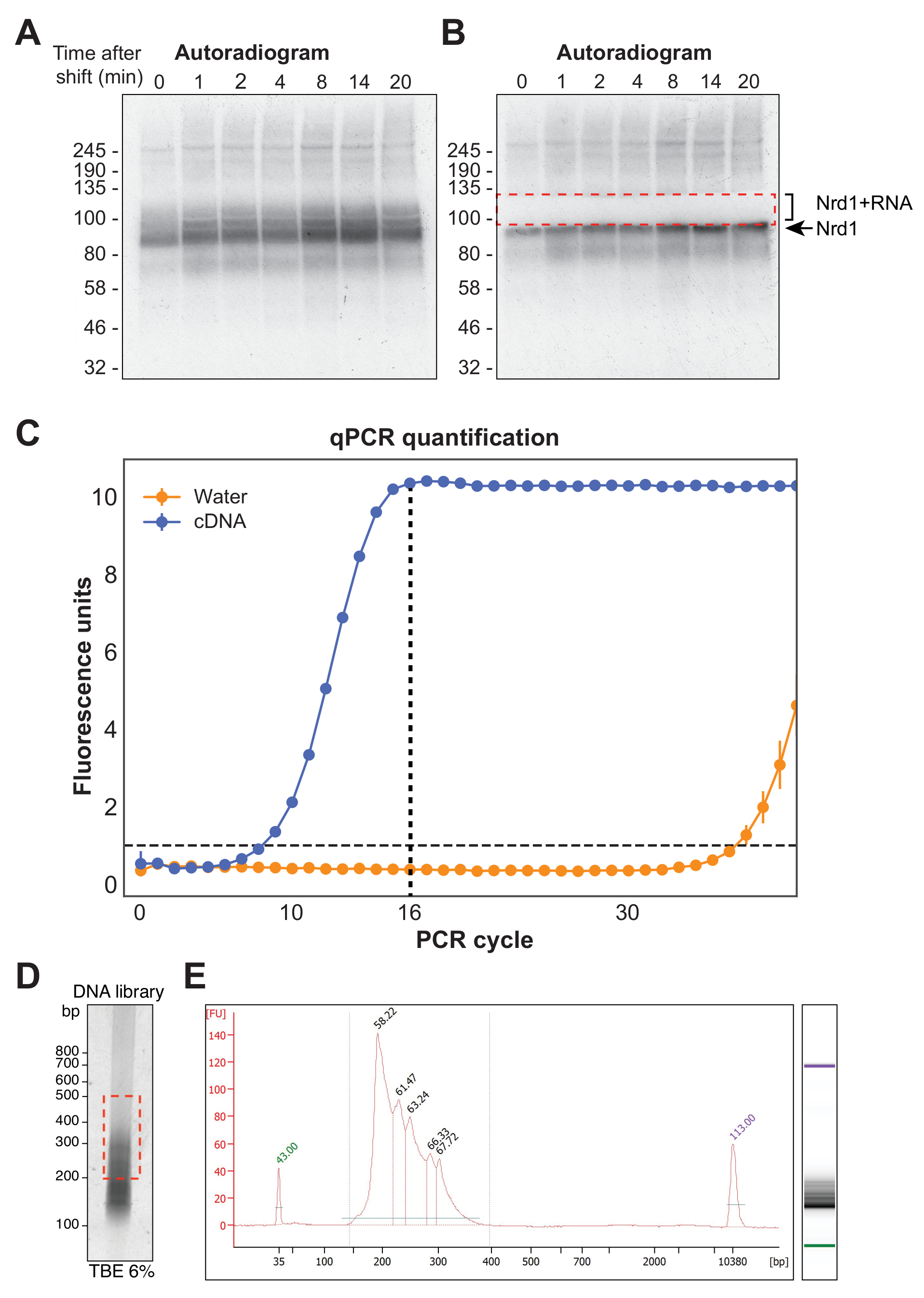
La figura 4A mostra un'autoradiografia rappresentativa di un esperimento χCRAC. Si noti che in questo esempio gli esempi non sono stati raggruppati insieme. Invece, ognuno è stato eseguito individualmente sul gel. Questo è raccomandato per i test sperimentali iniziali per dimostrare che la proteina si collega efficacemente all'RNA in tutti i punti temporali testati. Un segnale particolarmente intenso è stato osservato al peso molecolare atteso del RBP, che rappresenta la proteina legata a RNA radiomarcati molto corti non suscettibili di sequenziamento. Pertanto, il segnale di striscio sopra questa banda, che è la proteina reticolata a frammenti di RNA più lunghi, è stato isolato. Il frammento è stato tagliato appena sopra la banda proteica più circa 30 kDa. La figura 4B mostra un autoradiogramma dopo l'escissione, con la proteina reticolata a brevi RNA rimasti nel gel e il segnale precedentemente strisciante ora asportato.
Dopo la trascrizione inversa, la libreria di cDNA deve essere amplificata utilizzando la PCR. Tuttavia, la sovraamplificazione della libreria deve essere evitata in quanto ciò può introdurre pregiudizi verso sequenze preferenzialmente amplificate dalla polimerasi e generare artefatti PCR. Le librerie sovraamplificate contengono anche un gran numero di sequenze duplicate che sprecano letture sul sequencer. Al fine di calcolare il numero ideale di cicli di PCR per l'amplificazione della libreria finale, un'aliquota del cDNA è stata amplificata attraverso qPCR utilizzando gli oligonucleotidi P5 e BC. Il primo ciclo in cui la libreria ha raggiunto il picco di fluorescenza è stato scelto come conteggio del ciclo PCR. La Figura 4C fornisce un esempio di qPCR da una tipica libreria di cDNA, che ha prodotto un conteggio del ciclo di picco di 16. Questo valore è stato poi utilizzato per la PCR χCRAC finale. Per elaborare i dati sequenziati, abbiamo utilizzato software precedentemente sviluppato nel nostro laboratorio (pyCRAC) e la pipeline corrispondente per l'analisi dei dati cinetici CRAC (Nues et al., 2017; https://git.ecdf.ed.ac.uk/sgrannem/pycrac, https://bitbucket.org/sgrann/kinetic_crac_pipeline/src/default/). Questi strumenti software open source consentono il demultiplexing e il taglio dei dati, la rimozione dei duplicati della PCR, l'identificazione di picchi statisticamente significativi, le letture dei cluster in sequenze contigue e l'identificazione di motivi di legame39. Ulteriori dettagli su come funzionano questi strumenti sono disponibili sulle rispettive pagine web.
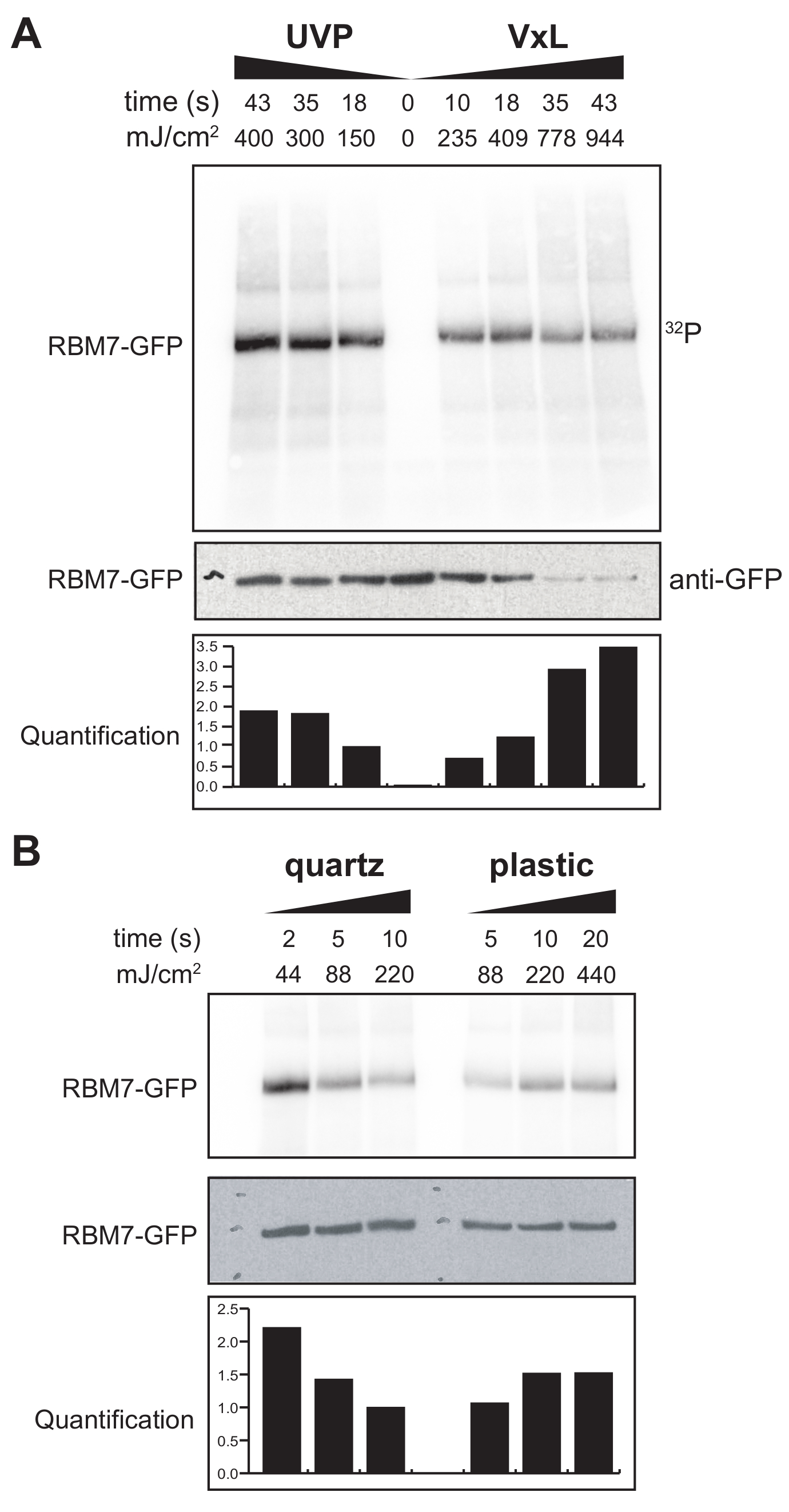
Abbiamo anche iniziato a sviluppare un protocollo χCRAC per le cellule di mammifero. La maggior parte delle linee cellulari di mammifero sono coltivate come monostrato e il vassoio nel nostro reticolante con la sacca permeabile ai raggi UV non è adatto per esperimenti con cellule aderenti. Per ovviare a questo problema, abbiamo sviluppato uno stadio in cui gli utenti possono irradiare UV 1-2 piastre di Petri (150 mm di diametro e 25 mm di profondità) con celle aderenti (Figura 3B). Come primo test, l'efficienza del cross-linker per le cellule di mammifero è stata misurata attraverso la reticolazione e la cattura di GFP-RBM7 stabilmente marcati utilizzando anticorpi anti-GFP e una tradizionale purificazione basata su CLIP. Come mostrato nella Figura 5A, il cross-linker è stato in grado di recuperare complessi proteina-RNA da cellule di mammifero cresciute come monostrato utilizzando l'irradiazione UV a 254 nm con efficienze paragonabili a un dispositivo di irradiazione UV ampiamente utilizzato. Tuttavia, le stoviglie standard per colture cellulari normalmente utilizzate per esperimenti di reticolazione UV sono impenetrabili ai raggi UV a 254 nm. Pertanto, nel nostro reticolante le cellule riceverebbero solo irradiazione dal banco superiore delle lampade UV. Per ovviare a questo, abbiamo sviluppato una capsula di Petri al quarzo permeabile ai raggi UV per la crescita cellulare e la reticolazione. L'uso della coltura al quarzo ha mostrato un robusto recupero di complessi proteina-RNA con appena 2 s di irradiazione UV (Figura 5B). Se combinati con metodi di cattura RBP per cellule di mammifero come le tecnologie CLIP, questi brevi tempi di cross-linking sono suscettibili con i tempi di recupero dei profili spaziotemporali di legame all'RNA degli RBP in risposta a stress genotossici o rapidi esaurimenti di fattori proteici, o in parallelo con la sincronizzazione trascrizionale o del ciclo cellulare.
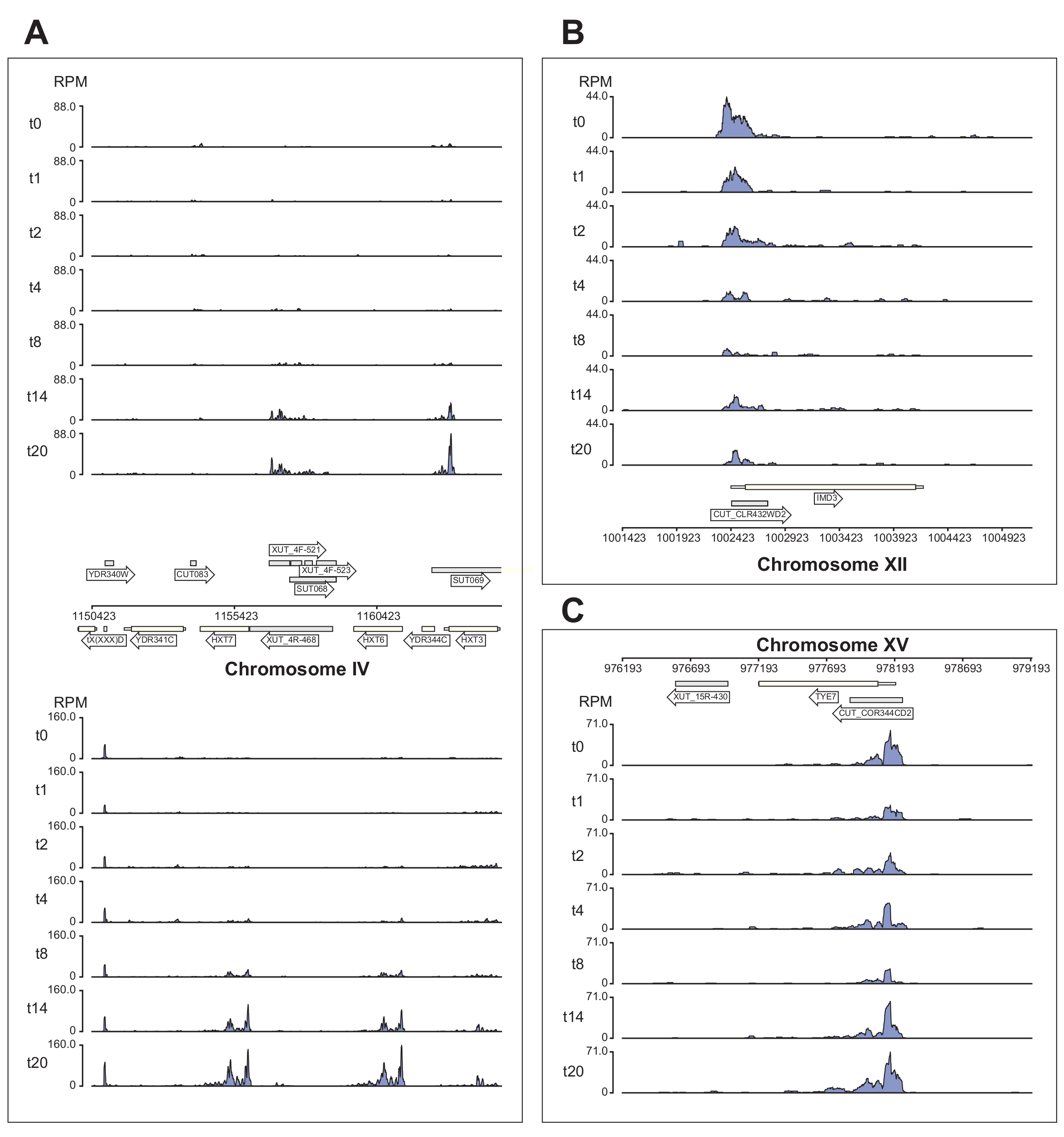
La figura 6 mostra diversi esempi dei dati Nrd1 elaborati dalla pipeline χCRAC. Questa figura è stata preparata utilizzando i file bedgraph generati dalla pipeline e il pacchetto python GenomeBrowser (https://pypi.org/project/GenomeBrowser/1.6.3/), che abbiamo progettato per semplificare la creazione di immagini del browser del genoma di qualità di pubblicazione dei dati. I rettangoli grigi rappresentano regioni genomiche che esprimevano RNA non codificanti, come il trascritto criptico instabile (CUT), i trascritti stabili non caratterizzati (SUT)40 e i trascritti instabili sensibili a Xrn1 (XUT)41. I dati in Figura 6 mostrano che Nrd1 si lega a molti di questi trascritti di RNA non codificanti, coerentemente con l'idea che questa proteina sia coinvolta nella degradazione di questa classe di trascritti42. La figura 6A mostra una regione di ~15 kb sul cromosoma IV. Qui c'è stato un aumento significativo del legame di Nrd1 ai trascritti che codificano i trasportatori di glucosio ad alta affinità HXT6 e HXT7, entrambi sovraregolati durante la fame di glucosio. È probabile che la terminazione della trascrizione da parte del complesso NNS possa influenzare la cinetica di induzione di questi geni durante la carenza di glucosio. La Figura 6B mostra un esempio di cross-linking Nrd1 alla trascrizione Imd3, che è nota per essere regolata da Nab343. In questo caso i dati hanno dimostrato una significativa riduzione del legame in caso di carenza di glucosio. Il lavoro precedente ha mostrato una diminuzione del legame di Nab3 alla trascrizione Tye7 durante la fame di glucosio44. Coerentemente con questa osservazione, i dati χCRAC suggeriscono che il legame di Nrd1 è diminuito durante la carenza di glucosio e il cross-linking di Nrd1 a Tye7 era al suo minimo dopo 8 minuti di stress (Figura 4C). Tuttavia, sembra che questo effetto fosse solo transitorio, perché dopo 14 minuti di fame di glucosio, il legame Nrd1 è tornato ai livelli iniziali.

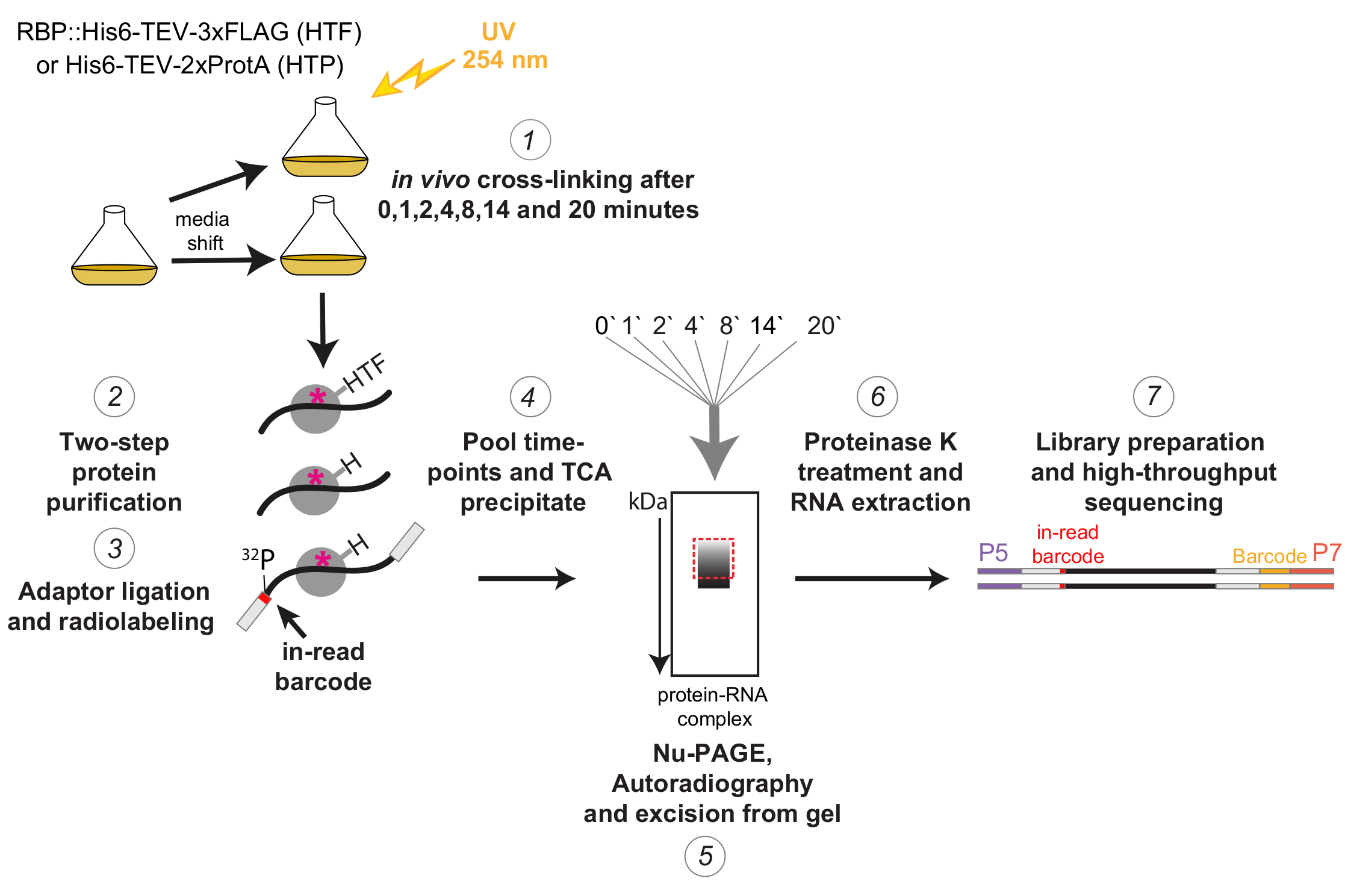
Figura 1: Rappresentazione schematica del protocollo χCRAC. I ceppi etichettati sono stati coltivati fino alla densità desiderata. RBP indica la proteina legante l'RNA. Successivamente, è stato prelevato un campione di riferimento e reticolato con luce UV a 254 nm. Le cellule rimanenti sono state raccolte mediante filtrazione e poi rapidamente spostate sul mezzo che induce stress. Per l'esperimento χCRAC qui descritto, sono stati prelevati campioni e reticolati 1, 2, 4, 8, 14 e 20 minuti dopo lo spostamento (1). L'RBP di interesse è stato quindi purificato utilizzando una purificazione di affinità a due fasi molto rigorosa (2). Successivamente, gli RNA reticolati catturati sono stati parzialmente digeriti con RNasi, radiomarcati all'estremità 5' e adattatori sono stati legati su di essi (3). Gli adattatori da 5' contenevano sequenze di codici a barre "in-read" uniche in modo che i singoli campioni potessero essere separati bioinformaticamente dopo il sequenziamento. I complessi RBP-RNA sono stati poi eluiti, raggruppati e precipitati insieme (4), risolti mediante SDS-PAGE e visualizzati attraverso l'autoradiografia (5). Successivamente, una singola fetta di gel contenente il segnale radioattivo appena sopra la banda principale, illustrata con una scatola rossa tratteggiata nell'immagine dell'autoradiografia, è stata tagliata dal gel (5). Le fette di gel sono state trattate con proteasi K e l'RNA è stato successivamente estratto (6), convertito in cDNA e amplificato mediante PCR (7). La fase PCR ha introdotto codici a barre aggiuntivi (blocco giallo introdotto da P7 oligo) in modo che molte librerie potessero essere multiplexate in una singola corsia. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 2: Reticolazione e filtrazione sotto vuoto. (A) Il reticolante. La sospensione cellulare viene versata in un imbuto situato in alto a destra della macchina (vedere anche la Figura 3A per un primo piano) e tenuta in un sacchetto trasparente ai raggi UV situato nel vassoio centrale. Questo sacchetto è affiancato da due persiane che rimangono chiuse fino a quando l'utente non istruisce la macchina per avviare la fase di irradiazione. Le celle vengono irradiate con luce UV dai vassoi sia sopra che sotto. La macchina viene fornita con lampade UV da 254 e 365 nm, quest'ultima applicabile per esperimenti PAR-CLIP. La macchina viene azionata attraverso un pannello touchscreen situato in alto a destra che consente di controllare il dosaggio UV o il tempo di esposizione. (B) Dopo la reticolazione, le cellule vengono drenate dal lato sinistro della macchina. Le sospensioni cellulari vengono recuperate sotto vuoto e scaricate in un pallone di vetro dove possono essere successivamente versate in un dispositivo di filtrazione sotto vuoto per la raccolta. (C) Dispositivi di filtrazione sotto vuoto. Questi vengono aperti e chiusi tramite una clip e un filtro viene inserito tra di esso. Quattro dispositivi di filtrazione sono stati utilizzati in parallelo per serie temporali molto brevi per non perdere tempo a causa della sostituzione dei filtri. D) Dopo la filtrazione, il supernatante del mezzo è stato aspirato in palloni per il successivo smaltimento. Le valvole sono state installate sotto i dispositivi di filtrazione del vuoto per mantenere il vuoto nel sistema quando il filtro viene rimosso. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 3: Cross-linking di cellule sospese vs. adiacenti. (A) Il reticolante con la camera Vari-X-linker per le celle di sospensione. La coltura cellulare viene versata nell'ingresso del campione (imbuto) situato in alto a destra del vassoio. (B) Vassoio che può contenere piastre di Petri di plastica o quarzo per reticolare celle aderenti o piccoli volumi di celle di sospensione. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 4: Preparazione della libreria. (A) Esempio di autoradiogramma da un esperimento Nrd1-HTP χCRAC. Il segnale forte e concentrato rappresenta la proteina reticolata a RNA molto corti, mentre lo striscio sopra rappresenta la proteina reticolata a RNA di lunghezza sufficiente per il sequenziamento. (B) Lo striscio è stato asportato come mostrato in un autoradiogramma prelevato dopo l'escissione del gel. (C) Una qPCR rappresentativa da una libreria di cDNA χCRAC. In questo esempio, l'amplificazione massima del cDNA è stata raggiunta a 16 cicli. Pertanto, sono stati utilizzati 16 cicli per l'amplificazione finale. La barra di errore rappresenta la deviazione standard di tre repliche tecniche qPCR. (D) Esempio di un'immagine di fosforo da una libreria di cDNA su un gel TBE al 6%. (E) analisi della lunghezza e della qualità del cDNA da un'elettroforesi capillare basata su chip. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 5: Test ad alta RNasi Esperimento iCLIP per testare la reticolazione in cellule di mammifero. Sono mostrati gli autoradiogrammi degli esperimenti GFP-RBM7 iCLIP che hanno testato l'efficienza del recupero di RNP attraverso varie energie di reticolazione. Le immunoprecipitazioni sono state eseguite utilizzando anticorpi anti-GFP accoppiati a sfere magnetiche su cellule reticolate che esprimevano stabilmente GFP-RBM7. Gli immunoprecipitati sono stati incubati con alte concentrazioni di RNasi I al fine di tagliare gli RNA associati a lunghezze corte e uniformi. Gli RNP sono stati visualizzati mediante marcatura 32P e SDS-PAGE e migrano come una banda definita, vicino alla migrazione della proteina non reticolata. La quantificazione indica i risultati delle analisi densitometriche del segnale RBM7-RNA radiomarcato normalizzato al segnale western blot anti-GFP. (A) Cross-linking time-course del cross-linker UVP comunemente usato rispetto al nostro cross-linker (Vari-X-linker; VxL). (B) Reticolazione del nostro reticolante su articoli di coltura al quarzo (a sinistra) e plastica (a destra). Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 6: Esempi di grafici del browser del genoma che mostrano la potenza di χCRAC per mostrare il legame differenziale e temporale di Nrd1 ai suoi obiettivi. Ogni riquadro mostra grafici per singole regioni genomiche. Le frecce indicano su quale filamento sono codificati i geni (freccia rivolta a sinistra = filo meno; freccia rivolta a destra = filo più). I punti temporali (min) sono indicati da t0, t1, t2, ecc. sugli assi y di ogni sottotrama. Vengono mostrati i numeri romani che indicano i cromosomi e le coordinate. (A) In caso di deprivazione di glucosio, Nrd1 lega due trasportatori del glucosio ad alta affinità, HXT6 e HXT7, che sono entrambi sovraregolati in questa condizione. (B) Si osserva che Nrd1 si lega a Imd3, un bersaglio già validato di Nab344, con intensità riducente in seguito alla carenza di glucosio. (C) Il legame Nrd1 di Tye7 presenta una natura dinamica e transitoria, diminuendo dopo la fame di glucosio al minimo dopo 8 minuti di stress. Tuttavia, il legame ritorna successivamente ai livelli basali dopo 14 minuti. Le letture sono state normalizzate a "letture per milione" (RPM; asse y). Le caselle grigie indicano regioni che codificano RNA non codificanti. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Discussione
Il metodo χCRAC, combinato con i nuovi dispositivi di reticolazione e di prelievo cellulare, ha un grande potenziale perché è applicabile a una vasta gamma di organismi modello e quindi dovrebbe essere di interesse generale per il campo dell'RNA. Ci sono molte aree in cui χCRAC può essere utilizzato. Ad esempio, il metodo potrebbe essere utilizzato per misurare l'assemblaggio gerarchico di proteine in grandi complessi macromolecolari, come lo spliceosoma e il ribosoma, che spesso comporta interazioni dinamiche tra proteine e molecole di RNA. Ora lo usiamo anche abitualmente per monitorare le interazioni tra i fattori di decadimento dell'RNA e i loro substrati quando le cellule sono sottoposte a diversi tipi di stress. Questo ci permette di determinare in quale fase della risposta adattiva questi fattori sono più attivi, a quali substrati si legano e quanto sono dinamiche queste interazioni. Tali dati dovrebbero consentire ai ricercatori di determinare il contributo relativo di ciascun fattore nell'adattamento ai cambiamenti ambientali.
χCRAC utilizza tag di purificazione a doppia affinità (HTF o HTP) per purificare la proteina in condizioni altamente rigorose e denaturanti. Ciò garantisce che l'RNA copurificato sia altamente arricchito per gli RNA che sono stati covalentemente reticolati alla proteina di interesse. Tuttavia, fare affidamento sui tag di affinità presenta degli svantaggi. Ad esempio, il tag potrebbe interferire con la funzione proteica, che potrebbe dare una lettura distorta del suo interattoma legante l'RNA. Inoltre, per alcuni organismi modello potrebbe non essere sempre possibile utilizzare i tag perché gli strumenti genetici per integrare frammenti di DNA nel genoma o per trasformare i plasmidi di espressione non sono ancora disponibili. Tuttavia, è semplice modificare alcune parti del protocollo χCRAC per renderlo compatibile con i protocolli basati su CLIP che si basano su anticorpi per la purificazione del RBP. In effetti, questo studio ha dimostrato che è possibile combinare purificazioni basate su iCLIP con il nostro reticolante. Siamo ora in procinto di sviluppare protocolli CLIP per studiare l'associazione temporale delle proteine leganti l'RNA umano con i trascritti nascenti dell'RNA.
Quando si esegue χCRAC su una nuova proteina, l'esposizione ai raggi UV deve essere ottimizzata per indurre la massima reticolazione. Questo è importante perché alte esposizioni ai raggi UV possono ridurre il recupero di RNA durante la fase di purificazione. Le cellule che esprimono il RBP ricombinante sono state esposte a varie dosi UV, 100 mJ / cm 2, 250 mJ / cm 2, 500 mJ / cm 2 e 1 J / cm 2. Gli RNP sono stati quindi catturati e gli RNA sono stati frammentati e radiomarcati. Successivamente, gli RNP sono stati risolti mediante SDS-PAGE ed è stato preso un autoradiogramma per dedurre quale esposizione ha dato il segnale più intenso (cioè il cross-linking massimo).
Una volta ottimizzate le condizioni sperimentali, si raccomandano diversi esperimenti di controllo quando si esegue χCRAC. In primo luogo, un campione irradiato UV e senza tag può essere utilizzato per monitorare il legame di fondo alle sfere di purificazione. In secondo luogo, quando si applica χCRAC durante un esperimento di spostamento, una seconda serie temporale in cui le cellule vengono spostate indietro nel mezzo originale consente di indagare se la filtrazione delle cellule stesse induce cambiamenti nei livelli di RNA o nelle interazioni proteina-RNA.
Come accennato nell'introduzione, numerosi articoli pubblicati di recente suggeriscono una serie di ottimizzazioni del protocollo CLIP. Ciò include l'uso di adattatori marcati con fluorescenza per rilevare il complesso proteina-RNA attraverso la scansione a infrarossi10, nonché ottimizzazioni per varie fasi di purificazione degli acidi nucleici e selezione delle dimensioni mostrate per aumentare la complessità delle librerie risultanti12,45. Attualmente stiamo implementando alcuni di questi miglioramenti per perfezionare ulteriormente il protocollo χCRAC. Il protocollo qui presentato contiene già una serie di miglioramenti ai protocolli CRAC e χCRAC originali che aumentano la complessità dei dati. Ad esempio, in precedenza, dopo aver risolto i complessi proteina-RNA radioattivi reticolati sui gel SDS-PAGE, sono stati trasferiti su una membrana di nitrocellulosa e l'RNA reticolato è stato isolato dalla macchia. Tuttavia, il trasferimento dell'RNP e la successiva estrazione dell'RNA possono essere molto inefficienti, in particolare quando si tratta di grandi RBP come le subunità della RNA polimerasi. Ciò può comportare una significativa riduzione del recupero dell'RNA reticolato. Nel protocollo attuale, l'RNA reticolato viene estratto direttamente dalle fette di gel SDS-PAGE, come illustrato nella Figura 1. Ciò ha aumentato il recupero di RNA reticolati. Inoltre, dopo l'amplificazione PCR dei cDNA, il prodotto è stato originariamente risolto su gel di agarosio al 3%, a bassa temperatura di fusione, e quindi sono stati estratti prodotti PCR da 175-300 bp dal gel. Tuttavia, questi gel possono essere facilmente sovraccaricati, con conseguente scarsa separazione del DNA. La sostituzione dei gel di agarosio con gel prefabbricati TBE ha portato a una separazione dimensionale più coerente e a un migliore recupero dei prodotti PCR.
Divulgazioni
A. Langford e W. Worboys sono affiliati con UVO3, una società commerciale. Non hanno avuto alcun ruolo nella progettazione dello studio, nella raccolta e nell'interpretazione dei dati o nella decisione di presentare il lavoro per la pubblicazione.
Riconoscimenti
Questo lavoro è stato sostenuto da sovvenzioni del Wellcome Trust (091549 a S.G e 109093 / Z / 15 / A a SM), il Wellcome Trust Centre for Cell Biology core grant (092076) e Medical Research Council Non-Clinical Senior Research Fellowship (MR / R008205 / 1 a S.G.), l'Organizzazione europea di biologia molecolare nell'ambito di una borsa di studio post-dottorato a lungo termine (ALTF 1070-2017 a R.A.C), e l'Independent Research Fund Denmark (T.H.J).
Materiali
| Name | Company | Catalog Number | Comments |
| 1,4-dithioreitol | Merck | 10708984001 | Buffer component in mammalian cell lysis |
| 1.5 mL tubes | Eppendorf | 0030 120.086 | General reaction tube |
| 2 mL tubes | Eppendorf | 0030 123.344 | For holding columns and collection of waste |
| 32P-yATP | Perkin Elmer | NEG502Z-250 | For radiolabelling the 5' end of the RNA |
| 4-12% Bis-Tris gel | Invitrogen | NP0321BOX | SDS-PAGE gel |
| 4X loading buffer | Novex | NP0008 | Protein loading dye concentrate |
| 50 bp ladder | New England Biolabs | N3236 | Reference ladder for excising region of interest from the amplified cDNA library |
| 50% PEG | NEB | B100045 | For the L5 linker ligation |
| 6% TBE gel | Invitrogen | EC6265BOX | For separation and purification of the cDNA library |
| Acetone | ACROS Organics | 423245000 | Washing of TCA-precipitated proteins |
| anti-FLAG beads | Sigma Aldrich | M8823-1ML | For purifcation of FLAG-tagged RBPs |
| ATP (100 mM) | Thermo Fisher Scientific | R0441 | For ligation of the L5 linker onto the 5' end of captured RNAs |
| Beta-mercaptoethanol | Sigma Aldrich | M3148-100ML | Buffer component |
| Biomax MS intensifying screen | Sigma Aldrich | Z363162-1EA | For intensifying the autoradiogram signal |
| Chloroform | Thermo Fisher Scientific | 1010219 | For phenol-chloroform extraction following RNA purification |
| cOmplete EDTA-free protease inhibitor cocktail | Roche | 11873580001 | For inhibition of cellular proteases after lysis |
| Complete supplement mixture -TRP | Formedium | DCS0149 | For preparation of synthetic defined medium |
| Costar Spin-X 0.22 µm filters | Sigma Aldrich | CLS8160 | For isolating the excised cDNAs following gel extraction |
| DNase RQ1 | Promega | M6101 | For DNA digest following cell lysis |
| dNTPs (10 mM) | Sigma Aldrich | 4638956001 | For reverse transcription and PCR |
| Ethanol | Thermo Fisher Scientific | 10041814 | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Ethylenediaminetetraacetic acid | Invitrogen | AM9261 | For protease K buffer |
| Exonuclease I | New England Biolabs | M0293 | For degradation of primers following PCR |
| Glass microfiber filters | Whatman | 1823-010 | For isolating the excised cDNAs following gel extraction |
| Glucose | Formedium | GLU03 | For preparation of glucose-containing, synthetic defined medium |
| Glycogen (20 mg/mL) | Roche | 10901393001 | Precipitation of proteins, RNA and DNA |
| GST-TEV | Homemade | Construct and purification protocol is available upon request | |
| Guanidium hydrochloroide | Thermo Fisher Scientific | 10071503 | Required for pulldown denaturing conditions and washing buffer |
| IgG beads | GE Healthcare | 17-0969-01 | For purification of protein A-tagged RBPs |
| Imidazole | Sigma Aldrich | I2399-100G | For elution of captured proteins from Nickel beads |
| Isoamyl alcohol | Thermo Fisher Scientific | A393-500 | For phenol-chloroform extraction following RNA purification |
| Luna Universal One-Step RT-qPCR | NEB | E3005S | For qPCR of the cDNA in order to calculate required number of PCR cycles |
| Magnesium chloride | Fluka Analytical | 63020-1L | For PNK buffer |
| Membrane filters | Millipore | AAWP09000 for yeast or HAWP09000 for bacteria | For vacuum filtration of cells |
| Micro bio-spin columns | Biorad | 732-6204 | For collecting eluate after gel extraction |
| Ni-NTA beads | Qiagen | 30210 | For secondary protein capture |
| NP-40 | Sigma Aldrich | I8896-100ML | Buffer component |
| Pfu polymerase | Promega | M7741 | For amplification of the cDNA library |
| Phenol | Sigma Aldrich | P4682-400ML | For phenol-chloroform extraction following RNA purification |
| Pierce spin columns | Thermo Fisher Scientific | 69725 | For on-column enzymatic reactions |
| Protease K | Roche | 3115887001 | For degradation of the RBP following gel extraction |
| Quartz Petri dish | UVO3 | N/A | For cross-linking of adherent cells. Available from https://www.vari-x-link.com for 400 GBP |
| Radiography films | Amersham | 28906843 | For autoradiography visualisation |
| RNAClean XP beads | Beckmann | A63987 | SPRI beads for clean up of RNAs and cDNAs |
| RNase H | New England Biolabs | M0297 | For degradation of RNAs following reverse transcription |
| RNase-It | Agilent | 400720 | For RNA digestion |
| rRNasin | Promega | N2511 | For inhibition of any contaminating RNases during enzymatic reaction |
| Sodium acetate | Sigma Aldrich | S2889-1KG | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Sodium chloride | Thermo Fisher Scientific | 7647-14-5 | Buffer component |
| Sodium deoxycholate | Sigma Aldrich | D6750-100G | Buffer component in mammalian cell lysis |
| Sodium dodecylsulfate | Sigma Aldrich | L3771-1KG | For protease K buffer |
| SUPERase-In | Invitrogen | AM2694 | For inhibition of cellular RNases after lysis |
| SuperScript IV | Thermo Fisher Scientific | 18090010 | For reverse transcription |
| T4 PNK | New England Biolabs | M0201 | For radiolabelling the 5' end of the RNA |
| T4 RNA ligase 1 | New England Biolabs | M0204 | For ligation of the L5 adaptor onto the RNA 5' end |
| T4 RNase ligase 2, truncated K222Q | NEB | M0351S | For ligation of the App_PE linker onto the 3' end of captured RNAs |
| TBE buffer (10X) | Invitrogen | 15581-028 | For running TBE gels |
| TEV protease | Homemade | For eluting captured proteins following FLAG capture | |
| Thermosensitive alkaline phosphatase | Promega | M9910 | For 5' and 3' dephosphorylation of RNAs |
| Trichloroacetic acid (100%) | Sigma Aldrich | T0699-100ML | For precipitation of RBP-RNA complexes |
| Tris hydrochloride | Invitrogen | 15504-020 | Buffer component |
| Triton X-100 | Sigma Aldrich | T8787-100ML | Buffer component in mammalian cell lysis |
| Vari Filter | UVO3 | N/A | Device for vacuum harvesting cells. Available from https://www.vari-x-link.com for 100 GBP |
| Vari-X-Linker | UVO3 | N/A | Cross-linker for cross-linking cells. Available from https://www.vari-x-link.com for 16,000 GBP |
| Yeast nitrogen base | Formedium | CYN0410 | For preparation of synthetic defined medium |
| Zirconia beads | Thistle | 11079105Z for yeast or 11079101Z for bacteria | For cell lysis via bead beating |
Riferimenti
- Ule, J., et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 302 (5648), 1212-1215 (2003).
- Granneman, S., Kudla, G., Petfalski, E., Tollervey, D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proceedings of the National Academy of Sciences. 106 (24), 9613-9618 (2009).
- Licatalosi, D. D., et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 456 (7221), 464-469 (2008).
- König, J., et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nature Structural & Molecular Biology. 17 (7), 909-915 (2010).
- Hafner, M., et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell. 141 (1), 129-141 (2010).
- Aktaş, T., et al. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature. 544 (7648), 115-119 (2017).
- Huppertz, I., et al. iCLIP: Protein–RNA interactions at nucleotide resolution. Methods. 65 (3), 274-287 (2014).
- Li, X., et al. Comprehensive in vivo RNA-binding site analyses reveal a role of Prp8 in spliceosomal assembly. Nucleic Acids Research. 41 (6), 3805-3818 (2013).
- Rosenberg, M., et al. Denaturing CLIP, dCLIP, Pipeline Identifies Discrete RNA Footprints on Chromatin-Associated Proteins and Reveals that CBX7 Targets 3′ UTRs to Regulate mRNA Expression. Cell Systems. 5 (4), 368-385 (2017).
- Zarnegar, B. J., et al. irCLIP platform for efficient characterization of protein–RNA interactions. Nature Methods. 13 (6), 489-492 (2016).
- Kargapolova, Y., Levin, M., Lackner, K., Danckwardt, S. sCLIP—an integrated platform to study RNA–protein interactomes in biomedical research: identification of CSTF2tau in alternative processing of small nuclear RNAs. Nucleic Acids Research. 45 (10), 6074-6086 (2017).
- Van Nostrand, E. L., et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nature Methods. 13 (6), 508-514 (2016).
- Flynn, R. A., et al. Dissecting noncoding and pathogen RNA–protein interactomes. RNA. 21 (1), 135-143 (2015).
- Brugiolo, M., Botti, V., Liu, N., Müller-McNicoll, M., Neugebauer, K. M. Fractionation iCLIP detects persistent SR protein binding to conserved, retained introns in chromatin, nucleoplasm and cytoplasm. Nucleic Acids Research. 45 (18), 10452-10465 (2017).
- Sanford, J. R., et al. Identification of Nuclear and Cytoplasmic mRNA Targets for the Shuttling Protein SF2/ASF. PLOS ONE. 3 (10), e3369 (2008).
- Garzia, A., Meyer, C., Morozov, P., Sajek, M., Tuschl, T. Optimization of PAR-CLIP for transcriptome-wide identification of binding sites of RNA-binding proteins. Methods. 118-119, 24-40 (2017).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Research. 22 (10), 2031-2042 (2012).
- Chen, K., et al. High-Resolution N6-Methyladenosine (m6A) Map Using Photo-Crosslinking-Assisted m6A Sequencing. Angewandte Chemie International Edition. 54 (5), 1587-1590 (2015).
- Ke, S., et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes & Development. 29 (19), 2037-2053 (2015).
- Linder, B., et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature Methods. 12 (8), 767-772 (2015).
- Kudla, G., Granneman, S., Hahn, D., Beggs, J. D., Tollervey, D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proceedings of the National Academy of Sciences. 108 (24), 10010-10015 (2011).
- Sugimoto, Y., et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature. 519 (7544), 491-494 (2015).
- Hwang, H. W., et al. cTag-PAPERCLIP Reveals Alternative Polyadenylation Promotes Cell-Type Specific Protein Diversity and Shifts Araf Isoforms with Microglia Activation. Neuron. 95 (6), 1334-1349 (2017).
- Hwang, H. W., et al. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Reports. 15 (2), 423-435 (2016).
- Lee, F. C. Y., Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Molecular Cell. 69 (3), 354-369 (2018).
- Beckmann, B. M. RNA interactome capture in yeast. Methods. 118-119, 82-92 (2017).
- Granneman, S., Petfalski, E., Tollervey, D. A cluster of ribosome synthesis factors regulate pre-rRNA folding and 5.8S rRNA maturation by the Rat1 exonuclease. The EMBO Journal. 30 (19), 4006-4019 (2011).
- Schaughency, P., Merran, J., Corden, J. L. Genome-Wide Mapping of Yeast RNA Polymerase II Termination. PLOS Genetics. 10 (10), e1004632 (2014).
- Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S., Cohen, S. N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proceedings of the National Academy of Sciences. 99 (15), 9697-9702 (2002).
- Kresnowati, M. T. A. P., et al. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Molecular Systems Biology. 2, 49 (2006).
- Marguerat, S., Lawler, K., Brazma, A., Bähler, J. Contributions of transcription and mRNA decay to gene expression dynamics of fission yeast in response to oxidative stress. RNA Biology. 11 (6), 702-714 (2014).
- van Nues, R., et al. Kinetic CRAC uncovers a role for Nab3 in determining gene expression profiles during stress. Nature Communications. 8 (1), 12 (2017).
- Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M., Rosenow, C. Global RNA Half-Life Analysis in Escherichia coli Reveals Positional Patterns of Transcript Degradation. Genome Research. 13 (2), 216-223 (2003).
- Tudek, A., Candelli, T., Libri, D. Non-coding transcription by RNA polymerase II in yeast: Hasard or nécessité?. Biochimie. 117, 28-36 (2015).
- Lingaraju, M., et al. The MTR4 helicase recruits nuclear adaptors of the human RNA exosome using distinct arch-interacting motifs. Nature Communications. 10 (1), 1-11 (2019).
- Lubas, M., et al. Interaction Profiling Identifies the Human Nuclear Exosome Targeting Complex. Molecular Cell. 43 (4), 624-637 (2011).
- Conrad, N. K., et al. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics. 154 (2), 557-571 (2000).
- Darby, M. M., Serebreni, L., Pan, X., Boeke, J. D., Corden, J. L. The Saccharomyces cerevisiae Nrd1-Nab3 Transcription Termination Pathway Acts in Opposition to Ras Signaling and Mediates Response to Nutrient Depletion. Molecular and Cellular Biology. 32 (10), 1762-1775 (2012).
- Webb, S., Hector, R. D., Kudla, G., Granneman, S. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biology. 15 (1), R8 (2014).
- Jensen, T. H., Jacquier, A., Libri, D. Dealing with Pervasive Transcription. Molecular Cell. 52 (4), 473-484 (2013).
- van Dijk, E. L., et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 475 (7354), 114-117 (2011).
- Thiebaut, M., et al. Futile Cycle of Transcription Initiation and Termination Modulates the Response to Nucleotide Shortage in S. cerevisiae. Molecular Cell. 31 (5), 671-682 (2008).
- Merran, J., Corden, J. L. Yeast RNA-Binding Protein Nab3 Regulates Genes Involved in Nitrogen Metabolism. Molecular and Cellular Biology. 37 (18), e00154-e00117 (2017).
- Bresson, S., Tuck, A., Staneva, D., Tollervey, D. Nuclear RNA Decay Pathways Aid Rapid Remodeling of Gene Expression in Yeast. Molecular Cell. 65 (5), 787-800 (2017).
- Buchbender, A., et al. Improved library preparation with the new iCLIP2 protocol. Methods. , (2019).
Erratum
Formal Correction: Erratum: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC
Posted by JoVE Editors on 2/17/2023. Citeable Link.
An erratum was issued for: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC. The Authors section was updated from:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Synthetic and Systems Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
to:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Mehak Chauhan1
Niki Christopoulou1
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Engineering Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati