
Method Article
Fluoreszenzaktivierte Kerne negative Sortierung von Neuronen kombiniert mit Einzelkern-RNA-Sequenzierung zur Untersuchung der neurogenen Hippocampus-Nische
In diesem Artikel
Zusammenfassung
Hier wird eine Methode zur Sequenzierung einzelner Kerne, die aus dem Gyrus dentatus der Maus isoliert wurden, vorgestellt, die die meisten Neuronen durch Fluoreszenz-aktivierte Kerne (FAN)-Sortierung ausschließt. Dieser Ansatz generiert qualitativ hochwertige Expressionsprofile und erleichtert die Untersuchung der meisten anderen Zelltypen, die in der Nische vertreten sind, einschließlich seltener Populationen wie neuraler Stammzellen.
Zusammenfassung
Die adulte Hippocampus-Neurogenese (AHN), die aus einer lebenslangen Aufrechterhaltung proliferativer und ruhender neuraler Stammzellen (NSCs) innerhalb der subgranulären Zone (SGZ) des Gyrus dentatus (DG) und ihrer Differenzierung von neugeborenen Neuronen in Körnerzellen in der Körnerzellschicht besteht, ist in zahlreichen Studien gut validiert. Die Verwendung genetisch veränderter Tiere, insbesondere Nagetiere, ist ein wertvolles Werkzeug, um Signalwege zu untersuchen, die AHN regulieren, und um die Rolle jedes Zelltyps zu untersuchen, aus dem sich die neurogene Nische des Hippocampus zusammensetzt. Um letzteres anzugehen, haben Methoden, die die Isolierung einzelner Kerne mit der Sequenzierung der nächsten Generation kombinieren, einen signifikanten Einfluss auf das Gebiet der AHN gehabt, um Gensignaturen für jede Zellpopulation zu identifizieren. Eine weitere Verfeinerung dieser Techniken ist jedoch erforderlich, um seltenere Zellpopulationen innerhalb der DG phänotypisch zu profilieren. Hier stellen wir eine Methode vor, die Fluorescence Activated Nuclei Sorting (FANS) verwendet, um die meisten neuronalen Populationen aus einer Einzelkernsuspension auszuschließen, die aus frisch seziertem DG isoliert wurde, indem ungefärbte Kerne für das NeuN-Antigen ausgewählt werden, um eine Einzelkern-RNA-Sequenzierung (snRNA-seq) durchzuführen. Diese Methode ist ein potenzielles Sprungbrett, um die interzelluläre Regulation des AHN weiter zu untersuchen und neue zelluläre Marker und Mechanismen über Spezies hinweg aufzudecken.
Einleitung
Die kontinuierliche Erzeugung von Hippocampus-Neuronen im Erwachsenenalter, auch bekannt als adulte Hippocampus-Neurogenese (AHN), ist mit kognitiven Funktionen wie Lernen, Gedächtniserwerb / -clearance und Mustertrennung verbunden und scheint ein wichtiger Mechanismus der Resilienz bei Alterung und neurodegenerativen Erkrankungen zu sein, um kognitive Defizite zu verhindern 1,2,3 . Nagetiere waren das Modell der Wahl, um AHN mit verschiedenen Methoden zu untersuchen, einschließlich Immunzytochemie und Next-Generation-Sequencing (NGS) -Methoden. Die Übertragung dieser Ergebnisse auf andere Arten bleibt umstritten. Tatsächlich wurde AHN bei den meisten Arten beobachtet, aber das Ausmaß, in dem es während des gesamten Lebens fortbesteht, insbesondere beim Menschen 4,5,6,7,8, wird regelmäßig diskutiert.
Bisher wurde bestätigt, dass verschiedene intrinsische und extrinsische Signalwege AHN1 modulieren. Die Auswirkungen der interzellulären Kommunikation auf AHN zeichnen sich jedoch gerade erst ab9. Dies konnte zunächst auf eine unzureichende Spezifität der derzeit bekannten Zellmarker zurückgeführt werden, um in vivo Analysen mit gentechnisch veränderten Tieren durchzuführen. Tatsächlich haben sich viele Studien auf Marker wie Doublecortin oder Gliafibrilläres saures Protein (GFAP) gestützt, die in mehreren Zelltypen exprimiertwerden 1. Zweitens bringt die Komplexität und der hohe Grad an Zelldiversität in der adulten Hippocampus-Nische10 technische Herausforderungen für die Profilierung jedes Zelltyps mit sich. Dies gilt insbesondere für die bioinformatische Analyse mit überlappenden zellulären Markern, die in analytischen Pipelines für verschiedene Populationen wie NSCs oder Gliazellen verwendet werden, was zu kontroversen Schlussfolgerungen bei der Bewertung von AHN 7,11 führt. Drittens untergräbt die große Anzahl von Neuronen die Untersuchung von weniger häufigen Zellpopulationen wie Astrozyten, Oligodendrozyten oder Ependymzellen, obwohl ihre Rolle bei der Feinabstimmung der AHN-Regulation immer wichtiger wird9. Zusammengenommen wirken sich diese Einschränkungen auf die Fähigkeit aus, Ergebnisse von Nagetieren auf andere Arten zu übertragen. Dies wird besonders verstärkt durch die Schwierigkeit, ein komplexes Gewebe wie die neurogene Nische des Hippocampus in vitro zu rekapitulieren, und durch die vielen Hürden für den Zugang zu qualitativ hochwertigem Gewebe zusammen mit dem Fehlen standardisierter Protokolle für die Gewebeverarbeitung in Studien mit menschlichem Gewebe12,13. Es ist daher wichtig, neue Ansätze zur Profilierung von Zellpopulationen zu entwickeln und neue zelluläre Marker innerhalb des Gyrus dentatus (DG) zu identifizieren, die letztendlich zu einem besseren Verständnis der verschiedenen Beiträge jedes Zelltyps zur AHN-Regulation führen.
Um dies zu erreichen, ist die Isolierung einzelner Zellen (sc) und einzelner Kerne (sn) in Kombination mit RNA-Sequenzierung entscheidend geworden, um komplexe Gewebe wie die DG14 zu untersuchen. Daher wurden Strategien der zellulären Anreicherung zur Isolierung einzelner Zellen aus der erwachsenen Hippocampus-Nische der Maus hauptsächlich zur Untersuchung von NSCsdurchgeführt 15,16. Eine interessante Strategie zur Anreicherung nicht-neuronaler Zellen aus der DG wurde durch die Sequenzierung von GluR1/Cd24 doppelnegativen Einzelzellen angewendet, was dazu führte, dass 1.408 Zellen ohne eindeutige Cluster zwischen Astrozyten und NSCs nach bioinformatischer Analyse sequenziert wurden17. Dies könnte auf die harte enzymatische Verdauung zurückzuführen sein, die für die Einzelzellpräparation erforderlich ist und die Zellintegrität und RNA schädigt. Um dieses technische Problem zu umgehen, wurden mehrere Methoden entwickelt, die stattdessen Einzelkernisolierung verwenden und besonders für komplizierte Gewebe geeignet sind11,18. Die Dominanz von Neuronen innerhalb der DG oder allgemeiner innerhalb des Hippocampus-Entorhinal-Systems erzeugt jedoch eine Stichprobenverzerrung, um die Gesamtheit der Zellpopulationen in diesen Gehirnbereichen zu untersuchen. Darüber hinaus akzentuiert die begrenzte Anzahl von Zellen, die für die Herstellung von Einzelzellbibliotheken geladen werden müssen, das Vorhandensein der Hauptzellpopulation in analytischen Pipelines sequenzierter Einzelkerne. Tatsächlich werden große neuronale Cluster oft annotiert und analysiert, während andere Zellpopulationen unterrepräsentiert sind oder übersehen werden 5,11.
In einem Versuch, diese Verzerrungen zu überwinden und in der Lage zu sein, andere Zelltypen als Neuronen zu profilieren, die in der Maus-DG vorhanden sind, wurde in dieser Studie eine Methode entwickelt, die das Prinzip der Fluorescence Activated Nuclei Sorting (FANS)18 verwendet, die die meisten neuronalen Populationen durch negative Selektion von gefärbten Einzelkernen mit neuronalem Kernantigen (NeuN, auch bekannt als Rbfox3). Diese Wahl des Antigens wurde von der Literatur geleitet, die NeuN als zuverlässigen neuronalen Marker19 beschreibt, und von der Notwendigkeit, ein Kernprotein für diesen Ansatz zu verwenden. NeuN-negative FACS-sortierte Zellen wurden dann auf einer 10-fachen Genomics-Plattform für die RNA-Sequenzierung vorbereitet. Die Ergebnisse zeigen, dass der Ausschluss von NeuN-exprimierenden Zellen ein zelltypspezifisches, qualitativ hochwertiges transkriptomisches Profiling von Glia- und seltenen Zellpopulationen ermöglicht.
Protokoll
Tierpflege und experimentelle Verfahren wurden in Übereinstimmung mit den Richtlinien des Francis Crick Institute sowie den Richtlinien und Gesetzen des britischen Innenministeriums durchgeführt.

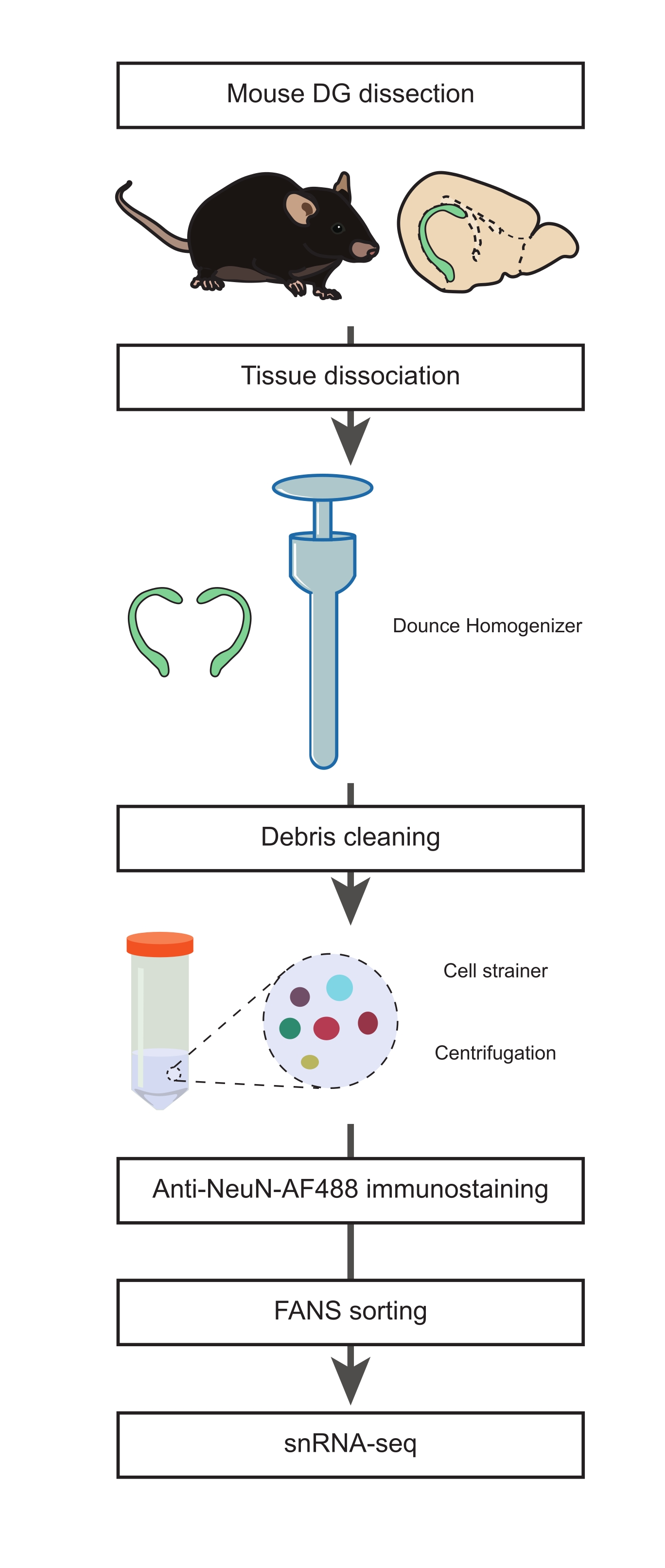
Abbildung 1: Herstellung einer Einzelkernsuspension aus der sezierten DG erwachsener Mäuse für snRNA-seq nicht-neuronaler Populationen. Flussdiagramm, das die wichtigsten Schritte des Protokolls beschreibt, einschließlich Dissektion der Maus-DG, Vorbereitung einer Suspension einzelner Kerne, NeuN-Immunfärbung und negative NeuN-FANS-Sortierung, bevor mit der snRNA-seq fortgefahren wird. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
1. Dissektion der DG (Dauer: 15 min)
- Bereiten Sie die Kernisolierungsmedien 1 und 2 (NIM1 und NIM2), den Homogenisierungspuffer (HB) und das Waschmedium (WM) vor (Zusatztabelle 1). Legen Sie alle Puffer, Medien, Reagenzien und Werkzeuge auf Eis, bis sie benötigt werden. Legen Sie den Dounce-Homogenisator (siehe Materialtabelle) während der Vorbereitung (mindestens 1 Stunde vor dem Homogenisierungsschritt) auf Eis.
HINWEIS: NIM1 kann zubereitet und bei 4 °C bis zu 6 Monate gelagert werden. NIM2, HB und WM sollten frisch zubereitet werden.
VORSICHT: Manipulieren Sie DTT, den Proteasehemmer und Triton X-100 mit Vorsicht. Diese Verbindungen sind haut- und augenreizend, akut toxisch und gefährlich für die aquatische Umwelt. Tragen Sie während der Verwendung dieser Chemikalien Schutzhandschuhe, Kleidung, Augen- und Gesichtsschutz, waschen Sie sich nach der Handhabung gründlich die Hände und vermeiden Sie die Freisetzung in die Umwelt. - Euthanasieren Sie eine 22 Monate alte männliche C57Bl / 6J-Maus durch zervikale Dislokation nach dem Home Office Schedule 1-Verfahren20.
HINWEIS: Siehe Diskussion für die Begründung bezüglich der Verwendung von 22 Monate alten Mäusen in dieser Studie. Dieses Protokoll kann jedoch in jedem Alter über die gesamte Lebensdauer hinweg durchgeführt werden. - Sezieren Sie das Gehirn aus einer eingeschläferten Maus und übertragen Sie es in eine 10 cm große Petrischale, die mit eiskaltem 1x PBS gefüllt ist (Abbildung 1). Die Petrischale auf Eis legen. Entfernen Sie das Kleinhirn mit einem Skalpell und schneiden Sie das Gehirn zwischen beiden Hemisphären (entlang der sagittalen Achse) in zwei Hälften.
- Füllen Sie eine neue 10 cm Petrischale mit eiskaltem PBS und legen Sie sie auf Eis. Übertragen Sie eine Hälfte des Gehirns auf die neue Petrischale. Sezieren Sie mit einem Fernglas die DG und wiederholen Sie diesen Schritt, um die zweite DG aus der zweiten Hälfte des Gehirns zu erhalten.
HINWEIS: Diese Schritte (Schritte 1.2-1.4) wurden von einem zuvor beschriebenen Verfahren21 übernommen. Es ist wichtig, in diesem Stadium so schnell wie möglich vorzugehen, um die Integrität der Zellen zu erhalten. - Die beiden DGs werden in den vorgekühlten Dounce-Homogenisator gegeben und 1 mL kaltes HB hinzugefügt.
2. Gewebedissoziation, Einzelkernisolierung und Anti-NeuN-Immunfärbung (Timing: 2 h)
- Homogenisieren Sie das Gewebe mit 10 Schlägen des losen "A" -Stößels, gefolgt von 15 Schlägen des engen "B" -Stößels.
HINWEIS: Die Dounce-Homogenisierung sollte mit dem Mörtel auf Eis mit sanften Hüben durchgeführt werden, um die durch Reibung und Schaumbildung verursachte Wärme zu reduzieren. Alle Puffer und Geräte sollten vorgekühlt und während des Verfahrens auf Eis gehalten werden. - Das Homogenat in ein vorgekühltes 15-ml-Röhrchen überführen; Spülen Sie den Dounce-Homogenisator mit 1 ml kaltem HB aus und kombinieren Sie ihn zum selben Röhrchen. 3 ml HB in das 15 ml Röhrchen geben und 5 min auf Eis inkubieren. Mischen Sie 2x, indem Sie die Tube vorsichtig umdrehen.
- Eine 70-μm-Siebkappe mit 0,5 mL HB über einem 50-ml-Reagenzglas vorbenetzen. Die Kernsuspension aus Schritt 2.2 abseihen, indem Sie das 15-ml-Röhrchen vorsichtig in das Zellsieb kippen. Waschen Sie das Zellsieb mit 0,5 ml HB.
- Entfernen Sie das Zellsieb und zentrifugieren Sie das Reagenzglas bei 500 x g für 5 min bei 4 °C mit einer Schaufelzentrifuge. Verwerfen Sie den Überstand.
HINWEIS: Das Belasten des Homogenats hilft, die Ablagerungen zu reduzieren, was für die Durchflusszytometrie und die nachgeschalteten Schritte der snRNA-Seq entscheidend ist. - Resuspendieren Sie das Pellet vorsichtig in 4 ml HB mit einer P1000-Pipette. Auf Eis 5 min inkubieren. Bei 500 x g 10 min bei 4 °C schleudern. Verwerfen Sie den Überstand und resuspendieren Sie das Pellet in 3 ml WM.
- Eine 35-μm-Siebkappe über einem 15-ml-Reagenzglas mit 0,5 ml WM vorbenetzen. Die Kernsuspension ab Schritt 2.5 durch das Zellsieb abseihen, wobei jeweils 0,5 ml mit einer P1000-Pipette vorsichtig pipettiert werden.
- Waschen Sie die Siebkappe mit 0,5 ml WM und legen Sie das Röhrchen auf Eis. Das Filtrat in ein neues 15-ml-Röhrchen überführen und 5 min und 4 °C bei 500 x g zentrifugieren. Verwerfen Sie den Überstand und resuspendieren Sie das Pellet in 3 ml WM.
- Bei 500 x g 5 min bei 4 °C schleudern. Verwerfen Sie den Überstand und resuspendieren Sie das Pellet in 1 ml WM mit dem Maus-Anti-NeuN, Alexa Fluor 488-konjugierten Antikörper (Anti-NeuN-AF488, 1:32.000) und 1 μg/ml DAPI. 45 min auf Eis im Dunkeln inkubieren.
HINWEIS: Um die Immunfärbung isolierter Kerne zu optimieren, wird empfohlen, den Antikörper zu titrieren, um die optimale Verdünnung für die Durchflusszytometrie-Analyse und -Sortierung zu bestimmen. Führen Sie dann geeignete Kontrollen durch, um zu bestätigen, dass die Färbebedingungen optimal sind. Zum Beispiel wurden mit dem konjugierten Anti-NeuN-AF488-Antikörper eine Negativkontrolle (d.h. keine Addition des Antikörpers, ergänzende Abbildung 1A) und eine Positivkontrolle (d.h. Färbung mit dem Antikörper, ergänzende Abbildung 1B) durchgeführt, um die Segregation von ungefärbten und gefärbten Populationen zu beurteilen. Wenn Sie mit der Arbeit mit einem AF488-konjugierten Antikörper beginnen, wird empfohlen, eine AF488-konjugierte Isotypkontrolle durchzuführen, um die Spezifität zu beurteilen. Wenn ein nicht konjugierter Antikörper verwendet wird, kann eine zusätzliche Kontrolle erforderlich sein, wie z. B. die Zugabe eines sekundären Antikörpers nur zu einem Kernpräparat, um die unspezifische Bindung des sekundären Antikörpers zu beurteilen.
3. Fluoreszenzaktivierte Kernsortierung (FANS) zum Ausschluss neuronaler Populationen (Dauer: 45 min)
- Die immungefärbte Kernsuspension wird in ein 5-ml-Reagenzglas überführt und bis zum Beginn der Durchflusszytometrie auf Eis gehalten.
HINWEIS: Wenn mit größeren Gewebestücken als zwei Maus-DG gearbeitet wird, kann eine weitere Verdünnung mit WM-Puffer erforderlich sein, um eine Verstopfung des FACS zu vermeiden, wenn die Kerndichte in der Lösung hoch wird. - Wirbeln Sie die Proben 3 s lang bei niedriger Geschwindigkeit durch, bevor die Röhrchen in das FACS-Instrument eingesetzt werden (siehe Materialtabelle).
HINWEIS: (FACS-Setup) Sortiermaschinen müssen zu Beginn des Verfahrens mit Kalibrierpartikeln gemäß den Empfehlungen des Herstellers ausgerichtet werden. Die Fallverzögerung wurde mit Perlen oder Mikrokugeln (siehe Materialtabelle) nach dem FACS-Modell kalibriert. Die Proben wurden bei 4 °C im Reinheitsmodus sortiert. Um das Sammelvolumen zu reduzieren, wurden die Kerne durch eine 70 μm Düse mit dem für das Durchflusszytometer empfohlenen Druck sortiert. Die Kerne wurden in 1,5 mL Low-Binding-Röhrchen sortiert (siehe Materialtabelle), die 50 μL WM enthielten. Alle Sammelröhrchen wurden über Nacht bei 4 °C mit PBS + 5% BSA beschichtet, um das Risiko zu verringern, dass Kerne an den Wänden der Röhre haften bleiben. - Um die Daten aus einer Probe der gefärbten Kernsuspension zu erhalten, setzen Sie die Gatter in DAPI-Höhe und DAPI-Bereich, um die Zelltrümmer und die aggregierten Kerne auszuschließen (Abbildung 2A). Darüber hinaus trennen Sie einzelne Kerne von allen verbleibenden DAPI-gefärbten Aggregaten oder Zelltrümmern, indem Sie die Gatter im log Side Scatter (SSC)-Bereich und im log Forward scatter (FCS)-Bereich setzen (Abbildung 2B).
- Stellen Sie die Gates für den Anti-NeuN-AF488 und den FSC-Bereich ein, um die NeuN-AF488-negative Population zu isolieren, wie in Abbildung 2C dargestellt.
- Sortieren Sie nach der Analyse mit der oben beschriebenen Gating-Strategie die NeuN-AF488-negative Population in ein 1,5-ml-Sammelröhrchen, das mit 50 μL WM gefüllt ist.
HINWEIS: Nach der oben beschriebenen Gating-Strategie und dem Dissektionsverfahren zur Isolierung der DG aus dem Gehirn einer erwachsenen Maus wird erwartet, dass die NeuN-AF488-negative Population ~14% der einzelnen Kerne ausmacht.

Abbildung 2: Isolierung und transkriptomisches Profiling von nicht-neuronalen Zellpopulationen aus der DG. (A-C) Gating-Strategie zur Isolierung von NeuN-AF488-negativen Einzelkernen und zum Ausschluss von Zelltrümmern. (A) FANS-Punktdiagramm einer repräsentativen Stichprobe isolierter Kerne, das die Gate-Einstellung für die Auswahl von DAPI+-Kernen und den Ausschluss von Zelltrümmern und -aggregaten darstellt. (B) Weitere Selektion relevanter Einzelkerne unter Verwendung von FSC-Bereich und SSC-Bereich. (C) Die Tore für NeuN-AF488, um die positive Population auszuschließen und nach den negativen Einzelkernen zu sortieren. (D) Schliffaufnahme einer guten Einzelkernsuspension mit minimaler Menge an Trümmern und höherem Anteil an Kernen guter Qualität (runde Form, schwarzer Pfeil) im Vergleich zu Kernen schlechter Qualität (weißer Pfeil). Maßstäbe = 50 μm, 10 μm (Einschub). (E,F) Analyse von snRNA-seq-Daten und Profilierung der verschiedenen Zellpopulationen, die aus der DG von 22 Monate alten männlichen C57BL/6J-Mäusen isoliert wurden. UMAP-Diagramme (Uniform Manifold Approximation and Projection for Dimension Reduction) von Einzelkernprofilen aus den (E) nicht FACS-sortierten Zellen und (F) NeuN-negativen FACS-sortierten Zellen, eingefärbt nach Zelltyp. (G) Kreisdiagramme, die die Häufigkeit identifizierter Zelltypen in beiden Stichproben vergleichen. (H) Entsprechende Metriken für die sequenzierten Proben: Anzahl der Kerne, mediane Anzahl der Gene und Transkripte pro Kern. (I) Geigendiagramme, die die Verteilung der Anzahl der Gene und Transkripte zeigen, die für jeden Zelltyp in beiden Proben nachgewiesen wurden. Astr. = Astrozyten, Olig. = Oligodendrozyten, Vasc. = Gefäßzellen, CRCs = Cajal-Retzius-Zellen, Neur. = Neuronen, Imm. = Immunzellen, OPCs = Oligodendrozyten-Vorläuferzellen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
4. Vorbereitung der Einzelkernsuspension zur Durchführung einer Einzelkern-RNA-Sequenzierung (Dauer: 30 min)
- Nach dem Sortieren 1 ml PBS mit 1% BSA in das Auffangröhrchen geben, um Tröpfchen an der Wand des Röhrchens aufzufangen, und 5 min bei 4 °C mit 500 x g schleudern. Verwerfen Sie den Überstand und lassen Sie 50 μL zurück.
HINWEIS: Behandeln Sie die zentrifugierten Kerne mit Vorsicht, da es schwierig sein kann, Pellets am Boden des Röhrchens zu beobachten. Die Verwendung einer Schaufelzentrifuge hilft, den Überstand zu verwerfen, ohne das Pellet zu stören. - Pipeten Sie vorsichtig, um zentrifugierte Kerne zu resuspendieren. Man gibt 5 μL der Kernsuspension zu 5 μL Trypanblau in einem 0,5 ml Mikroröhrchen.
VORSICHT: Behandeln Sie Trypan blue mit Vorsicht, da es gesundheitsgefährdend ist, Krebs verursachen kann und im Verdacht steht, die Fruchtbarkeit oder das ungeborene Kind zu schädigen. Tragen Sie Schutzhandschuhe, Kleidung sowie Augen- und Gesichtsschutz. Erst handhaben, wenn alle Sicherheitsvorkehrungen gelesen und verstanden wurden. - Messung der Konzentration und Beurteilung der Lebensfähigkeit der Einzelzellsuspension mit einem Hämozytometer oder einem automatisierten Zellzähler (siehe Materialtabelle). Führen Sie die Bibliotheksvorbereitung und Sequenzierung der Kerne durch, wie in Schritt 5 beschrieben.
HINWEIS: Proben, die für die Sequenzierung als von guter Qualität erachtet wurden, zeigten unter dem Mikroskop eine runde und regelmäßige Kernform ohne Zelltrümmer (Abbildung 2D). Das Vorhandensein eines Halos um die Kernmembran oder die Aggregation mehrerer Kerne zusammen sind Anzeichen für beschädigte Kerne, und solche Zellsuspensionen sollten für snRNA-seq nicht in Betracht gezogen werden (Abbildung 2D). Die gemessene Konzentration der Kerne lag im Bereich von 300-700 Kernen/μL.
5. Bibliotheksvorbereitung und -ablauf
HINWEIS: Die Beschreibung der folgenden Schritte basiert auf der in dieser Studie verwendeten internen Sequenzierungsplattform (siehe Materialtabelle). Daher können einige Einstellungen abweichen, wenn Sie eine andere Plattform verwenden. Hier werden nur die wichtigsten Schritte beschrieben und jeder Parameter sollte nach Anleitung und Protokollen des ausgewählten Herstellers bestimmt werden, wenn auch mit Optimierung vor der ersten Verwendung. Es muss unbedingt sichergestellt werden, dass die Vorbereitung der Bibliotheken so schnell wie möglich nach der Konzentration sortierter Kernsuspensionen erfolgt, um einen RNA-Abbau zu vermeiden und eine optimale Qualität der Sequenzierung zu gewährleisten.
- Laden Sie zwischen 7.000 und 10.000 Kerne in einen Mikrofluidik-Einzelzellchip.
- Partition geladene Kerne in Tröpfchen im Nanoliterbereich mit dem mitgelieferten Controller und den Reagenzien des ausgewählten Lieferanten. Lysieren Sie Kerne innerhalb jedes Tröpfchens und transkribieren Sie RNA.
HINWEIS: Innerhalb eines Tröpfchens teilte sich die gesamte resultierende cDNA denselben Zellbarcode. - Bereiten Sie Bibliotheken für snRNA-seq gemäß den Richtlinien des ausgewählten Anbieters vor und stellen Sie die Kompatibilität mit der Sequenzierungsplattform sicher. Überprüfen Sie die Qualität und Konzentration der endgültigen Bibliotheken mit Elektrophorese, Fluorometrie oder qPCR-basierten Methoden und bündeln Sie sie gegebenenfalls äquimolar vor der Sequenzierung.
- Denaturierung gepoolte 3ʹ Genexpressionsbibliotheken und verdünnte sie gemäß der Empfehlung des Herstellers.
- Führen Sie eine Paired-End-, Single- oder Dual-Indexing-Sequenzierung auf einer Sequenzierungsplattform der nächsten Generation mit einer Sequenzierungstiefe von 50.000 Lesepaaren pro Zelle durch.
Ergebnisse
Das hier vorgestellte Protokoll beschreibt eine Methode zur Herstellung einer Suspension von nicht-neuronalen Einzelkernen, die aus der DG isoliert wurden, um snRNA-seq durchzuführen. Mit oder ohne FANS zeigte das bioinformatische Clustering gut getrennte Gruppen von Kernen, die bekannten Zelltypen innerhalb der DG entsprechen (Abbildung 2E,F). Innerhalb der nicht FACS-sortierten Probe bestand die Mehrheit der hochwertigen Kerne, die sequenziert wurden, aus drei Gruppen von Neuronen (84,9% der gesamten Kerne für diese Probe, Abbildung 2E,G,H). Solche Ergebnisse werden erwartet, wenn man bedenkt, dass die am stärksten vertretenen Zellpopulationen in der DG granuläre Neuronen, andere erregende Neuronen (markierte exzitatorische Neuronen) und inhibitorische Neuronensind 10. Die identifizierten nicht-neuronalen Cluster bestanden hauptsächlich aus Gliazelltypen (11,1%), darunter Astrozyten, Oligodendrozyten und Oligodendrozytenvorläuferzellen (OPCs), Immunzellen (3,3%) und Cajal-Retzius-Zellen (0,6%). Bei der Durchführung von FANS zum Ausschluss von NeuN-positiven Populationen (NeuN-negative FACS-sortierte Stichprobe; Abbildung 2F,G,H) dominierten Cluster von Gliazellen (81,3%). Die Isolierung einer größeren Anzahl von Gliakernen ermöglicht eine bessere Segmentierung verschiedener Populationen, die sich ohne FANS zusammenballen würden. Tatsächlich trennten sich bei der erneuten Clusterung und Analyse spezifischer Gene, die entweder in NSCs oder in Astrozyten exprimiert wurden, vier Subcluster (ergänzende Abbildung 2A,B). Unter Berücksichtigung spezifischerer zellulärer Marker und der Bewertung der Genexpressionsniveaus über die Zelltypen hinweg wurde eine kleine Gruppe von NSCs nachgewiesen, die getrennt von den astrozytären Hauptpopulationen getrennt waren, mit einer höheren Expression von Hopx und Notch2 und fast keiner Expression von Aldh1a1 oder Aqp4 (ergänzende Abbildung 2C). Aufgrund der Überlappung der Genexpression zwischen Astrozyten und NSCs wären jedoch weitere Analysen erforderlich, um verschiedene Subtypen von Zellen spezifisch zu profilieren und zu identifizieren. Darüber hinaus hatte die NeuN-negative FANS-Stichprobe zusätzliche Cluster, die als vaskuläre Zellen markiert waren (2,3%), die Endothelzellen, Perizyten und vaskuläre leptomeningeale Zellen umfassen, wenn sie für die Expression zellspezifischer Marker querreferenziert wurden (Daten nicht gezeigt).
Nach der Anleitung für das gewählte Protokoll zur Generierung von Bibliotheken für die Sequenzierung wurden qualitativ hochwertige Expressionsprofile mit oder ohne FANS erhalten. Für Proben, die mit 50.000 Reads/Kern sequenziert wurden, wurden durchschnittlich 2.510 Gene pro Kern für die nicht FACS-sortierte Probe (5.578 Transkripte, Abbildung 2H) und 1.665,5 Gene (3.508 Transkripte) für NeuN-negative FANS-Probe nachgewiesen, nachdem sie minderwertige Kerne herausgefiltert hatten (Abbildung 2H, I). Diese Metriken bestätigen, dass dieses Protokoll ein qualitativ hochwertiges transkriptomisches Profiling von Einzelkernen erzeugt, vergleichbar mit Studien mit verschiedenen Ansätzen22,23 und dass der Prozess der FACS-Sortierung die Kerne für nachfolgende snRNA-seq nicht schädigt. Bemerkenswerterweise ist der Unterschied in der Anzahl der Gene und Transkripte pro Kern zwischen den beiden Proben nicht auf eine geringere Datenqualität zurückzuführen, sondern auf den hohen Anteil an Neuronen in der nicht-FACS-sortierten Stichprobe (84,9% im Vergleich zu 1,7% in der NeuN-negativen FANS-Probe), die eine höhere Transkriptionsaktivität aufweisen (2.660 Gene/Kern und 6.170 Transkripte/Kern in nicht FACS-sortierten Proben) als die durchschnittliche Transkriptionsaktivität aller nicht-neuronalen Zelltypen (1.090 Gene/Zellkern und 1.785 Transkripte/Zellkern, Abbildung 2I).
Zusammengenommen zeigen diese repräsentativen Ergebnisse, dass die Selektion von NeuN-negativen Kernen mit FANS ein leistungsfähiges Werkzeug ist, um Zelltypen mit geringer Häufigkeit aus frisch seziertem Hirngewebe zu isolieren und qualitativ hochwertige Einzelkern-Transkriptom-Profile dieser unterschiedlichen Zellpopulationen mittels snRNA-seq-Methoden durchzuführen.
Ergänzende Abbildung 1: Validierung der Immunfärbung für FANS. Die Kernsuspension wurde (A) ohne den Anti-NeuN-AF 488-Antikörper als Negativkontrolle oder (B) mit dem Antikörper inkubiert und durch den FACS-Sortierer geführt, um die Immunfärbungsbedingungen zu validieren. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung 2: Genexpressionsanalyse und Re-Clustering des Astrozytenhaufens. (A) UMAP-Diagramm (Uniform Manifold Approximation and Projection for Dimension Reduction), das die Clusterbildung von 4968 Kernen basierend auf genomweiten Expressionsprofilen aus Abbildung 2F zeigt. Zelltypaufrufe wurden basierend auf Zelltypmarkern durchgeführt. (B) Astrozytencluster bestehend aus 2579 Kernen, ausgewählt aus (A) für weitere Sub-Setting zur Untersuchung potenzieller zellulärer Subtypen. Vier Subtypen wurden durch Seurat (0-3) Clustering detektiert, dargestellt durch verschiedene Farben. (C) Genexpressionsniveaus spezifischer zellulärer Marker in den vier Zelltypen. Alle Parzellen wurden mit dem Seurat R-Paket24 erhalten. Kurz gesagt, die RNA-seq-Zahlen wurden für jede Zelle durch die Gesamtexpression normalisiert und mit dem Skalenfaktor (10.000) multipliziert. Dieses Ergebnis wurde dann log-transformiert. Die transformierten Werte wurden innerhalb jeder Zelle skaliert (Varianz auf eins skaliert) und zentriert (Mittelwert auf Null gesetzt), bevor UMAP angewendet wurde, um die Einbettungen zu berechnen, die als Werte auf der x- und y-Achse verwendet wurden. Graphen stellen die Ausgabe einer dimensionalen Reduktionstechnik in einem 2D-Streudiagramm dar, wobei jeder Punkt eine Zelle mit entsprechenden x- und y-Koordinaten basierend auf den durch die Reduktionstechnik bestimmten Zelleinbettungen darstellt. Zellen mit ähnlichen Gensignaturen werden durch die Einbettungen nahe beieinander positioniert. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Abbildung 3: Genexpressionsanalyse von NeuN in der neurogenen Linie. (A) IMAP-Diagramm, das die Clusterbildung der neurogenen Abstammungslinie aus dem öffentlich zugänglichen Datensatz15 zeigt. UMAPs wurden wie in der ergänzenden Abbildung 2 generiert. (B) Genexpressionsniveaus spezifischer zellulärer Marker in der neurogenen Linie mit Astrozyten (Aquaporin 4 = Aqp4), NSCs (Homöodomänenprotein = Hopx), NeuN / Rbfox3 (NSCs und intermediäre Vorläuferzellen [IPCs]) und zyklischen Zellen (Cyclin-abhängige Kinase 6 = Cdk6). Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Tabelle 1: Zusammensetzung der in der Studie verwendeten Medien und Puffer. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Um dieses Protokoll erfolgreich auszuführen, ist die Dissektion der DG der erste kritische Schritt, der etwas Übung erfordert, um es unbeschädigt zu halten und die Kontamination durch das umgebende Gewebe zu begrenzen. Erfahrungsgemäß konnte die Trennung der DG vom Hippocampus sehr schnell von einem erfahrenen Forscher erreicht werden, der dann an der Verfeinerung seiner Technik arbeiten konnte, um die Geschwindigkeit der Dissektion zu erhöhen und somit die Frische des Gewebes zu verbessern, um qualitativ hochwertige Daten zu generieren. In ähnlicher Weise erfordert die Vorbereitung und Resuspension einzelner Kerne Konsistenz über die verschiedenen Bedingungen hinweg, die in einem einzigen Experiment verwendet werden, aber auch die Vermeidung von übermäßigem Pipettieren, das die Kernmembran stören könnte, die Umgebungs-RNAs freisetzt, was die Sequenzierungsergebnisse verzerrt. Zusätzlich zu den bereits erwähnten Empfehlungen zur Herstellung hochwertiger Kerne ist auch die Konzentration der Einzelkernsuspension zu berücksichtigen, bevor mit der Sequenzierung fortgefahren wird. Nach den Richtlinien des Herstellers sollte ein Präparat mit einer Konzentration von mehr als 1.200 Nuc/μL verdünnt werden, da diese Kernkonzentration ein höheres Risiko für die Bildung mehrerer Multiplets hat, die sich auf nachgeschaltete bioinformatische Analysen auswirken. Bemerkenswert ist, dass sich die Sequenzierung von Proben mit Kernkonzentrationen unter 500 nuc/μL aufgrund der damit verbundenen Kosten möglicherweise nicht lohnt. Es wird auch empfohlen, den Rat eines fortgeschrittenen FACS-Benutzers zu befolgen, um das gesamte Gating einzurichten und mit den Einstellungen über Proben und biologische Replikate hinweg konsistent zu bleiben. Ebenso erfordert die Vorbereitung von Bibliotheken für die RNA-Sequenzierung einige Schulungen, um qualitativ hochwertige Ergebnisse zu erzielen, und die meisten Anbieter verfügen über hervorragende Unterstützung, um dies effizient zu erreichen. Diese Methode wurde in dieser Studie nur mit frischem Gewebe getestet; FANS wurde jedoch auch mit gefrorenem Gewebe25 durchgeführt. Es ist daher vernünftig anzunehmen, dass dieses Protokoll mit gefrorenem Gewebe durchgeführt werden könnte, wenn auch mit geringfügiger Optimierung.
Dieses Protokoll wurde mit Blick auf eine bestimmte nachgelagerte Anwendung entwickelt, die darin besteht, andere Zellpopulationen als Neuronen in der neurogenen Hippocampus-Nische zu untersuchen. In der Tat deuten zunehmende Evidenzlinien darauf hin, dass eine Beeinträchtigung der AHN im Alterungsalter auf die umgebenden Zellen innerhalb der Nische 1,2,3,9 zurückzuführen ist. Insbesondere Astrozyten und Oligodendrozyten erweisen sich als Schlüsselregulatoren von AHN; Ihre Isolierung aus der DG in Verbindung mit RNA-Sequenzierung hat jedoch zu gemischten Ergebnissen geführt, so dass diese Hypothese mit dieser Technik schwer zu beurteilen ist 1,17. Dieser Ansatz der FACS-Sortierung von NeuN-negativen Kernen ermöglichte die Isolierung von mehr Astrozyten und Oligodendrozyten im Vergleich zu Proben, die nicht FACS-sortiert waren, was eine bessere bioinformatische Analyse ermöglicht. Dieses Protokoll ist in allen Altersgruppen über die gesamte Lebensspanne anwendbar, und die repräsentativen Daten, die hier mit Geweben von alten Tieren präsentiert werden, liefern einen Machbarkeitsnachweis, dass diese Methode robust ist, um die alternde neurogene Nische des Hippocampus zu untersuchen. Um den Einsatz dieser Methode zu erweitern und für verschiedene biologische Fragestellungen anzupassen, ist es wichtig zu berücksichtigen, dass andere neuronale Kernmembranantigene zusammen mit einer gründlichen Titration der besten validierten Antikörper für diese Marker getestet werden könnten. Wenn man beispielsweise den Prozess der neuronalen Differenzierung von NSCs in der DG untersucht, beginnen einige Zelltypen wie Typ-2-Zellen oder Neuroblasten, NeuN zu exprimieren (ergänzende Abbildung 3). Daher wäre ein weiteres Antigen erforderlich, um diese Zelltypen gezielt zu untersuchen. Umgekehrt wurden in dieser Studie noch einige Neuronen nach NeuN-negativer FACS-Sortierung identifiziert, möglicherweise aufgrund einer geringen oder keinen Expression von NeuN in diesen Populationen (z. B. kortikale Cajal-Retzius-Neuronen19). Darüber hinaus wurde berichtet, dass NeuN in Subpopulationen von Oligodendrozyten26 exprimiert wird, was zu verzerrten Ergebnissen führen könnte, wenn diese Subpopulationen von Interesse wären. Daher sollte die Wahl des Antigens zu Beginn der Anwendung von FANS sorgfältig abgewogen werden, um den Ein- oder Ausschluss von Zellpopulationen zu vermeiden, die eine genaue Antwort auf eine bestimmte biologische Frage ausschließen würden. In Übereinstimmung damit wird auch empfohlen, dass jedes Sequenzierungsergebnis durch orthogonale Assays (z. B. Immunhistochemie oder RNA-Scope) weiter validiert wird, bevor die getestete Hypothese mit diesem Protokoll validiert oder widerlegt wird. Schließlich könnte der Schritt mit FANS weiterentwickelt werden, um mehr als einen Antikörper mit einer ausgefeilteren Sortierstrategie zum Ausschluss und/oder zur Einbeziehung gewünschter Zellpopulationen einzubeziehen.
Letztendlich könnten die in diesem Protokoll beschriebenen Technologien einige Einschränkungen haben, wenn sie mit anderen Arten verwendet werden. Zum Beispiel ist die Nische bei Nagetieren mit dem Vorhandensein von proliferativen und ruhenden NSCs oder neugeborenen Neuronen, die in bestimmten Subregionen der DG eingeschränkt sind, sehr gut definiert, aber es ist immer noch nicht klar, wie die neurogene Hippocampus-Nische in anderen Arten abgegrenzt werden sollte. Tatsächlich sind proliferative Zellen bei nichtmenschlichen Primaten und Menschen nicht innerhalb einer kontinuierlichen Zone der DG ausgerichtet, sondern um sie herum verstreut und könnten auch in der Amygdala7 vorhanden sein. Daher würde sich die Zerlegung und Isolierung breiterer Gebiete als die DG bei anderen Arten möglicherweise auf die Anwendung dieses Protokolls auswirken. Insbesondere müssen die Dissoziations- und Verreibungsschritte für die Herstellung von Gewebe optimiert werden, während mit größeren Gewebestückengearbeitet wird 27,28. In Bezug auf die bioinformatische Analyse haben Nagetiere in Inzucht zwar ein sehr homogenes und sehr gut annotiertes Genom, die genetische Variabilität des menschlichen Genoms in Kombination mit einer unzureichenden Anzahl zellulärer Marker, um verschiedene Zellpopulationen (z. B. NSCs und Astrozyten) eindeutig zu unterscheiden, erfordert jedoch eine hohe Normalisierung für die Analyse, die zu unterschiedlichen Schlussfolgerungen führen könnte, wenn ein kleiner Zellhaufen identifiziert wird7, 11. In solchen Situationen könnte die Zellanreicherung immer noch eine bevorzugte Option sein oder sollte zusammen mit anderen Strategien verwendet werden, um die Analyseleistung zu erhöhen.
Nichtsdestotrotz kann der aktuelle Ansatz es ermöglichen, die Rolle von wenig untersuchten, wenn auch potenziell wichtigen Zellpopulationen bei der Regulation von AHN zu untersuchen. Dies könnte insbesondere für Populationen von Astrozyten der Fall sein, die eine zentrale Rolle bei der Entstehung und dem Fortschreiten neurodegenerativer Erkrankungen spielen29,30. Diese Studie zeigte, dass Astrozyten und andere seltene Zellpopulationen identifiziert und profiliert werden können, indem einfach die überwiegende Mehrheit der in der DG vorhandenen Neuronen ausgeschlossen wird. Andere Studien, die andere Ansätze verwendeten, waren nicht in der Lage, eine ähnliche Erholung von Kernen aus dem gleichen Bereich von Zellpopulationenzu erreichen 5,11,17. Darüber hinaus zeigen die Ergebnisse dieser Studie, dass es mit diesem Ansatz möglich ist, einen NSC-Cluster ohne spezifische Anreicherung dieser Zellpopulation zu isolieren15.
Zusammenfassend wäre die Befolgung und Verbesserung dieser Methode ein Schritt nach vorne, um offene Fragen im Zusammenhang mit der kontextuellen Rolle der neurogenen Hippocampus-Nische für die Modulation von AHN zu beantworten. Insbesondere könnte es neue Einblicke in die Genexpressionsniveaus in gealterten und kranken Gehirnen in Zellpopulationen liefern, die mit der Regulation von AHN9 assoziiert sind, die Identifizierung einer potenziellen Heterogenität der NSCs1 unterstützen oder die Rolle des Gefäßsystems bei AHN untersuchen. Letztendlich könnte diese Methode für andere adulte Stammzellnischen mit ähnlichen Fragen und Problemen angepasst werden.
Offenlegungen
SG, TL und SK sind Mitarbeiter von Merck Sharp & Dohme LLC, einer Tochtergesellschaft von Merck & Co., Inc., Rahway, NJ, USA, außerhalb der USA und Kanadas als MSD bekannt. SG ist Gesellschafter von Merck & Co., Inc., Rahway, NJ, USA.
Danksagungen
Die Autoren danken Lachlan Harris und Piero Rigo für die technische Unterstützung sowie Jason M. Uslaner und Ditte Lovatt für das Feedback zum Manuskript. Diese Arbeit wurde durch Zuschüsse des MRC und eine vorwettbewerbliche Forschungskooperation mit MSD, dem Francis Crick Institute, unterstützt, das von Cancer Research UK (FC0010089), dem UK Medical Research Council (FC0010089), dem Wellcome Trust (FC0010089) und einem Wellcome Trust Investigator Award an FG (106187/Z/14/Z) finanziert wird. Wir entschuldigen uns bei den vielen Autoren, deren Arbeit wir aus Platzmangel nicht diskutieren und zitieren konnten.
Materialien
| Name | Company | Catalog Number | Comments |
| 0.5ml microtube | Eppendorf | 30124537 | |
| 10.00µm Flouresbrite YG Carboxylate Microspheres | Polysciences | 15700-10 | |
| 15 mL polypropylene centrifuge tubes | Corning | 430052 | |
| 2 pairs of sterile Dumont #5 forceps | Fine Science Tools | 11252-30 | |
| 4′,6-diamidino-2-phenylindole (DAPI) | Sigma Aldrich | D9564-10MG | |
| 4150 TapeStation System | Agilent | N/A | |
| 5 mL round bottom high clarity polypropylene test tube with snap cap | Falcon | 352063 | |
| 5 mL round bottom polystyrene test tube with cell strainer snap cap | Falcon | 352235 | |
| 50 mL polypropylene centrifuge tubes | Corning | 430829 | |
| 70 µm cell strainer | Falcon | 352350 | |
| 8 peak SPHERO Rainbow Calibration Particles | BD Biosciences | RCP-30-5A | |
| Accudrop Beads | BD Biosciences | N/A | |
| Allegra X-30R Centrifuge | Beckman Coulter | N/A | |
| Anti-NeuN antibody, clone A60, Alexa Fluor 488 conjugated | Millipore | MAB377X | |
| BD FACSAria Fusion Flow Cytometer | BD Biosciences | N/A | |
| Beckman Coulter MoFlo XDP | Beckman Coulter | N/A | |
| Chromium Controller | 10x Genomics | N/A | |
| Chromium Next GEM Single Cell 3' Reagent Kits v3.1 | 10x Genomics | PN-1000121; PN-1000120; PN-1000213 | |
| BSA 7.5% | Gibco | 15260037 | |
| Dithiothreitol (DTT) | Thermo Scientific | R0861 | |
| Dounce tissue grinder set: mortar, loose pestle (A) and tight pestle (B) | KIMBLE | D8938-1SET | |
| Eppendorf Tubes Protein LoBind 1.5ml | Eppendorf | 30108116 | |
| Halt, 100x Protease inhibitor | ThermoFisher | 78429 | |
| HiSeq 4000 Sequencing System | Illumina | N/A | Sequencing configuration: 28-8-0-91 |
| KCl | Any chemical supplier | Laboratory made | |
| LUNA-FX7 Automated Cell counter | Logos Biosystems | N/A | |
| MgCl2 | Any chemical supplier | Laboratory made | |
| N°10 guarded sterile disposable scalpels | Swann-Morton | 6601 | |
| Nuclease-free water | Sigma Aldrich | W4502-1L | |
| Pair of sterile student surgical scissors | Fine Science Tools | 91401-12 | |
| PBS | Any chemical supplier | Laboratory made | |
| RNase Inhibitor 40 U µl-1 | Ambion | AM2684 | |
| RNasin 40 U µl-1 | Promega | N211A | |
| Sterile Petri dish | Corning | 430167 | |
| Sucrose | Sigma Aldrich | 59378-500G | |
| Tris buffer, pH 8.0 | Any chemical supplier | Laboratory made | |
| Triton X-100 10% (v/v) | Sigma Aldrich | T8787-250ML | |
| Trypan blue | Invitrogen | T10282 |
Referenzen
- Gillotin, S. Targeting impaired adult hippocampal neurogenesis in ageing leveraging intrinsic mechanisms regulating neural stem cell activity. Ageing Research Reviews. 71, 101447 (2021).
- Urban, N., Blomfield, I. M., Guillemot, F. Quiescence of adult mammalian neural stem cells: A highly regulated rest. Neuron. 104 (5), 834-848 (2019).
- Hanspal, M. A., Gillotin, S. A new age in understanding adult hippocampal neurogenesis in Alzheimer's disease. Neural Regeneration Research. 17 (12), 2615-2618 (2022).
- Zhang, H., et al. Single-nucleus transcriptomic landscape of primate hippocampal aging. Protein & Cell. 12 (9), 695-716 (2021).
- Franjic, D., et al. Transcriptomic taxonomy and neurogenic trajectories of adult human, macaque, and pig hippocampal and entorhinal cells. Neuron. 110 (3), 452-469 (2022).
- Moreno-Jimenez, E. P., Terreros-Roncal, J., Flor-Garcia, M., Rabano, A., Llorens-Martin, M. Evidences for adult hippocampal neurogenesis in humans. Journal of Neuroscience. 41 (12), 2541-2553 (2021).
- Sorrells, S. F., et al. Positive controls in adults and children support that very few, if any, new neurons are born in the adult human hippocampus. Journal of Neuroscience. 41 (12), 2554-2565 (2021).
- Zhou, Y., et al. Molecular landscapes of human hippocampal immature neurons across lifespan. Nature. 607, 527-533 (2022).
- Bonafina, A., Paratcha, G., Ledda, F. Deciphering new players in the neurogenic adult hippocampal niche. Frontiers in Cell and Developmental Biology. 8, 548 (2020).
- Amaral, D. G., Scharfman, H. E., Lavenex, P. The dentate gyrus: fundamental neuroanatomical organization (dentate gyrus for dummies). Progress in Brain Research. 163, 3-22 (2007).
- Habib, N., et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nature Methods. 14 (10), 955-958 (2017).
- Flor-Garcia, M., et al. Unraveling human adult hippocampal neurogenesis. Nature Protocols. 15 (2), 668-693 (2020).
- Moreno-Jimenez, E. P., et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer's disease. Nature Medicine. 25 (4), 554-560 (2019).
- Kalinina, A., Lagace, D. Single-cell and single-nucleus RNAseq analysis of adult neurogenesis. Cells. 11 (10), 1633 (2022).
- Harris, L., et al. Coordinated changes in cellular behavior ensure the lifelong maintenance of the hippocampal stem cell population. Cell Stem Cell. 28 (5), 863-876 (2021).
- Shin, J., et al. Single-cell RNA-seq with Waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell. 17 (3), 360-372 (2015).
- Artegiani, B., et al. A single-cell RNA sequencing study reveals cellular and molecular dynamics of the hippocampal neurogenic niche. Cell Reports. 21 (11), 3271-3284 (2017).
- Nott, A., Schlachetzki, J. C. M., Fixsen, B. R., Glass, C. K. Nuclei isolation of multiple brain cell types for omics interrogation. Nature Protocols. 16 (3), 1629-1646 (2021).
- Sarnat, H. B., Nochlin, D., Born, D. E. Neuronal nuclear antigen (NeuN): a marker of neuronal maturation in early human fetal nervous system. Brain Development. 20 (2), 88-94 (1998).
- . Guidance on the Operation of ASPA Available from: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_dat/file/662364/Guidance_on_the_Operation_of_ASPA.pdf (2022)
- Hagihara, H., Toyama, K., Yamasaki, N., Miyakawa, T. Dissection of hippocampal dentate gyrus from adult mouse. Journal of Visualized Experiments. (33), e1543 (2009).
- Habib, N., et al. Disease-associated astrocytes in Alzheimer's disease and aging. Nature Neuroscience. 23 (6), 701-706 (2020).
- Ding, J., et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nature Biotechnology. 38 (6), 737-746 (2020).
- Hao, Y., et al. Integrated analysis of multimodal single-cell data. Cell. 184 (13), 3573-3587 (2021).
- Mussa, Z., Tome-Garcia, J., Jiang, Y., Akbarian, S., Tsankova, N. M. Isolation of adult human astrocyte populations from fresh-frozen cortex using fluorescence-activated nuclei sorting. Journal of Visualized Experiments. (170), e62405 (2021).
- Zhang, Y., et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. Journal of Neuroscience. 34 (36), 11929-11947 (2014).
- Marti-Mengual, U., Varea, E., Crespo, C., Blasco-Ibanez, J. M., Nacher, J. Cells expressing markers of immature neurons in the amygdala of adult humans. European Journal of Neuroscience. 37 (1), 10-22 (2013).
- Zhang, X. M., et al. Doublecortin-expressing cells persist in the associative cerebral cortex and amygdala in aged nonhuman primates. Frontiers in Neuroanatomy. 3, 17 (2009).
- Ding, Z. B., et al. Astrocytes: a double-edged sword in neurodegenerative diseases. Neural Regeneration Research. 16 (9), 1702-1710 (2021).
- Phatnani, H., Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harbor Perspectives in Biology. 7 (6), 020628 (2015).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten