
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Ein "Dual-Addition"-Calciumfluoreszenz-Assay für das Hochdurchsatz-Screening von rekombinanten G-Protein-gekoppelten Rezeptoren
In diesem Artikel
Zusammenfassung
In dieser Arbeit wird ein intrazellulärer Hochdurchsatz-Calciumfluoreszenz-Assay für 384-Well-Platten beschrieben, um kleine Molekülbibliotheken auf rekombinanten G-Protein-gekoppelten Rezeptoren (GPCRs) zu screenen. Das Ziel, der Kinin-Rezeptor aus der Rinderpestzecke Rhipicephalus microplus, wird in CHO-K1-Zellen exprimiert. Dieser Assay identifiziert Agonisten und Antagonisten unter Verwendung der gleichen Zellen in einem "Dual-Addition"-Assay.
Zusammenfassung
G-Protein-gekoppelte Rezeptoren (GPCRs) stellen die größte Superfamilie von Rezeptoren dar und sind das Ziel zahlreicher Humanmedikamente. Das Hochdurchsatz-Screening (HTS) von zufälligen niedermolekularen Bibliotheken gegen GPCRs wird von der pharmazeutischen Industrie für die zielgerichtete Wirkstoffforschung eingesetzt. In dieser Studie wurde ein HTS eingesetzt, um neuartige niedermolekulare Liganden von wirbellosen-spezifischen Neuropeptid-GPCRs als Sonden für physiologische Untersuchungen von Vektoren tödlicher human- und veterinärmedizinischer Krankheitserreger zu identifizieren.
Der wirbellose Kinin-Rezeptor wurde als Ziel gewählt, weil er viele wichtige physiologische Prozesse bei Wirbellosen reguliert, einschließlich Diurese, Fütterung und Verdauung. Darüber hinaus ist die Pharmakologie vieler wirbelloser GPCRs schlecht oder gar nicht charakterisiert; Daher fügt die differentielle Pharmakologie dieser Rezeptorgruppen in Bezug auf die verwandten GPCRs in anderen Metazoen, insbesondere beim Menschen, Erkenntnisse über die Struktur-Wirkungs-Beziehungen von GPCRs als Superfamilie hinzu. Ein HTS-Assay wurde für Zellen in 384-Well-Platten entwickelt, um Liganden des Kinin-Rezeptors aus der Rinderpestzecke oder der südlichen Rinderzecke Rhipicephalus microplus zu entdecken. Der Zecken-Kinin-Rezeptor wurde in CHO-K1-Zellen stabil exprimiert.
Der Kinin-Rezeptor löst, wenn er durch endogene Kinin-Neuropeptide oder andere niedermolekulare Agonisten aktiviert wird, die Freisetzung von Ca2+ aus den Kalziumspeichern in das Zytoplasma aus. Dieser Calciumfluoreszenz-Assay in Kombination mit einem "Dual-Addition"-Ansatz kann funktionelle Agonisten- und Antagonisten-"Hit"-Moleküle in derselben Assayplatte nachweisen. Jeder Assay wurde unter Verwendung von Wirkstoffplatten durchgeführt, die eine Anordnung von 320 zufälligen kleinen Molekülen enthielten. Es wurde ein zuverlässiger Z'-Faktor von 0,7 erhalten, und drei Agonisten- und zwei Antagonisten-Hit-Moleküle wurden identifiziert, wenn das HTS eine Endkonzentration von 2 μM aufwies. Der hier beschriebene Calciumfluoreszenz-Assay kann angepasst werden, um andere GPCRs zu screenen, die die Ca2+ -Signalkaskade aktivieren.
Einleitung
G-Protein-gekoppelte Rezeptoren (GPCRs), die von der Hefe bis zum Menschen vorhanden sind, stellen die größte Superfamilie von Rezeptoren in vielen Organismen dar1. Sie spielen eine entscheidende Rolle bei der Regulierung fast aller biologischen Prozesse bei Tieren. Es gibt 50-200 GPCRs im Genom von Arthropoden, was bedeutet, dass sie die größte Membranrezeptor-Superfamilie2 darstellen. Sie werden in sechs Hauptklassen, A-F, eingeteilt, basierend auf ihrer Sequenzähnlichkeit und ihren Funktionen3. GPCRs transduzieren verschiedene extrazelluläre Signale, wie z.B. die von Hormonen, Neuropeptiden, biogenen Aminen, Glutamat, Protonen, Lipoglykoproteinen und Photonen4. GPCRs koppeln an heterotrimere G-Proteine (Gα, Gβ und Gγ), um nachgeschaltete Signale zu übertragen. GPCRs, die an Gαs oder Gαi/o-Proteine gekoppelt sind, erhöhen bzw. senken die intrazellulären 3', 5'-zyklischen Adenosinmonophosphat (cAMP)-Spiegel durch Aktivierung oder Hemmung der Adenylylcyclase. GPCRs, die an Gαq/11 gekoppelt sind, induzieren die Freisetzung von Kalzium aus den Kalziumspeichern des endoplasmatischen Retikulums, indem sie den Phospholipase C (PLC)-Inositol-1,4,5-triphosphat (IP3) -Weg aktivieren. GPCRs, die an Gα12/13 gekoppelt sind, aktivieren die RhoGTPase-Nukleotidaustauschfaktoren 5,6. GPCRs sind das Ziel von mehr als 50% der Humanarzneimittel und eines Akarizids, Amitraz4. Da GPCRs so unterschiedliche Signale übertragen, sind sie vielversprechende Ziele für die Entwicklung neuartiger Pestizide, die wirbellose physiologische Funktionen stören.
Das Ziel von HTS ist es, Hit-Moleküle zu identifizieren, die Rezeptorfunktionen modulieren können. HTS umfasst Assay-Entwicklung, Miniaturisierung und Automatisierung7. Arthropoden-Neuropeptid-GPCRs sind an den meisten physiologischen Funktionen wie Entwicklung, Häutung und Ekdysis, Ausscheidung, Energiemobilisierung und Fortpflanzungbeteiligt 4. Die meisten Neuropeptid-GPCRs von Arthropoden und Metazoen signalisieren durch die Calcium-Signalkaskade 2,6,8,9,10, wie z.B. in den myoinhibitorischen Peptid- und SIFamid-Rezeptoren der schwarzbeinigen Zecke Ixodes scapularis; Ihre Liganden sind in den Motilitätstests des Hinterdarms antagonistisch, wobei SIF eine Kontraktion auslöst und MIP sie hemmt11,12. Ein NPY-ähnlicher Rezeptor der Gelbfiebermücke, Aedes aegypti, reguliert die weibliche Wirtssuche13. Im Vergleich zu anderen alternativen Calcium-Mobilisierungsassays, wie dem Aequorin-Calcium-Biolumineszenz-Assay14, ist der Calcium-Fluoreszenz-Assay einfach durchzuführen, erfordert keine Transfektion anderer rekombinanter Calcium-Detektionsproteine und ist kostengünstig. Der Calcium-Fluoreszenz-Assay erzeugt ein verlängertes Signal im Vergleich zu dem schnellen kinetischen Signal, das im Aequorin-Calcium-Biolumineszenz-Assay14,15 erhalten wurde.
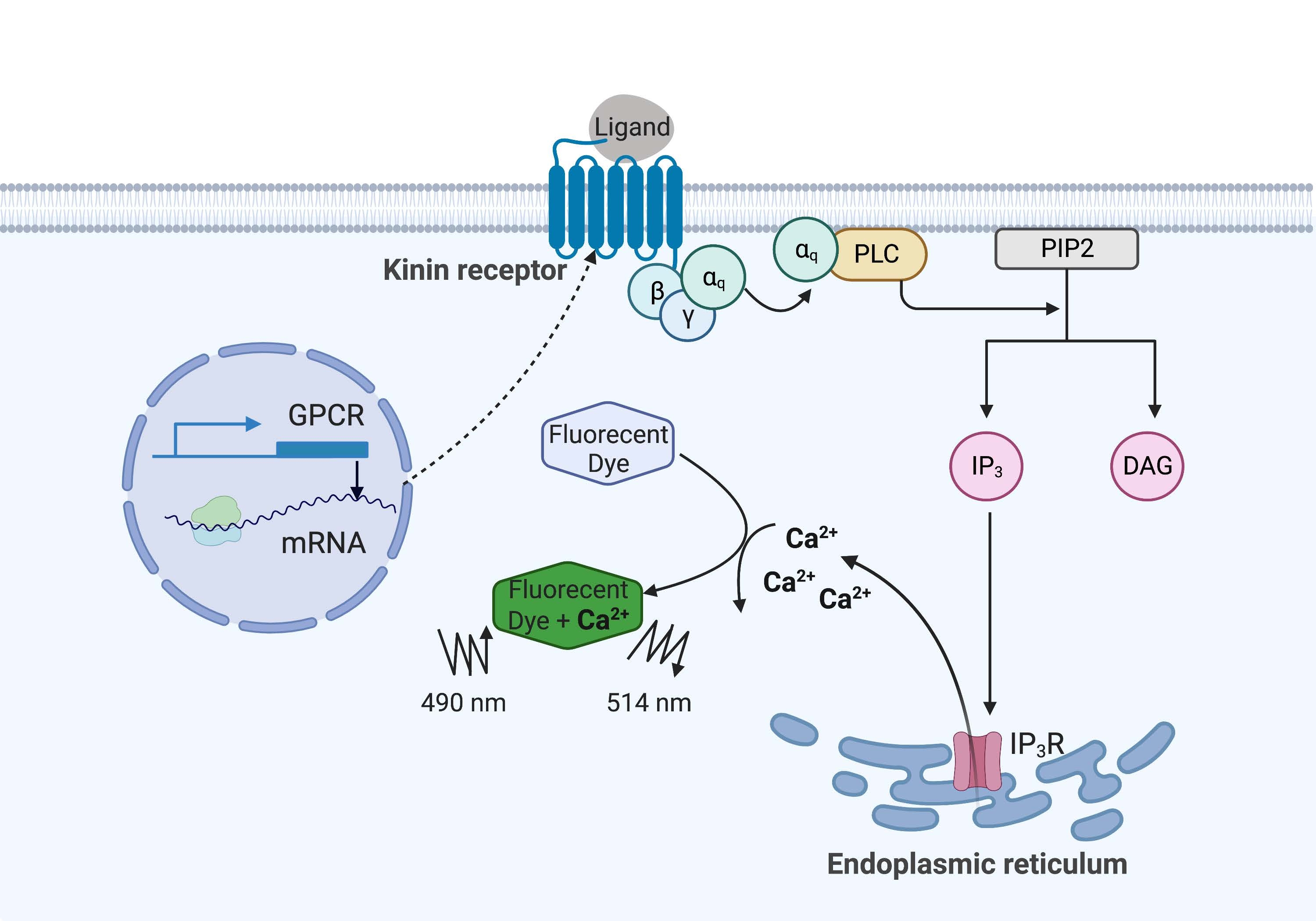
Im vorliegenden Beispiel wurde der Kinin-Rezeptor aus der Rinderpestzecke Rhipicephalus microplus rekombinant in der CHO-K1-Zelllinie exprimiert und für den Calciumfluoreszenz-Assay verwendet. Es gibt nur ein Kinin-Rezeptor-Gen in R. microplus; Der Rezeptor signalisiert über einen Gq-Protein-abhängigen Signalweg und löst den Ausfluss von Ca2+ aus Calciumspeichern in den intrazellulären Raum16 aus. Dieser Prozess kann durch ein Fluorophor nachgewiesen und quantifiziert werden, das bei der Bindung von Calciumionen ein Fluoreszenzsignal auslöst (Abbildung 1).
Der Kinin-Rezeptor ist ein wirbellosen-spezifischer GPCR, der zu den Klasse-A-Rhodopsin-ähnlichen Rezeptoren gehört. Kinin ist ein uraltes Signal-Neuropeptid, das in Mollusca, Crustacea, Insecta und Acari 4,17,18 vorkommt. Coleopteren (Käfer) fehlt das Kinin-Signalsystem; In der Mücke Aedes aegypti gibt es nur einen Kinin-Rezeptor, der drei Aedeskinine bindet, während Drosophila melanogaster einen Kinin-Rezeptor mit Drosokinin als einzigartigem Ligandenhat 19,20,21. Bei Wirbeltieren gibt es keine homologen Kinine oder Kininrezeptoren. Obwohl die genaue Funktion von Kinin bei Zecken unbekannt ist, zeigen die Kininrezeptor-RNAi-stillgelegten Weibchen von R. microplus eine signifikant reduzierte Fortpflanzungsfähigkeit22. Kinine sind pleotrope Peptide in Insekten. Bei Drosophila melanogaster sind sie sowohl am zentralen als auch am peripheren Nervensystembeteiligt 23, an der Präekdysis24, an der Nahrungsaufnahme25, am Stoffwechsel 26 und an den Schlafaktivitätsmustern 26,27 sowie an der Fortbewegung der Larven 28. Kinine regulieren die Kontraktion des Hinterdarms, die Diurese und die Fütterung der Mücke A. aegypti 29,30,31. Die Kininpeptide weisen ein konserviertes C-terminales Pentapeptid Phe-X1-X2-Trp-Gly-NH2 auf, das die minimal erforderliche Sequenz für die biologische Aktivität32 darstellt. Die Arthropodenspezifität, die geringe Größe des endogenen Liganden, die sie für kleinmolekulare Interferenzen empfänglich macht, und die pleiotropen Funktionen bei Insekten machen den Kininrezeptor zu einem vielversprechenden Ziel für die Schädlingsbekämpfung4.
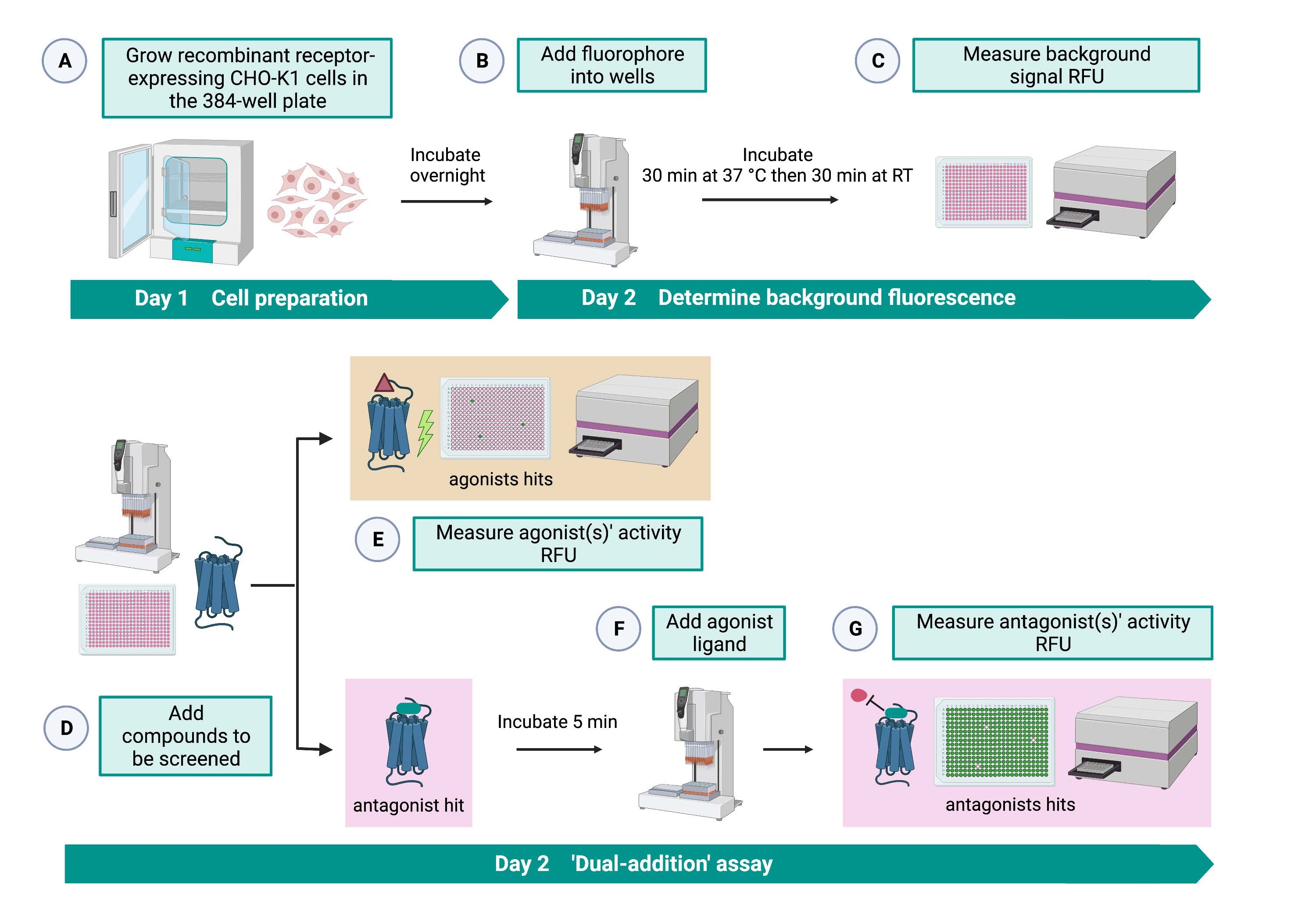
Der "Dual-Addition"-Assay (Abbildung 2) ermöglicht die Identifizierung von Agonisten oder Antagonisten im selben HTS-Assay15. Es basiert auf einem "Dual-Addition"-Assay, der in der pharmazeutischen Industrie häufig für die Wirkstoffforschung verwendet wird33. Kurz gesagt, die erste Zugabe von Medikamenten in die Zellplatte ermöglicht die Identifizierung potenzieller Agonisten in der chemischen Bibliothek, wenn ein höheres Fluoreszenzsignal im Vergleich zur Anwendung der Lösungsmittelkontrolle detektiert wird. Nach 5 Minuten Inkubation mit diesen kleinen Molekülen wird ein bekannter Agonist (Kinin-Peptid) auf alle Vertiefungen aufgetragen. Diejenigen Vertiefungen, die zufällig einen Antagonisten von der Wirkstoffplatte erhielten, zeigen ein niedrigeres Fluoreszenzsignal bei der Agonistenzugabe im Vergleich zu den Kontrolltöpfen, die das Lösungsmittel bei der ersten Zugabe erhielten. Dieser Assay ermöglicht dann die Identifizierung potenzieller Agonisten und Antagonisten mit den gleichen Zellen. In einem Standard-HTS-Projekt würden diese Hit-Moleküle durch Dosis-Wirkungs-Assays und durch zusätzliche biologische Aktivitätsassays, die hier nicht gezeigt werden, weiter validiert.

Abbildung 1: Illustration des Calciumfluoreszenz-Assay-Mechanismus. Das Gq-Protein löst den intrazellulären Kalzium-Signalweg aus. Der Kinin-Rezeptor (G-Protein-gekoppelter Rezeptor) wurde in CHO-K1-Zellen rekombinant exprimiert. Wenn der Agonistenligand an den Rezeptor bindet, aktiviert das mit dem Kininrezeptor assoziierte Gq-Protein PLC, das die Umwandlung eines PIP-2-Moleküls in IP3 und DAG katalysiert. IP 3 bindet dann an die IP3R auf der Oberfläche des endoplasmatischen Retikulums, was zur Freisetzung von Ca 2 + in das Zytoplasma führt, wo Ca2 + -Ionen an die Fluorophore binden und ein Fluoreszenzsignal auslösen. Das Fluoreszenzsignal kann durch Anregung bei 490 nm erhalten und bei 514 nm detektiert werden. Abkürzungen: GPCR = G-Protein-gekoppelter Rezeptor; PLC = phospholipase C; PIP2 = Phosphatidylinositol-4,5-bisphosphat; IP3 = Inositoltrisphosphat; DAG = Diacylglycerin; IP 3 R =IP-3-Empfänger. Erstellt mit BioRender.com. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 2: Der Arbeitsablauf für das Hochdurchsatz-Screening von kleinen Molekülen auf einem G-Protein-gekoppelten Rezeptor, der in CHO-K1-Zellen exprimiert wird. (A) Rekombinante CHO-K1-Zellen, die den Kinin-Rezeptor stabil exprimieren, wurden unter Verwendung eines Liquid-Handling-Systems (25 μL/Well) in die 384-Well-Platte (10.000 Zellen/Well) gegeben und in einem befeuchteten CO2 -Inkubator für 12-16 h inkubiert. ) Der Assay-Puffer, der den Fluoreszenzfarbstoff (25 μl/Well) enthielt, wurde unter Verwendung eines Liquid-Handling-Systems in die Zellplatte gegeben. Die Platte wurde für 30 min bei 37 °C für 30 min inkubiert und bei RT für weitere 30 min äquilibriert. (C) Das Hintergrundfluoreszenzsignal der Zellen in jeder Vertiefung wurde mit einem Plattenleser gemessen. (D) Arzneimittellösungen aus einer Bibliotheksplatte mit 384 Vertiefungen und ein Leerlösungsmittel (alle mit 0,5 μl/Well) wurden unter Verwendung eines Liquid-Handling-Systems in die zelluläre Assayplatte gegeben. (E) Die zellulären Calciumfluoreszenzreaktionen wurden unmittelbar nach der Zugabe der Arzneimittellösungen mit dem Plattenleser gemessen; Verbindung(en), die überdurchschnittliche Fluoreszenzsignale hervorruft, wurden als Agonisten-Treffer ausgewählt. Antagonisten-Treffer, die die GPCR blockieren (Symbol unten), wurden nach der Zugabe des Peptidagonisten während Schritt G aufgedeckt. (F) In der gleichen Testplatte wurde nach 5 min Inkubation der Zellen mit Screening-Verbindungen ein endogenes Agonistenpeptid Rhimi-K-1 (QFSPWGamid) des Zeckenkininrezeptors zu jeder Vertiefung (1 μM) gegeben. (G) Zelluläre Fluoreszenzreaktionen nach der Agonisten-Peptidaddition wurden vom Plattenleser sofort gemessen. Verbindung(en), die das Fluoreszenzsignal hemmen, wurden als Antagonisten-Treffer ausgewählt. Abkürzungen: GPCR = G-Protein-gekoppelter Rezeptor; RT = Raumtemperatur; RFU = relative Fluoreszenzeinheiten. Erstellt mit BioRender.com. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protokoll
1. Zellerhaltung
HINWEIS: EINE CHO-K1-Zelllinie, die den Kinin-Rezeptor von R. microplus stabil exprimiert, genannt BMLK3, wurde von Holmes et al. entwickelt.16 Die Einzelheiten der Entwicklung der Zelllinie werden an anderer Stelle14 dargestellt. Alle folgenden Schritte werden unter sterilen Bedingungen in einer Biosicherheitswerkbank der Klasse II durchgeführt.
- Züchten Sie die rekombinante Zelllinie in selektiven Medien (F-12K-Medium mit 10% fötalem Rinderserum [FBS] und 800 μg/ml G418sulfat), um die Expression des Zielrezeptors sicherzustellen. Lagern Sie die Zellen, die den Rezeptor stabil exprimieren, in 1 ml Gefriermedium (90% FBS und 10% Dimethylsulfoxid [DMSO]) in einem 2 mL Kryovie in einem Gefrierschrank von -80 °C.
HINWEIS: Für die langfristige Lagerung der gefrorenen Zellen lagern Sie sie in flüssigem Stickstoff. - Vor der Zellkultur alle Medien vorwärmen. In einer Biosicherheitswerkbank werden 13 ml vorgewärmtes selektives Medium (F-12K-Medium mit 10 % FBS und 800 μg/ml G418-Sulfat) in einen T-75-Kolben umgefüllt und in der Biosicherheitswerkbank aufbewahrt. Eine Durchstechflasche mit den gefrorenen BMLK 3 Zellen (~1,5 × 106 Zellen) in einem 37 °C warmen Wasserbad für2-3 min auftauen. Die aufgetauten Zellen werden in den T-75-Kolben überführt. Die Zellen werden in einem befeuchteten Inkubator bei 37 °C und 5 %CO2 gehalten, sofern nicht anders angegeben.
- Wenn die Zellen eine 90%ige Konfluenz erreicht haben (1-2 Tage), erwärmen Sie alle Medien auf 37 °C, mit Ausnahme der phosphatgepufferten Kochsalzlösung (DPBS) von Dulbecco, die bei Raumtemperatur gehalten wird. In einer Biosicherheitswerkbank wird das verbrauchte Medium aus dem T-75-Kolben entnommen, die Zellen 5 s lang mit 10 ml DPBS gewaschen, indem der Kolben vorsichtig geschwenkt wird, und anschließend das DPBS mit einer serologischen Pipette entfernt.
HINWEIS: Ein Bild von CHO-K1-Zellen bei 90% Konfluenz ist in Lu et al.14 gezeigt. - Die Zellen werden durch Zugabe von 2 ml 0,25%igem Trypsin-EDTA aus dem T-75-Kolben gelöst und 3-5 min bei 37 °C im Inkubator inkubiert. Dann fügen Sie 8 ml selektives Medium hinzu und mischen Sie gut durch Pipettieren und vorsichtiges 2x-3x mit derselben serologischen Pipette.
- 2 ml der Zellsuspension (~ 1 × 106 Zellen) aus Schritt 1.4 in einen neuen T-75-Kolben mit 10 ml warmem selektivem Medium überführen. Züchten Sie die Zellen für 1-2 Tage im Inkubator, bis sie 90% Konfluenz erreichen.
- Verwenden Sie die Zellen für den Test gemäß den nächsten Schritten oder wiederholen Sie die Schritte 1.4 bis 1.5 ein- oder zweimal, bevor Sie die Zellen im Test verwenden.
HINWEIS: Überschreiten Sie nicht drei bis vier Passagen, da das Assay-Signal bei bestimmten Zelllinien mit weiteren Passagen schwächer werden kann.
2. Calciumfluoreszenz-Assay
- Beschichten Sie die Zellplatte.
- Verwenden Sie ein Liquid-Handling-System, das sich in einer Biosicherheitswerkbank befindet, für alle Pipettierschritte in den 384-Well-Platten. Erstellen Sie benutzerdefinierte Programme für das Pipettieren in die 384-Well-Platten31 (Zusatztabelle S1). Beschichten Sie die sterilen 384-Well-Platten im Voraus. In einer Biosicherheitswerkbank werden 10 μl/Well einer wässrigen Lösung von Poly-D-Lysin (PDL) mit 0,05 mg/ml in jede Platte geladen und 5 min bei Raumtemperatur inkubiert.
- Entleeren Sie den Teller, indem Sie ihn schnell umdrehen und vorsichtig auf sterile Papiertücher tupfen. Spülen Sie dann jede Vertiefung mit 10 μL Wasser ab, leeren Sie die Platte und lassen Sie die Platte über Nacht ohne Deckel in der Biosicherheitswerkbank trocknen. Den Teller mit dem Deckel verschließen und bei 4 °C im Kühlschrank aufbewahren.
HINWEIS: Die beschichteten Platten können bis zu 6 Monate bei 4 °C gelagert werden.
- Tag 1
- Nehmen Sie die im Gefrierschrank von −20 °C gelagerte Arzneimittelplatte (100 μM in 90 % DPBS + 10 % DMSO, Endkonzentration in der Vertiefung für HTS beträgt 2 μM) heraus und stellen Sie sie bei Raumtemperatur.
HINWEIS: Anordnung der Arzneimittelplatte: Jede 384-Well-Platte (24 Spalten x 16 Reihen) enthält 320 Wells mit verschiedenen Bibliotheksverbindungen und 64 Wells mit Blank-Lösungsmittel (DPBS mit 10% DMSO), die in vier Spalten angeordnet sind, mit zwei Spalten am Rand jeder Seite. Siehe Ergänzungstabelle S2 für das Plattenlayout . - Wenn die Zellen im T-75-Kolben eine Konfluenz von ~70 %-90 % erreichen, werden die Zellen wie unten beschrieben vom T-75-Kolben getrennt. Vorwärmen Sie alle Medien bis auf die DPBS (Raumtemperatur) auf 37 °C vor.
- Entfernen Sie das verbrauchte Medium, waschen Sie die Zellen mit 10 ml DPBS und entfernen Sie dann das DPBS. Die Zellen werden aus dem T-75-Kolben mit 2 ml 0,25%igem Trypsin-EDTA für 3-5 min bei 37 °C im Inkubator gelöst, 8 mL selektives Medium hinzugefügt und die Zellsuspension in ein konisches 15-ml-Röhrchen überführt, um sie bei 1.000 × g für 3 min zu zentrifugieren.
- Verwerfen Sie den Überstand und resuspendieren Sie das Zellpellet in 10 ml F-12K-Medium, das 1% FBS und 400 μg/ml G418-Sulfat enthält. Bewahren Sie die Suspension in der Biosicherheitswerkbank auf, während Sie die Zellzahl bestimmen.
- Bestimmen Sie die Zelldichte der Suspension für eine weitere Verdünnung: Mischen Sie 20 μl Zellsuspension in 20 μl 0,4% Trypanblau und laden Sie dann 20 μl der Mischung in eine Zellzählkammer, um von einem Zellzähler auf Zelldichte abgelesen zu werden.
- Die Zellsuspension wird im gleichen Medium (F-12K-Medium mit 1 % FBS und 400 μg/ml G418sulfat) auf ein Endvolumen von mindestens 15 mL bei einer Dichte von 4 × 105 Zellen/ml verdünnt.
- Säen Sie die Zellen in die PDL-beschichtete 384-Well-Platte. Übertragen Sie 15 ml der oben genannten Zellsuspension (4 × 105 Zellen/ml) in ein autofreundliches 150-ml-Reagenzreservoir. Dosieren Sie 25 μL der Zellsuspension (~ 10.000 Zellen/Well) in jede Vertiefung der 384 Vertiefungen der Platte mit einem Liquid-Handling-System in zwei Schritten. Laden Sie 384/12,5 μL-Spitzen mit geringer Retention auf den Liquid-Handler-Kopf, saugen Sie 12,5 μL aus dem Reservoir ab (Geschwindigkeit 5,2 μL/s) und dosieren Sie in jede Vertiefung (Geschwindigkeit: 3,1 μL/s).
- Wiederholen Sie das Pipettieren wie im vorherigen Schritt 2.2.5, um 25 μl pro Vertiefung zu erreichen. Anschließend wird die Platte über Nacht (14-16 h) bei 37 °C und 5 %CO2 im befeuchteten Inkubator inkubiert.
HINWEIS: Da das maximale Pipettiervolumen für diesen speziellen Kopf 12,5 μl beträgt, wird das Pipettieren für 25 μl in zwei Schritten durchgeführt.
- Nehmen Sie die im Gefrierschrank von −20 °C gelagerte Arzneimittelplatte (100 μM in 90 % DPBS + 10 % DMSO, Endkonzentration in der Vertiefung für HTS beträgt 2 μM) heraus und stellen Sie sie bei Raumtemperatur.
- Tag 2
- Überprüfen Sie am nächsten Morgen die abgedeckte Zellplatte unter einem Mikroskop. Wenn nicht konfluent, warten Sie, bis die Zellen 90% Konfluenz erreicht haben.
- Bereiten Sie eine Stammlösung des Fluoreszenzfarbstoffs vor: Resuspendieren Sie lyophilisierten Fluoreszenzfarbstoff in 100 μl DMSO und vermeiden Sie direktes Licht auf die Stammlösung. Wickeln Sie die Tube mit Alufolie ein, um ein Lichtbleichen zu verhindern.
ANMERKUNG: Der Stamm kann für jeden Plattentest in 15-μl-Aliquots aliquotiert werden, um wiederholtes Einfrieren und Auftauen zu vermeiden. die Aliquots können bis zu 1 Monat bei −80 °C gelagert werden. - Bereiten Sie in der Biosicherheitswerkbank bei ausgeschaltetem Licht den Ladefarbstoff (1x) in einem konischen 15-ml-Röhrchen vor, das mit Aluminiumfolie umwickelt ist, und kombinieren Sie dabei 15 μl der Fluoreszenzfarbstoff-Stammlösung (aus Schritt 2.3.2) und die restlichen Komponenten des vorgewärmten Kits (37 °C): 13,5 mL 1x HHBS (Hank-Puffer mit 20 mM HEPES) und 1,5 mL Reagenz B (Farbstoff-Efflux-Hemmer).
HINWEIS: Die Raumbeleuchtung leuchtet während dieses Schritts. - Schließen Sie das Röhrchen und mischen Sie gut, indem Sie das Röhrchen mehrmals (typischerweise 3-5x) vorsichtig umdrehen.
HINWEIS: Bei Raumtemperatur aufbewahren und die Beladungsfarbstoffmischung innerhalb von 30 Minuten verwenden. - Wenn die Zellen eine Konfluenzrate von 90 % erreicht haben, entfernen Sie das verbrauchte Medium von der 384-Well-Assay-Platte, indem Sie die Platte schnell umdrehen und vorsichtig auf sterile Papiertücher tupfen. Wiederholen Sie diese Bewegung 2x-3x, um die gesamte Flüssigkeit von der Platte zu entfernen. Entsorgen Sie die nassen Handtücher.
- Schalten Sie die meisten künstlichen Direktlichter im Raum (lassen Sie eine weiche, schwache Schreibtischlampe oder ähnliches eingeschaltet, um sichtbare Arbeitsbedingungen zu ermöglichen) und in der Biosicherheitswerkbank für alle Schritte bis zum Ende des Tests (ca. 1,5 h) aus.
- Geben Sie 15 ml des Beladungsfarbstoffs (1x) aus Schritt 2.3.3 in ein autofreundliches 150-ml-Reservoir.
- Übertragen Sie 25 μL des Beladungsfarbstoffs (1x) aus dem Reservoir in jedes Bohrloch unter Verwendung eines Liquid-Handling-Systems mit 384/12,5 μL mit niedrigen Rückhaltespitzen, indem Sie 12,5 μL in jede Vertiefung der Platte dosieren (Aspirations- und Dosiergeschwindigkeit: 3,8 μL/s).
- Wiederholen Sie den Pipettierschritt wie in Abschnitt 2.3.7.1 beschrieben, um ein Endvolumen von 25 μL in jeder Vertiefung der Platte zu erreichen. Decken Sie die Platte ab und wickeln Sie sie mit Alufolie ein, um sie vor Umgebungslicht zu schützen.
- Die abgedeckte Zellplatte wird bei 37 °C im CO2 -befeuchteten Inkubator für 30 min inkubiert, aus dem Inkubator entnommen und bei Raumtemperatur im Plattenleser oder auf dem Tisch ausgeglichen, wobei die Platte für weitere 30 Minuten abgedeckt und mit Folie umwickelt gehalten wird. Die Zellen sind dann bereit für das Hochdurchsatz-Screening (HTS).
- Chemische Vorbereitung: Die Arzneiplatte aus Schritt 2.2.1 bei 1.200 × g in einer Plattenzentrifuge für 1 min bei Raumtemperatur drehen.
- Bereiten Sie eine 10-fache Agonisten-Peptidlösung von Rhimi-K-1 (QFSPWGamid) (10 μM) vor, indem Sie 100 nmol lyophilisiertes Peptid in 10 ml 1x HHBS mit 0,1% DMSO resuspendieren und die Lösung in ein autofreundliches 150-ml-Reservoir überführen.
- Messen Sie das Hintergrundsignal: Wählen Sie für den gesamten HTS-Assay im Plattenleser unter Protokolle den Endpunkt-Fluoreszenzmodus aus.
HINWEIS: Das Fluoreszenzsignal wird von der Unterseite der Platte bei Anregungs-/Emissionswellenlängen von 495 nm/525 nm gelesen.- Setzen Sie die Zellplatte in das Plattenlesegerät ein. Wählen Sie in der Instrumententafel die Option Verstärkung anpassen, wählen Sie eine zufällige Vertiefung auf der Platte aus, weisen Sie sie 5 % bis 10 % des maximal messbaren Fluoreszenzwerts zu, und wählen Sie Höhe anpassen (Messwert) aus.
- Klicken Sie auf Messung starten , um die gesamte Platte für das Hintergrundfluoreszenzsignal in relativen Fluoreszenzeinheiten (RFU) zu lesen.
- "Dual-Addition"-Assay
- Verwenden Sie das Liquid-Handling-System mit 384/12,5 μL-Spitzen, um die Arzneimittellösung in jeder Vertiefung der Arzneimittelplatte zu mischen, 10 μl der Arzneimittellösung (in DPBS mit 10% DMSO) 3x auf und ab zu pipettieren (Pipettiergeschwindigkeit: 5,2 μl / s) und "saugen" Sie 1,5 μl aus jeder Vertiefung der Arzneimittelplatte (Aspirationsgeschwindigkeit: 1,0 μl / s).
- "Dosieren" Sie 0,5 μL der Verbindungen in die Zellassayplatte (Pipettiergeschwindigkeit: 1 μL/s), um eine Endkonzentration von 2 μM in 0,2% DMSO zu erreichen.
- Setzen Sie die Assay-Platte sofort in den Plattenleser ein, nachdem Sie die Screening-Verbindungen hinzugefügt haben. Lesen Sie die gleiche Platte sowohl in Vorwärts- als auch in Rückwärtsleserichtung. Um diese Messwerte zu erhalten, definieren Sie ein Programm, das von "well 1-384" und sofort von "384-1" liest.
HINWEIS: Das Auslesen in beide Richtungen dauert insgesamt 2 Minuten. Dieses Auslesedesign soll die Abnahme der Signalstärke kompensieren, die während des Plattenlesens in beide Richtungen auftritt. Siehe Schritt 3.1 für die Ausleseanalysen. - Entsorgen Sie die verbleibenden 1 μL der Arzneimittellösung in den Spitzen, indem Sie die Spitzen in ein 150-ml-Abfallreservoir mit ~ 50 mL DPBS (Dosiergeschwindigkeit: 1,0 μl / s) eintauchen.
- Inkubieren Sie die Siebmassen mit den Zellen für insgesamt 5 Minuten (einschließlich der 2 Minuten Plattenablesung) bei Raumtemperatur in der Biosicherheitswerkbank bei ausgeschaltetem Licht. Geben Sie 3 μL des Agonistenpeptids Rhimi-K-1 (QFSPWGamid) aus dem Reservoir (Aspirationsgeschwindigkeit: 3,1 μL/s) unter Verwendung des Liquid-Handling-Systems (Dosiergeschwindigkeit: 3,1 μL/s) mit 384/12,5 μL-Spitzen in die Assay-Platte.
- Legen Sie die Platte unmittelbar nach der Zugabe des Agonistenpeptids in den Plattenleser. Lesen Sie die Platte sowohl in Vorwärts- als auch in Rückwärtsrichtung mit dem gleichen Programm wie in Schritt 2.3.12.3
3. Datenanalyse
- Berechnen Sie mit der Analysesoftware, die mit dem auf einem Computer installierten Plattenleser verbunden ist, die zellulären Fluoreszenzreaktionen (in RFU) für beide Messwerte nach der ersten Verbindungszugabe (Erstes Lesen; RFUvor [Ergänzungstabelle S2]) und nach den Agonisten-Additionsschritten (Zweiter Lesevorgang = RFU-Ameise [Ergänzungstabelle S2]). Jeder Messwert wird durch Mittelung der beiden Werte ermittelt, die sich aus den Vorwärts- und Rückwärtsablesungen aus Schritt 2.3.12.3 bzw. Schritt 2.3.12.6 ergeben (nicht dargestellt).
ANMERKUNG: RFUago bezieht sich auf die relativen Fluoreszenzeinheiten der potenziellen Agonistentreffer (siehe Schritt 2.3.12.3) bzw. der potenziellen Antagonistentreffer (siehe Schritt 2.3.12.6). - Exportieren Sie alle drei Datensätze (RFU bg, RFU ago und RFUant) aus der Analysesoftware in drei separate Tabellenkalkulationen. Jede Tabellenkalkulation hat nur zwei Spalten: Well-Position und Raw-RFU (Dateien werden hier nicht angezeigt).
HINWEIS: RFU bg bezieht sich auf die RFU des Hintergrunds, der in Schritt 2.3.11.2 gelesen wurde. - Formatieren Sie die Daten aus den drei Tabellenkalkulationen und organisieren Sie die obigen Daten in einer CSV-Datei (siehe Beispiel in Ergänzungstabelle S2). Subtrahieren Sie das Hintergrundsignal, das von der RFU ago bzw. der RFUant gelesen wird (Spalten G und H in der Zusatztabelle S2).
- Importieren Sie die csv-Datei in eine kommerziell erhältliche "Online-HTS-Datenplattform" (siehe Materialverzeichnis) zur nachgelagerten Datenanalyse (Tabelle 1) und Speicherung.
- Manuelles Berechnen des Z'-Faktors für die Qualitätskontrolle jedes Plattentests mit Gleichung (1):
Gleichung (1): (1)
(1)
ANMERKUNG: μ c- und μc+ stellen die durchschnittlichen RFUs der Messwerte der gleichen Vertiefungen dar, die bei der ersten Zugabe nach Zugabe des Lösungsmittels als Negativkontrollen dienen (Leerlösungsmittel, n = 64; Zahlen in blau in der Zusatztabelle S2) und als Positivkontrollen nach Zugabe des Agonisten (Leerlösungsmittel + Agonist, n = 64; Zahlen in Magenta). beziehungsweise. Darüber hinaus stellen σ c- und σc+ die entsprechenden Standardabweichungen (SDs) dar. - Wählen Sie die Treffermoleküle manuell aus den Heatmaps auf der "Online-HTS-Datenplattform" aus und berechnen Sie die normalisierte prozentuale Aktivierung (NPA) und die inhibitorische Aktivität (Io) für die Agonisten-Treffer und Antagonisten-Treffer unter Verwendung von Gleichung (2) und Gleichung (3):
 (2)
(2) (3)
(3)
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Eine hauseigene Wirkstoffplatte (SAC2-34-6170), die aus 320 zufälligen kleinen Molekülen besteht, wurde als Beispiel für die Demonstration dieses HTS-Assays verwendet. Das HTS hatte eine ausgezeichnete Assay-Qualität mit einem Z'-Faktor von 0,7 (Tabelle 1). Dieser Z'-Faktor spiegelt die Assay-Qualität unabhängig von den getesteten Verbindungenwider 34. Ein Z'-Faktor von 0,5 oder höher zeigt einen guten Assay-Signaldynamikbereich zwischen den RFUs der P...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Das Ziel von HTS ist es, Treffermoleküle durch das Screening einer großen Anzahl kleiner Moleküle zu identifizieren. Daher stellen die Ergebnisse dieses Beispiels nur einen kleinen Teil eines konventionellen HTS-Experiments dar. Darüber hinaus müssen die identifizierten Treffermoleküle in nachgeschalteten Assays validiert werden, wie z. B. einem dosisabhängigen Assay an derselben rekombinanten Zelllinie und an einer CHO-K1-Zelllinie, die nur den leeren Vektor trägt, was gleichzeitig durchgeführt werden kann, um ...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben keinen Interessenkonflikt offenzulegen.
Danksagungen
Diese Arbeit wurde durch den USDA-NIFA-AFRI Animal Health and Well-Being Award (Preisnummer 2022-67015-36336, PVP [Projektleiter]) und aus wettbewerbsfähigen Mitteln des Texas A&M AgriLife Research Insect Vector Diseases Grant Program (FY'22-23) an P.V.P. unterstützt. Die A.W.E.S.O.M.E.-Fakultätsgruppe des College of Agriculture and Life Sciences, TAMU, wird für die Hilfe bei der Bearbeitung des Manuskripts gedankt. Die Zusatztabelle S2 enthält Daten aus einer internen, zufälligen, kleinmolekularen Bibliothek, die aus dem Labor von Dr. James Sacchettini an der Texas A&M University und Texas A&M AgriLife Research stammt.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| 0.25% trypsin-EDTA | Gibco Invitrogen | 15050-065 | with phenol red |
| 0.4% trypan blue | MilliporeSigma | T8154 | liquid, sterile |
| 1.5 mL microcentrifuge tubes | Thermo Fisher | AM12400 | RNase-free Microfuge Tubes |
| 5 mL serological pipette | Corning | 29443-045 | Corning Costar Stripette individually wrapped |
| 10 mL serological pipette | Corning | 29443-047 | Corning Costar Stripette individually wrapped |
| 15 mL conical tubes | Falcon | 352196 | sterile |
| 20 µL filter tips | USA Scientifc Inc. | P1121 | sterile, barrier |
| 25 mL serological pipette | Corning | 29443-049 | Corning Costar Stripette individually wrapped |
| 50 mL conical tubes | Corning | 430828 | graduated, sterile |
| 150 mL auto-friendly reservior | Integra Bioscience | 6317 | sterile, individually wrapped for cell seeding in day 1 |
| 150 mL auto-friendly reservior | Integra Bioscience | 6318 | sterile, stacked, for loading dye in day 2 |
| 384/ 12.5 µL low retention tips | Integra Bioscience | 6405 | long, sterile filter |
| 384/ 12.5 µL tips | Integra Bioscience | 6404 | long, sterile filter |
| 384-well plate | Greiner | 781091 | CELLSTAR, clear polystyrene, µClear, Black/Flat |
| Aluminum plate seals | Axygen Scientific | PCR-AS-200 | polyester-based |
| Aluminum foil wrap | Walmart | ||
| Biosafty cabinet II | NuAire | NU-540-300 | |
| Cell counter | Nexcelom | AutoT4 | |
| cell counting slides | Nexcelom | SD-100 | 20 µL chamber |
| CO2 humidified incubator | Thermo Fisher | Forma Series II | |
| Desk Lamp | SunvaleeyTEK | RS1000B | |
| Dimethyl sulfoxide | MilliporeSigma | 276855 | anhydous, >99.9% |
| Drug plate | Corning | 3680 | |
| Dulbecco's phosphate-buffered saline | Corning | 21-031-CV | DPBS, 1x without calcium amd magnesium |
| Ethanol | Koptec | 2000 | |
| F-12K Nutrient Mixture | Corning | 45000-354 | (Kaighn's Mod.) with L-glutamine |
| Fetal bovine serum | Equitech-Bio | SFBU30 | |
| Fluorescent calcium assay kit | ENZO Lifescience | ENZ-51017 | 10x96 tests |
| G418 sulfate | Gibco Invitrogen | 10131-027 | Geneticin selective antibiotic 50 mg/mL |
| Hank's buffer | MilliporeSigma | 55037C | HBSS modified, with calcium, with magnesium, without phenol read |
| HEPES buffer | Gibco Invitrogen | 15630-080 | 1 Molar |
| HTS data storage plateform | CDD vault | https://www.collaborativedrug.com/ | |
| Liquid handling system | Integra Bioscience | Viaflo | 384/12.5 µL |
| Plate centrifuge | Thermo Fisher | Sorvall ST8 | |
| Plate reader | BMG technology | Clariostar | |
| Poly-D-lysine | MilliporeSigma | P6407 | |
| Rhimi-K-1 agonist peptide | Genscript | custom order | QFSPWGamide |
| T-75 flask | Falcon | 353136 |
Referenzen
- Hanlon, C. D., Andrew, D. J. Outside-in signaling - A brief review of GPCR signaling with a focus on the Drosophila GPCR family. Journal of Cell Science. 128 (19), 3533-3542 (2015).
- Liu, N., Li, T., Wang, Y., Liu, S. G-protein coupled receptors (GPCRs) in insects-A potential target for new insecticide development. Molecules. 26 (10), 2993(2021).
- Pierce, K. L., Premont, R. T., Lefkowitz, R. J. Seven-transmembrane receptors. Nature Reviews Molecular Cell Biology. 3, 639-650 (2002).
- Pietrantonio, P. V., Xiong, C., Nachman, R. J., Shen, Y. G protein-coupled receptors in arthropod vectors: Omics and pharmacological approaches to elucidate ligand-receptor interactions and novel organismal functions. Current Opinion in Insect Science. 29, 12-20 (2018).
- Hilger, D., Masureel, M., Kobilka, B. K. Structure and dynamics of GPCR signaling complexes. Nature Structural & Molecular Biology. 25 (1), 4-12 (2018).
- Liu, N., Wang, Y., Li, T., Feng, X. G-protein coupled receptors (GPCRs): Signaling pathways, characterization, and functions in insect physiology and toxicology. International Journal of Molecular Sciences. 22 (10), 5260(2021).
- Hansen, K. B., Bräuner-Osborne, H. FLIPR® assays of intracellular calcium in GPCR drug discovery. G Protein-Coupled Receptors in Drug Discovery. Leifert, W. , Humana Press. Totowa, NJ. (2009).
- Bauknecht, P., Jekely, G. Large-scale combinatorial deorphanization of Platynereis neuropeptide GPCRs. Cell Reports. 12 (4), 684-693 (2015).
- Frooninckx, L., et al. Neuropeptide GPCRs in C. elegans. Frontiers in Endocrinology. 3, 167(2012).
- Caers, J., et al. More than two decades of research on insect neuropeptide GPCRs: An overview. Frontiers in Endocrinology. 3, 151(2012).
- Šimo, L., Koči, J., Park, Y. Receptors for the neuropeptides, myoinhibitory peptide and SIFamide, in control of the salivary glands of the blacklegged tick Ixodes scapularis. Insect Biochemistry and Molecular Biology. 43 (4), 376-387 (2013).
- Šimo, L., Park, Y. Neuropeptidergic control of the hindgut in the black-legged tick Ixodes scapularis. International Journal for Parasitology. 44 (11), 819-826 (2014).
- Liesch, J., Bellani, L. L., Vosshall, L. B. Functional and genetic characterization of neuropeptide Y-like receptors in Aedes aegypti. PLoS Neglected Tropical Diseases. 7 (10), 2486(2013).
- Lu, H. L., Kersch, C. N., Taneja-Bageshwar, S., Pietrantonio, P. V. A calcium bioluminescence assay for functional analysis of mosquito (Aedes aegypti) and tick (Rhipicephalus microplus) G protein-coupled receptors. Journal of Visualized Experiments. (50), e2732(2011).
- Xiong, C., Baker, D., Pietrantonio, P. V. The cattle fever tick, Rhipicephalus microplus, as a model for forward pharmacology to elucidate kinin GPCR function in the Acari. Frontiers in Physiology. 10, 1008(2019).
- Holmes, S. P., Barhoumi, R., Nachman, R. J., Pietrantonio, P. V. Functional analysis of a G protein-coupled receptor from the Southern cattle tick Boophilus microplus (Acari: Ixodidae) identifies it as the first arthropod myokinin receptor. Insect Molecular Biology. 12 (1), 27-38 (2003).
- Cox, K. J., et al. Cloning, characterization, and expression of a G-protein-coupled receptor from Lymnaea stagnalis and identification of a leucokinin-like peptide, PSFHSWSamide, as its endogenous ligand. Journal of Neuroscience. 17 (4), 1197-1205 (1997).
- Dircksen, H. Chapter 32 - Crustacean bioactive peptides. Handbook of Biologically Active Peptides (Second Edition). Kastin, A. J. , Academic Press. Cambridge, MA. 209-221 (2013).
- Halberg, K. A., Terhzaz, S., Cabrero, P., Davies, S. A., Dow, J. A. Tracing the evolutionary origins of insect renal function. Nature Communications. 6, 6800(2015).
- Pietrantonio, P. V., Jagge, C., Taneja-Bageshwar, S., Nachman, R. J., Barhoumi, R. The mosquito Aedes aegypti (L.) leucokinin receptor is a multiligand receptor for the three Aedes kinins. Insect Molecular Biology. 14 (1), 55-67 (2005).
- Radford, J. C., Davies, S. A., Dow, J. A. Systematic G-protein-coupled receptor analysis in Drosophila melanogaster identifies a leucokinin receptor with novel roles. Journal of Biological Chemistry. 277 (41), 38810-38817 (2002).
- Brock, C. M., et al. The leucokinin-like peptide receptor from the cattle fever tick, Rhipicephalus microplus, is localized in the midgut periphery and receptor silencing with validated double-stranded RNAs causes a reproductive fitness cost. International Journal for Parasitology. 49 (3-4), 287-299 (2019).
- Nässel, D. R. Leucokinin and associated neuropeptides regulate multiple aspects of physiology and behavior in Drosophila. International Journal of Molecular Sciences. 22 (4), 1940(2021).
- Kim, Y. -J., et al. Central peptidergic ensembles associated with organization of an innate behavior. Proceedings of the National Academy of Sciences of the United States of America. 103 (38), 14211-14216 (2006).
- Al-Anzi, B., et al. The leucokinin pathway and its neurons regulate meal size in Drosophila. Current Biology. 20 (11), 969-978 (2010).
- Yurgel, M. E., et al. A single pair of leucokinin neurons are modulated by feeding state and regulate sleep-metabolism interactions. PLoS Biology. 17 (2), 2006409(2019).
- Nässel, D. R., Zandawala, M. Recent advances in neuropeptide signaling in Drosophila, from genes to physiology and behavior. Progress in Neurobiology. 179, 101607(2019).
- Okusawa, S., Kohsaka, H., Nose, A. Serotonin and downstream leucokinin neurons modulate larval turning behavior in Drosophila. Journal of Neuroscience. 34 (7), 2544-2558 (2014).
- Kersch, C. N., Pietrantonio, P. V. Mosquito Aedes aegypti (L.) leucokinin receptor is critical for in vivo fluid excretion post blood feeding. FEBS letters. 585 (22), 3507-3512 (2011).
- Kwon, H., et al. Leucokinin mimetic elicits aversive behavior in mosquito Aedes aegypti (L.) and inhibits the sugar taste neuron. Proceedings of the National Academy of Sciences of the United States of America. 113 (25), 6880-6885 (2016).
- Xiong, C., Baker, D., Pietrantonio, P. V. A random small molecule library screen identifies novel antagonists of the kinin receptor from the cattle fever tick, Rhipicephalus microplus (Acari: Ixodidae). Pest Management Science. 77 (5), 2238-2251 (2021).
- Torfs, P., et al. The kinin peptide family in invertebrates. Annals of the New York Academy of Sciences. 897 (1), 361-373 (1999).
- Ma, Q., Ye, L., Liu, H., Shi, Y., Zhou, N. An overview of Ca2+ mobilization assays in GPCR drug discovery. Expert Opinion on Drug Discovery. 12 (5), 511-523 (2017).
- Zhang, J. -H., Chung, T. D., Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 4 (2), 67-73 (1999).
- Zhang, R., Xie, X. Tools for GPCR drug discovery. Acta Pharmacologica Sinica. 33 (3), 372-384 (2012).
- Offermanns, S., Simon, M. I. Gα15 and Gα16 couple a wide variety of receptors to phospholipase C. Journal of Biological Chemistry. 270 (25), 15175-15180 (1995).
- Murgia, M. V., et al. High-content phenotypic screening identifies novel chemistries that disrupt mosquito activity and development. Pesticide Biochemistry and Physiology. 182, 105037(2022).
- Lismont, E., et al. Can BRET-based biosensors be used to characterize G-protein mediated signaling pathways of an insect GPCR, the Schistocerca gregaria CRF-related diuretic hormone receptor. Insect Biochemistry and Molecular Biology. 122, 103392(2020).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten