
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Isolierung kleiner präantraler Follikel aus dem Rindereierstock durch eine Kombination aus Fragmentierung, Homogenisierung und serieller Filtration
In diesem Artikel
Zusammenfassung
Um die Untersuchung der präantralen Follikulogenese voranzutreiben, sind effiziente Methoden der Follikelisolierung aus einzelnen Eierstöcken erforderlich. Hier wird ein optimiertes, mechanisches Protokoll zur Follikelisolierung aus Rinderovarien mit einem Gewebezerkleinerer und Homogenisator vorgestellt. Diese Methode ermöglicht die Entnahme einer großen Anzahl lebensfähiger präantraler Follikel aus einem einzigen Eierstock.
Zusammenfassung
Das Verständnis des gesamten Prozesses der Follikulogenese von Säugetieren ist entscheidend für die Verbesserung der assistierten Reproduktionstechnologien bei Nutztieren, Menschen und gefährdeten Arten. Die Forschung beschränkte sich meist auf antrale und große präantrale Follikel aufgrund von Schwierigkeiten bei der Isolierung kleinerer präantraler Follikel, insbesondere bei großen Säugetieren wie Rindern. Diese Arbeit stellt einen effizienten Ansatz dar, um eine große Anzahl kleiner präantraler Follikel aus einem einzelnen Rinderstock zu gewinnen. Der Kortex einzelner Rinderovarien wurde mit einem Gewebechopper in 500 μm Würfel geschnitten und 6 min bei 9.000-11.000 U/min mit einer 10 mm Sonde homogenisiert. Große Ablagerungen wurden mit einem Käsetuch vom Homogenat getrennt, gefolgt von einer seriellen Filtration durch 300 μm und 40 μm Zellsieb. Der im 40-μm-Sieb zurückgehaltene Inhalt wurde in eine Suchschale gespült, wo Follikel identifiziert und zu einem Tropfen Medium gesammelt wurden. Die Lebensfähigkeit der gesammelten Follikel wurde mittels Trypanblau-Färbung getestet. Diese Methode ermöglicht die Isolierung einer großen Anzahl lebensfähiger kleiner präantraler Follikel aus einem einzigen Rindereierstock in ca. 90 min. Wichtig ist, dass diese Methode vollständig mechanisch ist und die Verwendung von Enzymen zur Dissoziation des Gewebes vermeidet, was die Follikel schädigen kann. Die mit diesem Protokoll erhaltenen Follikel können für nachgeschaltete Anwendungen wie die Isolierung von RNA für RT-qPCR, Immunlokalisierung spezifischer Proteine und In-vitro-Kultur verwendet werden.
Einleitung
Eierstockfollikel sind die funktionellen Einheiten des Eierstocks, die für die Produktion der Gameten (Eizellen) sowie von Hormonen verantwortlich sind, die für die Fortpflanzungsfunktion und die allgemeine Gesundheit entscheidend sind. Primordiale Follikel bilden sich im Eierstock während der fetalen Entwicklung oder in der Neugeborenenperiode, abhängig von der Art1, und sie bilden die ovarielle Reserve einer Frau. Das follikuläre Wachstum beginnt mit der Aktivierung von primordialen Follikeln, die das Ruhebecken verlassen und in die Wachstumsphase eintreten. Die präantrale Follikulogenese, die alle Follikelstadien vor der Entwicklung des Antrums umfasst, ist ein hochdynamischer Prozess, der synchrone morphologische und metabolische Veränderungen in der Eizelle und den umgebenden Granulosazellen erfordert, die durch eine enge Kommunikation zwischen diesen beiden Zelltypen angetriebenwerden 2,3. Präantralfollikel bilden die Mehrheit der follikulären Einheiten, die zu einem bestimmten Zeitpunkt im Eierstock gefunden werden4. Es wird geschätzt, dass die Entwicklung durch die präantralen Stadien der Follikulogenese mehrere Wochen länger dauert als die antrale Entwicklung 5,6, und diese Zeit ist notwendig, damit die Eizellen und die Körperzellen eine ausreichende Reife erlangen, um in das Endstadium der Entwicklung (d.h. das Antralstadium) einzutreten und sich auf Eisprung, Befruchtung und Embryonalentwicklung vorzubereiten 7,8,9.
Ein Großteil des aktuellen Wissens über die präantrale Follikulogenese der Eierstöcke stammt aus Mausmodellen10,11,12,13, was zum Teil auf die Leichtigkeit zurückzuführen ist, eine große Anzahl dieser Follikel aus einem kleineren und weniger faserigen Eierstock zu gewinnen. Obwohl Berichte über die Isolierung einer großen Anzahl präantraler Follikel aus Rinderovarien etwa 30 Jahre zurückreichen14, ist ein vollständigeres Verständnis der Prozesse, die die Entwicklung dieser Follikel im Frühstadium regulieren, unrealisiert geblieben, hauptsächlich aufgrund des Mangels an optimierten, effizienten und wiederholbaren Methoden, um eine ausreichende Anzahl lebensfähiger präantraler Follikel zu gewinnen, insbesondere in frühen Entwicklungsstadien. Mit dem zunehmenden Interesse, die Eierstockreserve für die zukünftige Verwendung in der assistierten Reproduktion beim Menschen zu erhalten, werden Kühe aufgrund ihrer ähnlicheren Eierstockstruktur zu einem attraktiven Modell15. Der Rinderstock ist jedoch deutlich kollagenreicher als der Eierstock der Maus16, was die mechanische Isolierung mit den für die Maus beschriebenen Methoden sehr ineffizient macht. Die Bemühungen zur Erweiterung der Techniken zur Erhaltung der Fruchtbarkeit umfassen das vollständige In-vitro-Wachstum der präantralen Follikel in das Antralstadium, gefolgt von der In-vitro-Reifung (IVM) der eingeschlossenen Eizellen, der In-vitro-Fertilisation (IVF) sowie der Embryonenproduktion und -übertragung17. Bisher wurde dieser gesamte Prozess nur bei Mäusen18 erreicht. Bei Rindern beschränkt sich der Fortschritt in Richtung Follikelwachstum in vitro auf wenige Berichte mit variablen Follikelstadien zu Beginn der Kultur sowie variabler Kulturlänge zwischen den Protokollen17,19.
Die in der Literatur beschriebenen Methoden zur Gewinnung präantraler Follikel aus dem Rindereierstock haben meist mechanische und enzymatische Techniken verwendet, entweder isoliert oder in Kombination 2,14,17,20. Der erste Bericht über ein Protokoll zur präantralen Follikelisolierung von Rindern verwendete einen Gewebehomogenisator und eine serielle Filtration, um ganze Eierstöcke zu verarbeiten20. Dieser Studie folgten Berichte, die mechanische und enzymatische Verfahren kombinierten, die Kollagenase14 verwendeten. Ein wiederkehrendes Thema bei der Verwendung von Kollagenase zur Verdauung des Eierstockgewebes ist das potenzielle Risiko einer Schädigung der follikulären Basalmembran, die die Lebensfähigkeit der Follikel beeinträchtigen kann 14,21,22,23. Daher wurden verschiedene Kombinationen von mechanischen Methoden verwendet, wie die Verwendung eines Gewebehackers und wiederholtes Pipettieren oder eines Gewebehackers in Kombination mit Homogenisierung20,24,25,26. Eine andere mechanische Technik, die beschrieben wurde, verwendet Nadeln, um präantrale Follikel direkt aus dem Eierstockgewebe zu sezieren, was besonders nützlich ist, um größere (>200 μm) sekundäre Follikel zu isolieren. Dieser Prozess ist jedoch zeitaufwendig, ineffizient für die Isolierung kleinerer präantraler Follikel und abhängig von Fähigkeiten, wenn er in Rinderovarien versuchtwird 19,27,28.
Unter Ausnutzung der verschiedenen in der Literatur beschriebenen Techniken zielte dieses Protokoll darauf ab, die Isolierung präantraler Follikel aus einzelnen Rinderovarien auf einfache, konsistente und effiziente Weise zu optimieren, die eine Inkubation in enzymatischen Lösungen vermeidet. Die Verbesserung der Methoden zur Isolierung präantraler Follikel bietet die Möglichkeit, das Verständnis dieses Stadiums der Follikulogenese zu verbessern und die Entwicklung effektiver Kultursysteme zur Entwicklung präantraler Follikel bis zum Antralstadium zu ermöglichen. Die hier beschriebenen detaillierten Verfahren zur Isolierung präantraler Follikel von einem großen Säugetier wie der Rinderart werden für Forscher, die die frühe Follikulogenese bei einer nicht-murinen Art untersuchen wollen, die auf den Menschen übertragbar ist, von entscheidender Bedeutung sein.
Access restricted. Please log in or start a trial to view this content.
Protokoll
Die Eierstöcke von Rindern (Bos taurus) wurden aus einem örtlichen Schlachthof bezogen und innerhalb von 6 Stunden nach der Entnahme ins Labor transportiert. Aufgrund der großen Anzahl von Tieren, die in der Einrichtung verarbeitet werden, sind Alter, Rasse und Stadium des Brunstzyklus der Tiere unbekannt. Da in diesen Versuchen keine lebenden Tiere verwendet wurden, war kein genehmigtes Tierpflege- und Anwendungsprotokoll erforderlich.
1. Vorbereitung von Geräten und Reagenzien
- Decken Sie einen 2 Fuß breiten Abschnitt eines Labortisches mit Tischpapier ab.
- Besorgen Sie sich einen Skalpellgriff, eine sterile Skalpellklinge, einen Hämostaten, eine Dissektionszange, eine 20-ml-Luer-Lock-Spritze, eine 18-G-Nadel, zwei 200-ml-Bechergläser, einen 500-ml-Erlenmeyerkolben, einen Kunststofftrichter mit 104 mm Durchmesser, ein Kunststoffschneidebrett, ein 22 cm2-Schicht Käsetuch (das Käsetuch kann vor Gebrauch durch Autoklavieren sterilisiert werden) pro zu verarbeitendem Eierstock, ein 300-μm-Zellsieb und ein 40-μm-Zellsieb (siehe Materialtabelle).
- Übertragen Sie alle Geräte auf das Bankpapier.
- Verwenden Sie den Hämostat, um die Skalpellklinge auf den Skalpellgriff zu stecken. Richten Sie die abgewinkelte Basis der Klinge an der abgewinkelten Anzeige am Griff aus, und schieben Sie die Klinge dann in die Nut des Griffs.
- Den Trichter in den Erlenmeyerkolben geben und die Trichteröffnung mit dem Käsetuch abdecken.
- Geben Sie ein 50-ml-konisches Röhrchen pro Eierstock zur Verarbeitung in ein Wasser- oder Perlenbad auf 38,5 °C.
- Legen Sie eine 100 mm x 15 mm große quadratische Petrischale pro zu verarbeitendem Eierstock auf einen Objektträgerwärmer, der auf 38,5 °C eingestellt ist.
- Fügen Sie 10 ml Penicillin-Streptomycin (PenStrep; 10.000 U / ml Penicillin und 10.000 μg / ml Streptomycin) zu 1 L 1x phosphatgepufferter Kochsalzlösung (PBS) hinzu. Erwärmen Sie den PBS + PenStrep mindestens 2 h vor der Eierstockverarbeitung in einem auf 38,5 °C eingestellten Wasser- oder Perlenbad.
HINWEIS: PBS + PenStrep-Lösung ist unerlässlich für das Waschen von Eierstöcken, wenn isolierte Follikel kultiviert werden, und es wird immer noch für alle nachgeschalteten Experimente empfohlen, um mikrobielle Kontamination zu mildern. - Verwenden Sie zum Sammeln von verarbeitetem Eierstockfiltrat Follicle Wash Medium (FWM), bestehend aus TCM199 mit Hank-Salzen (siehe Materialtabelle), das 3 mg/ml Rinderserumalbumin (BSA), 25 mM HEPES-Puffer, 100 UI Penicillin / 100 μg / ml Streptomycin, 1 mM Natriumpyruvat (NaPyr) und 100 nM nicht-essentielle Aminosäuren (NEAA) enthält.
- Steriles TCM199, eine 250-ml-Flasche und ein 100-ml-Messzylinder in eine Biosicherheitswerkbank (BSC) überführen. 194 ml TCM199 in die Flasche geben.
- Nehmen Sie das Becherglas von TCM199 aus dem BSC und bringen Sie es zu einer Rührplatte. 600 mg BSA, 1,19 g HEPES-Puffer und einen autoklavierten Rührbalken in die Flasche geben und umrühren, bis sie aufgelöst ist.
- Sobald sich der BSA- und HEPES-Puffer vollständig aufgelöst haben, fügen Sie dem Medium 1 N Natronlauge (NaOH) hinzu, bis es einen pH-Wert von 7,6-7,8 erreicht, gemessen mit einem pH-Meter.
- Wischen Sie die Flasche mit dem Medium, ein Vakuumfiltergerät, vier 50-ml-konische Röhrchen und die Flaschen von PenStrep, NaPyr und NEAA mit 70% Ethanol ab, bevor Sie es in die BSC bringen.
- Fügen Sie je 2 ml PenStrep (10.000 U / ml Penicillin und 10.000 μg / ml Streptomycin), 100 mM NaPyr und 100x NEAA in die Flasche TCM199 + 3 mg / ml BSA + 25 mM HEPES hinzu. Sterilfiltrieren Sie das Endprodukt und aliquot in die 50 ml konischen Röhrchen. Lagern Sie das Medium bis zu 2 Wochen bei 4 °C.
- Erwärmen Sie ein 50-ml-konisches Röhrchen Medium pro zwei Eierstöcke mindestens 1 h vor der Eierstockverarbeitung in einem auf 38,5 °C eingestellten Perlbad.
2. Tissue Chopper Setup
- Stellen Sie sicher, dass der Tissue-Chopper (siehe Materialtabelle) eingesteckt und eingeschaltet ist.
- Stellen Sie die Scheibendicke auf 500 μm, den Messerkraftregler auf 20° und den Drehzahlregler auf 90° nach Herstellerangaben ein.
- Stecken Sie eine 60 mm große Kunststoff-Petrischale in den Plattenhalter und setzen Sie den Plattenhalter auf die Bühne.
- Heben Sie den Hackarm so hoch wie möglich an, indem Sie den manuellen Bedienknopf im Uhrzeigersinn drehen.
- Legen Sie mit einer Pinzette eine zweischneidige Rasierklinge (siehe Materialtabelle) auf die in den Hackarm eingesetzte Schraube. Legen Sie den Klingenverschluss über die Klinge und sichern Sie ihn mit der Unterlegscheibe und der Mutter. Lassen Sie die Mutter ein Viertel locker.
- Drehen Sie den manuellen Bedienknopf, bis der Hackarm die Klinge flach auf die Petrischale schnappt. Ziehen Sie die Mutter den Rest des Weges mit dem Mutternschrauber an.
- Heben Sie den Hackarm mit dem manuellen Bedienknopf so hoch an, wie es geht. Bewegen Sie den Tischauslöseknopf ganz nach links, bis er einrastet.
3. Eierstockvorbereitung
- Eierstöcke im Labor in warmes (38,5 °C) steriles PBS + PenStrep überführen.
HINWEIS: Es wird empfohlen, die Eierstöcke zur Follikelisolierung zu verarbeiten, sobald dies nach der Entfernung vom Tier möglich ist. In diesem Protokoll wurden Eierstöcke innerhalb von 6 Stunden nach der Ernte verarbeitet. Eierstöcke wurden in Thermoskannen mit steriler 0,9%iger Kochsalzlösung bei ca. 38,5 °C vom Schlachthof ins Labor transportiert. - Wenn möglich, wählen Sie kleine Eierstöcke (≤ 4 cm x 3 cm x 3 cm) aus, die kleine (3-5 mm) Antralfollikel, keine großen (≥8 mm) Antralfollikel und keinen prominenten Corpus luteum enthalten (Abbildung 1). Diese Kriterien werden empfohlen, um sicherzustellen, dass eine minimale Menge an Nichtfollikeltrümmern, wie Stromazellen und extrazelluläre Matrix, in der resultierenden quadratischen Schale enthalten ist, die isolierte Follikel enthält.
HINWEIS: Antralfollikel können als kugelförmige, flüssigkeitsgefüllte vesikuläre Strukturen auf der Oberfläche des Eierstocks identifiziert werden. Corpora lutea kann als rote, orange oder gelbe steife Strukturen identifiziert werden, die aus der Oberfläche des Eierstocks herausragen. - Verwenden Sie eine Schere, um überschüssiges Bindegewebe und Fett aus den Eierstöcken zu entfernen.
- Waschen Sie die Eierstöcke für 30 s in 70% Ethanol in einem Becherglas.
- Waschen Sie die Eierstöcke 3x für jeweils 2 min in Bechergläsern aus warmem (38,5 °C) PBS + PenStrep, wobei Sie für jede Wäsche frisches PBS + PenStrep verwenden.
- Bewahren Sie die Eierstöcke in warmem (38,5 °C) PBS + PenStrep auf, bis sie für die Verarbeitung bereit sind.
HINWEIS: Der Abstand zwischen Labor und Eierstockquelle kann variabel sein. Daher ist es wichtig, das Protokoll rechtzeitig zu vervollständigen, um die Aufrechterhaltung der Follikellebensfähigkeit zu gewährleisten.

Abbildung 1: Anatomie des Rindereierstocks. Der Rinderstock besteht aus zwei Hauptregionen, die in einer Epithelschicht eingeschlossen sind. Der Kortex, bestehend aus dem Gewebe links von der gestrichelten Linie, enthält Eierstockfollikel vom Urstadium bis zum Antralstadium. Präantralfollikel sind zu klein, um sie mit bloßem Auge zu sehen; Antralfollikel sind mit Sternchen gekennzeichnet. Die Medulla, bestehend aus dem Gewebe rechts von der gestrichelten Linie, enthält Blutgefäße, Lymphgefäße und Nerven. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
4. Chop-Verfahren
HINWEIS: Verarbeiten Sie jeweils nur einen Eierstock. Verarbeiten Sie die Eierstöcke schnell, um Temperaturabnahmen zu vermeiden, die die Lebensfähigkeit der Follikel beeinträchtigen können.
- Übertragen Sie einen Eierstock auf das Schneidebrett auf dem Tischpapier (Abbildung 2A) und bereiten Sie den Tissue-Chopper vor (Abbildung 2B).
- Schneiden Sie den Eierstock mit einer Pinzette und einem Skalpell in zwei Hälften und entfernen Sie die Medulla aus jeder Hälfte, wobei nur der Kortex eine Dicke von etwa 1 mm aufweist, wie in Abbildung 2C gezeigt.
- Schneiden Sie den Eierstock in Längsrichtung von einer Bandansatzstelle zur gegenüberliegenden Befestigungsstelle.
- Halten Sie eine Hälfte des Eierstocks auf dem zu bearbeitenden Schneidebrett und legen Sie die andere Hälfte des Eierstocks wieder in warme (38,5 °C) PBS + PenStrep.
- Schneiden Sie mit der exponierten Medulla nach oben entlang der Krümmung des Eierstocks etwa 2 mm von der Oberfläche des Eierstocks entfernt, ohne den Kortex zu durchschneiden.
- Verwenden Sie die Scheibe entlang der Krümmung des Eierstocks als Leitfaden, um die Scheibe zu vertiefen, wobei Sie immer noch der Krümmung des Eierstocks folgen, um den Kortex von der Medulla zu trennen.
- Sezieren und verwerfen Sie alle Corpora lutea aus dem Eierstock, indem Sie entlang der Grenze des Corpus luteum schneiden.
- Drehen Sie den Eierstock zur Hälfte um, so dass das Epithel nach oben zeigt, und verwenden Sie das Skalpell, um das Schneiden der Medulla vom Kortex wegzuschneiden. Schneiden Sie das verbleibende weiße Bindegewebe um den Rand des Eierstockstücks ab, das mit den Bändern verbunden war.
- Sobald der Großteil der Medulla entfernt ist, schneiden Sie den Kortex mit dem Skalpell auf etwa 1 mm Dicke. Manipulieren Sie das Skalpell mit kleinen Hin- und Herbewegungen, um den Rest der Medulla wegzurasieren.
HINWEIS: Die Medulla ist der innere Teil des Eierstocks, der große Blutgefäße enthält. Der Kortex ist der äußere Teil des Eierstocks, der direkt unter dem äußersten Oberflächenepithel liegt. Der Kortex ist etwa 1 mm dick im Rindereierstock, und so wird das Schneiden des Eierstocks auf eine Dicke von 1 mm die Medulla entfernen.
- Schneiden Sie den Kortex in Stücke, die nicht größer als 2,5 cm x 2,5 cm sind. Bewahren Sie die Cortex-Stücke in warmem (38,5 °C) PBS + PenStrep auf, bis sie zum Hacken bereit sind.
- Füllen Sie ein Becherglas mit mindestens 50 ml warmem (38,5 °C) PBS + PenStrep und erhalten Sie eine Transferpipette aus Kunststoff.
- Übertragen Sie ein einzelnes Stück Kortex auf die Petrischale auf dem Gewebehacker und befeuchten Sie das Gewebe mit drei oder vier Tropfen warmem (38,5 °C) PBS + PenStrep.
- Halten Sie das Stück Gewebe mit einer Pinzette ruhig und drücken Sie einmal die Reset-Taste, um den Tissue-Häcksler zu starten. Stabilisieren Sie die Petrischale mit einer Hand, während Sie das Gewebe mit der Pinzette weiter stabilisieren. Bewegen Sie die Pinzette nach Bedarf entlang des Gewebes, um zu vermeiden, dass die Klinge die Pinzette trifft. Die resultierenden Streifen werden ca. 500 μm lang sein.
- Sobald das gesamte Stück Kortex in Streifen geschnitten wurde, heben Sie die Klinge mit dem Klingenhalterknopf von der Petrischale und der Pinzette ab, um Gewebe von der Klinge zu entfernen.
- Drehen Sie den Plattenhalter um 90°.
- Drücken Sie einmal die Reset-Taste. Stabilisieren Sie die Petrischale mit einer Hand, während Sie die Gewebestreifen mit der Pinzette in den Pfad der Klinge drücken.
- Führen Sie die Klinge vollständig durch die Gewebestreifen. Verwenden Sie den Klingenhalterknopf, um die Klinge von der Petrischale zu heben, und die Pinzette, um Gewebe von der Klinge zu entfernen.
- Verwenden Sie die Transferpipette und warmes (38,5 °C) PBS + PenStrep, um das gehackte Gewebe (Endgröße des Gewebes: 500 μm x 500 μm x 1 mm Würfel) in ein vorgewärmtes (38,5 °C) 50 ml konisches Röhrchen zu waschen. Bringen Sie den konischen Schlauch in das Wasser- oder Perlenbad zurück, um das gehackte Gewebe warm zu halten (38,5 °C).
- Verwenden Sie den Mutterndreher, um die Mutter vom Schneidarm zu entfernen, und entfernen Sie die Unterlegscheibe und den Klingenverschluss. Entfernen Sie die Klinge mit einer Pinzette vom Hackarm, drehen Sie sie um, so dass die unbenutzte Kante der Petrischale zugewandt ist, und legen Sie sie wieder auf den Hackarm. Ersetzen Sie die Messerschließe, die Unterlegscheibe und die Mutter, und setzen Sie den Entriegelungsknopf des Tisches wie in den Schritten 2.5-2.7 beschrieben zurück.
- Wiederholen Sie die Schritte 4.5-4.12 für alle verbleibenden Kortexstücke aus dem Eierstock und ersetzen Sie die Klingen durch neue, nachdem jede Schneide verwendet wurde.
- Entsorgen Sie alle gebrauchten Klingen in einem hartwandigen, scharfen Kunststoffbehälter.
5. Homogenisierungsverfahren
- Stellen Sie sicher, dass die Homogenisatoreinheit (siehe Materialtabelle) angeschlossen ist und die Drehzahl auf den zweiten Balken (9.000-11.000 U/min) eingestellt ist. Setzen Sie die 10-mm-Generatorsonde gemäß den Herstellerangaben in das Gerät ein.
- Stellen Sie einen Timer auf 1 min und führen Sie die Sonde in das 50 ml konische Röhrchen ein, das das gehackte Kortexgewebe aus einem Eierstock enthält (Schritt 4.11) und genügend PBS + PenStrep, um das Röhrchen bis zur 25-ml-Leitung zu füllen. Die Tiefe, in die die Sonde eingeführt wird, muss 1/3 der Höhe der Flüssigkeit betragen, die vom Boden der Kammer aus gemessen wird. Positionieren Sie die Sonde leicht außerhalb der Mitte, um den Wirbel zu minimieren.
- Starten Sie den Timer und schalten Sie den Homogenisator ein. Stellen Sie sicher, dass der Boden der Sonde das Röhrchen nicht berührt, und halten Sie das Röhrchen still, während der Homogenisator eingeschaltet ist.
- Nach 1 min Homogenisierung die Sonde aus dem Röhrchen nehmen. Entfernen Sie mit einer Pinzette das Bindegewebe, das die Entlüftungslöcher und den Raum zwischen Rotormesser und Rotorrohr verstopft. Wenn irgendwelche Kortexstücke in der Sonde stecken, entfernen Sie sie mit einer Pinzette und legen Sie sie wieder in die Röhre.
- Wiederholen Sie die Schritte 5.2-5.4 zusätzlich 5x für insgesamt 6 Minuten Homogenisierung.
- Legen Sie den Schlauch mit homogenisiertem Gewebe in das Wasser- oder Perlenbad, um das Gewebe warm zu halten (38,5 °C). Nach der Verarbeitung des letzten Eierstocks sofort die Generatorsonde zerlegen, reinigen und trocknen Sie sie gemäß den Herstellerangaben.
6. Filtrationsverfahren
- Gießen Sie das dispergierte Gewebe in den mit Käsetuch bedeckten Trichter, der in den Erlenmeyerkolben eingeführt wird. Spülen Sie den Inhalt des Röhrchens mit warmem PBS + PenStrep (38,5 °C) in den Trichter, bis keine Gewebefragmente mehr im Röhrchen verbleiben.
- Zwingen Sie die Gewebefragmente, durch die Löcher des Tuchs zu gelangen, indem Sie das Käsetuch um die Gewebefragmente drehen und zusammendrücken, bis alle überschüssige Flüssigkeit und Gewebe aus dem Käsetuch entfernt sind.
- Öffnen Sie das Käsetuch wieder über dem Trichter, spülen Sie das Käsetuch mit PBS + PenStrep mit einer Transferpipette ab und drücken Sie alle verbleibenden Gewebefragmente erneut durch das Tuch.
- Verwenden Sie ein Hämostat, um das 300-μm-Zellsieb über ein 200-ml-Becherglas zu halten. Gießen Sie das Filtrat in den Erlenmeyerkolben durch das Zellsieb. Der Inhalt des Kolbens wird mit warmem PBS + PenStrep (38,5 °C) in das Zellsieb gespült, bis keine Gewebefragmente mehr vorhanden sind.
- Wenn das Zellsieb mit Gewebe verstopft ist, klopfen Sie das Zellsieb vorsichtig gegen das Becherglas, um sicherzustellen, dass die gesamte Flüssigkeit in das Becherglas gefiltert wurde, drehen Sie dann das Zellsieb auf den Kopf und klopfen Sie die großen Gewebereste auf das Bankpapier. Geben Sie das Zellsieb über das Becherglas zurück und gießen Sie das Filtrat weiter hindurch. Erforderlichenfalls wiederholen, bis das gesamte Filtrat aus dem Erlenmeyerkolben durchfiltriert ist.
- Verwenden Sie ein Hämostat, um das 40-μm-Zellsieb über ein zweites 200-ml-Becherglas zu halten. Gießen Sie das Filtrat in das erste 200-ml-Becherglas durch das Zellsieb. Spülen Sie den Inhalt des Becherglases mit warmem PBS + PenStrep (38,5 °C) in das Zellsieb aus, bis keine Gewebefragmente mehr übrig sind. Entsorgen Sie nicht den Inhalt des 40-μm-Zellsiebs.
- Passen Sie die 18-G-Nadel an die 20-ml-Spritze an. Füllen Sie die Spritze mit FWM. Drehen Sie das 40 μm große Zellsieb über einer quadratischen Petrischale auf den Kopf und waschen Sie mit der Spritze den Inhalt des Zellsiebs in die Schale aus. Füllen Sie die Spritze wieder auf und spülen Sie das Zellsieb nach Bedarf aus, bis keine Gewebefragmente mehr übrig sind.
HINWEIS: Typischerweise reichen 25 ml FWM aus, um den Inhalt des 40-μm-Zellsiebs vollständig auszuspülen.

Abbildung 2: Arbeitsbereichseinrichtung für Eierstockverarbeitung und Protokoll-Workflow . (A) Tischaufbau zum Schneiden der Eierstöcke vor dem Hacken und zum Filtern des Eierstockhomogenats. (B) Gewebezerkleinerer und Homogenisator mit Styroporunterstützung zur Verringerung der Vibrationen der Homogenisatorstufe. (C) Schematische Darstellung des Workflows für die Verarbeitung eines ganzen Eierstocks. Eierstöcke werden von überschüssigem Bindegewebe beschnitten und dann halbiert, und das Medulla wird entfernt, bis eine ~ 1 mm dicke Scheibe des Kortex übrig bleibt. Der Kortex wird in 2,5 cm x 2,5 cm große Stücke geschnitten und in einem auf einen Schnittabstand von 500 μm eingestellten Gewebehäcksler gehackt. Die Stücke werden dann homogenisiert, und das Homogenat wird durch Käsetuch filtriert, gefolgt von einer Filtration durch 300 μm und 40 μm Zellsieb. Der Inhalt des 40 μm großen Zellsiebs wird in eine quadratische Petrischale gespült, die mit einem Stereomikroskop nach Follikeln durchsucht wird. Erstellt mit BioRender.com. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
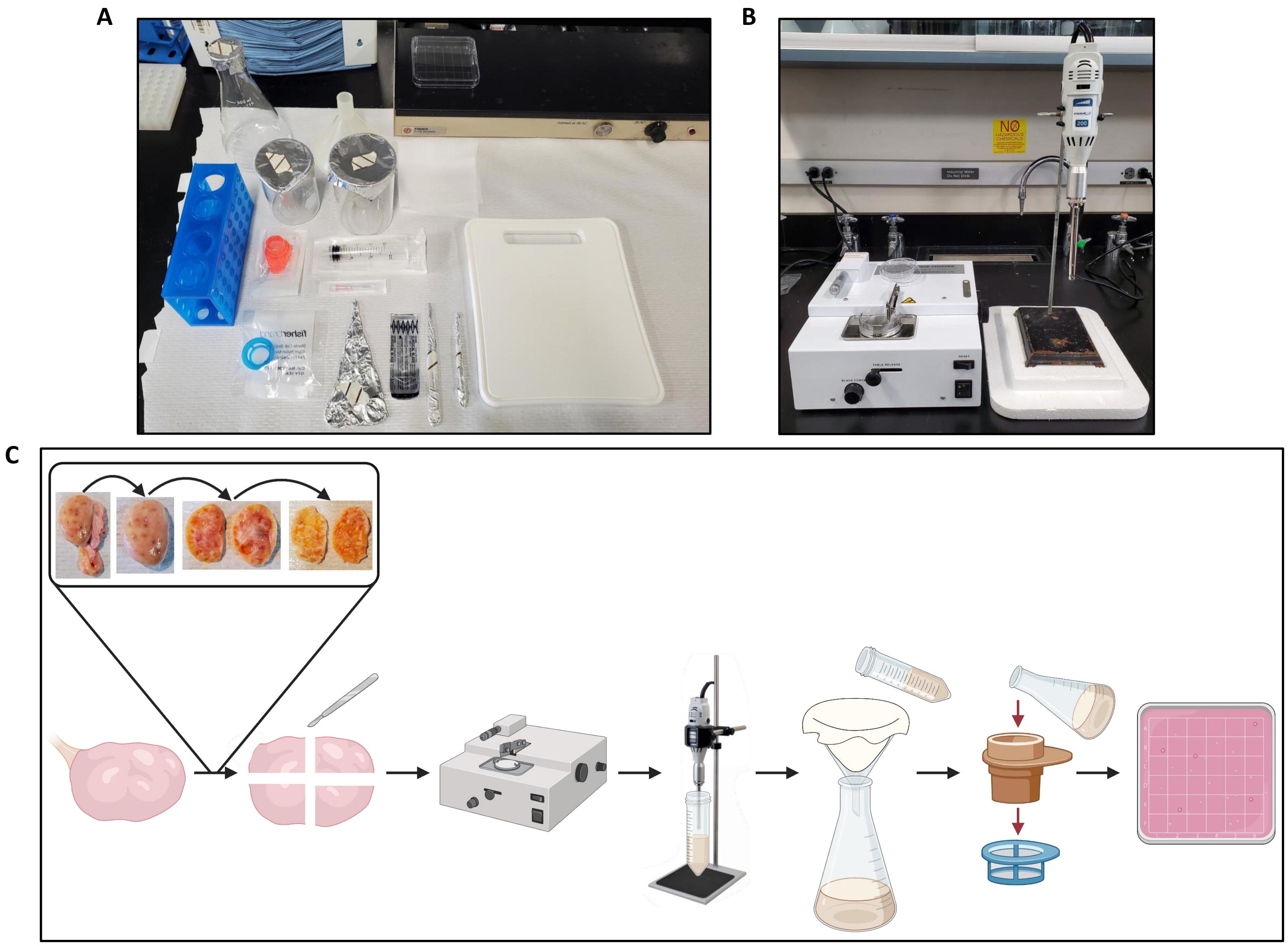{kind=link}
7. Follikel suchen und sammeln
- Übertragen Sie die quadratische Petrischale (Schritt 6.6) auf ein Stereoskop mit einer auf 38,5 °C eingestellten erwärmten Bühne. Die Stereoskopvergrößerung sollte je nach Vorliebe des Suchenden zwischen 1,25x und 3,2x eingestellt werden.
- 10 μL FWM-Tropfen in eine 60 mm Petrischale pipettieren und die Tropfen mit Mineralöl abdecken, um ein Austrocknen zu verhindern. Stellen Sie die Petrischale mit Medientropfen auf eine auf 38,5 °C eingestellte Wärmeplatte.
HINWEIS: Eine 4-Well-Platte kann verwendet werden, um Follikel zu sammeln. Geben Sie 500 μL Waschmedien in eine oder zwei Vertiefungen. Auf die auf 38,5 °C eingestellte Wärmeplatte stellen. - Besorgen Sie sich einen Mikropipettenkolben und eine Spitze.
HINWEIS: Eine 1-5 μL Glas-Mikropipette (siehe Materialtabelle) wird empfohlen, da die Follikel weniger wahrscheinlich an der Glaspipette haften und beim Transfer zwischen Lösungen verloren gehen. Es ist auch ein Instrument, das klein genug ist, um einfachere und präzisere Mikromanipulationen der Follikel zu ermöglichen. - Identifizieren Sie Follikel aus der quadratischen Petrischale und übertragen Sie sie mit der Mikropipette auf Medientropfen (FWM). Viele Follikel sind wahrscheinlich in Gewebetrümmer eingebettet und können mit einer von zwei Methoden wie unten beschrieben abgerufen werden.
HINWEIS: Follikel sind eher länglich als perfekte Kugeln und haben typischerweise eine Eizelle, die sich als fester weißer Kreis in dunkleren Kontrasten zur Mitte des Follikels präsentiert (Abbildung 3A-C). Achten Sie darauf, Follikel nicht mit entblößten Eizellen zu verwechseln. Eizellen neigen dazu, perfekte Kugeln zu sein und sind von einer dicken, klaren Membran (der Zona pellucida) umgeben. Ein inverses Mikroskop mit einer Vergrößerung von 10x (oder mehr) kann zur genaueren Untersuchung von Follikeln verwendet werden (Abbildung 3D).- Trennen Sie die Follikel vorsichtig von Ablagerungen mit der Spitze der Mikropipette oder feinen (27 g) Nadeln.
- Alternativ können Sie eine Pasteur-Glaspipette mit einem Gummikolben verwenden, um die Ablagerungen in der Schale mehrmals aufzunehmen und auszuspritzen, um Follikel von den Trümmern zu entfernen.
- Arbeiten Sie schnell und dauern Sie nicht länger als 30 Minuten, um die Petrischale zu durchsuchen, um die Lebensfähigkeit des Follikels zu erhalten.
- Platzieren Sie maximal fünf Follikel pro 10 μL Tropfen, da eine höhere Dichte die Wahrscheinlichkeit erhöhen kann, dass Follikel aneinander haften.
8. Trypanblau-Ausschluss-Lebensfähigkeitstest
HINWEIS: Verwenden Sie den Deckel einer Petrischale oder eine 4-Well-Platte für alle folgenden Schritte, da die Follikel weniger am Kunststoff des Deckels haften als am Kunststoff der eigentlichen Schale.
- PBS + 0,2% Polyvinylpyrrolidon (PVP) durch Auflösen von 100 mg PVP in 50 mL PBS herstellen.
HINWEIS: PVP wird hier verwendet, um die Wahrscheinlichkeit zu verringern, dass Follikel an der Schale haften. - Verwenden Sie die Mikropipette, um alle Follikel (durchschnittlich 40) aus den Medientropfen in einen 50 μL Tropfen PBS + 0,2% PVP zu übertragen.
- Waschen Sie die Follikel 2x, indem Sie sie nacheinander in frische 50 μL Tropfen PBS + 0,2% PVP überführen.
- Übertragen Sie die Follikel auf einen 285 μL Tropfen PBS + 0,2% PVP.
- Fügen Sie 15 μL Trypanblau zu dem 285 μL Tropfen PBS + 0,2% PVP (Endkonzentration von 0,05% Trypanblau) hinzu und mischen Sie den Tropfen vorsichtig mit einer 200 μL Pipettenspitze auf 100 μL.
HINWEIS: Wenn Sie die 4-Well-Platte für den Trypan-Lebensfähigkeitstest verwenden, fügen Sie 475 μL PBS + 0,2% PVP und 25 μL Trypanblau zu einer Vertiefung hinzu. - Inkubieren Sie die Follikel für 1 Minute im Trypanblauen Tropfen und übertragen Sie dann die Follikel in einen 50 μL Tropfen (oder 500 μL Well) PBS + 0,2% PVP.
- Waschen Sie die Follikel 3x gemäß Schritt 8.3 mit frischen 50 μL Tropfen (oder 500 μL pro Vertiefung) PBS + 0,2% PVP.
- Verwerfen Sie alle Follikel, die nach drei Wäschen in PBS + 0,2% PVP immer noch blau erscheinen, da diese nicht lebensfähig sind. Alle Follikel, die nach drei Wäschen keine blaue Färbung behalten, sind lebensfähig und können für Immunfluoreszenz, Kultur oder andere Verfahren verwendet werden (Abbildung 3E). Die Follikel in flüssigem Stickstoff einfrieren und bei Bedarf bis zur weiteren Verwendung bei -80 °C lagern.
- Führen Sie eine RT-qPCR-Analyse und Immunfluoreszenzfärbung der Follikel durch, wie in den Schritten 9 und 10 beschrieben.

Abbildung 3: Isolierte Follikel und Trypanblau-Ausschlusstest. (A-C) Isolierte Follikel wurden durch ein Stereomikroskop in mehreren Vergrößerungen abgebildet. (A) Isolierte Follikel zwischen Trümmern innerhalb der ursprünglichen Suchschale. Einzelne Follikel sind rot eingekreist. Maßstabsbalken = 2.000 μm. (B) Isolierte Follikel und Trümmer in einem mit Mineralöl bedeckten Follikelwaschmediumtröpfchen. Maßstabsbalken = 1.000 μm. (C) Isolierte Follikel ohne Trümmer bei höherer Vergrößerung. Maßstabsbalken = 1.000 μm. (D) Isolierte Follikel, aufgenommen mit einem inversen Hellfeldmikroskop. Maßstabsbalken = 100 μm. (E) Repräsentative Bilder von lebensfähigen (ungefärbten) und nicht lebensfähigen (blau gefärbten) Follikeln, aufgenommen mit einem inversen Hellfeldmikroskop und einem 20x-Objektiv. Maßstabsbalken = 100 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
9. RT-qPCR-Analyse
- Isolieren Sie RNA aus lebensfähigen Follikeln (aus Schritt 8.8) unter Verwendung eines RNA-Isolierreagenzes (siehe Materialtabelle). Reinigen Sie die RNA und behandeln Sie sie mit DNase mit einem handelsüblichen Reinigungskit (siehe Materialtabelle) gemäß den Anweisungen des Herstellers.
- Eluieren Sie die RNA mit 14 μL RNase-freiem Wasser und quantifizieren Sie sie mit einem Spektralphotometer. Die RNA kann bei -80 °C bis zur cDNA-Synthese gelagert werden.
- Führen Sie die cDNA-Synthese aus gleichen Mengen an RNA durch, die aus primären und frühen sekundären Follikeln extrahiert wurden, wobei ein kommerziell erhältliches cDNA-Synthesekit (siehe Materialtabelle) gemäß den Anweisungen des Herstellers verwendet wird. Das Reaktionsgemisch 5 min bei 25 °C anschließend 60 min bei 42 °C inkubieren, dann die Reaktion durch Erhitzen bei 70 °C für 5 min beenden.
- RT-qPCR mit der synthetisierten cDNA (5 ng pro Reaktion) und Primern (Tabelle 1) unter Verwendung einer kommerziell erhältlichen Reaktionsmischung (siehe Materialtabelle) durchzuführen. Verwenden Sie Temperaturwechselbedingungen: 30 s bei 95 °C für die Polymeraseaktivierung, gefolgt von 40 Amplifikationszyklen, wobei jeder Zyklus 15 s bei 95 °C für die Denaturierung und 30 s bei 60 °C für das Glühen/Ausdehnen umfasste. Analysieren Sie die RT-qPCR durch Quantifizierung von Zyklusschwellenwerten (Ct) und/oder betrachten Sie PCR-Produkte mittels Agarose-Gelelektrophorese.
HINWEIS: Die Transkriptexpression des Granulosazellmarkers FSHR und des Keimzellmarkers DAZL wurde in dieser Studie ausgewertet. Referenzgene waren H2A und ACTB. - Führen Sie eine Schmelzkurvenanalyse durch, indem Sie die Temperatur alle 5 s von 65 °C in Schritten von 0,5 °C auf 95 °C erhöhen.
10. Immunfluoreszenzanalyse
- Fixieren Sie lebensfähige Follikel (ab Schritt 8.8) für 15 min in einem 100 μL Tropfen von 4% (v/v) Paraformaldehyd (PFA) bei Raumtemperatur (RT), gefolgt von 3x Waschen in 100 μL Tropfen PBS + 0,1% BSA + 0,1% Tween 20.
- Blockieren Sie die Follikel für 1 h bei RT in einem Blockierpuffer bestehend aus 1x PBS + 5% (v/v) normalem Eselserum (NDS). Nach der Blockierung inkubieren Sie die Follikel über Nacht bei 4 °C in einem 100 μL Tropfen 4 μg/ml Kaninchen-Anti-Human-CX37-Antikörper oder 4 μg/ml Kaninchen-Isotyp IgG (Negativkontrolle), verdünnt in Blockierpuffer.
- Waschen Sie die Follikel 3x in 100 μL Tropfen PBS + 0,1% BSA + 0,1% Tween 20 und inkubieren Sie sie dann für 1 Stunde bei RT im Dunkeln in einem 100 μL Tropfen 2 μg/ml Esels-Anti-Kaninchen-AlexaFluor 488 sekundären Antikörper, verdünnt in Blockierungspuffer.
- Inkubieren Sie die Follikel für 5 min bei RT im Dunkeln in einem 100 μL Tropfen von 1 μg/ml Hoechst 33342, verdünnt in Blockierpuffer, um DNA zu markieren.
- Die Follikel werden auf einen 5 μL Tropfen Montagemedien (siehe Materialtabelle) auf einem glasmikroskopischen Objektträger überführt und mit einem Deckglas abgedeckt. Lassen Sie die Objektträger über Nacht bei RT aushärten, gefolgt von der Versiegelung mit Nagellack. Bewahren Sie sie bis zur Bildgebung bei 4 °C auf.
- Stellen Sie alle Dias innerhalb von 48 Stunden nach dem Verrutschen der Abdeckung dar. Führen Sie die Bildgebung mit einem inversen Epifluoreszenzmikroskop (siehe Materialtabelle) unter den Filtern DAPI (Anregung 380 nm und Emission 450 nm) und FITC (Anregung 470 nm und Emission 525 nm) durch.
- Legen Sie die Belichtungszeit für beide Kanäle fest. Passen Sie die FITC-Belichtungszeit (CX37) basierend auf der Negativkontrolle des Kaninchenisotyps an. Verwenden Sie ein 20x-Objektiv und den DAPI-Kanal auf 50 ms Belichtungszeit, um Kaninchen-Isotyp-markierte Follikel zu identifizieren.
- Stellen Sie diese Follikel unter dem FITC-Kanal dar und verringern Sie die Belichtungszeit, bis alle grünen Hintergrundsignale abgeschafft sind. Beachten Sie diese Belichtungszeit.
- Bilden Sie alle CX37-Antikörper-markierten Follikel mit der für den Isotyp-FITC-Kanal eingestellten Belichtungszeit und der Belichtungszeit von 50 ms für den DAPI-Kanal ab.
- Verarbeiten Sie die Signalintensität, gemessen als mittlere Grauzone nach dem Schwellwert, mit einem Computerbildverarbeitungsprogramm29 (siehe Materialtabelle).
- Passen Sie die TIFF-Datei des DAPI-Bildes für jeden Follikel so an, dass der gesamte Follikel umrissen wird. Verwenden Sie die Funktion Partikel analysieren des Programms, um den gesamten Follikel als Region of Interest (ROI) auszuwählen.
- Öffnen Sie die TIFF-Datei des FITC-Bildes für den entsprechenden Follikel und überlagern Sie den aus dem DAPI-Bild generierten ROI über das FITC-Bild. Verwenden Sie die Measure-Funktion des Programms, um den mittleren Graubereich des FITC-Bildes zu quantifizieren, der die Signalintensität darstellt.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Überblick und kritische Schritte
Mit diesem Protokoll können kleine präantrale Rinderfollikel zuverlässig aus einzelnen Eierstöcken in experimentell relevanter Anzahl isoliert werden. Von insgesamt 30 Replikaten wurden durchschnittlich 41 Follikel pro Replikat mit einem Bereich von 11 bis 135 Follikeln erhalten (Abbildung 4A). In 14 Replikaten wurden die Follikel für das Entwicklungsstadium wie zuvor beschrieben26 durch Messung des Follikeldu...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Das vorliegende Protokoll beschreibt eine reproduzierbare Methode zur Gewinnung präantraler Follikel im Frühstadium, insbesondere in primären und frühen sekundären Stadien, aus dem Rindereierstock. Dieses Protokoll baut auf früheren Berichten 20,25,30,34,35,36 auf und bietet Optimierungen, die zur Isolierung einer sinnvollen Anza...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben nichts offenzulegen.
Danksagungen
Dieses Projekt wurde teilweise durch das USDA Multi-State-Projekt W4112 und den UC Davis Jastro Shields Award an SM finanziert.
Die Autoren möchten Central Valley Meat, Inc. für die Bereitstellung der in allen Experimenten verwendeten Rindereierstöcke danken. Die Autoren danken auch Olivia Silvera für die Unterstützung bei der Eierstockverarbeitung und Follikelisolierung.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| 5-3/4" Soda Lime Disposable Glass Pasteur Pipette | Duran Wheaton Kimble | 63A54 | Pasteur pipette that can be used to dislodge follicles from debris while searching within the petri dish |
| 16% Paraformaldehyde | Electron Microscopy Sciences | 15710 | Diluted to 4%; fixation of follicles for immunostaining |
| 20 mL Luer-lock Syringe | Fisher Scientific | Z116882-100EA | Syringe used with the 18 G needle to dislodge follicles from the 40 μm cell strainer |
| #21 Sterile Scalpel Blade | Fisher Scientific | 50-365-023 | Used to cut the ovaries and remove the medula |
| 40 μm Cell Strainer | Fisher Scientific | 22-363-547 | Used to filter the filtrate from the 300 μm cell strainer |
| 104 mm Plastic Funnel | Fisher Scientific | 10-348C | Size can vary, but ensure the cheese cloth is cut appropriately and that the ovarian homogenate will not spill over |
| 300 μm Cell Strainer | pluriSelect | 43-50300-03 | Used to filter the filtrate from the cheese cloth |
| 500 mL Erlenmeyer Flask | Fisher Scientific | FB500500 | Funnel and flask used to catch filtrate from the cheese cloth |
| Air-Tite Sterile Needles 18 G | Thermo Fisher Scientific | 14-817-151 | 18 G offers enough pressure to dislodge follicles from the 40 μm cell strainer |
| Air-Tite Sterile Needles 27 G 13 mm | Fisher Scientific | 14-817-171 | Needles that can be used to manipulate any debris in which follicles are stuck |
| BD Hoechst 33342 Solution | Fisher Scientific | BDB561908 | Fluorescent DNA stain |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A7030-100G | Component of follicle wash media |
| Cheese Cloth | Electron Microscopy Sciences | 71748-00 | First filtering step of the ovarian homogenate meant to remove large tissue debris |
| Classic Double Edge Safety Razor Blades | Wilkinson Sword | N/A | Razor blades that fit the best in the McIlwain Tissue Chopper and do not dull quickly |
| Donkey-Anti-Rabbit Secondary Antibody, Alexa Fluor 488 | Fisher Scientific | A-21206 | Secondary antibody for immunostaining |
| Eisco Latex Pipette Bulbs | Fisher Scientific | S29388 | Rubber bulb to use with Pasteur pipettes |
| HEPES Buffer | Sigma-Aldrich | H3375 | Component of follicle wash media |
| Homogenizer | VWR | 10032-336 | Homogenize the ovarian tissue to release follicles |
| ImageJ/Fiji | NIH | v2.3.1 | Software used for analysis of fluorescence-immunolocalization |
| McIlwain Tissue Chopper | Ted Pella | 10184 | Used to cut ovarian tissue small enough for homogenization |
| Microscope - Stereoscope | Olympus | SZX2-ILLT | Dissection microscope used for searching and harvesting follicles from the filtrate |
| Microscope - Inverted | Nikon | Diaphot 300 | Inverted microscope used for high magnification brightfield visualization of isolated follicles |
| Microscope - Inverted | ECHO | Revolve R4 | Inverted microscope used for high magnification brightfield and epifluorescence visualization of isolated follicles |
| Mineral Oil | Sigma-Aldrich | M8410-1L | Oil to cover the drops of follicle wash medium to prevent evaporation during searching |
| Non-essential Amino Acids (NEAA) | Gibco | 11140-050 | Component of follicle wash medium |
| Normal Donkey Serum | Jackson ImmunoResearch | 017-000-001 | Reagent for immunostaining blocking buffer |
| Nunc 4-well Dishes for IVF | Thermo Fisher Scientific | 144444 | 4-well dishes for follicle isolation and washing |
| Penicillin-Streptomycin Solution 100x | Gibco | 15-140-122 | Component of follicle wash medium |
| Petri Dish 60 mm OD x 13.7 mm | Ted Pella | 10184-04 | Petri dish that fits the best in the McIlwain Tissue Chopper |
| Phosphate Buffered Saline (PBS) | Fisher Scientific | BP665-1 | Washing buffer for ovaries and follicles |
| Plastic Cutting Board | Fisher Scientific | 09-002-24A | Cutting board of sufficient size to safely cut ovaries |
| Polyvinylpyrrolidone (PVP) | Fisher Scientific | BP431-100 | Addition of PVP (0.1% w/v) to PBS prevents follicles from sticking to the plate or each other |
| ProLong Gold Antifade Mountant | Thermo Fisher Scientific | P36930 | Mounting medium for fluorescently labeled cells or tissue |
| Qiagen RNeasy Micro Kit | Qiagen | 74004 | RNA column clean-up kit |
| R | The R Foundation | v4.1.2 | Statistical analysis software |
| Rabbit-Anti-Human Cx37/GJA4 Polyclonal Antibody | Abcam | ab181701 | Cx37 primary antibody for immunostaining |
| RevertAid RT Reverse Transcription Kit | Thermo Fisher Scientific | K1691 | cDNA synthesis kit |
| Rstudio | RStudio, PBC | v2021.09.2 | Statistical analysis software |
| Sodium Hydroxide Solution (1N/Certified) | Fisher Scientific | SS266-1 | Used to increase media pH to 7.6-7.8 |
| Sodium Pyruvate (NaPyr) | Gibco | 11360-070 | Component of follicle wash medium |
| Square Petri Dish 100 mm x 15 mm | Thermo Fisher Scientific | 60872-310 | Gridded petri dishes allow for more efficient identification of follicles |
| SsoAdvanced Universal SYBR Green Supermix | BioRad | 1725271 | Mastermix for PCR reaction |
| Steritop Threaded Bottle Top Filter | Sigma-Aldrich | S2GPT02RE | Used to sterilize follicle wash medium |
| SYBR-safe DNA gel stain | Thermo Fisher Scientific | S33102 | Staining to visual PCR products on agarose gel |
| TCM199 with Hank’s Salts | Gibco | 12-350-039 | Component of follicle wash medium |
| Triton X-100 | Fisher Scientific | BP151-100 | Detergent for immunostaining permeabilization buffer |
| Trizol reagent | Thermo Fisher Scientific | 15596026 | RNA isolation reagent |
| Trypan Blue Solution, 0.4% | Gibco | 15-250-061 | Used for testing viability of isolated follicles |
| Tween 20 | Detergent for immunostaining wash buffer | ||
| Warmer Plate Universal | WTA | 20931 | Warm plate to keep follicles at 38.5 °C while searching under the microscope |
| Wiretrol II Calibrated Micropipets | Drummond | 50002-005 | Glass micropipettes to manipulate follicles |
Referenzen
- Fortune, J. E., Yang, M. Y., Allen, J. J., Herrick, S. L. Triennial reproduction symposium: The ovarian follicular reserve in cattle: What regulates its formation and size. Journal of Animal Science. 91 (7), 3041-3050 (2013).
- Fair, T., Hulshof, S. C., Hyttel, P., Greve, T., Boland, M. Oocyte ultrastructure in bovine primordial to early tertiary follicles. Anatomy and Embryology. 195 (4), 327-336 (1997).
- Jaffe, L. A., Egbert, J. R. Regulation of mammalian oocyte meiosis by intercellular communication within the ovarian follicle. Annual Review of Physiology. 79, 237-260 (2017).
- Driancourt, M. A., Reynaud, K., Cortvrindt, R., Smitz, J. Roles of KIT and KIT LIGAND in ovarian function. Reviews of Reproduction. 5 (3), 143-152 (2000).
- Lussier, J. G., Matton, P., Dufour, J. J. Growth rates of follicles in the ovary of the cow. Journal of Reproductive Fertility. 81 (2), 301-307 (1987).
- Aerts, J. M. J., Bols, P. E. J. Ovarian follicular dynamics: a review with emphasis on the bovine species. Part I: Folliculogenesis and preantral follicle development. Reproduction in Domestic Animals. 45 (1), 171-179 (2010).
- Sugiura, K., Pendola, F. L., Eppig, J. J. Oocyte control of metabolic cooperativity between oocytes and companion granulosa cells: energy metabolism. Developmental Biology. 279 (1), 20-30 (2005).
- Eppig, J. J., Pendola, F. L., Wigglesworth, K., Pendola, J. K. Mouse oocytes regulate metabolic cooperativity between granulosa cells and oocytes: amino acid transport. Biology of Reproduction. 73 (2), 351-357 (2005).
- Sugimura, S., et al. Amphiregulin co-operates with bone morphogenetic protein 15 to increase bovine oocyte developmental competence: effects on gap junction-mediated metabolite supply. Molecular Human Reproduction. 20 (6), 499-513 (2014).
- Edson, M. A., Nagaraja, A. K., Matzuk, M. M. The mammalian ovary from genesis to revelation. Endocrine Reviews. 30 (6), 624-712 (2009).
- Matzuk, M. M., Burns, K. H. Genetics of mammalian reproduction: modeling the end of the germline. Annual Review of Physiology. 74, 503-528 (2012).
- McGee, E. A., Raj, R. S. Regulators of ovarian preantral follicle development. Seminars in Reproductive Medicine. 33 (3), 179-184 (2015).
- Chen, Y., et al. The factors and pathways regulating the activation of mammalian primordial follicles in vivo. Frontiers in Cell and Developmental Biology. 8, 575706(2020).
- Figueiredo, J. R., et al. Development of a combined new mechanical and enzymatic method for the isolation of intact preantral follicles from fetal, calf and adult bovine ovaries. Theriogenology. 40 (4), 789-799 (1993).
- Sirard, M. A. The ovarian follicle of cows as a model for human. Animal Models and Human Reproduction. , 127-144 (2017).
- Parkes, W. S., et al. Hyaluronan and collagen are prominent extracellular matrix components in bovine and porcine ovaries. Genes. 12 (8), 1186(2021).
- Araújo, V. R., Gastal, M. O., Figueiredo, J. R., Gastal, E. L. In vitro culture of bovine preantral follicles: a review. Reproductive Biology and Endocrinology. 12 (1), 1-14 (2014).
- Eppig, J. J., Schroeder, A. C. Capacity of mouse oocytes from preantral follicles to undergo embryogenesis and development to live young after growth, maturation, and fertilization in vitro. Biology of Reproduction. 41 (2), 268-276 (1989).
- McLaughlin, M., Telfer, E. E. Oocyte development in bovine primordial follicles is promoted by activin and FSH within a two-step serum-free culture system. Reproduction. 139 (6), 971-978 (2010).
- Nuttinck, F., Mermillod, P., Massip, A., Dessy, F. Characterization of in vitro growth of bovine preantral ovarian follicles: A preliminary study. Theriogenology. 39 (4), 811-821 (1993).
- Demeestere, I., et al. Effect of preantral follicle isolation technique on in-vitro follicular growth, oocyte maturation and embryo development in mice. Human Reproduction. 17 (8), 2152-2159 (2002).
- Fattahi, A., et al. Optimization of porcine ovarian follicle isolation methods for better developmental potential. Tissue Engineering Part A. 26 (13-14), 712-719 (2020).
- Nagashima, J. B., Hill, A. M., Songsasen, N. In vitro development of mechanically and enzymatically isolated cat ovarian follicles. Reproduction and Fertility. 2 (1), 35-46 (2021).
- Lucci, C. M., Rumpf, R., Figueiredo, J. R., Báo, S. N. Zebu (Bos indicus) ovarian preantral follicles: Morphological characterization and development of an efficient isolation method. Theriogenology. 57 (5), 1467-1483 (2002).
- Langbeen, A., et al. Characterization of freshly retrieved preantral follicles using a low-invasive, mechanical isolation method extended to different ruminant species. Zygote. 23 (5), 683-694 (2014).
- Candelaria, J. I., Denicol, A. C. Characterization of isolated bovine preantral follicles based on morphology, diameter and cell number. Zygote. 28 (2), 154-159 (2020).
- vanden Hurk, R., et al. Ultrastructure and viability of isolated bovine preantral follicles. Human Reproduction Update. 4 (6), 833-841 (1998).
- Paes, V. M., et al. Effect of heat stress on the survival and development of in vitro cultured bovine preantral follicles and on in vitro maturation of cumulus-oocyte complex. Theriogenology. 86 (4), 994-1003 (2016).
- Schindelin, J., et al. Fiji: An open-source platform for biological image analysis. Nature Methods. 9 (7), 676-682 (2012).
- de Aguiar, L. H., Hyde, K. A., Pedroza, G. H., Denicol, A. C. Heat stress impairs in vitro development of preantral follicles of cattle. Animal Reproduction Science. 213, 106277(2020).
- Kristensen, S. G., Ebbesen, P., Andersen, C. Y. Transcriptional profiling of five isolated size-matched stages of human preantral follicles. Molecular and Cellular Endocrinology. 401, 189-201 (2015).
- Candelaria, J. I., Rabaglino, M. B., Denicol, A. C. Ovarian preantral follicles are responsive to FSH as early as the primary stage of development. Journal of Endocrinology. 247 (2), 153-168 (2020).
- Nuttinck, F., et al. Comparative immunohistochemical distribution of Connexin 37 and Connexin 43 throughout folliculogenesis in the bovine ovary. Molecular Reproduction and Development. 57 (1), 60-66 (2000).
- Itoh, T., Hoshi, H. Efficient isolation and long-term viability of bovine small preantral follicles in vitro. In Vitro Cellular and Developmental Biology-Animal. 36 (4), 235-240 (2000).
- Saha, S., Shimizu, M., Geshi, M., Izaike, Y. In vitro culture of bovine preantral follicles. Animal Reproduction Science. 63 (1-2), 27-39 (2000).
- Bus, A., et al. Preservation of connexin 43 and transzonal projections in isolated bovine pre-antral follicles before and following vitrification. Journal of Assisted Reproduction and Genetics. 38 (2), 479-492 (2021).
- Gougeon, A., Ecochard, R., Thalabard, J. C. Age-related changes of the population of human ovarian follicles: increase in the disappearance rate of non-growing and early-growing follicles in aging women. Biology of Reproduction. 50 (3), 653-663 (1994).
- Xu, D., et al. Raf-ERK1/2 signaling pathways mediate steroid hormone synthesis in bovine ovarian granulosa cells. Reproduction in Domestic Animals. 54 (5), 741-749 (2019).
- Santos, R. R., et al. Cryopreservation of ovarian tissue: an emerging technology for female germline preservation of endangered species and breeds. Animal Reproduction Science. 122 (3-4), 151-163 (2010).
- Leonel, E. C. R., Lucci, C. M., Amorim, C. A. Cryopreservation of human ovarian tissue: a review. Transfusion Medicine and Hemotherapy. 46 (3), 173-181 (2019).
- Bus, A., Langbeen, A., Martin, B., Leroy, J. I. M. R., Bols, P. E. J. Is the pre-antral ovarian follicle the 'holy grail' for female fertility preservation. Animal Reproduction Science. 207, 119-130 (2019).
- Chen, J., et al. Optimization of follicle isolation for bioengineering of human artificial ovary. Biopreservation and Biobanking. , (2021).
- Chiti, M. C., et al. A modified and tailored human follicle isolation procedure improves follicle recovery and survival. Journal of Ovarian Research. 10 (1), 1-9 (2017).
- Kristensen, S. G., Rasmussen, A., Byskov, A. G., Andersen, C. Y. Isolation of pre-antral follicles from human ovarian medulla tissue. Human Reproduction. 26 (1), 157-166 (2011).
- Oktay, K., et al. Isolation and characterization of primordial follicles from fresh and cryopreserved human ovarian tissue. Fertility and Sterility. 67 (3), 481-486 (1997).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten