
Method Article
En Vivo Modelado del Genoma Humano Morbid usando Danio rerio
En este artículo
Resumen
A continuación, presentamos un enfoque sistemático para desarrollar fisiológicamente relevante, sensible y específico In vivo Ensayos para interpretar la variación en la patología humana. Manipulación genética transitoria a través de la microinyección de WT y mutante ARNm humano y morfolino (MO) oligonucleótidos antisentido aprovechar la trazabilidad del embrión de pez cebra en desarrollo para rápidamente mutaciones patógenas de ensayo, especialmente, pero no exclusivamente, en el contexto de trastornos del desarrollo humanos.
Resumen
A continuación, presentamos los métodos para el desarrollo de ensayos para consultar nonsynonymous cambios potencial y clínicamente significativos utilizando en complementación vivo en el pez cebra. El pez cebra (Danio rerio) son un sistema de animal útil debido a su tratabilidad experimental; embriones son transparentes para permitir la visualización fácil, rápido desarrollo someterse a ex vivo, y se pueden manipular genéticamente 1 Estos aspectos han permitido avances significativos en el análisis de la embriogénesis,. procesos moleculares, y morfogenética de señalización. Tomados en conjunto, las ventajas de este modelo de pez cebra vertebrado hacen altamente susceptibles a la modelización de los defectos de desarrollo en la enfermedad pediátrica, y en algunos casos, los trastornos de inicio adulto. Debido a que el genoma pez cebra está altamente conservada con la de los seres humanos (~ 70% ortólogos), es posible recapitular estados de enfermedad humanos en el pez cebra. Esto se logra ya sea a través de la inyección de m mutante humanaARN para inducir el aumento de dominante negativo o de los alelos de función o la utilización de morpholino (MO) oligonucleótidos antisentido para suprimir genes para imitar la pérdida de función de las variantes. A través de la complementación de fenotipos MO inducidas con ARNm humano capsulado, nuestro enfoque permite la interpretación de los efectos perjudiciales de las mutaciones en la secuencia de la proteína humana sobre la base de la capacidad de ARNm mutante para rescatar a un medible, fenotipo fisiológicamente relevante. Modelado de los alelos de enfermedad humana se produce por medio de microinyección de embriones de pez cebra con MO y / o ARNm humano en la etapa de células 1-4 y fenotípicos hasta siete días después de la fertilización (dpf). Esta estrategia general se puede extender a una amplia gama de fenotipos de la enfermedad, como se demuestra en el siguiente protocolo. Presentamos nuestros modelos establecidos para morfogenética de señalización, la integridad craneofacial, cardiaca, vascular, la función renal, y del músculo esquelético fenotipos trastorno, así como otros.
Introducción
La interpretación funcional de la información genética y la asignación del valor clínico predictivo para un genotipo representa un problema importante en genética médica y se está convirtiendo cada vez más punzante con la aceleración de la viabilidad técnica y económica de la secuenciación de todo el genoma. Por lo tanto, es necesario desarrollar y poner en práctica nuevos paradigmas para probar la patogenicidad de las variantes de significado desconocido (VUS) detectados en los pacientes. Estos ensayos deben entonces ser exacta, el tiempo-y rentable, y el puerto el potencial para catalizar una transición a la utilidad clínica.
Mientras que el ratón ha sido tradicionalmente la herramienta de elección en el campo de la modelización de enfermedades humanas, el pez cebra se perfila como sustituto científica y económicamente favorable. A diferencia del ratón, la biología pez cebra permite el acceso fácil y oportuno a todas las etapas de desarrollo, ayudados por la claridad óptica de embriones que permite la formación de imágenes en tiempo real de patologías en desarrollo. 1 El relativamente reciente generación de líneas de pez cebra mutante ha proporcionado pruebas adicionales y opciones de modelado, empleado por muchos en los estudios funcionales, pero esta tecnología sigue siendo limitada (revisado en 1,38). No sólo son mutantes genéticos con knock-ins de mutaciones específicas laboriosas para alcanzar, también son no susceptibles de análisis a medio o de alto rendimiento para el ensayo de una amplia gama de mutaciones en un solo gen. Es importante destacar que, un único conjunto de pruebas puede proporcionar información no sólo para el potencial patógeno de alelos, sino también para la dirección del efecto en el nivel celular (por ejemplo, pérdida de la función vs ganancia de función), lo cual es crítico para el modo de herencia informar en las familias, especialmente cuando pequeñas genealogías humanos albergan información limitada sobre el modo de transmisión genética. Para comparación adicional de los usos de los modelos de ratón y pez cebra disponibles, véase la Tabla 1.
También tomamos nota de que elre limitaciones inherentes al sistema de modelo de pez cebra. Aunque D. rerio tiene un rápido desarrollo inicial de los sistemas de órganos, la madurez sexual requiere de aproximadamente tres meses. Debido a esto, los trastornos prenatales y pediátricos de aparición son los más susceptibles a este modelo de expresión transitoria. Mientras ideal para la realización de grandes pantallas de compuestos químicos, el uso de mutantes genéticos no es factible para la prueba sistemática de miles de variantes no sinónimas que contribuyen a, y continuar para ser detectados en los trastornos pediátricos.
Los ensayos de complementación describen aquí tomar ventaja de esta tratabilidad experimental, alto grado de homología, y la preservación de la función entre las proteínas humanas y pez cebra, sobre todo por lo que para moléculas necesarias para los procesos de desarrollo conservadas. Figura 1 resume la estrategia de identificación de ensayos y de varios efectos alelo. Tanto la pérdida de la función (LOF) y ensayos dominantes se pueden realizar. Para LOF, el experimento se inicia con la supresión del gen de interés con una caída morfolino, y ensayando para fenotipos que pueden ser relevantes para el fenotipo clínico bajo investigación. Supresión se puede lograr ya sea mediante el bloqueo de la traducción por dirigidas a un MO en o cerca del sitio de inicio de la traducción del locus de pez cebra (traducción bloqueador de morfolino; tbMO) o al interferir con corte y empalme mediante la colocación de un MO en un nudo de empalme, típicamente inducir bien a la inclusión de un intrón o exón aberrante saltar (empalme de bloqueo morpholino; SBMO).
Posteriormente, ARNm capsulado de la transcripción ortólogos humanos se introduce y cuantificable rescate del fenotipo se mide. Una vez que se estableció el ensayo, las mutaciones candidatos en el mensaje humano pueden ser introducidos y se ensayaron para su capacidad de rescatar el fenotipo MO-inducida en la misma eficiencia que WT ARNm humano. Por el contrario, para el candidato alelos dominantes, ARNm humano (pero no MO) es de introduccióned con la expectativa de que el ARNm humano WT no afectará gravemente pez cebra anatomía y la fisiología, mientras que la introducción de mutaciones de prueba que tengan un efecto dominante induce fenotipos análogos a los observados en la condición clínica humana. Este experimento puede ser de grano fino para diseccionar aún más si el efecto dominante se produce por una ganancia de función (GOF) o un mecanismo dominante negativo mediante la mezcla de WT y mutante ARNm humano; para eventos GOF, además de ARNm humano WT se espera que sea irrelevante, mientras que para los alelos dominantes negativos, mezcla de ARNm WT y mutante debería alterar la gravedad del fenotipo mutante inducida por el mensaje. En todos los casos, se recomienda que todas las combinaciones de inyecciones (MO con WT ARNm humanos vs morfolino con mutante ARNm humanos, etc realizarse, de preferencia dentro de la misma nidada de embriones (ver Figura 1) La interpretación es la siguiente.:
Para las pruebas de LOF:
- Si la caídaproduce un fenotipo que puede ser rescatado por equivalente de mRNA mutante y WT, el alelo es probable benigna.
- Si el rescate del fenotipo mutante caída es indistinguible de la propia desmontables fenotipo, el alelo es un probable nula funcional. El experimento no puede discriminar entre los verdaderos valores nulos (sin proteína funcional) y los niveles de actividad de la proteína ultrabajas que no tienen capacidad de rescate.
- Si el rescate del fenotipo mutante caída es estadísticamente mejor que el MO, pero peor que el WT, es probable que el alelo de un hypomorph como este resultado demuestra la pérdida parcial de la función.
Para las pruebas dominantes:
- Si no hay desmontables fenotipo, pero la inyección de ARNm WT produce un fenotipo, un plan de contingencia debe ser utilizado si el experimento es proceder (ver más abajo).
- Si no hay desmontables fenotipo y la inyección de ARNm WT no produce ningún fenotipo, el experimento continúa de forma normal.
- Si la inyecciónde ARNm mutante es equivalente a la del ARNm de tipo salvaje, el alelo puede ser benignos o pérdida de la función, o el ensayo puede haber fallado. Esto requiere más experimentación para discriminar entre estas opciones.
- Si la inyección de ARNm mutante es indistinguible de MO desmontables, la función del producto del gen es probable alterado de alguna manera. Para discernir el cambio en la función, una titulación de ARNm mutante con de tipo salvaje ARNm debe llevarse a cabo.
- Si el resultado de esta valoración es indistinguible de tipo salvaje ARNm solos, se ha demostrado que el producto de la proteína mutante utiliza la proteína de tipo salvaje como un sustrato, ya que su efecto se varía con la cantidad de titulación. Esto indica un fenotipo dominante negativo.
- Si el resultado de esta valoración es indistinguible de ARNm mutante solo, se ha demostrado que el producto de la proteína mutante ya no tiene la misma función que la de tipo salvaje, y por lo tanto no se ve afectado por la cantidad de de tipo salvaje pre producto proteicoenviado. Esto demuestra que el alelo es probable ganancia de función.
Plan de contingencia:
- Si no se presenta el fenotipo de MO desmontables, pero hace presente con mRNA WT, la experimentación puede ocurrir, aunque destacamos que esta situación no es ideal. ARNm humano WT debe ajustarse para minimizar el fenotipo, y también se puede utilizar como un nuevo punto de ajuste. Además coinyección de WT y ARNm humano mutante puede evaluarse sobre la base de la capacidad de rescatar el mutante.
Protocolo
1. Bioinformática Análisis
- Determinar si el gen humano de interés tiene un ortólogo de pez cebra, y si es así, ¿cuántos ejemplares. Recomendamos BLAST recíproco ( http://blast.ncbi.nlm.nih.gov/ ) de la proteína humana en contra del genoma pez cebra, y la posterior BLAST de la mejor pez cebra golpear contra el genoma humano. Es cierto orthologs será el mejor éxito en cada caso.
- Determinar el tamaño del marco de lectura abierto (ORF) del gen humano. Si más de 6 kb, este modelo es intratable en la actualidad debido a limitaciones de alta calidad en la transcripción in vitro de plantillas de largo.
- Obtener o generar un constructo que contiene el ORF humano en el pCS2 + cadena principal del vector (o vector equivalente con un 5 'SP6 sitio de la transcripción y 3' poliA de la señal).
- Diseñar un MO bloquear empalme o traducción del gen de pez cebra de destino. Si existen varias copias en el genoma del pez cebra, there dos opciones: a) diseño de OM adicional, o b) la identificación de un sitio de empalme totalmente conservado entre las copias en contra de los que un MO pueden ser eficaces. Algunas secuencias de MO publicadas están disponibles en www.zfin.org .
2. Análisis de la expresión en el embrión de pez cebra en desarrollo
- Determinar si el ortólogo de pez cebra se expresa en un contexto espacio-temporal relevante para la lectura fenotípica. Si están disponibles para el gen de interés no hay datos de expresión, llevar a cabo la transcripción inversa (RT)-PCR usando ADNc a partir de embriones de pez cebra enteros o hibridación in situ. (Ver 2,3).
3. Site-Directed Mutagénesis
- Cebadores de mutagénesis Diseño 25-45 bases de longitud, con la mutación deseada en el centro. La temperatura de fusión imprimación debe ser mayor que o igual a 78 ° C. Diseñar un cebador de la mutagénesis de avance y retroceso para recocer a frentecadenas del plásmido.
- Obtener cebadores para la confirmación de la secuencia de la ORF después de la mutagénesis, los cuales deben de baldosas a través de ~ 300 pares de bases secciones para cubrir toda la ORF.
- Montar la reacción de mutagénesis con una alta fidelidad de la polimerasa y el ciclo de la siguiente manera (1: 95 º C 30 seg, 2: 95 º C 30 seg, 3: 55 ° C 1 min, 4: 68 ° C 6 min, 5: Ir a 2 18x, 6: 4 ° C para siempre, 7: end).
- Añadir 1 l de endonucleasa de restricción DpnI por reacción para eliminar presa plantilla metilado; se incuba a 37 ° C durante 2 horas.
- Transformar 2 l de reacción de mutagénesis en células competentes de 20 l de acuerdo con protocolos estándar.
- Elige 3-4 colonias e inocular 5 ml de medio LB que contenía antibiótico adecuado. Agitar a 37 ° C durante la noche a 225 rpm.
- ADN de minipreparaciones y determinar la concentración.
- Secuencia del sitio de mutación para confirmar la presencia de la mutación de interés.
- Toda la secuencia ORF para confirmar la integridad de secuencia.
4. La transcripción del mRNA in vitro
- El uso de un pCS2 + plantilla linealizado, generar ARNm utilizando el kit de capsulado SP6 mMessage mMachine (Ambion). Recomendamos el uso de la mitad de las cantidades de componentes de reacción.
- Purificar la muestra de ARN con la precipitación LiCl o cloroformo fenol como se describe en las instrucciones del kit.
- Determinar la concentración de ARNm utilizando la absorbancia, garantizar la integridad del ARNm mediante electroforesis en gel, y almacenar la muestra en tres o más partes alícuotas a -80 ° C hasta que esté listo para su uso. No recomendamos múltiples ciclos de congelación y descongelación de alícuotas de ARNm.
5. Ensayo in vivo de la variante de pérdida de la función
- Obtener embriones de pez cebra de apareamientos de pez cebra naturales, y mantenerlos a 28 ° C en agua de embriones en platos de 6 o 10 cm.
- Realizar una curva dosis-respuesta morfolino para evaluar la especificidad fenotipo, MO eficacia y toxicidad MO. Inyectar una curva de dosis de por lomenos tres concentraciones diferentes entre 1-10 ng MO en 50-100 (estadio 1-4 células), los embriones de pez cebra / lote. OM eficientes deberían dar lugar a un aumento dosis-dependiente de la proporción de los embriones afectados en un lote.
- Fenotipo embriones en etapa de desarrollo apropiada basan en la expresión de genes de pez cebra específica de interés y la etapa en la que se observó un fenotipo correspondiente. Esto puede ser o bien cuantitativa (por ejemplo, una medición entre las estructuras anatómicas) o cualitativos (basado en criterios fenotípicos normalizados). Para todas las inyecciones obtuvo> 24 HPF: embriones deben ser tratados con PTU (0,003% de 1-fenil-2-tiourea en los medios de comunicación de embrión) a las 24 HPF a la reducción máxima de la formación de los melanocitos.
- Para el bloqueo de empalme-OMs, MO prueba la eficiencia mediante la extracción de ARN total a partir de embriones toda lisados en el punto de puntuación fenotípica tiempo, generar cDNA y llevar a cabo la RT-PCR del gen diana usando los cebadores que flanquean el sitio de destino MO.
- Para comprobar la efic supresióniency de un tb-MO, recolección de embriones enteros proteína lisados y realizar inmunotransferencia para comparar los niveles de la proteína específica versus control. Sin embargo, este enfoque no es susceptible a todos los genes diana porque no hay reactividad cruzada limitada para muchos anticuerpos comerciales a las proteínas de pez cebra. Dos métodos indirectos de MO que muestra especificidad, incluyen: a) lo que demuestra que hay un efecto dependiente de la dosis en el fenotipo, y b) que muestra que la co-inyección de ARNm de tipo salvaje con TB-MO rescata el fenotipo de manera eficiente. Por estas razones, se recomienda un bloqueador de empalme cuando sea posible porque la eficiencia se puede controlar directamente.
- Si se observa un fenotipo continúe en el paso 5.7, si no se observa ningún fenotipo continúe en el paso 6.1.
- Para los caracteres cualitativos, seleccione una dosis MO en el que el 50-75% de los embriones se ven afectados, pues los caracteres cuantitativos, seleccione una dosis MO en que la medida fenotípica es significativamente diferente de la de tipo salvaje (p <0,001). Inyectar nuevos lotes de pez cebra embryos (etapa 1-4 de células, n = 50-100/batch) con un cóctel que contiene la dosis de "ensayo" de MO y una curva de dosis de ARNm WT humana (que van desde 10 hasta 200 pg ARNm; estas dosis asegurar la sobreexpresión sustancial por encima la línea de base de cualquier única transcripción, lo que representa 0,25-0,5% del total de ARNm poli A en un embrión de pez cebra). 4
- Realizar puntuación enmascarada de los lotes de inyección, elegir la dosis mRNA WT con el rescate más importante en comparación con solo MO, lo que es la dosis "ensayo" de ARNm.
- Inyectar nuevos lotes (estadio 1-4 células, n = 50-100/batch) con la dosis de ensayo de la MO y la dosis de ensayo de ARNm humano mutante. Fenotipo embriones en el momento oportuno y comparar los resultados a WT rescate mRNA humano usando una prueba estadística apropiada (t-test o chi-cuadrado). Ver Figura 1 para los resultados y vaya al paso 7. Inyectar las dosis de ensayo de WT y de ARNm mutante solos para controlar los efectos de toxicidad de ARNm.
6. Ensayo in vivo de Variant Domnante negativa o ganancia de función de los efectos
- Si no se observa pérdida de la función fenotipo (Paso 5.5) o si ARNm mutante da lugar a fenotipos no significativamente diferentes de solo MO (Paso 5.7), inyectar una curva de dosificación que varía desde 10 hasta 200 pg WT ARNm humano (recomendamos 25, 50 y 100 pg como prueba inicial) en lotes de embriones (estadio 1-4 células; 50-100 embriones / lote).
- En la etapa apropiada (ver 5.3 más arriba), llevar a cabo de puntuación fenotípica, y determinar la dosis más alta a la que no hay un número estadísticamente significativo de embriones muertos y / o afectada en comparación con los controles no inyectados. Esta es la dosis "ensayo".
- Inyectar la dosis de ensayo de ARNm humano mutante (estadio 1-4 células; 50-100 embriones / lote). Embriones fenotipo y comparar los resultados de puntuación de la dosis de ensayo de inyección WT mRNA humano o la concentración de MO ensayo. Ver Figura 1 para los resultados.
- Si los resultados son indistinguibles de la MO, se valorarán ARNm mutante humana con ARNm WTy comparar con el WT humana y las inyecciones de mRNA mutantes. Mejora de fenotipos con lotes inyectados con ARNm mutantes más WT indica un dominante negativo. No se mejora indica una ganancia de función.
7. Reproducir en vivo Resultados de las pruebas
- Repita los ensayos in vivo con un mínimo de tres veces.
8. Integrar pez cebra en vivo de datos de patogenicidad con otras líneas de evidencia
- Comparar los datos de patogenicidad obtenidos a partir de experimentos de pez cebra a: los datos genéticos dentro de un pedigree (si es aplicable), datos de frecuencia de control de la población, en los estudios in vitro (ensayos basados en células de la estabilidad de proteínas, localización celular, la producción de señalización, o de la actividad enzimática).
Resultados
Trastornos recesivos y Pseudorecessive
Cilios primarios son estructuras casi ubicuos en el plan del cuerpo vertebrado que desempeñan papeles de señalización celular en múltiples destinos celulares, incluyendo la proliferación, la polaridad, la diferenciación y mantenimiento de los tejidos. 5 La disfunción de estos orgánulos conduce a una amplia gama de trastornos genéticos humanos que se refiere colectivamente como ciliopatías. 6,7 Uno de tales entidad clínica es el síndrome de Bardet-Biedl (BBS), un trastorno pediátrica multisistémica caracterizada por degeneración de la retina, la obesidad, hipogonadismo, polidactilia, y disfunción renal. 7 El desarrollo de ensayos in vivo de patogenicidad alelo fue necesario para BBS porque a) es un trastorno genéticamente heterogéneo causada principalmente por cambios privados no sinónimas ocurren en al menos 17 genes 7-10, y b) la herencia oligogenic en> 25% de las familias del BBS, en el que la presencia de cambios heterocigotos en un segundoBBS gen (además de las mutaciones causales primarios recesivos) puede modular la penetrancia clínica y expresividad. Por lo general, estas terceras alelos son nonsynonymous cambios heterocigotos de, desde un punto de vista genético, el potencial patogénico poco claro, por lo que necesitarán una interpretación precisa de sus efectos biológicos en función de las proteínas. 11-13
Para investigar el potencial patogénico de las mutaciones que contribuyen a la carga mutacional en BBS, inicialmente a prueba todos los cambios missense identificadas en BBS1 - BBS14. Tanto nosotros y otros han demostrado que la pérdida de proteínas basal del cuerpo da lugar a la desregulación de la polaridad celular plana (PCP; señalización de Wnt no canónica). Manifestándose como defectos convergentes de extensión en embriones de pez cebra mediados de somíticos 14 El uso de este lectura fenotípica fisiológicamente relevante, nos han encontrado que la supresión de los genes BBS resultó en ejes acortadas del cuerpo, somitas más amplias y más delgado, y de ancho, notochords retorcidas. 15 </ Sup> (Figura 2) de supresión de MO-inducida producido defectos gastrulación, y co-inyección con ARNm humano rescatado significativamente (y reproducible) estos fenotipos como anotado por tres diferentes métodos in vivo. En primer lugar, los embriones fueron anotados viva de acuerdo con criterios fenotípicos cualitativos (Normal, Clase I y Clase II, para las definiciones detalladas de las clases fenotípicas ver 15). A continuación, su cuantificación migración celular durante epibolia (una etapa de desarrollo temprano que se caracteriza por el adelgazamiento y la difusión de las capas de células sobre la celda yema 16) mediante el empleo de un trazador de fluoresceína para visualizar las células que migran. Por último, se midió la longitud del tronco somite en embriones de nueve somite hibridadas in situ con un cóctel de Krox20, pax2 y ribosondas MyoD, que se han montado plana para el análisis morfológico.
Esta metodología se ha utilizado para probar en exceso de 500 alelos en el espacio mutacional ciliar. En unoestudiar solo, las pruebas in vivo de> 130 alelos en producido un rango de puntuaciones fenotípicas; como se indica por nuestro protocolo (Figura 1) completa rescates fueron clasificados como benignos (no significativamente diferente de WT rescate), parcial rescata fueron clasificados como hypomorphs (mejorado significativamente de MO, pero más grave que la WT rescate), la falta de rescate se clasificaron como nulo funcional (no significativamente diferente de MO), y los fenotipos inducidos por el mutante ARNm solos en comparación con MO fueron clasificados como dominantes negativos.
También hemos evaluado la sensibilidad y especificidad del ensayo de complementación in vivo en el pez cebra en. La especificidad fue confirmada por co-inyección de SNPs frecuentes (> 5% alelo menor frecuencia en las poblaciones sanas de control), los cuales se encontraron para dar fenotipos benignos en 14/17 prueba (> 82%), y la sensibilidad ha demostrado ser 98%, como se indica por la concordancia entre los datos in vivo y argumentos genéticos suficientes camisetaso atribuir un alelo como causal en un BBS pedigrí. 17 Además, los efectos fenotípicos observados utilizando los tres en medidas in vivo (puntuación en vivo, el seguimiento de epibolia, e ISH morfometría) fueron validados in vitro utilizando inmunotransferencia y estudios de localización celular. Si bien la interpretación de estos resultados requiere conocimiento previo de al menos uno de los mecanismos de la patogénesis de la enfermedad, este ejemplo proporciona pruebas para justificar la utilidad y solidez de nuestro protocolo. Hemos corroborado desde nuestra puntuación en vivo con varias otras líneas de evidencia experimental en la investigación y el estudio mutacional imparcial análisis funcional de TTC21B, una proteína de transporte intraflagellar retrógrada. 18
Trastornos dominantes
Distrofias musculares faja (LGMD) son una clase autosómica de distrofia muscular, que causa debilidad muscular progresiva lenta en las caderas y los hombros. Este genéticamente y mechanistically grupo heterogéneo de enfermedades es causada por dos mutaciones dominantes y recesivos a través del sarcolema múltiple, sarcoméricos, citoplasma y proteínas nucleares. Basado en la presentación de los fenotipos clínicos y la evidencia de afectación de los músculos de la resonancia magnética, se determinó la causa de la LGMD dominante se encuentra en Finlandia, EE.UU., y 19 familias italianas. La secuenciación de los candidatos posicionales en el locus asignada reveló mutaciones en DNAJB6, un gen que codifica un co-chaperona de la familia HSP70 expresa como al menos dos isoformas de empalme (nuclear y citoplásmica) en los seres humanos. Para obtener más conocimientos sobre DNAJB6 función y su importancia para la LGMD, hemos examinado su papel en la integridad muscular en el pez cebra. RT-PCR de la ortólogo de pez cebra (dnajb6b) detectó la expresión tan pronto como la etapa de cinco somite, que fue seguido por la inyección de embriones con un empalme morfolino-bloqueo. A las 48 h después de la fertilización, la puntuación enmascarado mostró desprendimiento lentofibras de sus puntos de inserción. La especificidad de este fenotipo fue probado con una segunda MO que no se superponen y rescatado posteriormente con WT DNAJB6 ARNm humano.
Para consultar cómo la pérdida de función DNAJB6 conduce a defectos en la integridad muscular, hemos introducido mutaciones sin sentido que se encuentran en los pacientes en transcripciones humanos de ambas isoformas y las inyectaron en embriones de pez cebra. Mientras que la inyección de ARNm humano WT no produjo ningún fenotipo apreciable, estos cambios phenocopied la pérdida de función de los efectos de la MO cuando diseñado en el citoplasma, pero no isoforma nuclear. Esto fue seguido por co-inyección de cantidades equimolares de ARNm mutante y WT, que mostró una mayor severidad del fenotipo muscular, lo que sugiere un efecto dominante. Para probar esta idea, más inyecciones se llevaron a cabo con la alteración de las relaciones molares de ARNm mutante y el WT. De acuerdo con la predicción, un exceso de ARNm mutante en comparación con la letalidad inducida WT en los embriones, mientras que unn superior a WT produce un aumento progresivo de rescate. Esto fue seguido por los experimentos in vitro para determinar las propiedades oligimerization y las posibles interacciones de proteínas. Estos mostraron que las mutaciones alteran la actividad antiagregación de DNAJB6 citoplasmática e interfieren con la rotación de ambos mutantes y WT, así como interactuar con otra molécula, BAG3, que también es relevante para el mecanismo patógeno de la LGMD, desde LOF BAG3 causa una forma pediátrica de distrofia muscular 20. A continuación pregunta si BAG3 podría modular el fenotipo inducido por DNAJB6b mutante en el pez cebra. Mientras que la inyección de WT BAG3 sola produjo ningún fenotipo, coinyección con DNAJB6 mutantes aumentado de manera significativa la gravedad fenotípica, lo que sugiere que BAG3 juega un papel en la mediación de la patogenicidad de tales mutantes 19.
| Modelo de pez cebra transitoria | pez cebra mutante Line | Ratones transgénicos Line | |
| La edad de aparición del fenotipo humano bajo investigación |
|
|
|
| Fenotipos viables |
|
|
|
| Tiempo hasta su uso (desde el nacimiento) | 1-7 días | > 3 meses | > 6 meses |
| Rendimiento | medio-alto | bajo | bajo |
| Ventajas |
|
|
|
Tabla 1. Comparación de modelos in vivo.

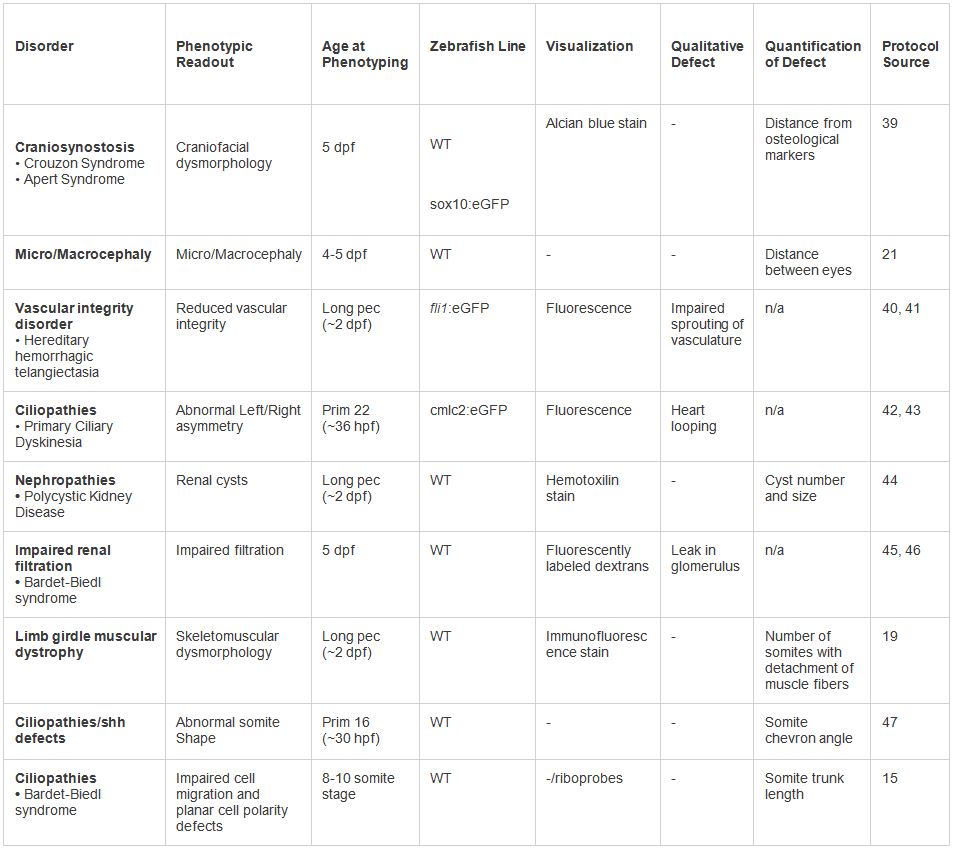
Tabla 2. Ejemplos de modelos en vivo de dysmorphologies humanos. Varios fenotipos probados en el protocolo presentado. Una serie de lecturas fenotípicas y técnicas de visualización se puede emplear en función del tipo de trastorno. Haga clic aquí para ver la tabla más grande.
{kind=link}

Figura 1. En las pruebas funcionales in vivo de las variantes no sinónimas. Un enfoque sistemático para la prueba de funcionamiento de todas laseles de significado desconocido o hipótesis. Gen desmontables a través de morfolino microinyección es seguido por una serie de (co) inyecciones de ambos WT y ARNm humano mutante. Los análisis estadísticos de los resultados fenotípicos informar a la patogenicidad alelo y la función molecular. En pocas palabras, la pérdida de las pruebas de función: Si la caída produce un fenotipo que puede ser rescatado por igual las mRNA mutantes y WT, el alelo probablemente benigna (caja verde). Si el rescate del fenotipo mutante caída es indistinguible de la desmontables fenotipo, el alelo es un nulo funcional probable (caja amarilla). Si el rescate del fenotipo mutante caída es estadísticamente mejor que el MO, pero peor que el WT, es probable que el alelo de un hypomorph (caja verde) Para las pruebas dominantes:. Si la inyección de ARNm mutante es equivalente a la de los de tipo salvaje ARNm , el alelo puede ser benignos o pérdida de la función, o el ensayo puede haber fallado (caja verde). Si la inyección de ARNm mutante es indis distinguible de MO desmontables, la función del producto del gen se altera probablemente de algún modo. Para discernir el cambio en la función, se valorarán ARNm mutante de tipo salvaje ARNm. Si el resultado de esta valoración es indistinguible de tipo salvaje ARNm solos, el producto de proteína mutante utiliza la proteína de tipo salvaje como sustrato, indicando de este modo un fenotipo dominante negativo (caja azul). Si el resultado de esta valoración es indistinguible de ARNm mutante solo, el producto de la proteína mutante ya no tiene la misma función que la de tipo salvaje, y por lo tanto es probable que una ganancia de función (caja azul). Si no hay ningún fenotipo presenta de MO desmontables, pero lo hace presente con ARNm WT, puede ocurrir la experimentación adicional, WT ARNm humano debe ajustarse para minimizar el fenotipo, y también se puede utilizar como un nuevo punto de ajuste. Además coinyección de WT y mutante ARNm humano puede ser evaluada sobre la base de la capacidad de rescate de la mutante (caja de color rosa).et = "_blank"> Haga clic aquí para ver más grande la figura.

Figura 2. La evaluación cuantitativa y cualitativa de MKS1 mutaciones detectadas en los seres humanos. Defectos del desarrollo en embriones MKS1 morphant. En base a la gravedad, los fenotipos se clasifican en tres grupos. Se muestran ejemplos de cada clase (a), y su prevalencia dentro de su cohorte de embriones (n = 100-160 embriones) fueron tabulados (no se muestra). MO inyectado embriones con Clase I fenotipos tenían groseramente morfología normal, pero eran más cortos, con tejido embrionario excesiva en la yema de huevo en comparación con el control de embriones inyectados a la misma edad somitic somitas (8-9). Morphants Clase II se adelgazan, corto y poco habían desarrollado estructuras de cabeza y la cola, y, además, carecían de definición somiticy la simetría. Embriones clase III fueron severamente retrasados somitas poco desarrollados y deforme, onduladas notochords, y por lo general no sobreviven más allá de la etapa 10-somite. Co-inyección de MKS1 ARNm humano rescatado cada uno de estos defectos, que demuestra la especificidad de los fenotipos de MKS1 supresión. La hibridación in situ de embriones en la etapa 11-somite (± 1 somite) teñidas con Krox20, pax2 y ribosondas MyoD (B, C ). Fenotipos se cuantificaron mediante mediciones de la primera a la última somite apreciable de cada embrión (flechas), cuantificado en c. Figura adaptada con autorización de 15.

Figura 3. Ejemplos de modelos en vivo de dismorfología humana. (A) Dismorfología craneofacial. MRNA de control inyectado embriones (izquierda) y el embrión inyectado mutante (derecha) teñidas con azul alcián a 5 dpf. Mutantes embriones inyectados con mRNA muestran cabezas sobre todo pequeñas y deformes con una desorganización general del esqueleto craneofacial cartilaginoso incluyendo extendidas arcos branquiales y faltantes o estructuras malformadas. (B) Micro / macrocefalia. Control no inyectado embriones (izquierda) y kctd13 embrión MO-inyectado (a la derecha) a 5 dpf. Morphants muestran ensanchamiento de la cabeza, como se ve por el espacio entre los ojos. 21 (c) la integridad vascular reducida. Control no inyectado embriones (arriba) y eng embrión MO-inyectado (abajo) fotografiada usando microscopía de fluorescencia a 2 dpf en un fli1: reportero línea transgénica eGFP. Morphants muestran deterioro crecimiento de vasos intersegmentales y otras estructuras vasculares. 41 (d) de bucle corazón alterado. Hibridación in situ de la uniembriones de tipo salvaje njected (izquierda) muestran una expresión spaw en el mesodermo de la placa lateral izquierdo, mientras que ccdc39 morphant embriones mostraron bilateral (derecha) o, en la mayoría de los casos, la expresión spaw indetectable (no se muestra). 43 (e) Los quistes renales. Embrión WT no inyectado (arriba) y ift80 morphant (abajo). Morphants muestran grandes quistes renales (flecha), edema pericárdico (punta de flecha) y una cola rizada. 44 (f) Reducción de la filtración glomerular. Visualización fluorescente de un embrión de control inyectado (parte superior) y ift80 morphant (parte inferior) 24 h después de la inyección de la rodamina dextrano en el corazón. Se dispersa de fluorescencia en todo el sistema vascular y es casi completamente evacuados por el riñón como se ve la ausencia completa de la fluorescencia en el control. El morphant muestra persistente dextrano fluorescente, lo que sugiere una caída del filtrado glomerular. 46 (g) Distrofia Muscular. WT DNAJB6 mRNA embriones inyectados (arriba) muestran fibras musculares lentas normales que abarcan los somitas normalmente entre myosepta adyacente como se determina por inmunotinción con anticuerpos anti-miosina lenta. Mutant DNAJBb (abajo) muestra parcial a completa separación de fibras musculares de myosepta en uno o varios somitas. 19 (h) Ángulo de somitas. Magnified vistas laterales en vivo del control no inyectado (arriba) o kif7 morphants (abajo) fotografiados en 30 HPF. Morphants muestran somitas forma anormal, atribuibles al embarazo ectópico señalización de Hedgehog en el pez cebra miotoma 47.
Todas las cifras adaptadas con permiso.
Discusión
Los métodos descritos aquí representan un protocolo general aplicable a la determinación de los cambios no sinónimos asociados con una variedad de fenotipos de la enfermedad genéticos humanos (Tabla 2, Figura 3). Nuestros métodos han demostrado ser útiles para evaluar el impacto potencial de la variación de fenotipos de la enfermedad, y para ayudar a diseccionar los mecanismos de la enfermedad (por ejemplo, la contribución de las mutaciones dominantes negativas para el síndrome de Bardet-Biedl, un trastorno autosómico recesivo, principalmente 17). Hasta la fecha, a través del desarrollo del árbol de decisión presentado, hemos modelado a un costo razonable y el tiempo en exceso de 200 genes causalmente asociados con los trastornos genéticos, a un exceso de alelos de 1000.
Aunque no se discute en detalle aquí, también hemos demostrado que estos métodos son apropiados para modelar otros tipos de lesiones genéticas, tales como variantes del número de copia (CNV), así como las interacciones y genética. Los análisis de este tipo de eventos están más allá del alcance demétodo de la presente descripción, a pesar de que fundamentalmente se basan en el mismo principio de la prueba sistemática de genes candidatos (incluyendo pares de genes inyectados al mismo tiempo) para determinar la inducción o exacerbación de fenotipos clínicamente apropiadas. Por ejemplo, para dilucidar cuál de los 29 genes en el 16p11.2 CNV podría ser relevante para la microcefalia observada observado en pacientes con la duplicación de un segmento genómico de 660 kb, ARNm correspondientes a cada uno de los 29 genes en el segmento de la cabeza y se inyectaron las mediciones de tamaño se realizaron a 2 dpf y 5 dpf, revelando una importante contribución de un único transcrito, KCTD13. 21 Además, hemos utilizado este modelo para las interacciones genéticas de ensayo de lesiones genómicas en pacientes tanto con síndrome de Bardet-Biedl y la enfermedad de Hirschsprung. 22 Por comparación de MO supresión de los genes causales de las dos identidades clínicos por separado y al mismo tiempo, hemos sido capaces de identificar el fenotipo resultante como being una interacción sinérgica en lugar de simplemente la gravedad aditivo.
A pesar de haber establecido una alta sensibilidad (98%) y especificidad (> 82%) para las variantes que contribuyen a ciliopatías 17, aún no disponemos de datos suficientes para determinar si son generalizables a todas las lecturas fenotípicas en los modelos de pez cebra. Para ello, un gran número de alelos, predijo genéticamente para ser benignos o patógenos, deben ser a prueba dentro de cada categoría fenotípica. Esto será particularmente importante para la aplicación de dichos ensayos en el ámbito clínico, en el que la interpretación funcional de VUSs puede informar diagnóstico y manejo sólo si una comprensión sólida de falsos positivos y falsos negativos puede acompañar a la entrega de estos resultados a los médicos y pacientes. No obstante, estos métodos pueden contribuir significativamente a una mejor comprensión del paisaje de la enfermedad genética humana. Anticipamos que estos modelos no sólo servirán de fundacionesdación para una mejor interpretación de la información genética clínica, pero también se emplea como modelos útiles para llevar a cabo pantallas terapéuticos. Los datos in vivo también puede ser comparado con in silico predicciones computacionales de fuentes tales como PolyPhen 23, estancos a 24, SNP y GO 25, o MutPred 26 para mostrar la concordancia. Tenga en cuenta que en un estudio anterior de la predicción de las bases de datos de SNPs y GO y MutPred resultaron ser los más precisos, con precisiones alcanzando sólo 0,82 y 0,81, respectivamente. 27
Aunque hemos descrito la robustez de estos métodos para un subconjunto de defectos anatómicos pediátricos (Tabla 2, Figura 3), ciertos fenotipos son menos manejables por estos métodos. Algunas excepciones pesar de ello, hay tres clases principales de trastornos no susceptibles de nuestro protocolo. Trastornos de aparición en adultos (tales como la enfermedad de Parkinson) representan un reto al modelo en un sistema embrionario. Lento progfenotipos degenerativos resión (tales como la demencia frontotemporal) pueden requerir más tiempo que la ventana dpf siete de actividad MO para producir un fenotipo. Otras tecnologías desmontables de genes tales como ARNi y ARNsi están disponibles para interferir con o degradar el gen diana, pero se ha demostrado que ninguno es tan específica, estable, no tóxico, o de larga duración como OMs 28, por tanto, también la limitación del periodo de tiempo de fenotipo. En tercer lugar, algunas estructuras de vertebrados, como el pulmón de mamíferos, no tienen una estructura lo suficientemente ortólogos en el embrión de pez cebra. También hemos proporcionado un plan de contingencia propuesto para la investigación de los casos en que la inyección de ARNm humano WT conduce a un fenotipo, aunque advertimos esto es una situación inusual e indeseable.
Algunos fenotipos de la enfermedad pueden entonces requerir un mayor grado de abstracción y subrogación. Es posible que la función del gen se ha alejado lo suficiente como para debilitar la similitud fenotípica entre el modelo y true fenotipo, o que el pez cebra fisiología complica inherentemente los efectos de la enfermedad inducida. En estos casos, se sugiere una nueva disección del fenotipo producido antes de la salida. Hemos dado algunos ejemplos de éxito en el que los fenotipos problemáticas de este ensayo han sido modelados en embriones de pez cebra. Por ejemplo, las mutaciones en TCF8, un gen asociado con distrofia de Fuchs córnea (FCD), se analizaron utilizando el protocolo mediante el uso de defectos gastrulación como una lectura fenotípica sustituto basado en las funciones conocidas de esta transcripción en el desarrollo temprano. 29 En otros casos, tales como la distrofia muscular adulta causada por mutaciones en DNAJB6, hemos sido capaces de generar fenotipos miofibrilar en embriones 5dpf a pesar del hecho de que los seres humanos son privados de la patología muscular apreciable en sus primeros tres a cuatro décadas de la vida. 19
Además de los modelos mutantes transitorios que se presentan aquí, otros también han Advantage de este sistema transitorio para modelar la enfermedad humana en una variedad de sistemas del cuerpo. En un ejemplo, la retinitis pigmentosa se modeló en el pez cebra por la caída de la RP2 gen, lo que resulta en la muerte celular retiniana y la disminución de la laminación de retina. Rescate con el ARNm humano de tipo salvaje dio como resultado en el desarrollo de todas las tres capas de laminación de la retina, mientras que cuatro de cada cinco ARNm mutantes no pudieron rescatar. 30 Aunque este modelo de un trastorno sensorial humana se basa en un fenotipo morfológico, también es posible ensayo de respuesta a los estímulos tales como sobresalto acústico o la inhibición prepulso. 47
Recientemente, se ha utilizado un modelo de pez cebra para investigar la patogénesis de la enfermedad de Alzheimer a través de la proteína precursora de amiloide. 31 Los autores mostraron que el gen desmontables causada deteriorado crecimiento de los axones de las neuronas motoras, lo que podría ser rescatados con ARNm humano. Este modelo ha demostrado ser especialmente informativo, como los modelos de ratón sólo se muestran phenot sutilipos (simple caída) o letalidad postnatal (doble caída). La capacidad para evaluar los embriones de pez cebra en vivo durante todo el desarrollo ayudó a discernir el efecto patógeno de la reducción de la proteína precursora de amiloide, así como la evidencia directa, siempre que la proteína requiere tanto un dominio extracelular y el intracelular para la función apropiada. Otros modelos notables incluyen el de distrofias musculares adicionales 32, Diamond Blackfan anemia 33, síndrome Axenfeld-Reiger (desarrollo ocular y craneofacial) 34, la enfermedad inflamatoria intestinal 35 (actividad antibacteriana), la enfermedad de Parkinson 36 (neurona y la pérdida de la locomoción), y la incautación 37 (hidrocefalia e hiperactividad).
Más comunes son las líneas de pez cebra mutantes que se han demostrado también para recapitular un fenotipo de la enfermedad humana. Opinión de 1,38, los modelos incluyen la leucemia, melanoma, miocardiopatías dilatadas, la distrofia muscular de Duchenne,y muchos otros.
Divulgaciones
Los autores declaran que no tienen intereses financieros en competencia.
Agradecimientos
Reconocemos el apoyo de la beca a la Universidad de Dean Duke Verano de Investigación (AN), la Asociación Americana del Corazón (AHA) beca 11POST7160006 (CG), los Institutos Nacionales de Salud (NIH) subvenciones R01-EY021872 del Instituto Nacional del Ojo (EED), R01HD04260 del Instituto Nacional de Salud Infantil y Desarrollo (NK), R01DK072301 y R01DK075972 del Instituto Nacional de Diabetes y Enfermedades Digestivas Riñón (NK), y la Unión Europea (Financiado por la UE 7 º PM con AG n ° 241955, proyecto SYSCILIA;. EED, NK) NK es un distinguido Jean y George W. Brumley profesor.
Materiales
| Name | Company | Catalog Number | Comments |
| Reagent | |||
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S, M0530L | |
| DpnI restriction endonuclease | NEB | R0176L, R0176S | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258-012 | |
| Big Dye Terminator | Applied Biosystems | 4337455 | |
| mMESSAGE mMACHINE Kit | Invitrogen | AM1340, AM1344, AM1348 | |
| Morpholino | Gene-Tools | n/a | |
| 1-phenyl-2-thiourea (PTU) | Sigma Aldrich | P7629 | Prepare as 0.003% PTU in embryo media |
| Paraformaldehyde (PFA) | Sigma Aldrich | P6148 | For embryos that must be fixed prior to phenotyping, prepare as 4% |
| Tricaine methane sulfonate | Western Chemical | N/A | For anesthetization and euthanasia |
| Equipment | |||
| PTC-225 Tetrad Thermal Cycler | BioRad | Any equivalent thermal cycler | |
| Nano Drop 2000 spectrophotometer | Thermo Scientific | ||
| SMZ 745T Stereomicroscope | Nikon | ||
| AZ100 Stereomicroscope | Nikon | ||
| DS Fi1 Digital Camera | Nikon | For color/fluorescent imaging | |
| DS QiMC Digital Camera | Nikon | For black/white imaging | |
| Advanced Resarch 3.2 Imaging Software | NIS- Elements | ||
Referencias
- Lieschke, G. J., Currie, P. D. Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353-367 (2007).
- Nolan, T., Hands, R. E., Bustin, S. A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559-1582 (2006).
- Thisse, C., Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69 (2008).
- Detrich, H. W., Westerfield, M., Zon, L. I. Overview of the Zebrafish system. Methods Cell Biol. 59, 3-10 (1999).
- Gerdes, J. M., Davis, E. E., Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 137, 32-45 (2009).
- Hildebrandt, F., Benzing, T., Katsanis, N. Ciliopathies. N. Engl. J. Med. 364, 1533-1543 (2011).
- Zaghloul, N. A., Katsanis, N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 119, 428-437 (2009).
- Marion, V. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet-Biedl syndrome with situs inversus and insertional polydactyly. J. Med. Genet. 49, 317-321 (2012).
- Otto, E. A. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 42, 840-850 (2010).
- Schaefer, E. Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur. J. Med. Genet. 54, 157-160 (2011).
- Katsanis, N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 13 Spec No 1, R65-R71 (2004).
- Badano, J. L. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 439, 326-330 (1038).
- Badano, J. L. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 12, 1651-1659 (2003).
- Gerdes, J. M. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 39, 1350-1360 (2007).
- Leitch, C. C. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 40, 443-448 (2008).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 203, 253-310 (1995).
- Zaghloul, N. A., et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 10602-10607 (2010).
- Davis, E. E. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189-196 (2011).
- Sarparanta, J., et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450-455 (2012).
- Selcen, D. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Annals of neurology. 65, 83-89 (2009).
- Golzio, C., et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 485, 363-367 (2012).
- de Pontual, L., et al. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13921-13926 (2009).
- Adzhubei, I. A., et al. A method and server for predicting damaging missense mutations. Nature Methods. 7, 248-249 (2010).
- Kumar, P., Henikoff, S., Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073-1081 (2009).
- Calabrese, R., Capriotti, E., Fariselli, P., Martelli, P. L., Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30, 1237-1244 (2009).
- Li, B., et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 25, 2744-2750 (2009).
- Thusberg, J., Olatubosun, A., Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 32, 358-368 (2011).
- Summerton, J. E. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 7, 651-660 (2007).
- Riazuddin, S. A., et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 86, 45-53 (2010).
- Shu, X., et al. Knockdown of the zebrafish ortholog of the retinitis pigmentosa 2 (RP2) gene results in retinal degeneration. Investigative Ophthalmology & Visual Science. 52, 2960-2966 (2011).
- Song, P., Pimplikar, S. W. Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PloS one. 7, e34209 (2012).
- Kawahara, G., Guyon, J. R., Nakamura, Y., Kunkel, L. M. Zebrafish models for human FKRP muscular dystrophies. Hum. Mol. Genet. 19, 623-633 (2010).
- Danilova, N., Sakamoto, K. M., Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 112, 5228-5237 (2008).
- Bohnsack, B. L., Kasprick, D. S., Kish, P. E., Goldman, D., Kahana, A. A zebrafish model of axenfeld-rieger syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Investigative Ophthalmology & Visual Science. 53, 7-22 (2012).
- Oehlers, S. H., et al. The inflammatory bowel disease (IBD) susceptibility genes NOD1 and NOD2 have conserved anti-bacterial roles in zebrafish. Disease Models & Mechanisms. 4, 832-841 (2011).
- Sheng, D., et al. Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS genetics. 6, e1000914 (2010).
- Teng, Y., et al. Loss of zebrafish lgi1b leads to hydrocephalus and sensitization to pentylenetetrazol induced seizure-like behavior. PloS one. 6, e24596 (2011).
- Santoriello, C., Zon, L. I. Hooked! Modeling human disease in zebrafish. J. Clin. Invest. 122, 2337-2343 (2012).
- Javidan, Y., Schilling, T. F. Development of cartilage and bone. Methods Cell Biol. 76, 415-436 (2004).
- Lawson, N. D., Weinstein, B. M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307-318 (2002).
- Lee, N. Y. Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Molecular Biology of the Cell. 23, 2412-2423 (2012).
- Huang, C. J., Tu, C. T., Hsiao, C. D., Hsieh, F. J., Tsai, H. J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 228, 30-40 (2003).
- Merveille, A. C., et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 43, 72-78 (2011).
- Beales, P. L. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 39, 727-729 (2007).
- Drummond, I. A., Davidson, A. J. Zebrafish kidney development. Methods Cell Biol. 100, 233-260 (2010).
- Tobin, J. L., Beales, P. L. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 23, 2095-2099 (2008).
- Putoux, A. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 43, 601-606 (2011).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados