
Method Article
Surveillance de la dynamique d’interaction protéine-ARN in vivo à haute résolution temporelle à l’aide de χCRAC
Dans cet article
Erratum Notice
Résumé
La réticulation cinétique et l’analyse de l’ADNc est une méthode qui permet d’étudier la dynamique des interactions protéine-ARN dans les cellules vivantes à haute résolution temporelle. Ici, le protocole est décrit en détail, y compris la croissance des cellules de levure, la réticulation UV, la récolte, la purification des protéines et les étapes de préparation de la bibliothèque de séquençage de nouvelle génération.
Résumé
L’interaction entre les protéines de liaison à l’ARN (RBP) et leurs substrats d’ARN présente fluidité et complexité. Au cours de sa durée de vie, un seul ARN peut être lié par de nombreux RBP différents qui réguleront sa production, sa stabilité, son activité et sa dégradation. En tant que tel, beaucoup a été fait pour comprendre la dynamique qui existe entre ces deux types de molécules. Une percée particulièrement importante a été réalisée avec l’émergencede l’encrage et de larécipitation (CLIP). Cette technique a permis d’étudier rigoureusement quels ARN sont liés par une RBP particulière. En bref, la protéine d’intérêt est réticulée par UV à ses substrats d’ARN in vivo, purifiée dans des conditions très strictes, puis les ARN réticulés par covalence à la protéine sont convertis en banques d’ADNc et séquencés. Depuis sa conception, de nombreuses techniques dérivées ont été développées afin de rendre CLIP adapté à des domaines d’étude particuliers. Cependant, la réticulation à l’aide de la lumière ultraviolette est notoirement inefficace. Il en résulte des temps d’exposition prolongés qui rendent impossible l’étude temporelle des interactions RBP-ARN. Pour surmonter ce problème, nous avons récemment conçu et construit des dispositifs d’irradiation UV et de collecte de cellules bien améliorés. À l’aide de ces nouveaux outils, nous avons développé un protocole d’analyses à résolution temporelle des interactions RBP-ARN dans les cellules vivantes à haute résolution temporelle : Kinetic CRoss-linking et Analysis of cDNA (χCRAC). Nous avons récemment utilisé cette technique pour étudier le rôle des RBP de levure dans l’adaptation au stress nutritif. Ce manuscrit donne un aperçu détaillé de la méthode χCRAC et présente les résultats récents obtenus avec le RBP Nrd1.
Introduction
Les ARN s’appuient souvent sur les RBP pour exercer leur fonction, ce qui a suscité un grand intérêt pour la compréhension de la dynamique entre ces molécules. De nombreuses pratiques de récupération des bénéfices durables ont été identifiées dans une grande variété d’organismes. Cependant, il a toujours été notoirement difficile d’étudier les interactions RBP-ARN in vivo. Une percée majeure dans l’étude de ces interactions est venue avec l’émergence de CLIP1. Cette méthode utilise l’irradiation ultraviolette (UV, 254 nm) pour induire des liaisons covalentes entre les RBP et leurs ARN directement liés (c.-à-d. réticulation à distance nulle). Par la suite, la RBP d’intérêt est immunopurifiée dans des conditions rigoureuses pour s’assurer que seuls les ARN réticulés de manière covalente aux protéines sont identifiés. Les ARN liés sont ensuite partiellement digérés avec des RNases et ensuite convertis en banques d’ADNc pour le séquençage. La rigueur élevée de la purification est importante car elle augmente considérablement la spécificité de la récupération des protéines et de l’ARN, qui est également renforcée par la purification SDS-PAGE du complexe ribonucléoprotéine réticulée (RNP). CLIP et les méthodes connexes fournissent également un aperçu de la résolution nucléotidique dans le site de liaison à la protéine, car lors de la préparation de la bibliothèque de séquençage, les acides aminés qui ont réticulé l’ARN terminent fréquemment la transcriptase inverse ou provoquent l’introduction de mutations par l’enzyme sur ce site 1,2,3.
Depuis son introduction, le protocole CLIP original a produit une variété remarquable de méthodologies dérivées. Une percée particulièrement importante est venue avec le développement de HITS-CLIP (ou CLIP-seq), qui fusionne le séquençage à haut débit avec l’approche CLIP3. Cela a depuis été adopté par toutes les méthodologies basées sur CLIP. iCLIP a introduit des améliorations dans les techniques de rognage et de ligature de l’adaptateur médiées par RNase qui facilitent une cartographie plus précise des sites de liaison RBP4. PAR-CLIP a combiné le marquage 4thio-uridine/uracile avec la réticulation à 365 nm, ce qui a permis de cartographier les sites de réticulation en analysant les substitutions T-C5. CRAC, urea-iCLIP, dCLIP et uvCLAP ont introduit des conditions de dénaturation et des étapes de purification à double affinité qui réduisent davantage la liaison de fond à la résine d’affinité et augmentent encore la spécificité de la capture des protéines 2,6,7,8,9. De plus, CRAC, uvCLAP et dCLIP ont introduit le marquage de la RBP d’intérêt avec une étiquette d’affinité, éliminant ainsi la nécessité de générer des anticorps spécifiques.
Plusieurs optimisations ont également été apportées pour accélérer la méthodologie CLIP. Le protocole CLIP original utilisait le radiomarquage des ARN capturés afin de visualiser les complexes RBP-ARN après SDS-PAGE. Cependant, l’utilisation de la radioactivité peut être problématique pour les laboratoires qui ne sont pas mis en place pour de tels travaux. irCLIP intègre un adaptateur couplé aux fluorophores qui facilite la visualisation par imagerie infrarouge10 et sCLIP utilise la biotinylation des ARN capturés afin de les visualiser à travers HRP11 conjugué à la streptavidine. De plus, eCLIP renonce complètement au marquage de l’ARN; Au lieu de cela, la protéine est excisée uniquement en fonction de sa taille connue12. La purification à base de streptavidine a également été utilisée pour accélérer le processus de préparation de la bibliothèque dans FAST-iCLIP, où un adaptateur 3' biotinylé est ligaturé sur les ARN et utilisé pour permettre la purification après transcription inverse et circularisation13. Des améliorations supplémentaires apportées au protocole iCLIP ont également considérablement accru la complexité des bibliothèques4.
Enfin, CLIP a été modifié pour permettre la capture de RBP à partir de différents sous-compartiments cellulaires 14,15, pour visualiser les ARN nouvellement transcrits en utilisant l’induction pulsée de ribonucléosides photoactivables 5,16,17, pour capturer les ARN méthylés18,19,20, pour examiner les interactions ARN-ARN 21,22, et pour cartographier les extrémités 3' 23,24.
Malgré les grandes contributions des techniques basées sur CLIP pour nous aider à comprendre les interactions entre les RBP et les ARN, elles ont été limitées par l’inefficacité de la réticulation UV. Bien que les cellules de culture cultivées dans une monocouche soient généralement relativement faciles à réticuler, cela est beaucoup plus difficile dans les tissus ou les cellules en solution. Les tissus peuvent nécessiter plusieurs cycles d’exposition aux UV pour pénétrer dans les couches cellulaires requises, tandis que les cellules microbiennes sont souvent cultivées dans des milieux riches contenant des composés aromatiques absorbant les UV25. En effet, des temps d’irradiation UV allant jusqu’à 30 min ont été utilisés pour générer une réticulation suffisante entre les RBP et leurs ARN liés pour de tels échantillons26,27,28. Cette exposition prolongée aux UV induit des réponses au stress dans la cellule, telles que des dommages à l’ADN induits par les UV, qui peuvent contaminer les données finales dans certaines applications.
La majorité des études CLIP se sont concentrées sur la génération d'« instantanés » uniques d’interactions protéine-ARN spécifiques dans une cellule. Cependant, les interactions protéine-ARN sont intrinsèquement dynamiques, en particulier lorsque les cellules sont sujettes à des changements dans leur environnement. Cela peut inclure une réduction soudaine de la disponibilité des nutriments essentiels ou des changements rapides de température. Ainsi, pour vraiment comprendre le rôle d’une RBP en période de stress, il est préférable d’effectuer des analyses résolues dans le temps, car elles peuvent saisir tout le spectre des cibles RBP pendant le stress et déterminer à quel stade de la réponse au stress la RBP choisie est active. En particulier, des études sur la levure ont montré que les premières minutes d’adaptation sont absolument cruciales pour la survie et que les demi-vies de l’ARN chez les bactéries peuvent varier de quelques minutes à quelques secondes 29,30,31,32,33. Par conséquent, de telles analyses résolues dans le temps devraient idéalement être effectuées à une résolution temporelle élevée. Cependant, les longs temps de réticulation rendent l’étude des réponses adaptatives à un stade précoce particulièrement difficile.
Afin de surmonter ces problèmes, nous avons récemment développé une méthode améliorée capable de réticuler et de récolter des cellules sur des échelles de temps d’une minute. Notre méthode χCRAC permet une mesure quantitative des changements dynamiques dans les interactions RBP-ARN à une résolution inédite. Le développement d’un nouveau dispositif d’irradiation UV32 qui réduit le temps de réticulation requis dans les levures et les bactéries en solution environ 10 fois, congelant efficacement les interactions RBP-ARN instantanément, a joué un rôle crucial dans cette méthode. De plus, afin de récolter rapidement les cellules après irradiation UV, nous avons développé un dispositif de filtration sous vide capable de récolter des levures à croissance exponentielle dans une culture de 0,5 L en environ 30 s32. Ces innovations technologiques permettent l’étude de la dynamique RBP-ARN à une résolution à l’échelle infime. De plus, nous avons également introduit plusieurs optimisations au protocole originalCRAC 2 afin d’augmenter sa praticité.
En utilisant χCRAC, nous avons récemment étudié le cibleome d’une RBP nucléaire de levure, Nab3, en réponse à la privation de glucose. Chez Saccharomyces cerevisiae, Nab3 peut former un complexe avec Nrd1, un RBP et l’hélicase à ARN Sen1 pour former le complexe NNS. La liaison de NNS à l’ARN polymérase et au transcrit naissant peut déclencher la terminaison transcriptionnelle34. Ce complexe est principalement impliqué dans l’élimination des transcrits d’ARN cryptiques non codants, mais il a également été démontré qu’il régule l’expression des gènes codant pour les protéines. L’étude a montré un ciblage différentiel de Nab3 par rapport aux transcriptions non codantes et codantes après seulement une minute de stress32. Nous avons démontré que la terminaison co-transcriptionnelle par Nab3 entraîne une expression très transitoire, semblable à une impulsion, des gènes rétrotransposons, ce qui aurait été difficile à détecter en utilisant des approches traditionnelles basées sur CLIP. De plus, les temps d’irradiation UV courts dans notre réticulation UV ont également augmenté de manière significative la récupération des ARN non codants à vie courte32. χCRAC sera probablement un outil crucial pour élucider non seulement comment les pratiques commerciales restrictives façonnent la réponse au stress sur des échelles de temps immédiates, mais aussi leurs rôles changeants tout au long du cycle de vie d’une intervention. Ce manuscrit donne un aperçu détaillé de toutes les étapes du protocole χCRAC. À des fins d’illustration, la méthode a été utilisée pour étudier la protéine de levure Nrd1, qui est impliquée dans la terminaison transcriptionnelle et la désintégration de l’ARN35,36, et son cibleome ARN en réponse à la privation de glucose sur une multitude de points temporels. Enfin, nous démontrons également que notre unité d’irradiation UV peut rapidement réticuler les RBP à l’ARN dans les cellules HeLa, ce qui permet également d’effectuer des analyses à haute résolution résolues dans le temps dans les cellules adhérentes.
Protocole
| TN150 |
| 50 mM Tris pH 7,8 |
| 150 mM NaCl |
| 0,1 % NP-40 |
| Inhibiteur de la protéase 1X |
| TN1000 |
| 50 mM Tris pH 7,8 |
| 1M NaCl |
| 0,1 % NP-40 |
| NP-PNK |
| 50 mM Tris-HCl pH 7,8 |
| 10 mM MgCl2 |
| 0,1 % NP-40 |
| 5 mM de bêta-mercaptoéthanol |
| 5 x PNK |
| 250 mM Tris-HCl pH 7,8 |
| 50 mM MgCl2 |
| 50 mM de bêta-mercaptoéthanol |
| BM I |
| 50 mM Tris-HCl pH 7,8 |
| 300 mM NaCl |
| 10 mM d’imidazole |
| 6M guanidine-HCl |
| 0,1 % NP-40 |
| 5 mM de bêta-mercaptoéthanol |
| WB II |
| 50 mM Tris-HCl pH 7,8 |
| 50 mM NaCl |
| 10 mM d’imidazole |
| 0,1 % NP-40 |
| 5 mM de bêta-mercaptoéthanol |
| Tampon d’élution |
| 50 mM Tris pH 7,8 |
| 50 mM NaCl |
| 250 mM d’imidazole |
| 0,1 % NP-40 |
| 5 mM de bêta-mercaptoéthanol |
| Tampon Protéase K |
| 50 mM Tris |
| 0,1 % NP-40 |
| 5 mM β-mercaptoéthanol |
| FDS de 1 % |
| 5 mM d’EDTA |
| 50 mM NaCl2 |
| Tampon de lyse chez les mammifères |
| 50 mM Tris-HCl pH 8 |
| 100 mM NaCl |
| 0,5 % v/v Triton X-100 |
| 0,25 % p/v de Na-désoxycholate |
| 0,1 % p/v FDS |
| 5 mM d’EDTA |
| 1 mM de TNT (ajouté frais) |
| Inhibiteur de la protéase 1X |
Tableau 1: Les tampons nécessaires pour χCRAC et leurs compositions.
1. Réticulation UV et production de lysat
- Micro-organismes en solution
- Inoculer 3,5 L du milieu désiré avec de la levure d’une culture de nuit à une DO de départde 600 de 0,05. Croissance à 30 °C avec agitation continue à 180 tr/min.
- Pendant la croissance, préparez d’autres matériaux nécessaires.
- Préparez un récipient d’azote liquide.
- Préparer 3 L de milieu stressant et chauffer à 30 °C au bain-marie.
- Installez l’appareil filtrant, allumez l’agent de réticulation (figure 2A) et étiquetez les tubes coniques de 50 mL, un pour chaque point temporel.
- Une fois que les cellules atteignent la DO600 souhaitée, versez 500 mL de cellules directement dans le réticulant et irradiez les UV avec 250 mJ d’UV de 254 nm. Reportez-vous à la figure 2A et à la figure 3A pour plus de détails sur l’utilisation de la réticulation.
REMARQUE: L’énergie d’irradiation UV doit être soigneusement optimisée pour chaque protéine d’intérêt. Voir la discussion pour plus de détails. - Après réticulation, filtrer les cellules à l’aide de l’un des dispositifs de filtration sous vide (Figure 2B,C). Enrouler la membrane avec les cellules filtrées, placer dans le tube conique t = 0 (temps zéro) de 50 mL et congeler dans de l’azote liquide.
- Filtrez les cellules restantes sur six filtres différents. Resuspendre les cellules recueillies dans les 3 L de milieu induisant le stress préalablement chauffé en laissant tomber les membranes dans le milieu et en mélangeant vigoureusement avec une stripette pendant 50 s. Après les années 50, préparez-vous à prélever l’échantillon t = 1.
- Après 1 min, réticuler 500 mL de cellules et récolter par filtration comme dans les étapes 1.1.3 à 1.1.4. Répétez l’opération après 2, 4, 8, 14 et 20 minutes, ou à des points de temps différents selon vos besoins.
- Conserver les tubes coniques contenant les cellules à -80 °C. Mettez la solution saline tamponnée au phosphate (PBS) à 4 °C pendant la nuit.
- Le lendemain, prélever chaque tube conique contenant un échantillon réticulé et remettre les cellules en suspension dans 25 mL de PBS froid en agitant vigoureusement.
- Transférer les suspensions cellulaires dans de nouveaux tubes coniques et faire tourner à 4 600 x g, 5 min à 4 °C.
- Versez le PBS, essorez rapidement à nouveau pour recueillir le PBS résiduel, puis décantez le liquide restant avec une pipette.
- Calculez le poids de la pastille dans le tube en le comparant à un tube vide.
- Ajouter deux volumes de pastilles de TN150 glacé, 60 μL de DNase 1 et 10 μL d’inhibiteur de RNase. Incuber sur la glace pendant 30 min.
- Par exemple, pour 400 mg de cellules, ajoutez 800 μL de TN150 glacé.
- L’ajout de la DNase n’est pas essentiel pour la plupart des protéines solubles, mais est très important lors de l’étude des protéines liées à la chromatine telles que l’ARN polymérase. De plus, il réduit la viscosité des lysats bactériens. Il est très important d’utiliser exactement deux volumes de pastilles du tampon de lyse, sinon l’efficacité de la lyse peut diminuer.
- Ajouter trois volumes de granulés (en mL) de billes de zircone à la suspension cellulaire. Pour la levure, utilisez des billes de 0,5 mm de diamètre et pour les bactéries, utilisez 0,1 mm.
- Par exemple, pour 400 mg de cellules, mesurez 1,2 mL de billes de zircone dans un tube de 1,5 mL et ajoutez-les aux cellules remises en suspension dans un tampon de lyse.
- Vortex les suspensions cellulaires pendant 1 min, puis placer sur la glace pendant 1 min. Répétez l’opération pour un total de 5x.
- Ajouter vigoureusement deux volumes de pastilles de tampon TN150 et de vortex pour mélanger.
- Centrifuger la suspension dans le tube conique à 4 600 g pendant 20 min à 4 °C dans une centrifugeuse de paillasse.
- Après centrifugation, prélever un échantillon de 50 μL du surnageant pour une analyse ultérieure par transfert Western afin d’examiner l’expression de la protéine cellulaire entière.
- Transférer les surnageants dans des tubes de 1,5 mL et faire tourner le lysat pendant 20 min à 20 000 x g à 4 °C, dans un microfuge.
- Sinon, si vous utilisez des tubes de 5 mL, centrifuger à 13 000 x g pendant 20 min.
- Après la centrifugation, prélever un échantillon de 50 μL du surnageant pour une analyse ultérieure par transfert Western afin d’examiner l’expression soluble de la protéine.
- Passer à la saisie RBP (section 2).
- Cellules adhérentes en culture
- Ensemencez suffisamment de cellules adhérentes dans une boîte de Petri 24 h avant la réticulation UV afin qu’elles puissent atteindre 80% de confluence le lendemain. Cultiver pendant la nuit dans le milieu désiré dans un incubateur de culture cellulaire à 37 °C, 5% de CO2.
REMARQUE: Si vous utilisez des boîtes de Petri en quartz, il est bénéfique de favoriser l’adhésion cellulaire par le traitement des articles de culture avec de la poly-D-lysine (70 000 à 140 000 poids à poids) et du sérum de veau fœtal (FCS) 2,5 heures avant l’ensemencement. Ajouter suffisamment de poly-D-lysine pour couvrir toute la surface de croissance et incuber à température ambiante (RT) pendant 5 min. Ensuite, la boîte de Petri en quartz doit être rincée abondamment à l’eau et séchée dans l’incubateur de culture cellulaire pendant 2 h ou jusqu’à ce qu’elle soit complètement sèche. Ensuite, ajoutez suffisamment de FCS pour couvrir complètement la surface de croissance et placez-le dans l’incubateur pendant au moins 30 minutes. Le FCS doit être complètement enlevé avant l’ensemencement des cellules. - Une fois que les cellules ont atteint 80% de confluence, retirez le média et lavez avec 15 ml de PBS glacé. Ensuite, retirez complètement tout le liquide restant et passez immédiatement à l’étape suivante.
- Transférer la boîte de Petri dans le plateau pour les cellules adhérentes (Figure 3B) et irradier les UV avec 300 mJ d’UV de 254 nm. Voir la figure 2A et la figure 3B pour plus de détails sur l’utilisation de la réticulation.
REMARQUE: L’énergie d’irradiation UV doit être soigneusement optimisée pour chaque protéine d’intérêt. Voir la discussion pour plus de détails. - Immédiatement après la réticulation, placer la boîte de Petri sur de la glace et ajouter 10 ml de PBS glacé. Recueillir les cellules par grattage et transfert dans un tube conique de 15 mL. Pelleter par centrifugation à 300 x g pendant 5 min à 4 °C.
- Retirer le PBS et remettre en suspension la pastille de cellule dans 1 mL de PBS glacé, puis transférer dans un tube microcentrifuge de 1,5 mL. Pelleter à nouveau les cellules par centrifugation pendant 5 min à 300 x g à 4 °C.
- Retirez le PBS et congelez les granulés de cellule sur de la glace sèche. Entreposer les granulés de la cellule à -80 °C jusqu’à ce qu’ils soient nécessaires.
- Répétez les étapes 1.2.3 à 1.2.6 pour chaque point temporel.
- Resuspendre les pastilles cellulaires dans 1 mL de tampon de lyse et transférer dans un tube conique de 15 mL. Ensuite, ajoutez 1 mL de tampon de lyse pour un total de 2 mL.
- Ajouter 5 μL d’inhibiteur de RNase de mammifère.
- Soniquer 5x pendant 10 s sur glace à 10 ampères. Attendez 30 s entre les cycles de sonication.
- Calculer la concentration en protéines de chaque échantillon et normaliser à la concentration la plus faible.
- Transférer 1,98 mL de lysat dans un tube de 2 mL.
- Ajouter 10 μL de DNase I et incuber à 37 °C pendant 5 min en agitant à 1 200 tr/min.
- Centrifuger le lysat à 16 000 x g pendant 20 min à 4 °C.
- Après centrifugation, prélever un échantillon de 50 μL du surnageant pour une analyse ultérieure par transfert Western afin d’examiner l’expression soluble de la protéine.
- Passer à la saisie RBP (section 2).
- Ensemencez suffisamment de cellules adhérentes dans une boîte de Petri 24 h avant la réticulation UV afin qu’elles puissent atteindre 80% de confluence le lendemain. Cultiver pendant la nuit dans le milieu désiré dans un incubateur de culture cellulaire à 37 °C, 5% de CO2.
2. Capture RBP
- Lavez les perles magnétiques anti-FLAG (75 μL de boue par échantillon) ou d’agarose IgG (500 μL de boue par échantillon) 3x avec 5 mL de TN150. Remettez en suspension dans un volume final de 700 μL de TN150 et ajoutez 100 μL de billes lavées à sept tubes coniques de 15 mL.
- Conserver sur de la glace jusqu’à ce que nécessaire.
- Une fois les lysats clarifiés, ajouter le surnageant au tube contenant les billes anti-FLAG/IgG.
- Nutate à 4 °C pendant 2 h.
REMARQUE: Certains protocoles décrivent des incubations nocturnes avec les billes, mais cela n’est pas recommandé, car de longues périodes d’incubation peuvent réduire considérablement la récupération des ARN réticulés.
3. Lavage des billes et du clivage TEV des étiquettes
- Récoltez les perles et retirez le lysat.
- Prélevez un échantillon de 50 μL du surnageant pour une analyse ultérieure par transfert Western afin d’examiner la protéine non capturée.
- Remettez les billes en suspension dans du TN1000 glacé et transférez-les dans un tube de 1,5 mL. Laver pendant 10 min, 4 °C, avec la nutation. Répétez pour un total de trois lavages.
- Si vous utilisez des billes d’agarose IgG, laver avec 5 mL de TN1000. Si vous utilisez des perles anti-FLAG, utilisez 2 mL.
- Ensuite, lavez les perles 3x avec TN150, avec le même volume que ci-dessus.
- Après le troisième lavage, remettre les billes en suspension dans 600 μL de TN150.
- Ajouter 30 U de protéase GST-TEV maison à la suspension du cordon et faire tourner pendant 2 h à TA.
REMARQUE : La protéase GST-TEV recombinante est maintenant également disponible dans le commerce, mais elle n’a pas été testée avec ce protocole.- Pendant la digestion, préparez-vous aux prochaines étapes en installant des colonnes de trois tubes de 1,5 mL pour chaque échantillon (c.-à-d. pour sept échantillons, ayez trois rangées de sept colonnes).
- À la dernière rangée de tubes, ajouter 0,4 g de chlorhydrate de guanidium, 27 μL de chlorure de sodium 5M et 3 μL d’imidazole 2,5 M (pH = 8). Notez que le pH de l’imidazole doit être de 8. Ceci est essentiel pour maintenir l’intégrité de l’ARN.
- De plus, lavez le volume requis de billes de nickel dans WB I 3x. Utilisez 100 μL de lisier par échantillon. Après le lavage final, remettre les perles en suspension dans le même volume d’origine de WB I et les conserver sur de la glace.
- Une fois la digestion TEV terminée, recueillir le surnageant à l’aide d’un support magnétique pour les billes anti-FLAG ou de la centrifugation pour les billes d’IgG, et transférer sur la première rangée des tubes préalablement installés.
- Prélever un échantillon de 50 μL de l’éluat TEV pour l’analyse par transfert Western.
- Réglez un incubateur thermobloc à 37 °C. À la deuxième rangée de tubes, ajouter 1 μL de cocktail RNase (dilution 1:50).
- Prélever 550 μL d’éluat TEV de la première rangée de tubes et ajouter à la deuxième rangée (contenant le cocktail RNase). Pipeter vigoureusement pour assurer le mélange.
- Après avoir terminé cela pour le premier échantillon, placez immédiatement le tube dans le thermobloc et démarrez une minuterie. Passez aux échantillons suivants, de sorte que chacun soit échelonné.
- Incuber pendant exactement 5 min. Une fois terminé, retirer le premier échantillon du thermobloc et transférer la solution dans la troisième rangée de tubes (contenant la poudre de chlorhydrate de guanidium).
REMARQUE: Une incubation de 5 minutes avec une dilution de 1:50 du cocktail RNase convient généralement à la plupart des protéines, mais cette étape devra être soigneusement optimisée avec des temps d’incubation ou des concentrations différents pour chaque protéine afin de s’assurer que les ARN réticulés sont de la bonne taille (30-100 nt). - Immédiatement vortex pendant quelques secondes à pleine vitesse pour dissoudre la poudre de guanidium, puis passer à l’échantillon suivant.
- Une fois que tous les échantillons ont été transférés dans la poudre de guanidium, vortex à nouveau pour s’assurer que toute la poudre est complètement dissoute.
- Ajouter 100 μL de billes de nickel lavées et faire tourner pendant la nuit à 4 °C. Cette incubation peut être raccourcie à 2 h.
4. Traitement de la phosphatase alcaline sur billes
- Réglez un thermobloc à 37 °C.
- Placer une colonne d’essorage de purification dans un tube de 2 mL, un pour chaque échantillon. Transférer les billes de nickel dans les colonnes et laisser le surnageant s’écouler à travers. Ensuite, assurez-vous que toutes les billes de nickel ont été retirées du tube de 1,5 mL en rinçant avec WB I et en appliquant sur la colonne.
- Mettre en place des tubes de 2 mL, six par échantillon (un pour prélever chaque lavage). Gardez l’extérieur des colonnes sec pour maintenir le débit. Laver les billes 3x avec 500 μL de WB I puis 3x avec 500 μL de NP-PNK.
- Fermez le couvercle de la colonne de rotation et faites tourner brièvement les perles pour éliminer l’excès de tampon.
- Placer le bouchon sur la colonne, placer les colonnes dans des tubes de 1,5 mL et ajouter 60 μL du mélange réactionnel vu dans le tableau 2.
| Composant | 1x | 7,5x |
| 5 x tampon PNK | 12 | 90 |
| Phosphatase alcaline | 4 | 30 |
| Inhibiteur de la RNase | 2 | 15 |
| H2O | 42 | 315 |
| Volume final | 60 μL | 450 μL |
Tableau 2 : Mélange réactionnel de phosphatase alcaline.
- Incuber les billes pendant 1 h à 37 °C.
- Laver les billes 1x avec 500 μL de WB I pour inactiver la phosphatase alcaline puis 3x avec 500 μL de tampon NP-PNK. Assurez-vous de bien rincer l’intérieur de la colonne avec le tampon NP-PNK pour éliminer toute trace de guanidium.
5. Ligature sur perle du linker App-PE à l’extrémité 3' de l’ARN
- Faire tourner le tampon restant et ajouter 60 μL du mélange spécifié dans le tableau 3 (voir le tableau 4 pour la séquence App-PE) dans les colonnes. Incuber la réaction pendant 6 h à 25 °C.
| Composant | 1x | 7,5x |
| 5 x tampon PNK | 12 | 90 |
| Adaptateur App-PE (100 μM) | 0.6 | 4.5 |
| ARN ligase T4 2 tronquée K227Q | 3 | 22.5 |
| Inhibiteur de la RNase | 1.5 | 11.25 |
| 50% PEG 8000 | 12 | 90 |
| H2O | 30.9 | 231.75 |
| Volume final | 60 μL | 450 μL |
Tableau 3 : mélange de réaction de ligature de liant App-PE.
| Nom de l’oligonucléotide | Séquence (5'-3') | |||
| L5Aa | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrNrUrArGrCrN-OH | |||
| L5Ab | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrArUrArGrCrN-OH | |||
| L5Ac | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrGrCrGrCrCrArGrCrN-OH | |||
| L5Ad | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrNrCrGrCrUrUrArGrCrN-OH | |||
| L5Ba | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrCrUrNrNrArGrArGrCrN-OH | |||
| L5Bb | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrGrUrGrArGrCrN-OH | |||
| L5Bc | invddT-ACACrGrArCrGrCrCrUrUrCrCrGrArUrCrUrNrNrNrCrArCrCrUrArGrCrN-OH | |||
| L5Bd | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrUrCrUrCrUrArGrCrN-OH | |||
| L5Ca | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrNrCrUrArGrCrN-OH | |||
| L5Cb | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrNrUrGrArGrCrN-OH | |||
| L5Cc | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrArCrCrCrCrArGrCrN-OH | |||
| L5Cd | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrRGrArCrUrUrArGrCrN-OH | |||
| L5Da | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrCrGrUrGrArUrN-OH | |||
| L5Db | invddT-ACACrGrArCrGrCrUrCrUrCrGrArUrCrUrNrNrGrCrArCrUrArN-OH | |||
| L5Dc | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrNrUrGrCrN-OH | |||
| L5Dd | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrArUrCrArCrGrN-OH | |||
| L5Ea | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrNrCrArCrUrGrUrN-OH | |||
| L5Eb | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrNrNrGrUrGrArCrArN-OH | |||
| L5Ec | invddT-ACACrGrArCrGrCrUrCrUrCrCrGrArUrCrUrNrNrRRUrGrUrCrArCrN-OH | |||
| L5Ed | invddT-ACACrGrArCrGrCrUrUrCrCrGrArUrCrUrUrCrUrNrNrArCrArGrUrGrN-OH | |||
| App_PE | App-NAGATCGGAAGAGCACACGTCTG-ddC | |||
Tableau 4 : Les séquences des adaptateurs d’ADN et d’ARN nécessaires à la ligature sur les extrémités 5' et 3' des ARN capturés. Ceux-ci ont été purifiés par HPLC sans RNase.
- Laver les billes 1x avec 500 μL de WB I et 3x avec 500 μL de tampon NP-PNK. Placez la colonne dans un nouveau tube et faites tourner le tampon restant.
6. Phosphorylation sur bille des extrémités 5' de l’ARN
- Ajouter 80 μL du mélange spécifié au tableau 5 dans les colonnes. Incuber la réaction pendant 40 min à 37 °C.
REMARQUE : Les échantillons seront maintenant hautement radioactifs. Ainsi, tous les travaux ultérieurs devraient être effectués derrière un écran de protection et les déchets devraient être éliminés conformément aux règles locales de santé et de sécurité.
| Composant | 1x | 7,5x |
| 5 x tampon PNK | 16 | 120 |
| 32P-ɣATP (10 μCi/μL) | 3 | 22.5 |
| T4 PNK | 3 | 22.5 |
| H2O | 58 | 435 |
| Volume final | 80 μL | 600 μL |
Tableau 5 : Mélange réactionnel de phosphorylation.
- Ajouter 1 μL d’ATP 100 mM et laisser la réaction se dérouler pendant encore 20 min. Cela permettra de s’assurer que presque toutes les extrémités 5' ont des phosphates pour faciliter la ligature de l’agent de liaison 5'.
- Mettre en place des tubes de 2 mL, cinq par échantillon.
- Laver les billes 1x avec 500 μL de WB I et 3x avec 500 μL de tampon NP-PNK. Notez que ces élutions seront très radioactives et doivent donc être éliminées de manière appropriée.
- Déplacez la colonne vers le tube final et faites tourner le tampon restant.
7. Ligature sur perle de l’éditeur de liaison 5'
REMARQUE: Les linkers 5' contiennent un code à barres ARN qui est utilisé pour l’identification de chaque échantillon après le séquençage. Ainsi, il est absolument crucial de noter quel éditeur de liens est utilisé pour quel échantillon.
- Ajouter 78 μL du mélange décrit au tableau 6 dans les colonnes. Ajouter 2 μL d’adaptateur 5' (100 μM; voir tableau 4) dans chaque tube et incuber pendant une nuit à 18 °C.
| Composant | 1x | 7,5x |
| 5 x tampon PNK | 16 | 120 |
| ATP (10 mM) | 8 | 60 |
| Inhibiteur de la RNase | 2 | 15 |
| ARN ligase T4 | 4 | 30 |
| H2O | 48 | 360 |
| Volume final | 78 μL | 585 μL |
Tableau 6 : Mélange de réaction de ligature de liaison 5'.
- Le lendemain, laver les billes 1x avec 500 μL de WB I et 3x avec 500 μL de WB II et transférer les colonnes dans un nouveau tube de 2 mL.
8. Extraction d’élution, de SDS-PAGE et d’ARN
- Réglez la centrifugeuse à 4 °C. Préparer deux rangées de tubes de 1,5 mL par échantillon pour élution.
- Faites tourner le volume vide des colonnes avec des perles de nickel avec une rotation rapide. Placer les colonnes dans la première rangée de tubes d’élution et ajouter 200 μL de tampon d’élution. Attendez 2 minutes, puis forcez la mémoire tampon à travers la colonne avec une rotation rapide.
- Déplacez les colonnes vers la deuxième rangée de tubes et répétez l’étape 8.2. Chaque échantillon contiendra désormais 400 μL d’éluat au total, répartis sur deux tubes de 1,5 mL.
- Prenez tous les éluats et transférez-les ensemble dans un tube de 5 mL. Ajouter 2 μL de 20 mg/mL de glycogène. Ainsi, si l’on utilise sept échantillons, il y aura maintenant 2,8 mL d’éluat groupé dans le tube de 5 mL.
- Ajouter 100 μL d’acide trichloracétique (TCA) par échantillon [p. ex. 700 μL de TCA pour 7 échantillons (2,8 mL d’éluat combiné)] dans le tube de 5 mL, et bien vorter pendant 30 s.
- Incuber sur glace pendant 20 min.
- Centrifuger pendant 30 min à 17 000 x g, 4 °C, dans une centrifugeuse de paillasse.
- Retirez délicatement le surnageant du tube conique, en vérifiant la pipette avec un compteur Geiger pour vous assurer que la pastille n’a pas été retirée accidentellement. Si c’est le cas, remettre le surnageant dans le tube et centrifuger pendant encore 10 minutes.
REMARQUE : Le surnageant peut encore être hautement radioactif. Assurez-vous d’utiliser un blindage approprié. - Remettez complètement la pastille en suspension dans 2 mL d’acétone glacée.
- Centrifuger pendant 15 min à 17 000 x g, 4 °C.
- Enlevez autant d’acétone que possible avec une pipette P1000. Ensuite, faites tourner brièvement le tube pour recueillir de petites gouttelettes d’acétone, puis retirez-le avec une pipette P10. Sécher pendant 2 min dans une hotte.
REMARQUE : Le surnageant à l’acétone peut encore être radioactif. Assurez-vous d’utiliser un blindage approprié. - Remettez l’échantillon en suspension dans 30 μL de 1x tampon de charge protéique. Pour s’assurer que la pastille est correctement remise en suspension, vérifiez que la grande majorité de la radioactivité est maintenant présente dans le tampon de chargement et non laissée dans le tube de 1,5 mL en retirant la solution dans une pipette P200 et en mesurant l’activité laissée dans le tube de 1,5 mL à l’aide d’un compteur Geiger.
- Chauffer l’échantillon pendant 10 min à 65 °C. Charger sur un gel Bis-Tris préfabriqué de 1 mm à 4 à 12 % et faire fonctionner pendant 1,5 h à 125 V dans un tampon MOPS.
- Une fois le gel terminé, ouvrez la cassette de gel. Le gel doit être retenu sur la plaque inférieure. Jetez le dessus.
- Enveloppez le gel dans un film alimentaire, puis fixez-le à l’intérieur d’une cassette étanche à la lumière à l’aide de ruban adhésif. Assurez-vous que la cassette dispose d’un écran amplificateur pour améliorer le signal.
- Exposer un film autoradiographique au gel et conserver la cassette à -80 °C pendant l’exposition. Le temps d’exposition variera entre les protéines avec différentes efficacités de réticulation.
- Lors de la mise en place du film, il doit y avoir un moyen de le réaligner sur la cassette afin de couper la bande d’intérêt à l’étape suivante. Pour ce faire, utilisez une règle fluorescente et assurez-vous également que le gel se trouve dans un coin de la cassette, qui est ensuite recouvert par le film également placé dans le coin le plus extrême.
REMARQUE: En règle générale, les éluates dans le tampon de chargement qui donnent une lecture d’au moins ~250 cps lorsqu’ils sont affichés à un compteur Geiger donnent un signal suffisant pour une exposition de 3 h. Sinon, l’exposition de nuit est effectuée.
- Lors de la mise en place du film, il doit y avoir un moyen de le réaligner sur la cassette afin de couper la bande d’intérêt à l’étape suivante. Pour ce faire, utilisez une règle fluorescente et assurez-vous également que le gel se trouve dans un coin de la cassette, qui est ensuite recouvert par le film également placé dans le coin le plus extrême.
- Développer le film. Coupez le film alimentaire qui recouvre le gel, mais ne déplacez pas le gel. Sinon, l’image sera décalée par rapport au gel.
REMARQUE: Le gel sera probablement très radioactif. Assurez-vous d’utiliser un blindage approprié lorsque vous découpez la tranche de gel. - Placez le film sur le gel et excisez la bande d’intérêt. Mettez la tranche de gel dans un tube de 2 mL.
- Écraser la tranche de gel à l’aide d’une pointe de pipette P1000 et ajouter 600 μL de tampon protéinase K plus 200 μg de protéinase K (ce protocole utilise 10 μL d’une solution de protéinase K à 20 mg/mL). Incuber pendant 2 h à 55 °C en agitant vigoureusement.
- Ensuite, coupez l’extrémité d’une pointe P1000 avec un scalpel propre et transférez le surnageant et les morceaux de gel dans une colonne de rotation placée dans un tube de 2 mL.
- Faire tourner la colonne pendant 1 min à 17 000 x g à TA. Recueillir le flux continu, qui contient les ARN radioactifs isolés.
- Effectuer une extraction phénol:chloroforme.
- Ajouter 50 μL d’acétate de sodium 3 M, pH = 5,2 et 500 μL de phénol:chloroforme et bien vortex. Faire tourner pendant 5 min à 17 000 x g. Retirer la couche supérieure aqueuse et la placer dans un nouveau tube de 1,5 mL.
- Ajouter 500 μL de chloroforme et vortex vigoureusement pendant 10–15 s. Faire tourner pendant 5 min à 17 000 x g à TA. Retirer la couche aqueuse et la placer dans un nouveau tube de 1,5 mL.
- Ajouter 1 μL de glycogène à 20 mg/mL et 1 mL d’éthanol glacé à 96 %. Précipiter pendant 30 min à -80 °C ou toute la nuit à -20 °C.
- Centrifuger pendant 30 min à 4 °C, 17 000 x g. Retirer le surnageant, ajouter 500 μL d’éthanol à 70 % et centrifuger pendant 5 min, 4 °C à 17 000 x g. Retirez tout l’éthanol, faites un tour rapide pour recueillir les résidus et enlevez l’excès avec une pipette P10.
- Sécher le granulé pendant ~3 min dans une hotte. Remettez en suspension dans 20 μL d’eau traitée au DEPC.
- Conserver l’ARN à -80 °C pendant une nuit ou passer immédiatement à l’étape de transcription inverse.
9. Transcription inverse
- Ajouter 2 μL d’oligo RT 10 μM (PE_reverse; voir tableau 7) et 4 μL de 5 mM dNTP aux 20 μL d’ARN.
| Nom de l’oligonucléotide | Séquence (5'-3') | |||
| P5 avant | AATGATACGGCGACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | |||
| BC1 | CAAGCAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC3 | CAAGCAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC4 | CAAGCAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC5 | CAAGCAGACGGCATACGAGATCACTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC7 | CAAGCAGACGGCATACGAGATCAGATCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC8 | CAAGCAGACGGCATACGAGATTAGCTTGTGACTGGAGTTCAGACGTGCTCTTCCGATCT | |||
| BC9 | CAAGCAGACGGCATACGAGATGATCAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| BC10 | CAAGCAGACGGCATACGAGATATCACGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |||
| PE_reverse | CAGACGTGTGCTCTTCCGATCT | |||
Tableau 7 : Les amorces de PCR (y compris les séquences de codes à barres) et l’amorce de transcription inverse.
- Transférer dans un thermobloc préchauffé à 85 °C pendant 3 min, puis refroidir sur glace pendant 5 min. Recueillir le contenu du tube par centrifugation brève, puis ajouter 8 μL de tampon transcriptase inverse 5x, 2 μL de DTT 100 mM et 2 μL d’inhibiteur de RNase.
- Incuber le mélange à 50 °C pendant 3 min, puis ajouter 2 μL de transcriptase inverse et incuber pendant 1 h à 50 °C.
- Inactiver la transcriptase inverse par incubation à 65 °C pendant 15 min.
- Transférer les tubes dans un thermobloc préchauffé à 37 °C et laisser s’acclimater pendant 3 minutes.
- Ajouter 2 μL de RNase H et incuber pendant 30 min à 37 °C.
- Isoler l’ADNc à l’aide de billes SPRI.
- Ajouter deux volumes de 84 μL de billes. Incuber pendant 15 min. Mettez les perles sur une grille magnétique et laissez reposer 1 min pour récolter les perles.
- Retirer et éliminer le surnageant et ajouter 200 μL d’éthanol à 70 %. Ne retirez pas les perles du support magnétique. Incuber les billes avec l’éthanol pendant 30 s.
- Retirez l’éthanol et répétez l’étape de lavage. Retirez tout l’éthanol résiduel à l’aide d’un embout P10.
- Mettez les perles dans une hotte pendant 2 min pour les sécher. Retirez les billes de la grille, remettez-les en suspension dans 12 μL d’eau, puis remettez les perles sur la grille. Retirer 11 μL de surnageant.
- Congeler l’ADNc à -20 °C ou passer immédiatement à l’étape de PCR.
10. Réaction qPCR
- Avant la PCR finale pour l’amplification des ADNc, une réaction en chaîne par polymérase quantitative (qPCR) est effectuée pour identifier le nombre optimal de cycles pour amplifier les ADNc afin d’éviter une suramplification de la bibliothèque.
- Mettre en place une réaction qPCR sur la glace conformément au tableau 8. Voir le tableau 7 pour tous les abécédaires.
| Composant | 1x |
| 2x mastermix de réaction qPCR | 5 |
| 0,1 μM d’amorce P5 (avant) | 0.8 |
| 0,1 μM d’amorce BC (inverse) | 0.8 |
| ADNc (ou l’eau comme témoin négatif) | 1 |
| H2O | 2.4 |
| Volume final | 10 μL |
Tableau 8 : mélange réactionnel qPCR.
- Pour une quantification correcte des cycles nécessaires à l’amplification, utiliser trois réplications techniques pour l’ADNc et trois témoins négatifs (c’est-à-dire de l’eau).
- Scellez les plaques avec un film optiquement transparent et exécutez la qPCR conformément aux instructions du fabricant du kit.
- Analyser les échantillons au moyen d’une méthode de quantification absolue pour identifier le nombre de cycles (n) auxquels le genou de croissance exponentielle est atteint (voir la figure 4C pour un exemple). Ce nombre de cycles est ensuite utilisé pour l’amplification finale du reste de l’ADNc.
11. Réaction PCR et extraction de gel
- Configurez la réaction de PCR sur la glace conformément au tableau 9. Voir le tableau 7 pour tous les abécédaires.
REMARQUE: Seulement 5 μL de la bibliothèque d’ADNc sont utilisés.
| Composant | 1x |
| 10 x tampon polymérase de relecture | 5 |
| 10 μM d’apprêt P5 (avant) | 1 |
| 10 μM BC primer (revers) | 1 |
| 5 mM dNTP | 2.5 |
| Relecture de l’enzyme polymérase | 1 |
| ADNc | 5 |
| H2O | 34.5 |
| Volume final | 50 μL |
Tableau 9 : Mélange réactionnel PCR.
- Exécutez la PCR comme suit : 95 °C pendant 2 min ; n cycles de 98 °C pendant 20 s, 52 °C pendant 30 s et 72 °C pendant 1 min; et 72 °C pendant 5 min. Le nombre (n) de cycles pour amplifier la bibliothèque χCRAC est déterminé par la qPCR décrite au point 10.
- Ajouter 1 μL d’exonucléase I et incuber à 37 °C pendant 60 min.
- Nettoyez l’ADNc amplifié à l’aide de billes SPRI comme décrit ci-dessus en utilisant deux volumes de billes (c.-à-d. 100 μL). Éluter dans 11 μL.
- Ajouter 3 μL de colorant de chargement 6x et faire fonctionner sur un gel préfabriqué 6% TBE à 100 V pendant 1 h dans 1x tampon TBE. Utilisez une échelle appropriée pour la quantification de courts fragments d’ADN.
- Une fois terminé, retirez le gel de la cassette et placez-le dans un récipient approprié et étanche aux liquides avec suffisamment de 1x TBE pour couvrir le gel (par exemple ~ 50 mL). Ajouter une quantité appropriée de colorant SYBR (p. ex. pour 50 mL, utiliser 5 μL d’un colorant 10 000x)
- Laisser le gel tacher en mélangeant doucement pendant 15 minutes à TA. Égoutter le 1x TBE contenant du SYBR et le remplacer par 1x TBE propre. Laver le gel pendant 10 min en secouant doucement à TA.
- Égoutter le 1x TBE et placer le gel dans une chemise transparente. Coupez le dossier à une taille appropriée.
- Imagez le gel par un moyen approprié tel qu’un phosphorimageur. Fragments d’ADN d’accise entre ~175 pb et ~400 pb. Mettez la tranche de gel dans un tube de 1,5 mL.
- Bien écraser la tranche de gel à l’aide d’une pointe P1000 et ajouter 400 μL deH2O. Incuber à 37 °C en agitant pendant 1 h dans un thermobloc.
- Congeler l’échantillon sur de la glace sèche pendant 10 min, puis remettre dans le thermobloc à 37 °C en agitant pendant 1 h.
- Créez une unité de filtration en prenant une colonne filtrante et en insérant deux filtres en microfibre de verre à l’intérieur. Placez l’appareil dans un tube de 1,5 mL.
- Coupez l’extrémité d’une pointe P1000 avec un scalpel propre et prenez la suspension de gel TBE brisée, puis distribuez dans l’unité de filtration créée à l’étape 11.12. Essorage à 17 000 x g pendant 30 s.
- Ajouter 1 μL de glycogène au surnageant, ainsi que 40 μL d’acétate de sodium, pH = 5,2, et 1 mL d’éthanol à 96 %. Incuber à -80 °C pendant 30 min.
- Centrifuger pendant 30 min à 17 000 x g, 4 °C. Jeter le surnageant et laver avec 500 μL d’éthanol à 70 %.
- Faire tourner pendant 5 min, retirer complètement l’éthanol puis sécher la pastille dans une hotte pendant 3 min.
- Resuspendre dans 10 μL deH2Oet mesurer la concentration d’ADN.
Résultats
Pour démontrer l’efficacité de la méthode χCRAC, une expérience temporelle avec des souches de levure exprimant une protéine Nrd1 marquée au HTP a été réalisée. Une représentation schématique détaillée décrivant le fonctionnement de la méthode est fournie à la figure 1. Comme Nab3, Nrd1 est impliqué dans la désintégration de l’ARN nucléaire d’une variété de transcrits d’ARN37. Des travaux antérieurs du laboratoire Corden ont suggéré que la liaison de Nrd1 à ses cibles d’ARN change de manière significative lorsque les cellules sont soumises à une privation de glucose28,38. En tant que tels, les cellules se développant exponentiellement dans un milieu contenant du glucose (SD-TRP) ont été déplacées vers le même milieu sans glucose (S-TRP) au cours d’une période de temps pour surveiller les changements dynamiques dans les interactions Nrd1-ARN. Des échantillons ont été prélevés et réticulés dans la chambre de liaison Vari-X (figure 3A) avant le poste, puis après 1, 2, 4, 8, 14 et 20 minutes. Le milieu utilisé pour la croissance cellulaire était délibérément déficient en tryptophane pour réduire l’absorption UV par cet acide aminé aromatique. Notez qu’il est préférable d’utiliser un milieu synthétique stérilisé par filtre car l’autoclavage du milieu peut entraîner une caramélisation des sucres. Cela réduit ensuite l’efficacité de la réticulation.
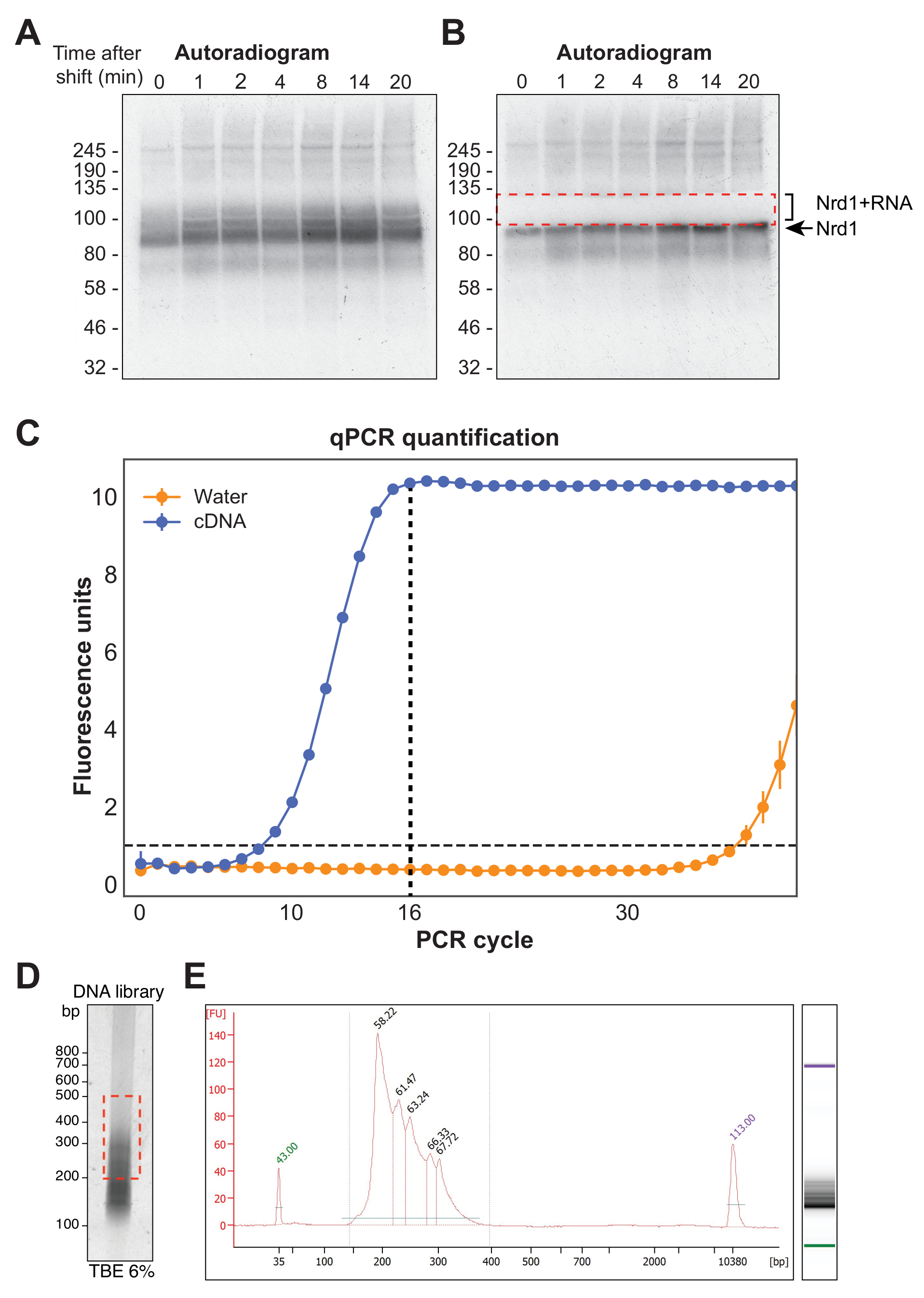
La figure 4A montre une autoradiographie représentative d’une expérience χCRAC. Notez que dans cet exemple, les échantillons n’ont pas été regroupés. Au lieu de cela, chacun a été exécuté individuellement sur le gel. Ceci est recommandé pour les tests expérimentaux initiaux afin de montrer que la protéine se lie efficacement à l’ARN à tous les points temporels testés. Un signal particulièrement intense a été observé au poids moléculaire attendu de la RBP, représentant la protéine liée à des ARN radiomarqués très courts et ne se prêtant pas au séquençage. Par conséquent, le signal de frottis au-dessus de cette bande, qui est la protéine réticulée à des fragments d’ARN plus longs, a été isolé. Le fragment a été coupé juste au-dessus de la bande protéique plus environ 30 kDa. La figure 4B montre un autoradiogramme après l’excision, avec la protéine réticulée aux ARN courts laissés dans le gel et le signal précédemment frottis maintenant excisé.
Après transcription inverse, la bibliothèque d’ADNc doit être amplifiée par PCR. Cependant, la suramplification de la bibliothèque doit être évitée car cela peut introduire un biais vers des séquences amplifiées préférentiellement par la polymérase et générer des artefacts de PCR. Les bibliothèques suramplifiées contiennent également un grand nombre de séquences dupliquées qui gaspillent les lectures sur le séquenceur. Afin de calculer le nombre idéal de cycles de PCR pour l’amplification de la bibliothèque finale, une partie aliquote de l’ADNc a été amplifiée par qPCR en utilisant les oligonucléotides P5 et BC. Le premier cycle auquel la bibliothèque a atteint le pic de fluorescence a été choisi comme nombre de cycles de PCR. La figure 4C donne un exemple de qPCR à partir d’une bibliothèque d’ADNc typique, qui a donné un nombre de cycles de pointe de 16. Cette valeur a ensuite été utilisée pour la PCR finale χCRAC. Afin de traiter les données séquencées, nous avons utilisé un logiciel précédemment développé dans notre laboratoire (pyCRAC) et le pipeline correspondant pour l’analyse des données CRAC cinétiques (Nues et al., 2017; https://git.ecdf.ed.ac.uk/sgrannem/pycrac, https://bitbucket.org/sgrann/kinetic_crac_pipeline/src/default/). Ces outils logiciels open source permettent le démultiplexage et le découpage des données, la suppression des doublons de PCR, l’identification de pics statistiquement significatifs, la lecture de clusters en séquences contiguës et l’identification de motifs de liaison39. De plus amples détails sur le fonctionnement de ces outils se trouvent sur leurs pages Web respectives.
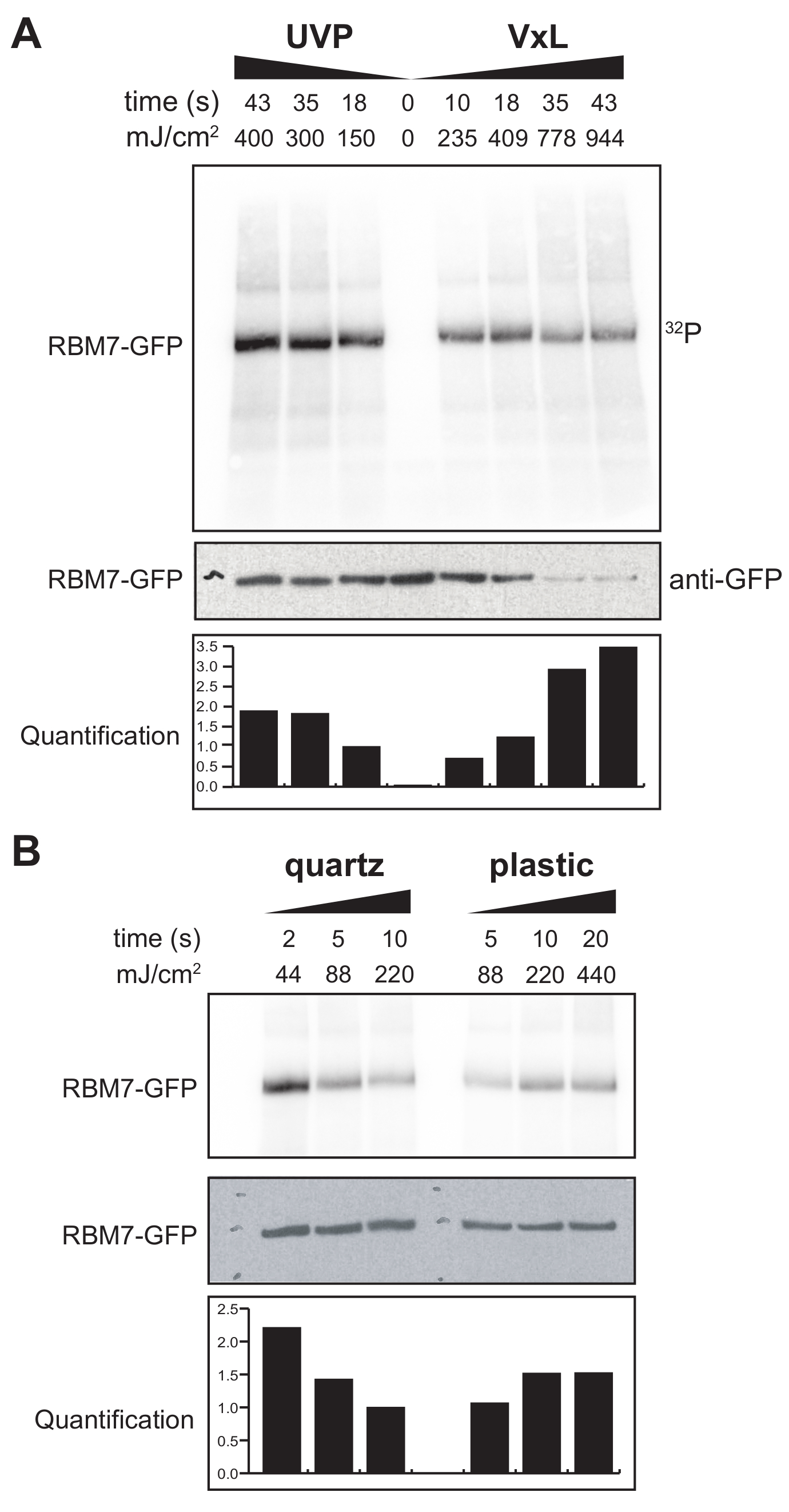
Nous avons également commencé à développer un protocole χCRAC pour les cellules de mammifères. La majorité des lignées cellulaires de mammifères sont cultivées comme monocouche et le plateau de notre réticulateur avec le sac perméable aux UV ne convient pas aux expériences avec des cellules adhérentes. Pour surmonter ce problème, nous avons développé une étape où les utilisateurs peuvent irradier UV 1 à 2 boîtes de Petri (150 mm de diamètre et 25 mm de profondeur) avec des cellules adhérentes (Figure 3B). Comme premier test, l’efficacité de la réticulation pour les cellules de mammifères a été mesurée par réticulation et capture de GFP-RBM7 marqués de manière stable à l’aide d’anticorps anti-GFP et d’une purification traditionnelle à base de CLIP. Comme le montre la figure 5A, l’agent de réticulation a pu récupérer des complexes protéine-ARN à partir de cellules de mammifères cultivées en monocouche en utilisant une irradiation UV de 254 nm avec des efficacités comparables à celles d’un dispositif d’irradiation UV largement utilisé. Cependant, les articles en plastique de culture cellulaire standard normalement utilisés pour les expériences de réticulation UV sont impénétrables aux UV de 254 nm. Par conséquent, dans notre réticulation, les cellules ne recevraient l’irradiation que de la banque supérieure de lampes UV. Pour surmonter ce problème, nous avons développé une boîte de Petri en quartz perméable aux UV pour la croissance cellulaire et la réticulation. L’utilisation de la culture de quartz a montré une récupération robuste des complexes protéine-ARN avec aussi peu que 2 s d’irradiation UV (Figure 5B). Lorsqu’ils sont combinés à des méthodes de capture de RBP pour les cellules de mammifères telles que les technologies CLIP, ces temps de réticulation courts se prêtent à des parcours temporels pour récupérer les profils spatio-temporels de liaison à l’ARN des RBP en réponse à des stress génotoxiques ou à des déplétions rapides de facteurs protéiques, ou parallèlement à la synchronisation transcriptionnelle ou du cycle cellulaire.
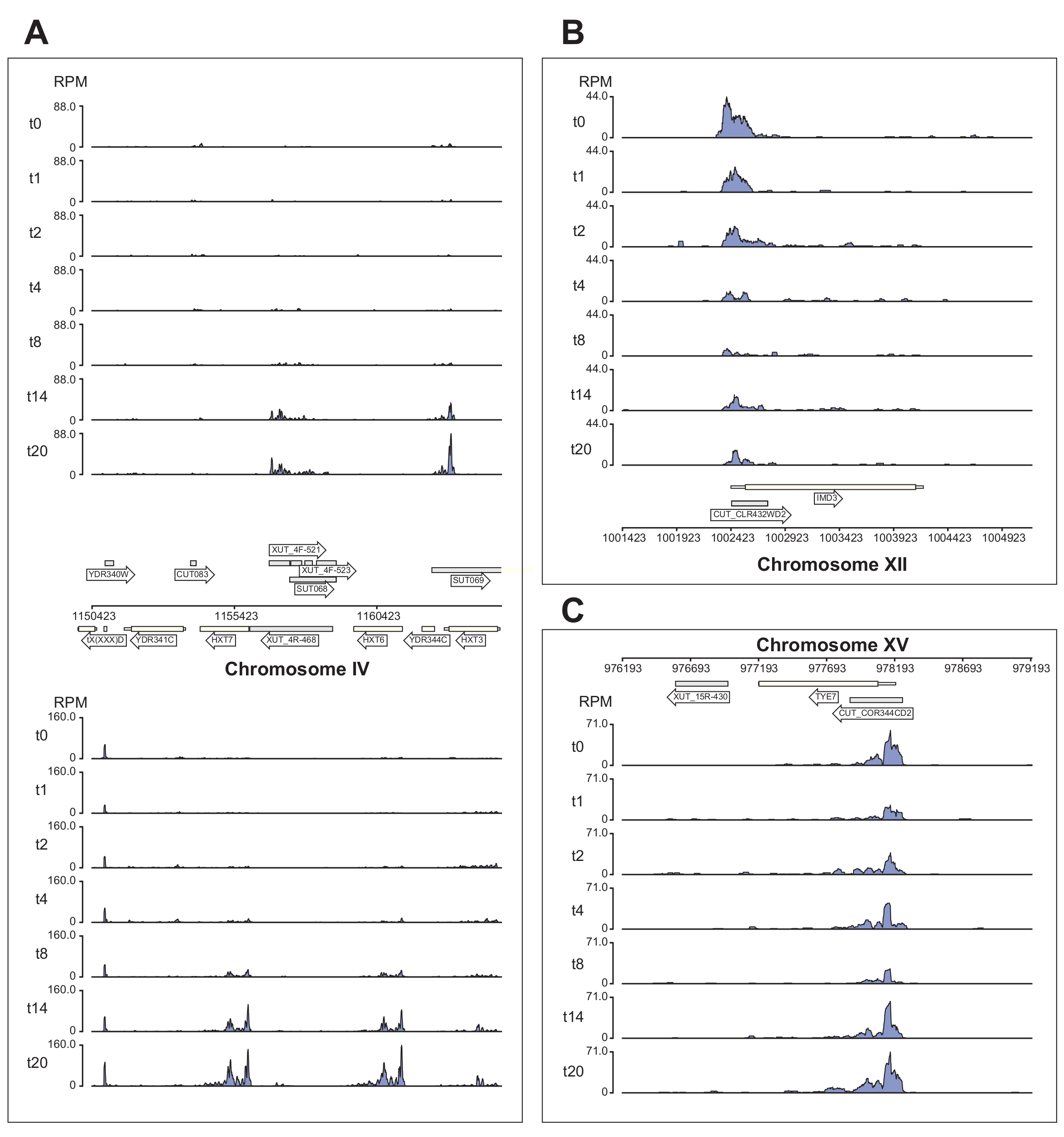
La figure 6 montre plusieurs exemples de données Nrd1 traitées par le pipeline χCRAC. Cette figure a été préparée à l’aide des fichiers bedgraph générés par le pipeline et le paquet python GenomeBrowser (https://pypi.org/project/GenomeBrowser/1.6.3/), que nous avons conçu pour simplifier la création d’images de navigateur génomique de qualité publication des données. Les rectangles gris représentent les régions génomiques qui expriment des ARN non codants, comme le transcrit instable cryptique (CUT), les transcrits stables non caractérisés (TUS)40 et les transcrits instables sensibles à Xrn1 (XUT)41. Les données de la figure 6 montrent que Nrd1 se lie à plusieurs de ces transcrits d’ARN non codants, ce qui est cohérent avec l’idée que cette protéine est impliquée dans la dégradation de cette classe de transcrits42. La figure 6A montre une région de ~15 kb sur le chromosome IV. Ici, il y a eu une augmentation significative de la liaison de Nrd1 aux transcrits codant pour les transporteurs de glucose de haute affinité HXT6 et HXT7, qui sont tous deux régulés à la hausse pendant la privation de glucose. Il est probable que la terminaison de transcription par le complexe NNS puisse influencer la cinétique d’induction de ces gènes pendant la privation de glucose. La figure 6B montre un exemple de réticulation Nrd1 avec le transcrit Imd3, qui est connu pour être régulé par Nab343. Dans ce cas, les données ont démontré une réduction significative de la liaison lors de la privation de glucose. Des travaux antérieurs ont montré une diminution de la liaison de Nab3 au transcrit Tye7 pendant la privation de glucose44. Conformément à cette observation, les données de χCRAC suggèrent que la liaison de Nrd1 a diminué pendant la privation de glucose et que la réticulation de Nrd1 avec Tye7 était à son plus bas après 8 minutes de stress (Figure 4C). Cependant, il semble que cet effet n’ait été que transitoire, car après 14 minutes de privation de glucose, la liaison Nrd1 est revenue aux niveaux de départ.

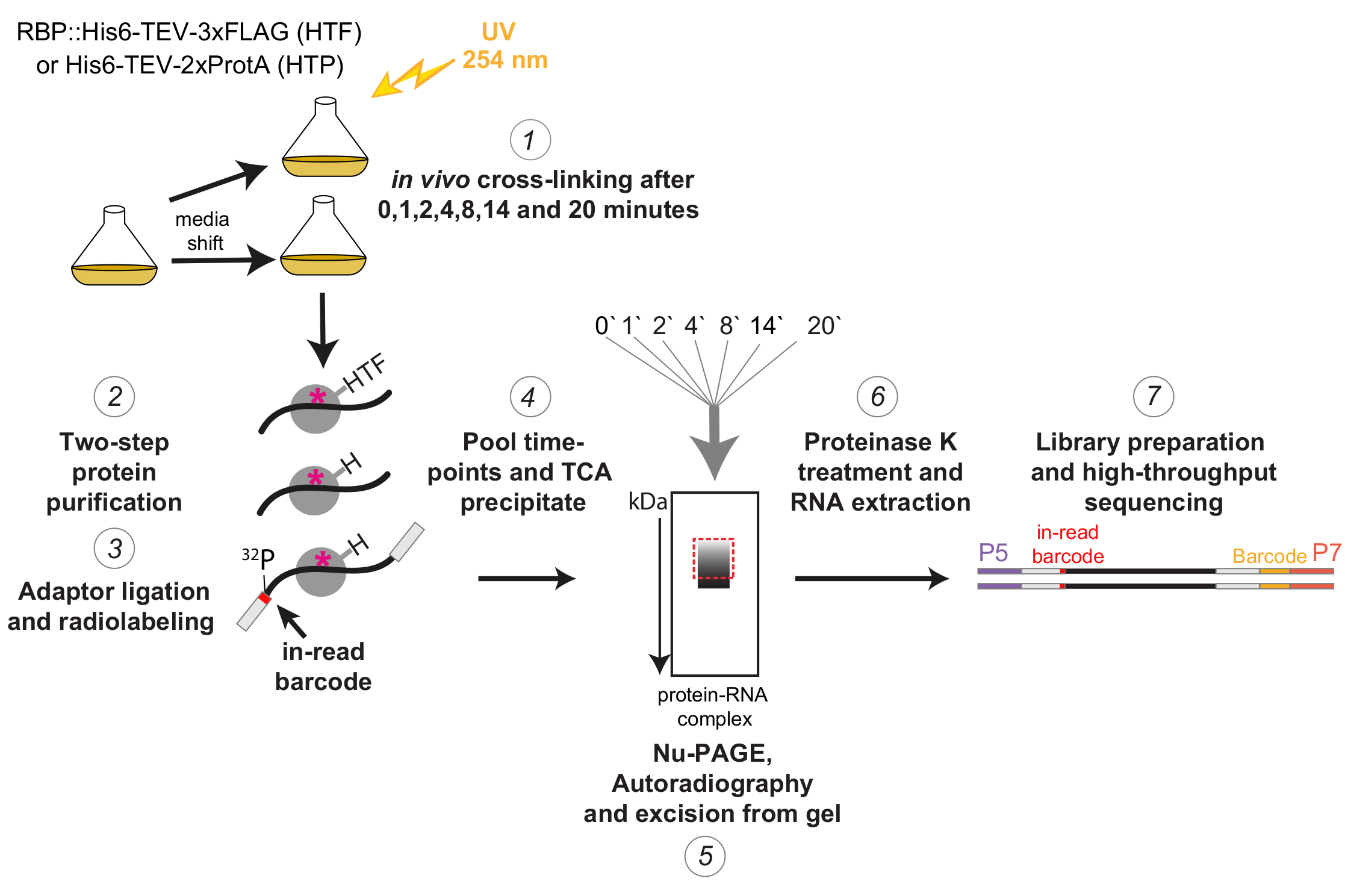
Graphique 1 : Représentation schématique du protocole χCRAC. Les souches marquées ont été cultivées jusqu’à la densité souhaitée. RBP indique une protéine de liaison à l’ARN. Ensuite, un échantillon de référence a été prélevé et réticulé avec une lumière UV de 254 nm. Les cellules restantes ont été récoltées par filtration, puis rapidement déplacées vers le milieu induisant le stress. Pour l’expérience χCRAC décrite ici, des échantillons ont été prélevés et réticulés 1, 2, 4, 8, 14 et 20 minutes après le poste (1). La RBP d’intérêt a ensuite été purifiée à l’aide d’une purification d’affinité en deux étapes très stricte (2). Ensuite, les ARN réticulés capturés ont été partiellement digérés avec des RNases, radiomarqués à l’extrémité 5' et des adaptateurs ont été ligaturés sur eux (3). Les adaptateurs 5' contenaient des séquences de codes-barres uniques « en lecture » afin que les échantillons individuels puissent être séparés bio-informatique après le séquençage. Les complexes RBP-ARN ont ensuite été élués, regroupés et précipités ensemble (4), résolus par SDS-PAGE et visualisés par autoradiographie (5). Par la suite, une seule tranche de gel contenant le signal radioactif juste au-dessus de la bande principale, illustrée par une boîte rouge en pointillés dans l’image d’autoradiographie, a été découpée du gel (5). Les tranches de gel ont été traitées avec de la protéase K et l’ARN a ensuite été extrait (6), converti en ADNc et amplifié par PCR (7). L’étape PCR a introduit des codes-barres supplémentaires (bloc jaune introduit par P7 oligo) afin que de nombreuses bibliothèques puissent être multiplexées en une seule voie. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Réticulation et filtration sous vide. (A) Le réticuleur. La suspension cellulaire est versée dans un entonnoir situé en haut à droite de la machine (voir également la figure 3A pour un gros plan) et maintenue dans un sac transparent aux UV situé dans le plateau du milieu. Ce sac est flanqué de deux volets qui restent fermés jusqu’à ce que l’utilisateur demande à la machine de démarrer l’étape d’irradiation. Les cellules sont irradiées avec la lumière UV des plateaux à la fois au-dessus et au-dessous. La machine est livrée avec des lampes UV de 254 et 365 nm, ces dernières étant applicables aux expériences PAR-CLIP. La machine est commandée par un écran tactile situé en haut à droite, ce qui permet de contrôler le dosage UV ou le temps d’exposition. (B) Après réticulation, les cellules sont drainées du côté gauche de la machine. Les suspensions cellulaires sont récupérées sous vide et drainées dans un flacon en verre où elles peuvent ensuite être versées dans un dispositif de filtration sous vide pour la récolte. C) Dispositifs de filtration sous vide. Ceux-ci sont ouverts et fermés via un clip et un filtre est inséré entre eux. Quatre dispositifs de filtration ont été utilisés en parallèle pour de très courtes séries chronologiques afin de ne pas perdre de temps à la suite du changement de filtre. (D) Après filtration, le surnageant du milieu a été égoutté dans des flacons en vue de leur élimination ultérieure. Des vannes ont été installées sous les dispositifs de filtration sous vide pour maintenir le vide dans le système lorsque le filtre est retiré. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Réticulation des cellules suspendues par rapport aux cellules adhérentes. (A) La réticulation avec la chambre de liaison Vari-X pour les cellules en suspension. La culture cellulaire est versée dans l’entrée de l’échantillon (entonnoir) située en haut à droite du plateau. (B) Plateau pouvant contenir des boîtes de Petri en plastique ou en quartz pour la réticulation des cellules adhérentes ou de petits volumes de cellules en suspension. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Préparation de la bibliothèque. (A) Exemple d’autoradiogramme issu d’une expérience Nrd1-HTP χCRAC. Le signal fort et concentré représente la protéine réticulée aux ARN très courts, tandis que le frottis ci-dessus représente la protéine réticulée aux ARN de longueur suffisante pour le séquençage. (B) Le frottis a été excisé comme le montre un autoradiogramme prélevé après l’excision sur gel. (C) Une qPCR représentative d’une bibliothèque d’ADNc χCRAC. Dans cet exemple, l’amplification maximale de l’ADNc a été atteinte à 16 cycles. Ainsi, 16 cycles ont été utilisés pour l’amplification finale. La barre d’erreur représente l’écart-type de trois répétitions techniques qPCR. (D) Exemple d’une image phosphorée provenant d’une bibliothèque d’ADNc sur un gel de TBE à 6%. (E) Analyse de la longueur et de la qualité de l’ADNc à partir d’une électrophorèse capillaire à base de puce. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5 : Expérience iCLIP de test de RNase élevée pour tester la réticulation dans des cellules de mammifères. Les autoradiogrammes des expériences GFP-RBM7 iCLIP qui ont testé l’efficacité de la récupération RNP à travers diverses énergies de réticulation sont montrés. Les immunoprécipitations ont été réalisées à l’aide d’anticorps anti-GFP couplés à des billes magnétiques sur des cellules réticulées qui exprimaient de manière stable la GFP-RBM7. Les immunoprécipités ont été incubés avec des concentrations élevées de RNase I afin de réduire les ARN associés à des longueurs courtes et uniformes. Les RNP ont été visualisés par marquage 32P et SDS-PAGE et migrent comme une bande définie, proche de la migration de la protéine non réticulée. La quantification indique les résultats des analyses densitométriques du signal d’ARN RBM7 radiomarqué normalisé au signal Western blot anti-GFP. (A) Réticulation temporelle de la réticulation UVP couramment utilisée par rapport à notre réticulation (Vari-X-linker; VxL). (B) Réticulation temporelle de notre agent de réticulation sur des articles de culture en quartz (à gauche) et en plastique (à droite). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 6 : Exemples de graphiques de navigateur génomique montrant la puissance de χCRAC pour montrer la liaison différentielle et temporelle de Nrd1 à ses cibles. Chaque boîte montre des graphiques pour les régions génomiques individuelles. Les flèches indiquent sur quel brin les gènes sont codés (flèche pointant vers la gauche = brin moins; flèche pointante droite = brin plus). Les points temporels (min) sont indiqués par t0, t1, t2, etc. sur les axes y de chaque sous-placette. Les chiffres romains indiquant les chromosomes et les coordonnées sont affichés. (A) Lors de la privation de glucose, Nrd1 lie deux transporteurs de glucose de haute affinité, HXT6 et HXT7, qui sont tous deux régulés à la hausse dans cette condition. (B) On observe que Nrd1 se lie à Imd3, une cible déjà validée de Nab344, avec une intensité réduite après une privation de glucose. (C) La liaison Nrd1 de Tye7 présente une nature dynamique et transitoire, diminuant après une privation de glucose à un minimum après 8 minutes de stress. Cependant, la liaison revient ensuite aux niveaux basaux après 14 min. Les lectures ont été normalisées en « lectures par million » (RPM; axe y). Les cases grises indiquent les régions codant pour des ARN non codants. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Discussion
La méthode χCRAC, combinée aux nouveaux dispositifs de réticulation et de prélèvement cellulaire, présente un grand potentiel car elle est applicable à un large éventail d’organismes modèles et devrait donc présenter un intérêt général pour le domaine de l’ARN. Il existe de nombreux domaines dans lesquels χCRAC peut être utilisé. Par exemple, la méthode pourrait être utilisée pour mesurer l’assemblage hiérarchique des protéines en grands complexes macromoléculaires, tels que le splicéosome et le ribosome, ce qui implique souvent des interactions dynamiques entre les protéines et les molécules d’ARN. Nous l’utilisons aussi couramment pour surveiller les interactions entre les facteurs de désintégration de l’ARN et leurs substrats lorsque les cellules sont soumises à divers types de stress. Cela nous permet de déterminer à quel stade de la réponse adaptative ces facteurs sont les plus actifs, à quels substrats ils se lient et à quel point ces interactions sont dynamiques. Ces données devraient permettre aux chercheurs de déterminer la contribution relative de chaque facteur à l’adaptation aux changements environnementaux.
χCRAC utilise des marqueurs de purification à double affinité (HTF ou HTP) pour purifier la protéine dans des conditions très strictes et dénaturantes. Cela garantit que l’ARN copurifié est hautement enrichi pour les ARN qui ont été réticulés de manière covalente à la protéine d’intérêt. Cependant, s’appuyer sur des balises d’affinité présente des inconvénients. Par exemple, l’étiquette pourrait interférer avec la fonction de la protéine, ce qui pourrait donner une lecture déformée de son interactome de liaison à l’ARN. De plus, pour certains organismes modèles, il n’est pas toujours possible d’utiliser des marqueurs parce que les outils génétiques pour intégrer des fragments d’ADN dans le génome ou pour transformer des plasmides d’expression ne sont pas encore disponibles. Cependant, il est facile de modifier certaines parties du protocole χCRAC pour le rendre compatible avec les protocoles basés sur CLIP qui reposent sur des anticorps pour la purification du RBP. En effet, cette étude a montré qu’il est possible de combiner des purifications à base d’iCLIP avec notre réticulation. Nous sommes actuellement en train de développer des protocoles CLIP pour étudier l’association temporelle des protéines de liaison à l’ARN humain avec les transcrits d’ARN naissants.
Lors de l’exécution de χCRAC sur une nouvelle protéine, l’exposition aux UV doit être optimisée afin d’induire une réticulation maximale. Ceci est important car des expositions élevées aux UV peuvent réduire la récupération de l’ARN pendant l’étape de purification. Les cellules exprimant la RBP recombinante ont été exposées à diverses doses d’UV, 100 mJ/cm2, 250 mJ/cm2, 500 mJ/cm2 et 1 J/cm2. Les RNP ont ensuite été capturés et les ARN ont été fragmentés et radiomarqués. Ensuite, les RNP ont été résolus par SDS-PAGE et un autoradiogramme a été pris afin de déduire quelle exposition donnait le signal le plus intense (c’est-à-dire la réticulation maximale).
Une fois les conditions expérimentales optimisées, plusieurs expériences de contrôle sont recommandées lors de l’exécution de χCRAC. Tout d’abord, un échantillon non marqué irradié par UV peut être utilisé pour surveiller la liaison de fond aux billes de purification. Deuxièmement, lors de l’application de χCRAC au cours d’une expérience de décalage, une deuxième série chronologique où les cellules sont replacées dans le milieu d’origine permet de déterminer si la filtration des cellules elle-même induit des changements dans les niveaux d’ARN ou les interactions protéine-ARN.
Comme mentionné dans l’introduction, de nombreux articles récemment publiés suggèrent un certain nombre d’optimisations du protocole CLIP. Cela comprend l’utilisation d’adaptateurs marqués par fluorescence pour détecter le complexe protéine-ARN par balayage infrarouge10 ainsi que l’optimisation de diverses étapes de purification des acides nucléiques et de sélection de la taille qui augmentent la complexité des bibliothèques résultantes 12,45. Nous mettons actuellement en œuvre certaines de ces améliorations afin d’affiner le protocole χCRAC. Le protocole présenté ici contient déjà un certain nombre d’améliorations par rapport aux protocoles originaux CRAC et χCRAC qui augmentent la complexité des données. Par exemple, auparavant, après avoir résolu les complexes protéine-ARN radioactifs réticulés sur des gels SDS-PAGE, ils étaient transférés dans une membrane de nitrocellulose et l’ARN réticulé était isolé du transfert. Cependant, le transfert du RNP et l’extraction ultérieure de l’ARN peuvent être très inefficaces, en particulier lorsqu’il s’agit de grands RBP tels que les sous-unités d’ARN polymérase. Cela peut entraîner une réduction significative de la récupération de l’ARN réticulé. Dans le protocole actuel, l’ARN réticulé est extrait directement des tranches de gel SDS-PAGE comme illustré à la figure 1. Cela a augmenté la récupération des ARN réticulés. De plus, après amplification par PCR des ADNc, le produit a été initialement résolu sur des gels d’agarose à 3% à basse température de fusion, puis des produits PCR de 175 à 300 pb ont été extraits du gel. Cependant, ces gels peuvent être facilement surchargés, ce qui entraîne une très mauvaise séparation de l’ADN. Le remplacement des gels d’agarose par des gels TBE préfabriqués a permis une séparation plus uniforme de la taille et une meilleure récupération des produits de PCR.
Déclarations de divulgation
A. Langford et W. Worboys sont affiliés à UVO3, une société commerciale. Ils n’ont joué aucun rôle dans la conception de l’étude, la collecte et l’interprétation des données, ni dans la décision de soumettre le travail pour publication.
Remerciements
Ce travail a été soutenu par des subventions du Wellcome Trust (091549 à S.G et 109093/Z/15/A à S.M.), de la subvention de base (092076) du Wellcome Trust Centre for Cell Biology et de la bourse de recherche senior non clinique du Conseil de recherches médicales (MR/R008205/1 à S.G.), de l’Organisation européenne de biologie moléculaire dans le cadre d’une bourse postdoctorale de longue durée (ALTF 1070-2017 à R.A.C), et le Fonds indépendant de recherche du Danemark (T.H.J).
matériels
| Name | Company | Catalog Number | Comments |
| 1,4-dithioreitol | Merck | 10708984001 | Buffer component in mammalian cell lysis |
| 1.5 mL tubes | Eppendorf | 0030 120.086 | General reaction tube |
| 2 mL tubes | Eppendorf | 0030 123.344 | For holding columns and collection of waste |
| 32P-yATP | Perkin Elmer | NEG502Z-250 | For radiolabelling the 5' end of the RNA |
| 4-12% Bis-Tris gel | Invitrogen | NP0321BOX | SDS-PAGE gel |
| 4X loading buffer | Novex | NP0008 | Protein loading dye concentrate |
| 50 bp ladder | New England Biolabs | N3236 | Reference ladder for excising region of interest from the amplified cDNA library |
| 50% PEG | NEB | B100045 | For the L5 linker ligation |
| 6% TBE gel | Invitrogen | EC6265BOX | For separation and purification of the cDNA library |
| Acetone | ACROS Organics | 423245000 | Washing of TCA-precipitated proteins |
| anti-FLAG beads | Sigma Aldrich | M8823-1ML | For purifcation of FLAG-tagged RBPs |
| ATP (100 mM) | Thermo Fisher Scientific | R0441 | For ligation of the L5 linker onto the 5' end of captured RNAs |
| Beta-mercaptoethanol | Sigma Aldrich | M3148-100ML | Buffer component |
| Biomax MS intensifying screen | Sigma Aldrich | Z363162-1EA | For intensifying the autoradiogram signal |
| Chloroform | Thermo Fisher Scientific | 1010219 | For phenol-chloroform extraction following RNA purification |
| cOmplete EDTA-free protease inhibitor cocktail | Roche | 11873580001 | For inhibition of cellular proteases after lysis |
| Complete supplement mixture -TRP | Formedium | DCS0149 | For preparation of synthetic defined medium |
| Costar Spin-X 0.22 µm filters | Sigma Aldrich | CLS8160 | For isolating the excised cDNAs following gel extraction |
| DNase RQ1 | Promega | M6101 | For DNA digest following cell lysis |
| dNTPs (10 mM) | Sigma Aldrich | 4638956001 | For reverse transcription and PCR |
| Ethanol | Thermo Fisher Scientific | 10041814 | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Ethylenediaminetetraacetic acid | Invitrogen | AM9261 | For protease K buffer |
| Exonuclease I | New England Biolabs | M0293 | For degradation of primers following PCR |
| Glass microfiber filters | Whatman | 1823-010 | For isolating the excised cDNAs following gel extraction |
| Glucose | Formedium | GLU03 | For preparation of glucose-containing, synthetic defined medium |
| Glycogen (20 mg/mL) | Roche | 10901393001 | Precipitation of proteins, RNA and DNA |
| GST-TEV | Homemade | Construct and purification protocol is available upon request | |
| Guanidium hydrochloroide | Thermo Fisher Scientific | 10071503 | Required for pulldown denaturing conditions and washing buffer |
| IgG beads | GE Healthcare | 17-0969-01 | For purification of protein A-tagged RBPs |
| Imidazole | Sigma Aldrich | I2399-100G | For elution of captured proteins from Nickel beads |
| Isoamyl alcohol | Thermo Fisher Scientific | A393-500 | For phenol-chloroform extraction following RNA purification |
| Luna Universal One-Step RT-qPCR | NEB | E3005S | For qPCR of the cDNA in order to calculate required number of PCR cycles |
| Magnesium chloride | Fluka Analytical | 63020-1L | For PNK buffer |
| Membrane filters | Millipore | AAWP09000 for yeast or HAWP09000 for bacteria | For vacuum filtration of cells |
| Micro bio-spin columns | Biorad | 732-6204 | For collecting eluate after gel extraction |
| Ni-NTA beads | Qiagen | 30210 | For secondary protein capture |
| NP-40 | Sigma Aldrich | I8896-100ML | Buffer component |
| Pfu polymerase | Promega | M7741 | For amplification of the cDNA library |
| Phenol | Sigma Aldrich | P4682-400ML | For phenol-chloroform extraction following RNA purification |
| Pierce spin columns | Thermo Fisher Scientific | 69725 | For on-column enzymatic reactions |
| Protease K | Roche | 3115887001 | For degradation of the RBP following gel extraction |
| Quartz Petri dish | UVO3 | N/A | For cross-linking of adherent cells. Available from https://www.vari-x-link.com for 400 GBP |
| Radiography films | Amersham | 28906843 | For autoradiography visualisation |
| RNAClean XP beads | Beckmann | A63987 | SPRI beads for clean up of RNAs and cDNAs |
| RNase H | New England Biolabs | M0297 | For degradation of RNAs following reverse transcription |
| RNase-It | Agilent | 400720 | For RNA digestion |
| rRNasin | Promega | N2511 | For inhibition of any contaminating RNases during enzymatic reaction |
| Sodium acetate | Sigma Aldrich | S2889-1KG | For phenol-chloroform extraction following RNA purification and DNA precipitation |
| Sodium chloride | Thermo Fisher Scientific | 7647-14-5 | Buffer component |
| Sodium deoxycholate | Sigma Aldrich | D6750-100G | Buffer component in mammalian cell lysis |
| Sodium dodecylsulfate | Sigma Aldrich | L3771-1KG | For protease K buffer |
| SUPERase-In | Invitrogen | AM2694 | For inhibition of cellular RNases after lysis |
| SuperScript IV | Thermo Fisher Scientific | 18090010 | For reverse transcription |
| T4 PNK | New England Biolabs | M0201 | For radiolabelling the 5' end of the RNA |
| T4 RNA ligase 1 | New England Biolabs | M0204 | For ligation of the L5 adaptor onto the RNA 5' end |
| T4 RNase ligase 2, truncated K222Q | NEB | M0351S | For ligation of the App_PE linker onto the 3' end of captured RNAs |
| TBE buffer (10X) | Invitrogen | 15581-028 | For running TBE gels |
| TEV protease | Homemade | For eluting captured proteins following FLAG capture | |
| Thermosensitive alkaline phosphatase | Promega | M9910 | For 5' and 3' dephosphorylation of RNAs |
| Trichloroacetic acid (100%) | Sigma Aldrich | T0699-100ML | For precipitation of RBP-RNA complexes |
| Tris hydrochloride | Invitrogen | 15504-020 | Buffer component |
| Triton X-100 | Sigma Aldrich | T8787-100ML | Buffer component in mammalian cell lysis |
| Vari Filter | UVO3 | N/A | Device for vacuum harvesting cells. Available from https://www.vari-x-link.com for 100 GBP |
| Vari-X-Linker | UVO3 | N/A | Cross-linker for cross-linking cells. Available from https://www.vari-x-link.com for 16,000 GBP |
| Yeast nitrogen base | Formedium | CYN0410 | For preparation of synthetic defined medium |
| Zirconia beads | Thistle | 11079105Z for yeast or 11079101Z for bacteria | For cell lysis via bead beating |
Références
- Ule, J., et al. CLIP identifies Nova-regulated RNA networks in the brain. Science. 302 (5648), 1212-1215 (2003).
- Granneman, S., Kudla, G., Petfalski, E., Tollervey, D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proceedings of the National Academy of Sciences. 106 (24), 9613-9618 (2009).
- Licatalosi, D. D., et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 456 (7221), 464-469 (2008).
- König, J., et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nature Structural & Molecular Biology. 17 (7), 909-915 (2010).
- Hafner, M., et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell. 141 (1), 129-141 (2010).
- Aktaş, T., et al. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature. 544 (7648), 115-119 (2017).
- Huppertz, I., et al. iCLIP: Protein–RNA interactions at nucleotide resolution. Methods. 65 (3), 274-287 (2014).
- Li, X., et al. Comprehensive in vivo RNA-binding site analyses reveal a role of Prp8 in spliceosomal assembly. Nucleic Acids Research. 41 (6), 3805-3818 (2013).
- Rosenberg, M., et al. Denaturing CLIP, dCLIP, Pipeline Identifies Discrete RNA Footprints on Chromatin-Associated Proteins and Reveals that CBX7 Targets 3′ UTRs to Regulate mRNA Expression. Cell Systems. 5 (4), 368-385 (2017).
- Zarnegar, B. J., et al. irCLIP platform for efficient characterization of protein–RNA interactions. Nature Methods. 13 (6), 489-492 (2016).
- Kargapolova, Y., Levin, M., Lackner, K., Danckwardt, S. sCLIP—an integrated platform to study RNA–protein interactomes in biomedical research: identification of CSTF2tau in alternative processing of small nuclear RNAs. Nucleic Acids Research. 45 (10), 6074-6086 (2017).
- Van Nostrand, E. L., et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nature Methods. 13 (6), 508-514 (2016).
- Flynn, R. A., et al. Dissecting noncoding and pathogen RNA–protein interactomes. RNA. 21 (1), 135-143 (2015).
- Brugiolo, M., Botti, V., Liu, N., Müller-McNicoll, M., Neugebauer, K. M. Fractionation iCLIP detects persistent SR protein binding to conserved, retained introns in chromatin, nucleoplasm and cytoplasm. Nucleic Acids Research. 45 (18), 10452-10465 (2017).
- Sanford, J. R., et al. Identification of Nuclear and Cytoplasmic mRNA Targets for the Shuttling Protein SF2/ASF. PLOS ONE. 3 (10), e3369(2008).
- Garzia, A., Meyer, C., Morozov, P., Sajek, M., Tuschl, T. Optimization of PAR-CLIP for transcriptome-wide identification of binding sites of RNA-binding proteins. Methods. 118-119, 24-40 (2017).
- Windhager, L., et al. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Research. 22 (10), 2031-2042 (2012).
- Chen, K., et al. High-Resolution N6-Methyladenosine (m6A) Map Using Photo-Crosslinking-Assisted m6A Sequencing. Angewandte Chemie International Edition. 54 (5), 1587-1590 (2015).
- Ke, S., et al. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes & Development. 29 (19), 2037-2053 (2015).
- Linder, B., et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nature Methods. 12 (8), 767-772 (2015).
- Kudla, G., Granneman, S., Hahn, D., Beggs, J. D., Tollervey, D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proceedings of the National Academy of Sciences. 108 (24), 10010-10015 (2011).
- Sugimoto, Y., et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature. 519 (7544), 491-494 (2015).
- Hwang, H. W., et al. cTag-PAPERCLIP Reveals Alternative Polyadenylation Promotes Cell-Type Specific Protein Diversity and Shifts Araf Isoforms with Microglia Activation. Neuron. 95 (6), 1334-1349 (2017).
- Hwang, H. W., et al. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Reports. 15 (2), 423-435 (2016).
- Lee, F. C. Y., Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Molecular Cell. 69 (3), 354-369 (2018).
- Beckmann, B. M. RNA interactome capture in yeast. Methods. 118-119, 82-92 (2017).
- Granneman, S., Petfalski, E., Tollervey, D. A cluster of ribosome synthesis factors regulate pre-rRNA folding and 5.8S rRNA maturation by the Rat1 exonuclease. The EMBO Journal. 30 (19), 4006-4019 (2011).
- Schaughency, P., Merran, J., Corden, J. L. Genome-Wide Mapping of Yeast RNA Polymerase II Termination. PLOS Genetics. 10 (10), e1004632(2014).
- Bernstein, J. A., Khodursky, A. B., Lin, P. H., Lin-Chao, S., Cohen, S. N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proceedings of the National Academy of Sciences. 99 (15), 9697-9702 (2002).
- Kresnowati, M. T. A. P., et al. When transcriptome meets metabolome: fast cellular responses of yeast to sudden relief of glucose limitation. Molecular Systems Biology. 2, 49(2006).
- Marguerat, S., Lawler, K., Brazma, A., Bähler, J. Contributions of transcription and mRNA decay to gene expression dynamics of fission yeast in response to oxidative stress. RNA Biology. 11 (6), 702-714 (2014).
- van Nues, R., et al. Kinetic CRAC uncovers a role for Nab3 in determining gene expression profiles during stress. Nature Communications. 8 (1), 12(2017).
- Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M., Rosenow, C. Global RNA Half-Life Analysis in Escherichia coli Reveals Positional Patterns of Transcript Degradation. Genome Research. 13 (2), 216-223 (2003).
- Tudek, A., Candelli, T., Libri, D. Non-coding transcription by RNA polymerase II in yeast: Hasard or nécessité? Biochimie. 117, 28-36 (2015).
- Lingaraju, M., et al. The MTR4 helicase recruits nuclear adaptors of the human RNA exosome using distinct arch-interacting motifs. Nature Communications. 10 (1), 1-11 (2019).
- Lubas, M., et al. Interaction Profiling Identifies the Human Nuclear Exosome Targeting Complex. Molecular Cell. 43 (4), 624-637 (2011).
- Conrad, N. K., et al. A yeast heterogeneous nuclear ribonucleoprotein complex associated with RNA polymerase II. Genetics. 154 (2), 557-571 (2000).
- Darby, M. M., Serebreni, L., Pan, X., Boeke, J. D., Corden, J. L. The Saccharomyces cerevisiae Nrd1-Nab3 Transcription Termination Pathway Acts in Opposition to Ras Signaling and Mediates Response to Nutrient Depletion. Molecular and Cellular Biology. 32 (10), 1762-1775 (2012).
- Webb, S., Hector, R. D., Kudla, G., Granneman, S. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biology. 15 (1), R8(2014).
- Jensen, T. H., Jacquier, A., Libri, D. Dealing with Pervasive Transcription. Molecular Cell. 52 (4), 473-484 (2013).
- van Dijk, E. L., et al. XUTs are a class of Xrn1-sensitive antisense regulatory non-coding RNA in yeast. Nature. 475 (7354), 114-117 (2011).
- Thiebaut, M., et al. Futile Cycle of Transcription Initiation and Termination Modulates the Response to Nucleotide Shortage in S. cerevisiae. Molecular Cell. 31 (5), 671-682 (2008).
- Merran, J., Corden, J. L. Yeast RNA-Binding Protein Nab3 Regulates Genes Involved in Nitrogen Metabolism. Molecular and Cellular Biology. 37 (18), e00154-e00117 (2017).
- Bresson, S., Tuck, A., Staneva, D., Tollervey, D. Nuclear RNA Decay Pathways Aid Rapid Remodeling of Gene Expression in Yeast. Molecular Cell. 65 (5), 787-800 (2017).
- Buchbender, A., et al. Improved library preparation with the new iCLIP2 protocol. Methods. , (2019).
Erratum
Formal Correction: Erratum: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC
Posted by JoVE Editors on 2/17/2023. Citeable Link.
An erratum was issued for: Monitoring Protein-RNA Interaction Dynamics in vivo at High Temporal Resolution using χCRAC. The Authors section was updated from:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Synthetic and Systems Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
to:
Stuart W. McKellar1
Ivayla Ivanova1
Robert W. van Nues2
Ross A. Cordiner3
Mehak Chauhan1
Niki Christopoulou1
Will Worboys4
Andrew Langford4
Torben Heick Jensen3
Sander Granneman1
1Centre for Engineering Biology, University of Edinburgh
2Institute of Cell Biology, University of Edinburgh
3Department of Molecular Biology and Genetics, Aarhus University, 4UVO3 Ltd.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.