
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Un test de fluorescence calcique « à double addition » pour le criblage à haut débit de récepteurs couplés à la protéine G recombinante
Dans cet article
Résumé
Dans ce travail, un test de fluorescence calcique intracellulaire à haut débit pour des plaques à 384 puits afin de cribler de petites banques de molécules sur des récepteurs couplés aux protéines G recombinantes (RCPG) est décrit. La cible, le récepteur kinine de la tique de la fièvre bovine, Rhipicephalus microplus, est exprimée dans les cellules CHO-K1. Ce test identifie les agonistes et les antagonistes utilisant les mêmes cellules dans un test de « double addition ».
Résumé
Les récepteurs couplés aux protéines G (RCPG) représentent la plus grande superfamille de récepteurs et sont la cible de nombreux médicaments humains. Le criblage à haut débit (HTS) de bibliothèques aléatoires de petites molécules par rapport aux RCPG est utilisé par l’industrie pharmaceutique pour la découverte de médicaments spécifiques à la cible. Dans cette étude, un HTS a été utilisé pour identifier de nouveaux ligands à petites molécules de RCPG neuropeptidiques spécifiques aux invertébrés comme sondes pour des études physiologiques de vecteurs d’agents pathogènes humains et vétérinaires mortels.
Le récepteur kinin spécifique aux invertébrés a été choisi comme cible parce qu’il régule de nombreux processus physiologiques importants chez les invertébrés, y compris la diurèse, l’alimentation et la digestion. De plus, la pharmacologie de nombreux RCPG invertébrés est mal caractérisée ou pas caractérisée du tout; par conséquent, la pharmacologie différentielle de ces groupes de récepteurs par rapport aux RCPG apparentés chez d’autres métazoaires, en particulier chez les humains, ajoute des connaissances aux relations structure-activité des RCPG en tant que superfamille. Un test HTS a été développé pour des cellules dans des plaques de 384 puits pour la découverte de ligands du récepteur kinine de la tique de la fièvre bovine, ou tique du bétail du sud, Rhipicephalus microplus. Le récepteur de la kinine à tiques était exprimé de manière stable dans les cellules CHO-K1.
Le récepteur des kinines, lorsqu’il est activé par des neuropeptides de kinine endogènes ou d’autres agonistes de petites molécules, déclenche la libération de Ca2+ des réserves de calcium dans le cytoplasme. Ce test de fluorescence calcique combiné à une approche de « double addition » peut détecter des molécules « hit » agonistes et antagonistes fonctionnelles dans la même plaque de dosage. Chaque essai a été réalisé à l’aide de plaques médicamenteuses portant un réseau de 320 petites molécules aléatoires. Un facteur Z' fiable de 0,7 a été obtenu, et trois molécules d’agonistes et deux molécules d’antagoniste ont été identifiées lorsque le HTS était à une concentration finale de 2 μM. Le test de fluorescence calcique rapporté ici peut être adapté pour dépister d’autres RCPG qui activent la cascade de signalisation Ca2+ .
Introduction
Les récepteurs couplés aux protéines G (RCPG), qui sont présents de la levure à l’homme, représentent la plus grande superfamille de récepteurs dans de nombreux organismes1. Ils jouent un rôle essentiel dans la régulation de presque tous les processus biologiques chez les animaux. Il y a 50 à 200 RCPG dans le génome des arthropodes, ce qui signifie qu’ils représentent la plus grande superfamille de récepteurs membranaires2. Ils sont classés en six classes principales, A-F, en fonction de leur similitude de séquence et de leurs fonctions3. Les RCPG transduisent divers signaux extracellulaires, tels que ceux des hormones, des neuropeptides, des amines biogènes, du glutamate, du proton, des lipoglycoprotéines et des photons4. Les RCPG se couplent aux protéines G hétérotrimères (Gα, Gβ et Gγ) pour transmettre des signaux en aval. Les RCPG couplés aux protéines Gαs ou Gαi/o augmentent ou diminuent, respectivement, les niveaux intracellulaires d’adénosine monophosphate (AMPc) 3', 5'-cyclique en activant ou en inhibant l’adénylylcyclase. Les RCPGs couplés à Gαq/11 induisent la libération de calcium à partir des réserves de calcium du réticulum endoplasmique en activant la voie phospholipase C (PLC)-inositol-1,4,5-triphosphate (IP3). Les RCPGs couplés à Gα12/13 activent les facteurs d’échange nucléotidiquesde la RhoGTPase 5,6. Les RCPG sont la cible de plus de 50% des médicaments humains et d’un acaricide, l’amitraze4. Comme les RCPG transduisent des signaux aussi divers, ils sont des cibles prometteuses pour le développement de nouveaux pesticides qui perturbent les fonctions physiologiques spécifiques aux invertébrés.
L’objectif de HTS est d’identifier les molécules de frappe qui peuvent moduler les fonctions des récepteurs. HTS implique le développement de tests, la miniaturisation et l’automatisation7. Les RCPG des neuropeptides arthropodes sont impliqués dans la plupart des fonctions physiologiques, telles que le développement, la mue et l’ecdysis, l’excrétion, la mobilisation énergétique et la reproduction4. La plupart des RCPG neuropeptidiques des arthropodes et des métazoaires signalent par la cascade de signalisation calcique 2,6,8,9,10, comme dans le peptide myoinhibiteur et les récepteurs SIFamide de la tique à pattes noires Ixodes scapularis; leurs ligands sont antagonistes dans les essais de motilité de l’intestin postérieur, le SIF provoquant une contraction et la MIP l’inhibant11,12. Un récepteur de type NPY du moustique de la fièvre jaune, Aedes aegypti, régule l’hôte femelle à la recherche13. Comparé à d’autres tests alternatifs de mobilisation du calcium tels que le test de bioluminescence calcique de l’équorine14, le test de fluorescence du calcium est facile à réaliser, ne nécessite pas la transfection d’autres protéines de détection du calcium recombinant et est rentable. Le test de fluorescence calcique produit un signal prolongé par rapport au signal cinétique rapide obtenu dans le test de bioluminescence calcique de l’équorine14,15.
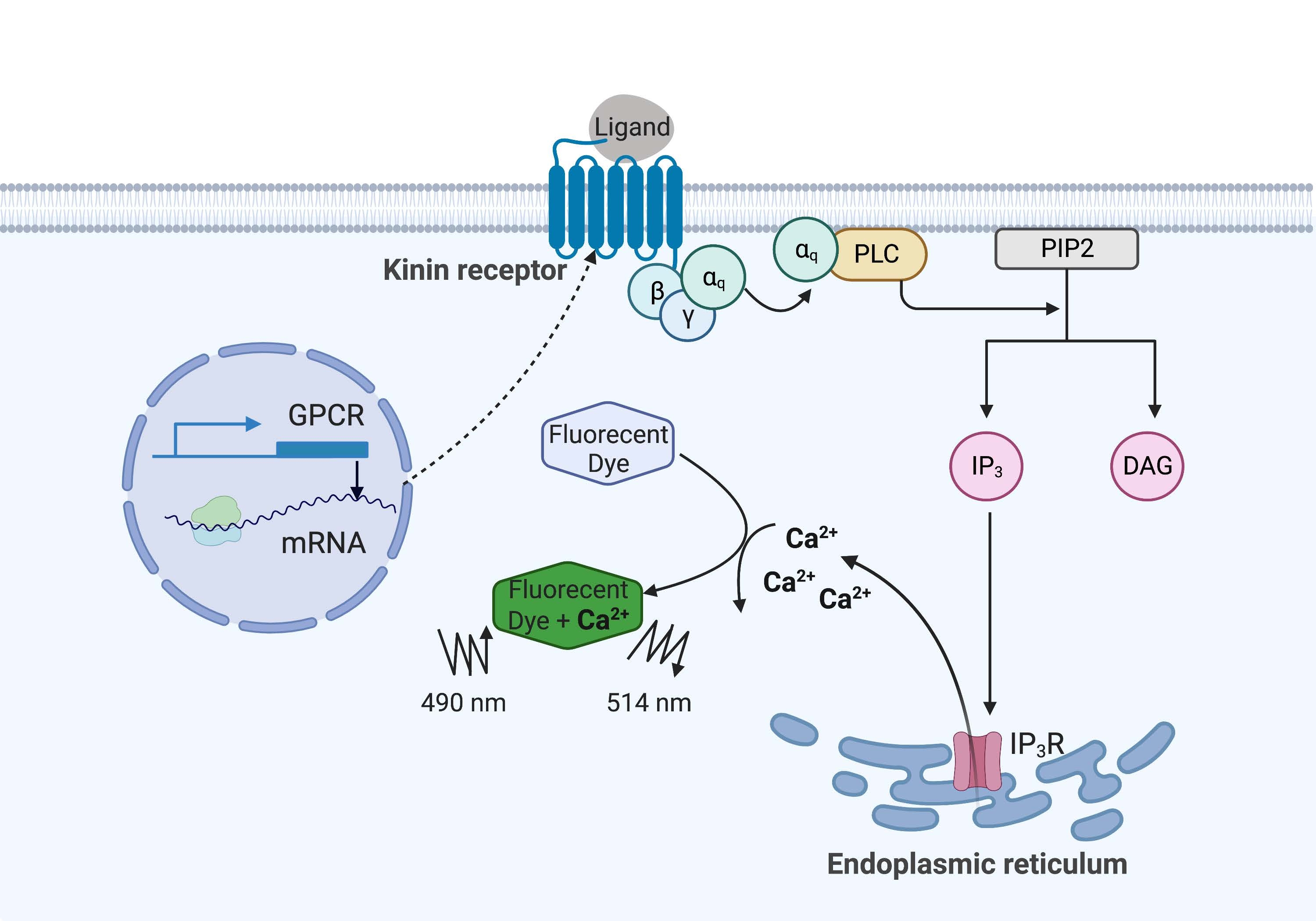
Dans l’exemple ici, le récepteur kinine de la tique de la fièvre bovine, Rhipicephalus microplus, a été exprimé de manière recombinante dans la lignée cellulaire CHO-K1 et utilisé pour le test de fluorescence calcique. Il n’y a qu’un seul gène récepteur de la kinine trouvé dans R. microplus; le récepteur signale par une voie de signalisation dépendante de la protéine Gq et déclenche l’efflux de Ca2+ des réserves de calcium dans l’espace intracellulaire16. Ce processus peut être détecté et quantifié par un fluorophore, qui provoque un signal de fluorescence lors de la liaison des ions calcium (Figure 1).
Le récepteur kinine est un RCPG spécifique aux invertébrés, qui appartient aux récepteurs de type rhodopsine de classe A. La kinine est un ancien neuropeptide de signalisation présent dans Mollusca, Crustacea, Insecta et Acari 4,17,18. Les coléoptères (coléoptères) n’ont pas le système de signalisation des kinines; chez le moustique Aedes aegypti, il n’y a qu’un seul récepteur de kinine qui se lie à trois aedeskinines, tandis que Drosophila melanogaster a un récepteur de kinine avec la drosokinine comme ligandunique 19,20,21. Il n’y a pas de kinines homologues ou de récepteurs de kinines chez les vertébrés. Bien que la fonction exacte de la kinine soit inconnue chez les tiques, les femelles de R. microplus silencieuses de R. microplus soumises au récepteur de la kinine sont réduites de manière significative22. Les kinines sont des peptides pléotropes chez les insectes. Chez Drosophila melanogaster, ils sont impliqués dans les systèmes de régulation nerveux central et périphérique23, la pré-ecdysis 24, l’alimentation25, le métabolisme 26 et les schémas d’activité du sommeil26,27, ainsi que la locomotionlarvaire 28. Les kinines régulent la contraction de l’intestin postérieur, la diurèse et l’alimentation du moustique A. aegypti 29,30,31. Les peptides kinines ont conservé un pentapeptide C-terminal Phe-X1-X2-Trp-Gly-NH2, qui est la séquence minimale requise pour l’activité biologique32. La spécificité des arthropodes, la petite taille du ligand endogène, qui les rend sujets à l’interférence des petites molécules, et les fonctions pléiotropiques chez les insectes font du récepteur kinine une cible prometteuse pour la lutte antiparasitaire4.
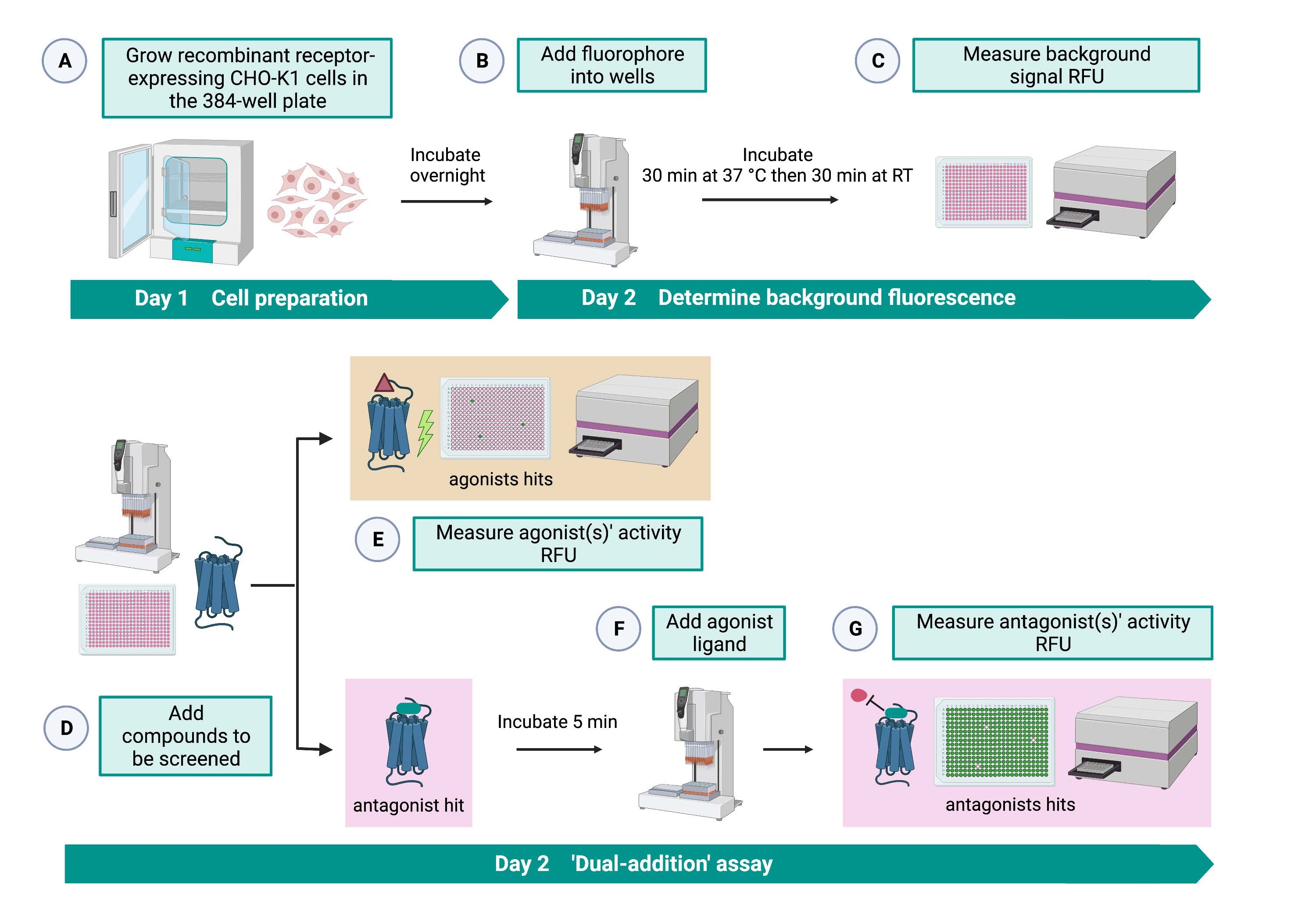
Le test de « double addition » (Figure 2) permet l’identification d’agonistes ou d’antagonistes dans le même test HTS15. Il est adapté d’un test de « double addition » couramment utilisé dans l’industrie pharmaceutique pour la découverte de médicaments33. En bref, le premier ajout de médicaments dans la plaque cellulaire permet d’identifier les agonistes potentiels dans la bibliothèque chimique lorsqu’un signal de fluorescence plus élevé est détecté par rapport à l’application du contrôle du solvant. Après 5 min d’incubation avec ces petites molécules, un agoniste connu (peptide kinine) est appliqué sur tous les puits. Les puits qui ont reçu au hasard un antagoniste de la plaque médicamenteuse affichent un signal de fluorescence plus faible lors de l’ajout d’agonistes par rapport aux puits témoins qui ont reçu le solvant lors de la première addition. Ce test permet ensuite d’identifier des agonistes et antagonistes potentiels avec les mêmes cellules. Dans un projet HTS standard, ces molécules frappées seraient validées par des tests dose-réponse et par des tests d’activité biologique supplémentaires, qui ne sont pas présentés ici.

Figure 1 : Illustration du mécanisme d’essai de fluorescence calcique. La protéine Gq déclenche la voie de signalisation intracellulaire du calcium. Le récepteur de la kinine (récepteur couplé à la protéine G) a été exprimé de manière recombinante dans les cellules CHO-K1. Lorsque le ligand agoniste se lie au récepteur, la protéine Gq associée au récepteur kinin active l’API, ce qui catalyse la conversion d’une molécule PIP2 en IP3 et DAG. IP 3 se lie ensuite à l’IP3R à la surface du réticulum endoplasmique, conduisant à la libération de Ca 2+ dans le cytoplasme, où les ions Ca2+ se lient aux fluorophores et provoquent un signal de fluorescence. Le signal de fluorescence peut être obtenu par excitation à 490 nm et détecté à 514 nm. Abréviations : RCPG = récepteur couplé aux protéines G; PLC = phospholipase C; PIP2 = phosphatidylinositol 4,5-bisphosphate; IP3 = inositol trisphosphate; DAG = diacylglycérol; IP3 R = récepteur IP3. Créé avec BioRender.com. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Flux de travail pour le criblage à haut débit de petites molécules sur un récepteur couplé à une protéine G exprimé dans des cellules CHO-K1. (A) Des cellules CHO-K1 recombinantes exprimant de manière stable le récepteur kinine ont été ajoutées à la plaque de 384 puits (10 000 cellules/puits) à l’aide d’un système de manipulation de liquide (25 μL/puits) et incubées dans un incubateur de CO2 humidifié pendant 12 à 16 h. (B ) Le tampon d’essai contenant le colorant fluorescent (25 μL/puits) a été ajouté dans la plaque cellulaire à l’aide d’un système de manipulation de liquide. La plaque a été incubée pendant 30 min à 37 °C pendant 30 min et équilibrée à TA pendant 30 minutes supplémentaires. (C) Le signal de fluorescence de fond des cellules de chaque puits a été mesuré à l’aide d’un lecteur de plaques. (D) Des solutions médicamenteuses provenant d’une plaque bibliothèque de 384 puits et d’un solvant à blanc (tous à 0,5 μL/puits) ont été ajoutées dans la plaque d’essai cellulaire à l’aide d’un système de manipulation de liquide. (E) Les réponses cellulaires de fluorescence calcique ont été mesurées avec le lecteur de plaque immédiatement après l’ajout des solutions médicamenteuses; Les composés suscitant des signaux de fluorescence supérieurs à la moyenne ont été choisis comme des coups agonistes. Des hits antagonistes qui bloquent le RCPG (icône ci-dessous) ont été révélés après l’ajout de l’agoniste peptidique à l’étape G. (F) Dans la même plaque d’essai, après 5 minutes d’incubation des cellules avec des composés de criblage, un peptide agoniste endogène Rhimi-K-1 (QFSPWGamide) du récepteur des tiques kinines a été ajouté à chaque puits (1 μM). (G) Les réponses de fluorescence cellulaire après l’ajout du peptide agoniste ont été mesurées immédiatement par le lecteur de plaques. Le(s) composé(s) inhibant(s) le signal de fluorescence a été sélectionné comme antagoniste(s). Abréviations : RCPG = récepteur couplé aux protéines G; RT = température ambiante; RFU = unités de fluorescence relative. Créé avec BioRender.com. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Protocole
1. Entretien des cellules
REMARQUE : UNE LIGNÉE CELLULAIRE CHO-K1 qui exprime de façon stable le récepteur des kinines de R. microplus, nommée BMLK3, a été développée par Holmes et al.16. Les détails du développement de la lignée cellulaire sont présentés ailleurs14. Toutes les étapes suivantes sont effectuées dans des conditions stériles dans une enceinte de biosécurité de classe II.
- Cultiver la lignée cellulaire recombinante dans un milieu sélectif (milieu F-12K contenant 10 % de sérum fœtal bovin [FBS] et 800 μg/mL de sulfate G418) pour assurer l’expression du récepteur cible. Entreposer les cellules exprimant de façon stable le récepteur dans 1 mL de milieu de congélation (FBS à 90 % et 10 % de diméthylsulfoxyde [DMSO]) dans un cryovial de 2 mL dans un congélateur à -80 °C.
REMARQUE: Pour le stockage à long terme des cellules congelées, stockez-les dans de l’azote liquide. - Préchauffer tous les milieux avant la culture cellulaire. Dans une enceinte de biosécurité, transférer 13 mL de milieu sélectif préchauffé (milieu F-12K contenant 10 % de FBS et 800 μg/mL de sulfate G418) dans une fiole T-75 et la conserver dans l’enceinte de biosécurité. Décongeler un flacon de3 cellules BMLK congelées (~1,5 × 106 cellules) dans un bain-marie à 37 °C pendant 2-3 min. Transvaser les cellules décongelées dans le ballon T-75. Maintenir les cellules dans un incubateur humidifié à 37 °C et 5 % de CO2 , sauf indication contraire.
- Lorsque les cellules atteignent 90% de confluence (1-2 jours), préchauffer tout le milieu à 37 ° C, à l’exception de la solution saline tamponnée au phosphate (DPBS) de Dulbecco, qui est maintenue à température ambiante. Dans une enceinte de biosécurité, retirer le milieu usé de la fiole T-75, laver les cellules pendant 5 s avec 10 mL de DPBS en remuant doucement la fiole, puis retirer la DPBS à l’aide d’une pipette sérologique.
REMARQUE: Une image de cellules CHO-K1 à 90% de confluence est montrée dans Lu et al.14. - Détacher les cellules du ballon T-75 en ajoutant 2 mL de trypsine-EDTA à 0,25 % et incuber pendant 3 à 5 minutes à 37 °C dans l’incubateur. Ensuite, ajouter 8 mL de milieu sélectif et bien mélanger en pipetant et en relâchant doucement 2x-3x avec la même pipette sérologique.
- Transvaser 2 mL de la suspension cellulaire (~1 × 106 cellules) de l’étape 1.4 dans une nouvelle fiole T-75 contenant 10 mL de milieu sélectif chaud. Cultivez les cellules pendant 1-2 jours dans l’incubateur jusqu’à ce qu’elles atteignent 90% de confluence.
- Utilisez les cellules pour le test en suivant les étapes suivantes, ou répétez les étapes 1.4-1.5 une ou deux fois avant d’utiliser les cellules dans le test.
REMARQUE: Ne pas dépasser trois à quatre passages, car le signal de dosage avec certaines lignées cellulaires peut s’affaiblir avec d’autres passages.
2. Dosage de fluorescence calcique
- Enduire la plaque cellulaire.
- Utiliser un système de manipulation des liquides placé à l’intérieur d’une enceinte de biosécurité pour toutes les étapes de pipetage dans les plaques de 384 puits. Créez des programmes personnalisés pour le pipetage dans les plaques 384 puits31 (tableau supplémentaire S1). Enduisez à l’avance les plaques stériles à 384 puits. Dans une enceinte de biosécurité, charger 10 μL/puits d’une solution aqueuse de Poly-D-lysine (PDL) à 0,05 mg/mL dans chaque plaque et incuber pendant 5 min à température ambiante.
- Videz l’assiette en la retournant rapidement et en l’épongeant doucement sur des serviettes en papier stériles. Ensuite, rincez chaque puits avec 10 μL d’eau, videz l’assiette et laissez l’assiette sécher dans l’enceinte de biosécurité pendant la nuit sans couvercle. Fermez la plaque avec le couvercle et conservez à 4 °C au réfrigérateur.
NOTE: Les plaques revêtues peuvent être conservées à 4 ° C jusqu’à 6 mois.
- Jour 1
- Sortez la plaque de médicament (100 μM dans 90% de DPBS + 10% de DMSO, la concentration finale dans le puits pour HTS sera de 2 μM) stockée dans le congélateur à -20 °C et placez-la à température ambiante.
NOTE: Disposition de la plaque médicamenteuse: Chaque plaque de 384 puits (24 colonnes x 16 rangées) contient 320 puits avec différents composés de bibliothèque et 64 puits avec solvant blanc (DPBS contenant 10% de DMSO), qui sont disposés en quatre colonnes, avec deux colonnes sur le bord de chaque côté. Voir le tableau supplémentaire S2 pour la disposition des plaques. - Lorsque les cellules atteignent une confluence de ~70 % à 90 % dans le ballon T-75, détacher les cellules du ballon T-75 comme décrit ci-dessous. Préchauffer tout le média à 37 °C à l’exception de la DPBS (température ambiante).
- Retirez le milieu usé, lavez les cellules avec 10 ml de DPBS, puis retirez le DPBS. Détacher les cellules du ballon T-75 à l’aide de 2 mL de trypsine-EDTA à 0,25 % pendant 3 à 5 min à 37 °C dans l’incubateur, ajouter 8 mL de milieu sélectif et transférer la suspension cellulaire dans un tube conique de 15 mL pour centrifuger à 1 000 × g pendant 3 min.
- Jeter le surnageant et remettre en suspension la pastille de cellule dans 10 mL de milieu F-12K contenant 1 % de FBS et 400 μg/mL de sulfate G418. Gardez la suspension dans l’enceinte de biosécurité tout en déterminant le nombre de cellules.
- Déterminer la densité cellulaire de la suspension en vue d’une dilution ultérieure : Mélanger 20 μL de suspension cellulaire dans 20 μL de bleu de trypan à 0,4 %, puis charger 20 μL du mélange dans une chambre de comptage de cellules pour être lu par un compteur de cellules pour la densité cellulaire.
- Diluer la suspension cellulaire dans le même milieu (milieu F-12K contenant 1 % de FBS et 400 μg/mL de sulfate de G418) jusqu’à obtenir un volume final d’au moins 15 mL à une densité de 4 × 105 cellules/mL.
- Ensemencez les cellules dans la plaque de 384 puits recouverte de PDL. Transférer 15 mL de la suspension cellulaire ci-dessus (4 × 105 cellules/mL) dans un réservoir de réactif auto-respectueux de 150 mL. Distribuer 25 μL de la suspension cellulaire (~10 000 cellules/puits) dans chaque puits des 384 puits de la plaque à l’aide d’un système de manipulation des liquides en deux étapes. Charger 384/12,5 μL de pointes à faible rétention sur la tête du manipulateur de liquides, aspirer 12,5 μL du réservoir (vitesse 5,2 μL/s) et distribuer dans chaque puits (vitesse : 3,1 μL/s).
- Répéter le pipetage comme à l’étape précédente 2.2.5 pour atteindre 25 μL par puits. Ensuite, incuber la plaque pendant une nuit (14-16 h) à 37 °C et 5% de CO2 dans l’incubateur humidifié.
REMARQUE: Comme le volume maximal de pipetage est de 12,5 μL pour cette tête spécifique, le pipetage pour 25 μL est effectué en deux étapes.
- Sortez la plaque de médicament (100 μM dans 90% de DPBS + 10% de DMSO, la concentration finale dans le puits pour HTS sera de 2 μM) stockée dans le congélateur à -20 °C et placez-la à température ambiante.
- Jour 2
- Le lendemain matin, vérifiez la plaque cellulaire couverte au microscope; Si elles ne sont pas confluentes, attendez que les cellules atteignent 90% de confluence.
- Préparez une solution mère de colorant fluorescent : Remettez en suspension le colorant fluorescent lyophilisé dans 100 μL de DMSO et évitez la lumière directe sur la solution mère. Enveloppez le tube avec du papier d’aluminium pour éviter le photoblanchiment.
NOTA : Le stock peut être aliquote en aliquotes de 15 μL pour chaque essai sur plaque afin d’éviter la congélation et la décongélation répétées; les aliquotes peuvent être conservées à −80 °C jusqu’à 1 mois. - Dans l’enceinte de biosécurité avec les lumières éteintes, préparer le colorant de chargement (1x) dans un tube conique de 15 mL enveloppé de papier d’aluminium, en combinant 15 μL de la solution mère de colorant fluorescent (à partir de l’étape 2.3.2) et le reste des composants préchauffés de la trousse (37 °C) : 13,5 mL de 1x HHBS (tampon de Hank avec 20 mM HEPES) et 1,5 mL de réactif B (inhibiteur d’efflux de colorant).
REMARQUE: La lumière de la pièce est allumée pendant cette étape. - Fermez le tube et mélangez bien en le retournant doucement plusieurs fois (généralement 3 à 5 fois).
NOTE: Conserver à température ambiante et utiliser le mélange de colorant de chargement dans les 30 minutes. - Lorsque les cellules atteignent 90 % de confluence, retirer le milieu usé de la plaque d’essai à 384 puits en retournant rapidement la plaque et en épongeant doucement sur du papier absorbant stérile; Répétez ce mouvement 2x-3x pour retirer tout le liquide de la plaque. Jetez les serviettes mouillées.
- Éteignez la plupart des lumières directes artificielles dans la pièce (en laissant une lampe de bureau douce et faible allumée ou similaire pour permettre des conditions de travail visibles) et dans l’armoire de biosécurité pour toutes les étapes jusqu’à la fin du test (environ pendant 1,5 h).
- Placer 15 mL du colorant de chargement (1x) de l’étape 2.3.3 dans un réservoir auto-friendly de 150 mL.
- Transférer 25 μL du colorant de chargement (1x) du réservoir dans chaque puits à l’aide d’un système de manutention de liquide avec 384/12,5 μL de pointes de faible rétention en distribuant 12,5 μL dans chaque puits de la plaque (vitesse d’aspiration et de distribution : 3,8 μL/s).
- Répéter l’étape de pipetage comme au point 2.3.7.1 pour atteindre un volume final de 25 μL dans chaque puits de la plaque. Couvrez et enveloppez la plaque avec du papier d’aluminium pour la protéger de la lumière ambiante.
- Incuber la plaque cellulaire couverte à 37 °C dans l’incubateur humidifié au CO2 pendant 30 min, la retirer de l’incubateur et l’équilibrer à température ambiante à l’intérieur du lecteur de plaques ou sur le banc, en gardant la plaque couverte et enveloppée de papier d’aluminium pendant encore 30 min. Les cellules sont alors prêtes pour le criblage à haut débit (HTS).
- Préparation chimique: Faire tourner la plaque de médicament de l’étape 2.2.1 à 1 200 × g dans une centrifugeuse à plaques pendant 1 min à température ambiante.
- Préparer une solution peptidique agoniste 10x de Rhimi-K-1 (QFSPWGamide) (10 μM) en remettant en suspension 100 nmoles de peptide lyophilisé dans 10 mL de 1x HHBS contenant 0,1% de DMSO, et transférer la solution dans un réservoir auto-friendly de 150 mL.
- Mesurer le signal de fond : Pour l’ensemble du test HTS dans le lecteur de plaques, sous Protocoles, choisissez le mode de fluorescence du point final.
NOTE: Le signal fluorescent est lu à partir du bas de la plaque à des longueurs d’onde d’excitation/émission de 495 nm/525 nm.- Insérez la plaque cellulaire dans le lecteur de plaque. Dans le tableau de bord, sélectionnez ajuster le gain, sélectionnez un puits aléatoire sur la plaque, attribuez-le à 5 % à 10 % de la valeur de fluorescence maximale mesurable et sélectionnez ajuster la hauteur (lecture).
- Cliquez sur Démarrer la mesure pour lire la plaque entière pour le signal de fluorescence de fond en unités de fluorescence relative (RFU).
- Essai de « double addition »
- À l’aide du système de manipulation des liquides avec des pointes de 384/12,5 μL, pour mélanger la solution médicamenteuse dans chaque puits de la plaque médicamenteuse, pipeter 3x de haut en bas de 10 μL de la solution médicamenteuse (dans DPBS contenant 10% de DMSO) (vitesse de pipetage: 5,2 μL / s) et « aspirer » 1,5 μL de chaque puits de la plaque médicamenteuse (vitesse d’aspiration: 1,0 μL / s).
- « Distribuer » 0,5 μL des composés dans la plaque d’essai cellulaire (vitesse de pipetage : 1 μL/s) pour atteindre une concentration finale de 2 μM dans du DMSO à 0,2 %.
- Placez immédiatement la plaque de dosage dans le lecteur de plaque après avoir ajouté les composés de criblage. Lisez la même plaque dans les directions de lecture avant et arrière. Pour obtenir ces lectures, définissez un programme à lire à partir de « puits 1-384 » et immédiatement à partir de « 384-1 ».
REMARQUE: La lecture dans les deux sens prend 2 min au total. Cette conception de lecture vise à compenser la diminution de l’intensité du signal qui se produit pendant la lecture de la plaque dans chaque direction. Voir l’étape 3.1 pour les analyses de lecture. - Éliminer le reste de 1 μL de solution médicamenteuse dans les embouts en immergeant les embouts dans un réservoir de déchets de 150 mL contenant ~50 mL de DPBS (vitesse de distribution : 1,0 μL/s).
- Incuber les composés de criblage avec les cellules pendant un total de 5 minutes (y compris les 2 minutes de lecture sur plaque) à température ambiante dans l’enceinte de biosécurité avec les lumières éteintes. Ajouter 3 μL du peptide agoniste, Rhimi-K-1 (QFSPWGamide), du réservoir (vitesse d’aspiration : 3,1 μL/s) dans la plaque d’essai à l’aide du système de manipulation du liquide (vitesse de distribution : 3,1 μL/s) avec des pointes de 384/12,5 μL.
- Placez la plaque dans le lecteur de plaque immédiatement après l’ajout du peptide agoniste. Lire la plaque dans les directions avant et arrière en utilisant le même programme qu’à l’étape 2.3.12.3
3. Analyse des données
- À l’aide du logiciel d’analyse associé au lecteur de plaques installé sur un ordinateur, calculer les réponses de fluorescence cellulaire (en RFU) pour les deux lectures après la première addition de composé (Première lecture; RFUago [Tableau supplémentaire S2]) et après les étapes d’addition agoniste (Deuxième lecture = RFU ant [Tableau supplémentaire S2]). Chaque lecture est obtenue en faisant la moyenne des deux valeurs obtenues (non représentées) par les lectures sur plaque avant et arrière de l’étape 2.3.12.3 et de l’étape 2.3.12.6, respectivement.
NOTE: Il y a RFU, les fourmis RFU désignent les unités de fluorescence relatives des hits agonistes potentiels (lire à l’étape 2.3.12.3) et des hits antagonistes potentiels (lire à l’étape 2.3.12.6), respectivement. - Exportez les trois ensembles de données, RFU bg, RFUago et RFUant, du logiciel d’analyse vers trois feuilles de calcul distinctes. Chaque feuille de calcul n’aura que deux colonnes: position du puits et RFU brute (fichiers non affichés ici).
NOTE: RFU bg fait référence à la RFU de l’arrière-plan lu à l’étape 2.3.11.2. - Mettez en forme les données des trois feuilles de calcul et organisez les données ci-dessus dans un fichier csv (voir l’exemple du tableau supplémentaire S2). Soustrayez le signal de fond lu de la RFU ago et de la RFU ant, respectivement (colonnes G et H dans le tableau supplémentaire S2).
- Importez le fichier csv dans une « plateforme de données HTS en ligne » disponible dans le commerce (voir le tableau des matériaux) pour l’analyse des données en aval (tableau 1) et le stockage.
- Calculer manuellement le facteur Z' pour le contrôle de la qualité de chaque essai sur plaque à l’aide de l’équation (1) :
Équation (1): (1)
(1)
NOTE: μ c- et μc+ représentent les UFR moyennes des lectures des mêmes puits, qui servent de témoins négatifs dans la première addition après l’ajout du solvant (solvant blanc, n = 64; nombres en bleu dans le tableau supplémentaire S2) et servent de témoins positifs après l’ajout de l’agoniste (solvant blanc + agoniste, n = 64; nombres en magenta), respectivement. De plus, σ c- et σc+ représentent leurs écarts-types (ET) correspondants. - Sélectionnez manuellement les molécules d’impact à partir des cartes thermiques sur la « plate-forme de données HTS en ligne » et calculez le pourcentage normalisé d’activation (NPA) et d’activité inhibitrice (Io) pour les hits agonistes et les hits antagonistes à l’aide de l’équation (2) et de l’équation (3):
 (2)
(2) (3)
(3)
Résultats
Une plaque médicamenteuse interne (SAC2-34-6170) composée de 320 petites molécules aléatoires a été utilisée pour démontrer ce test HTS à titre d’exemple. Le HTS avait une excellente qualité de dosage avec un facteur Z' de 0,7 (tableau 1). Ce facteur Z' reflète la qualité du dosage indépendamment des composés testés34. Un facteur Z de 0,5 ou plus indique une bonne plage dynamique du signal de test entre les UFR des témoins positifs et des t?...
Discussion
L’objectif de HTS est d’identifier les molécules frappées en criblant un nombre massif de petites molécules. Par conséquent, les résultats de cet exemple ne représentent qu’une petite partie d’une expérience HTS conventionnelle. De plus, les molécules hit, identifiées, doivent être validées dans des essais en aval tels qu’un dosage dose-dépendant sur la même lignée cellulaire recombinante et sur une lignée cellulaire CHO-K1 ne portant que le vecteur vide, qui peut être réalisé simultanément p...
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts à divulguer.
Remerciements
Ce travail a été soutenu par le prix USDA-NIFA-AFRI Animal Health and Well-Being Award (numéro de prix 2022-67015-36336, PVP [Project Director]) et par des fonds compétitifs du Texas A&M AgriLife Research Insect Vector Diseases Grant Program (FY'22-23) à P.V.P. Le groupe de professeurs A.W.E.S.O.M.E. du College of Agriculture and Life Sciences, TAMU, est reconnu pour son aide dans l’édition du manuscrit. Le tableau supplémentaire S2 contient des données provenant d’une bibliothèque interne aléatoire de petites molécules obtenues du laboratoire du Dr James Sacchettini à la Texas A&M University et de Texas A&M AgriLife Research.
matériels
| Name | Company | Catalog Number | Comments |
| 0.25% trypsin-EDTA | Gibco Invitrogen | 15050-065 | with phenol red |
| 0.4% trypan blue | MilliporeSigma | T8154 | liquid, sterile |
| 1.5 mL microcentrifuge tubes | Thermo Fisher | AM12400 | RNase-free Microfuge Tubes |
| 5 mL serological pipette | Corning | 29443-045 | Corning Costar Stripette individually wrapped |
| 10 mL serological pipette | Corning | 29443-047 | Corning Costar Stripette individually wrapped |
| 15 mL conical tubes | Falcon | 352196 | sterile |
| 20 µL filter tips | USA Scientifc Inc. | P1121 | sterile, barrier |
| 25 mL serological pipette | Corning | 29443-049 | Corning Costar Stripette individually wrapped |
| 50 mL conical tubes | Corning | 430828 | graduated, sterile |
| 150 mL auto-friendly reservior | Integra Bioscience | 6317 | sterile, individually wrapped for cell seeding in day 1 |
| 150 mL auto-friendly reservior | Integra Bioscience | 6318 | sterile, stacked, for loading dye in day 2 |
| 384/ 12.5 µL low retention tips | Integra Bioscience | 6405 | long, sterile filter |
| 384/ 12.5 µL tips | Integra Bioscience | 6404 | long, sterile filter |
| 384-well plate | Greiner | 781091 | CELLSTAR, clear polystyrene, µClear, Black/Flat |
| Aluminum plate seals | Axygen Scientific | PCR-AS-200 | polyester-based |
| Aluminum foil wrap | Walmart | ||
| Biosafty cabinet II | NuAire | NU-540-300 | |
| Cell counter | Nexcelom | AutoT4 | |
| cell counting slides | Nexcelom | SD-100 | 20 µL chamber |
| CO2 humidified incubator | Thermo Fisher | Forma Series II | |
| Desk Lamp | SunvaleeyTEK | RS1000B | |
| Dimethyl sulfoxide | MilliporeSigma | 276855 | anhydous, >99.9% |
| Drug plate | Corning | 3680 | |
| Dulbecco's phosphate-buffered saline | Corning | 21-031-CV | DPBS, 1x without calcium amd magnesium |
| Ethanol | Koptec | 2000 | |
| F-12K Nutrient Mixture | Corning | 45000-354 | (Kaighn's Mod.) with L-glutamine |
| Fetal bovine serum | Equitech-Bio | SFBU30 | |
| Fluorescent calcium assay kit | ENZO Lifescience | ENZ-51017 | 10x96 tests |
| G418 sulfate | Gibco Invitrogen | 10131-027 | Geneticin selective antibiotic 50 mg/mL |
| Hank's buffer | MilliporeSigma | 55037C | HBSS modified, with calcium, with magnesium, without phenol read |
| HEPES buffer | Gibco Invitrogen | 15630-080 | 1 Molar |
| HTS data storage plateform | CDD vault | https://www.collaborativedrug.com/ | |
| Liquid handling system | Integra Bioscience | Viaflo | 384/12.5 µL |
| Plate centrifuge | Thermo Fisher | Sorvall ST8 | |
| Plate reader | BMG technology | Clariostar | |
| Poly-D-lysine | MilliporeSigma | P6407 | |
| Rhimi-K-1 agonist peptide | Genscript | custom order | QFSPWGamide |
| T-75 flask | Falcon | 353136 |
Références
- Hanlon, C. D., Andrew, D. J. Outside-in signaling - A brief review of GPCR signaling with a focus on the Drosophila GPCR family. Journal of Cell Science. 128 (19), 3533-3542 (2015).
- Liu, N., Li, T., Wang, Y., Liu, S. G-protein coupled receptors (GPCRs) in insects-A potential target for new insecticide development. Molecules. 26 (10), 2993 (2021).
- Pierce, K. L., Premont, R. T., Lefkowitz, R. J. Seven-transmembrane receptors. Nature Reviews Molecular Cell Biology. 3, 639-650 (2002).
- Pietrantonio, P. V., Xiong, C., Nachman, R. J., Shen, Y. G protein-coupled receptors in arthropod vectors: Omics and pharmacological approaches to elucidate ligand-receptor interactions and novel organismal functions. Current Opinion in Insect Science. 29, 12-20 (2018).
- Hilger, D., Masureel, M., Kobilka, B. K. Structure and dynamics of GPCR signaling complexes. Nature Structural & Molecular Biology. 25 (1), 4-12 (2018).
- Liu, N., Wang, Y., Li, T., Feng, X. G-protein coupled receptors (GPCRs): Signaling pathways, characterization, and functions in insect physiology and toxicology. International Journal of Molecular Sciences. 22 (10), 5260 (2021).
- Hansen, K. B., Bräuner-Osborne, H., Leifert, W. FLIPR® assays of intracellular calcium in GPCR drug discovery. G Protein-Coupled Receptors in Drug Discovery. , (2009).
- Bauknecht, P., Jekely, G. Large-scale combinatorial deorphanization of Platynereis neuropeptide GPCRs. Cell Reports. 12 (4), 684-693 (2015).
- Frooninckx, L., et al. Neuropeptide GPCRs in C. elegans. Frontiers in Endocrinology. 3, 167 (2012).
- Caers, J., et al. More than two decades of research on insect neuropeptide GPCRs: An overview. Frontiers in Endocrinology. 3, 151 (2012).
- Šimo, L., Koči, J., Park, Y. Receptors for the neuropeptides, myoinhibitory peptide and SIFamide, in control of the salivary glands of the blacklegged tick Ixodes scapularis. Insect Biochemistry and Molecular Biology. 43 (4), 376-387 (2013).
- Šimo, L., Park, Y. Neuropeptidergic control of the hindgut in the black-legged tick Ixodes scapularis. International Journal for Parasitology. 44 (11), 819-826 (2014).
- Liesch, J., Bellani, L. L., Vosshall, L. B. Functional and genetic characterization of neuropeptide Y-like receptors in Aedes aegypti. PLoS Neglected Tropical Diseases. 7 (10), 2486 (2013).
- Lu, H. L., Kersch, C. N., Taneja-Bageshwar, S., Pietrantonio, P. V. A calcium bioluminescence assay for functional analysis of mosquito (Aedes aegypti) and tick (Rhipicephalus microplus) G protein-coupled receptors. Journal of Visualized Experiments. (50), e2732 (2011).
- Xiong, C., Baker, D., Pietrantonio, P. V. The cattle fever tick, Rhipicephalus microplus, as a model for forward pharmacology to elucidate kinin GPCR function in the Acari. Frontiers in Physiology. 10, 1008 (2019).
- Holmes, S. P., Barhoumi, R., Nachman, R. J., Pietrantonio, P. V. Functional analysis of a G protein-coupled receptor from the Southern cattle tick Boophilus microplus (Acari: Ixodidae) identifies it as the first arthropod myokinin receptor. Insect Molecular Biology. 12 (1), 27-38 (2003).
- Cox, K. J., et al. Cloning, characterization, and expression of a G-protein-coupled receptor from Lymnaea stagnalis and identification of a leucokinin-like peptide, PSFHSWSamide, as its endogenous ligand. Journal of Neuroscience. 17 (4), 1197-1205 (1997).
- Dircksen, H., Kastin, A. J. Chapter 32 - Crustacean bioactive peptides. Handbook of Biologically Active Peptides (Second Edition). , 209-221 (2013).
- Halberg, K. A., Terhzaz, S., Cabrero, P., Davies, S. A., Dow, J. A. Tracing the evolutionary origins of insect renal function. Nature Communications. 6, 6800 (2015).
- Pietrantonio, P. V., Jagge, C., Taneja-Bageshwar, S., Nachman, R. J., Barhoumi, R. The mosquito Aedes aegypti (L.) leucokinin receptor is a multiligand receptor for the three Aedes kinins. Insect Molecular Biology. 14 (1), 55-67 (2005).
- Radford, J. C., Davies, S. A., Dow, J. A. Systematic G-protein-coupled receptor analysis in Drosophila melanogaster identifies a leucokinin receptor with novel roles. Journal of Biological Chemistry. 277 (41), 38810-38817 (2002).
- Brock, C. M., et al. The leucokinin-like peptide receptor from the cattle fever tick, Rhipicephalus microplus, is localized in the midgut periphery and receptor silencing with validated double-stranded RNAs causes a reproductive fitness cost. International Journal for Parasitology. 49 (3-4), 287-299 (2019).
- Nässel, D. R. Leucokinin and associated neuropeptides regulate multiple aspects of physiology and behavior in Drosophila. International Journal of Molecular Sciences. 22 (4), 1940 (2021).
- Kim, Y. -. J., et al. Central peptidergic ensembles associated with organization of an innate behavior. Proceedings of the National Academy of Sciences of the United States of America. 103 (38), 14211-14216 (2006).
- Al-Anzi, B., et al. The leucokinin pathway and its neurons regulate meal size in Drosophila. Current Biology. 20 (11), 969-978 (2010).
- Yurgel, M. E., et al. A single pair of leucokinin neurons are modulated by feeding state and regulate sleep-metabolism interactions. PLoS Biology. 17 (2), 2006409 (2019).
- Nässel, D. R., Zandawala, M. Recent advances in neuropeptide signaling in Drosophila, from genes to physiology and behavior. Progress in Neurobiology. 179, 101607 (2019).
- Okusawa, S., Kohsaka, H., Nose, A. Serotonin and downstream leucokinin neurons modulate larval turning behavior in Drosophila. Journal of Neuroscience. 34 (7), 2544-2558 (2014).
- Kersch, C. N., Pietrantonio, P. V. Mosquito Aedes aegypti (L.) leucokinin receptor is critical for in vivo fluid excretion post blood feeding. FEBS letters. 585 (22), 3507-3512 (2011).
- Kwon, H., et al. Leucokinin mimetic elicits aversive behavior in mosquito Aedes aegypti (L.) and inhibits the sugar taste neuron. Proceedings of the National Academy of Sciences of the United States of America. 113 (25), 6880-6885 (2016).
- Xiong, C., Baker, D., Pietrantonio, P. V. A random small molecule library screen identifies novel antagonists of the kinin receptor from the cattle fever tick, Rhipicephalus microplus (Acari: Ixodidae). Pest Management Science. 77 (5), 2238-2251 (2021).
- Torfs, P., et al. The kinin peptide family in invertebrates. Annals of the New York Academy of Sciences. 897 (1), 361-373 (1999).
- Ma, Q., Ye, L., Liu, H., Shi, Y., Zhou, N. An overview of Ca2+ mobilization assays in GPCR drug discovery. Expert Opinion on Drug Discovery. 12 (5), 511-523 (2017).
- Zhang, J. -. H., Chung, T. D., Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 4 (2), 67-73 (1999).
- Zhang, R., Xie, X. Tools for GPCR drug discovery. Acta Pharmacologica Sinica. 33 (3), 372-384 (2012).
- Offermanns, S., Simon, M. I. Gα15 and Gα16 couple a wide variety of receptors to phospholipase C. Journal of Biological Chemistry. 270 (25), 15175-15180 (1995).
- Murgia, M. V., et al. High-content phenotypic screening identifies novel chemistries that disrupt mosquito activity and development. Pesticide Biochemistry and Physiology. 182, 105037 (2022).
- Lismont, E., et al. Can BRET-based biosensors be used to characterize G-protein mediated signaling pathways of an insect GPCR, the Schistocerca gregaria CRF-related diuretic hormone receptor. Insect Biochemistry and Molecular Biology. 122, 103392 (2020).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.