
Method Article
In Vivo modélisation du génome humain morbide utilisant Danio rerio
Dans cet article
Résumé
Ici, nous présentons une approche systématique de développement physiologiquement pertinente, sensible et spécifique In vivo Tests pour interpréter la variation en pathologie humaine. Manipulation génétique transitoire par microinjection d'ARNm humain WT et mutant et morpholino (MO) des oligonucléotides antisens exploiter la traçabilité du poisson-zèbre développement embryon rapidement mutations d'analyse de pathogènes, en particulier, mais pas exclusivement, dans le contexte des troubles du développement humain.
Résumé
Ici, nous présentons les méthodes pour le développement de tests pour interroger des changements non synonymes potentiellement cliniquement significatives à l'aide de complémentation in vivo chez le poisson zèbre. Le poisson zèbre (Danio rerio) sont un système animal utile en raison de leur docilité expérimentale; embryons sont transparents pour permettre la visualisation facile, subir ex développement rapide vivo, et peuvent être manipulées génétiquement 1 Ces aspects ont permis des avancées significatives dans l'analyse de l'embryogenèse. processus moléculaires et signalisation morphogénétique. Pris ensemble, les avantages de ce modèle vertébré font poisson zèbre prête très bien à la modélisation des défauts de développement dans les maladies pédiatriques, et dans certains cas, des troubles adulte. Parce que le génome du poisson zèbre est hautement conservée avec celle des humains (~ 70% orthologue), il est possible de récapituler les états de maladies humaines chez le poisson zèbre. Ceci est réalisé soit par l'injection de mutant m humaineARN à induire une prise dominant négatif ou d'allèles de fonction, ou l'utilisation de morpholino (MO) des oligonucléotides antisens pour supprimer les gènes d'imiter la perte de variantes de fonction. Grâce à la complémentarité des phénotypes MO-induite avec l'ARNm humain plafonné, notre approche permet à l'interprétation de l'effet délétère des mutations sur la séquence de la protéine humaine basée sur la capacité de l'ARNm mutant pour sauver un mesurable, phénotype physiologiquement pertinents. Modélisation des allèles humains de la maladie se fait par micro-injection d'embryons de poisson zèbre avec MO et / ou de l'ARNm humain au stade de cellules 1-4 et phénotypage jusqu'à sept jours après la fécondation (DPF). Cette stratégie générale peut être étendue à une large gamme de phénotypes de la maladie, comme le montre le protocole suivant. Nous présentons nos modèles établis pour la signalisation morphogénétique, l'intégrité craniofaciale, cardiaques, vasculaires, la fonction rénale et squelettiques phénotypes des troubles musculaires, ainsi que d'autres.
Introduction
L'interprétation fonctionnelle de l'information génétique et l'affectation de la valeur clinique prédictive à un génotype représente un problème majeur en génétique médicale et devient de plus en plus poignant avec l'accélération de la faisabilité technique et économique du séquençage du génome entier. Par conséquent, il est nécessaire de développer et mettre en œuvre de nouveaux paradigmes pour tester la pathogénicité des variants de signification inconnue (VUS) détectés chez les patients. Ces tests doivent alors être précis, le temps et le coût-efficacité, et le port le potentiel de catalyser une transition à l'utilité clinique.
Alors que la souris a été traditionnellement l'outil de choix dans le domaine de la modélisation des maladies humaines, le poisson-zèbre sont en train de devenir un substitut scientifiquement et économiquement favorable. Contrairement à la souris, poisson zèbre biologie permet un accès facile et rapide à tous les stades de développement, aidés par la clarté optique d'embryons qui permet l'imagerie en temps réel des pathologies en développement. 1 La génération relativement récente de lignes de poisson zèbre mutant a fourni des tests supplémentaires et des options de modélisation, utilisé par beaucoup dans les études fonctionnelles, mais cette technologie reste limité (revue dans 1,38). Non seulement les mutants génétiques avec Knock-in de mutations spécifiques laborieux à atteindre, ils ne sont pas favorable à l'analyse à moyen ou à haut débit pour l'essai d'une série de mutations dans un seul gène. Surtout, une seule suite de tests peut fournir des informations non seulement pour le potentiel pathogène des allèles, mais aussi pour la direction de l'effet au niveau cellulaire (par exemple, perte de la fonction vs gain de fonction), ce qui est essentiel pour le mode de transmission informer dans les familles, en particulier lorsque de petites pedigrees humaines recèlent peu d'informations sur le mode de transmission génétique. Pour une comparaison de l'utilisation des souris disponibles et les modèles de poisson-zèbre, voir tableau 1.
Nous notons également que lere des limites inhérentes au système de modèle de poisson zèbre. Bien D. rerio ont développement initial rapide des systèmes d'organes, la maturité sexuelle nécessite environ trois mois. Pour cette raison, les troubles prénataux et pédiatriques d'apparition sont les plus prêtent à ce modèle d'expression transitoire. Bien idéal pour faire des écrans composés de grandes chimiques, l'utilisation de mutants génétiques n'est pas possible pour le test systématique de milliers de variantes non synonymes qui contribuent à, et continuent d'être détectés dans les troubles pédiatriques.
Les tests de complémentation décrites ici profiter de cette docilité expérimental, un haut degré d'homologie, et la préservation de la fonction entre les protéines humaines et le poisson zèbre, particulièrement pour les molécules nécessaires pour les processus de développement conservées. Figure 1 présente le test et la stratégie d'identification des divers effets des allèles. Tant la perte de fonction (LOF) et des dosages dominantes peuvent être réalisées. Pour LOF, l'expérience commence avec la suppression du gène d'intérêt avec un knockdown morpholino, et dosage de phénotypes qui pourraient être pertinents pour le phénotype clinique sous enquête. Suppression peut être réalisée soit par blocage de la traduction en ciblant un MO à ou près du site d'initiation de traduction du poisson zèbre locus (traduction bloquant morpholino; TBMO) ou par interférence avec l'épissage en plaçant un MO sur une jonction d'épissage, ce qui induit généralement soit l'inscription d'une intron ou exon aberrante à sauter (épissure blocage morpholino; SBMO).
Par la suite, l'ARNm coiffé de la transcription humain orthologue est introduit et quantifiable sauvetage du phénotype est mesurée. Une fois que le test est établie, les mutations candidats dans le message humain peuvent être introduits et testés pour leur capacité à sauver le phénotype MO-induite à la même efficacité que WT ARNm humain. En revanche, pour le candidat allèles dominants, l'ARNm de l'homme (mais pas MO) est introed avec une attente que l'ARNm humain WT ne sera pas affecter gravement l'anatomie et la physiologie du poisson zèbre, alors que l'introduction de mutations de test qui ont un effet dominant va induire des phénotypes analogues à ceux observés dans l'état clinique humaine. Cette expérience peut être fine davantage de disséquer si l'effet dominant se fait par un gain de fonction (GOF) ou un mécanisme dominant négatif en mélangeant ARNm humain WT et mutant, pour les événements GOF, l'ajout d'ARNm humain WT devrait être pertinent, tandis que pour les allèles dominants négatifs, mélangeant de l'ARNm WT et mutant devrait modifier la sévérité du phénotype mutant induit par un message. Dans tous les cas, nous recommandons que toutes les combinaisons d'injections (MO avec WT ARNm humain vs morpholino avec mutant ARNm humain etc s'effectuer, de préférence dans le même embrayage d'embryons (voir Figure 1) L'interprétation est comme suit.:
Pour les tests LOF:
- Si le knockdownproduit un phénotype qui peut être sauvé de façon équivalente par l'ARNm mutant et WT, l'allèle est probablement bénigne.
- Si le sauvetage mutant du phénotype knockdown est indiscernable de la phénotype knockdown lui-même, l'allèle est un nul fonctionnel probable. L'expérience ne peut pas distinguer entre les vraies valeurs nulles (aucune protéine fonctionnelle) et les niveaux d'activité de protéines ultra-basse qui n'ont pas la capacité de sauvetage.
- Si le sauvetage mutant du phénotype knockdown est statistiquement meilleure que la MO, mais pire que le WT, l'allèle est probablement un hypomorph que ce résultat démontre la perte partielle de la fonction.
Pour les tests dominantes:
- S'il n'ya pas de phénotype knockdown, mais l'injection de l'ARNm WT produit un phénotype, un plan d'urgence doit être utilisée si l'expérience est de procéder (voir ci-dessous).
- S'il n'ya pas de phénotype knockdown et l'injection de l'ARNm WT ne produit pas de phénotype, l'expérience se déroule comme d'habitude.
- Si injectionl'ARNm mutant est équivalente à celle de l'ARNm de type sauvage, de l'allèle peut être bénignes ou la perte de fonction, ou le dosage peut être défaillant. Cela nécessite une expérimentation plus poussée de discriminer entre ces options.
- Si l'injection d'ARNm mutant est indiscernable de la MO knockdown, la fonction du produit du gène est probablement altéré en quelque sorte. Pour distinguer le changement de fonction, d'un titrage de l'ARNm mutant avec l'ARNm de type sauvage ne doit être effectuée.
- Si le résultat de ce titrage est indiscernable de type sauvage ARNm seul, il a été démontré que le produit de la protéine mutante utilise la protéine de type sauvage comme un substrat, comme son effet varie avec la quantité de titration. Cela indique un phénotype négatif dominant.
- Si le résultat de ce titrage est indiscernable à l'ARNm mutant seul, il a été démontré que le produit de la protéine mutante n'a plus la même fonction que le type sauvage, et donc n'est pas affecté par la quantité de type sauvage avant de produit protéiqueenvoyé. Cela démontre que l'allèle est le gain probable de fonction.
Plan d'urgence:
- Si aucun phénotype présente de MO knockdown, mais ne présente avec l'ARNm WT, une expérimentation plus poussée peut se produire, mais nous insistons sur le fait que cette situation n'est pas idéale. ARNm humain HT doit être adaptée pour minimiser le phénotype, et peut également être utilisé en tant que nouvelle valeur de consigne. En outre coinjection de WT et ARNm humain mutant peut être évaluée en fonction de la capacité de sauvetage du mutant.
Protocole
1. Bioinformatique Analyse
- Déterminer si le gène humain d'intérêt a un orthologue poisson zèbre, et si oui, combien de copies. Nous recommandons BLAST réciproque ( http://blast.ncbi.nlm.nih.gov/ ) de la protéine humaine contre génome du poisson zèbre, et BLAST ultérieure du meilleur poisson-zèbre frapper contre le génome humain. Vrai orthologues sera le meilleur succès dans chaque cas.
- Déterminer la taille de la phase ouverte de lecture (ORF) du gène humain. Si plus de 6 kb, ce modèle est intraitable à l'heure actuelle en raison des limitations de haute qualité dans la transcription in vitro de longues modèles.
- Obtenir ou générer une construction contenant l'ORF humains dans le pCS2 + squelette du vecteur (ou vecteur équivalent avec un 5 'SP6 site de transcription et 3 «signal polyA).
- Concevoir un MO pour bloquer l'épissage ou la traduction du gène de poisson zèbre ciblée. Si plusieurs copies existent dans le génome du poisson zèbre, there sont deux options: a) la conception d'OM supplémentaire, ou b) l'identification d'un site d'épissage entièrement conservée entre copies contre lequel un seul MO peuvent être efficaces. Certaines séquences MO publiés sont disponibles à www.zfin.org .
2. analyse de l'expression dans l'embryon de poisson zèbre développement
- Déterminer si le poisson zèbre orthologue est exprimé dans un contexte spatio-temporel pertinent pour la lecture phénotypique. S'il n'y a pas de données d'expression sont disponibles pour le gène d'intérêt, effectuer la transcription inverse (RT)-PCR en utilisant l'ADNc à partir de l'ensemble des embryons de poisson zèbre ou l'hybridation in situ. (Voir 2,3).
3. Mutagenèse dirigée
- Concevoir des amorces de mutagenèse 25-45 bases de longueur, avec la mutation désirée dans le centre. La température de fusion de l'amorce doit être supérieure ou égale à 78 ° C. Concevoir une amorce de mutagenèse avant et arrière pour recuire à facebrins du plasmide.
- Amorces pour obtenir confirmation de la séquence de l'ORF post-mutagenèse, qui devraient tuile à travers environ 300 sections de paires de bases pour couvrir l'ensemble de l'ORF.
- Monter la réaction de mutagenèse d'une polymérase haute fidélité et le cycle qui suit (1: 95 ° C 30 sec, 2: 95 ° C 30 sec, 3: 55 ° C 1 min, 4: 68 ° C 6 minutes, 5: Go à 2 x18, 6: 4 ° C pour toujours, 7: fin).
- Ajouter 1 ul endonucléase de restriction DpnI par réaction d'enlever barrage modèle méthylé; incuber à 37 ° C pendant 2 heures.
- Transformer 2 pi de réaction de mutagenèse en 20 cellules compétentes ul selon des protocoles standards.
- Choisissez 3-4 colonies et inoculer 5 ml de milieu LB contenant de l'antibiotique approprié. Agiter à 37 ° C pendant la nuit à 225 rpm.
- ADN Miniprep et déterminer la concentration.
- Séquence du site de mutation pour confirmer la présence de la mutation d'intérêt.
- Toute la séquence de l'orf pour confirmer l'intégrité de la séquence.
4. ARNm transcription in vitro en
- L'utilisation d'un linéarisé pCS2 + modèle, générer des ARNm coiffé en utilisant le kit SP6 mMachine mMessage (Ambion). Nous vous recommandons d'utiliser la moitié des quantités de composants de la réaction.
- Purifier l'échantillon d'ARN avec des précipitations LiCl ou phénol-chloroforme comme décrit dans les instructions du kit.
- Déterminer la concentration de l'ARNm en utilisant l'absorbance, d'assurer l'intégrité de l'ARNm par électrophorèse sur gel, et stocker l'échantillon en trois ou plusieurs portions à -80 ° C jusqu'au moment de son utilisation. Nous ne recommandons pas de multiples cycles de gel-dégel des portions d'ARNm.
5. Essai in vivo de la variante Perte fonctionnelle
- Obtenir des embryons de poisson zèbre à partir de croisements de poisson zèbre naturelles, et de les maintenir à 28 ° C dans l'eau de l'embryon dans 6 ou 10 plats cm.
- Réaliser une courbe dose-réponse morpholino d'évaluer la spécificité du phénotype, l'efficacité et la toxicité MO MO. Injecter une courbe de dosage d'autrois concentrations moindres différents entre 1-10 ng MO dans 50-100 (stade de la cellule 1-4) poisson zèbre embryons / batch. OM efficaces devraient donner lieu à une augmentation dose-dépendante de la proportion d'embryons affectés dans un lot.
- embryons phénotypiques au stade de développement approprié basé sur l'expression du gène de poisson zèbre ciblée d'intérêt et le stade auquel un phénotype pertinent sera observée. Cela peut être soit quantitative (comme une mesure entre les structures anatomiques) ou qualitative (basée sur des critères phénotypiques standardisés). Pour toutes les injections ont marqué> 24 HPF: embryons doivent être traités avec PTU (0,003% 1-phényl-2-thiourée dans les médias d'embryons) à 24 HPF pour réduire au maximum la formation des mélanocytes.
- Pour épissure-blocage OM, tester l'efficacité MO par extraction de l'ARN total de l'ensemble embryon lysats au point de marquer phénotypique de temps, générer et diriger ADNc par RT-PCR du gène cible en utilisant des amorces flanquant le site cible MO.
- Pour vérifier l'effic de suppressioniency d'un TB-MO, récolte lysats protéiques de l'embryon et de mener des immuno pour comparer les niveaux de la protéine par rapport à un contrôle ciblé. Cependant, cette approche ne se prête pas à tous les gènes cibles parce qu'il ya une réactivité croisée limitée pour de nombreux anticorps commerciaux aux protéines de poisson zèbre. Deux méthodes indirectes de montrer MO spécificité, notamment: a) démontre qu'il existe un effet dose-dépendant sur le phénotype, et b) montrant que la co-injection de l'ARNm de type sauvage avec TB-MO sauve le phénotype efficacement. Pour ces raisons, nous recommandons un bloqueur d'épissure lorsque cela est possible parce que l'efficacité peut être contrôlé directement.
- Si un phénotype est observé passez à l'étape 5.7, si aucun phénotype est observé passez à l'étape 6.1.
- Pour phénotypes qualitatifs, sélectionnez une dose MO où 50-75% des embryons sont touchés, car phénotypes quantitatifs, sélectionnez une dose MO dans lequel la mesure phénotypique est significativement différente de type sauvage (p <0,001). Injecter de nouveaux lots de poisson zèbre embryô (étape 4.1 de cellules, n = 50-100/batch) avec un cocktail contenant la dose "d'essai" de la MO et une courbe de dosage de l'ARNm de WT humaine (variant de 10 à 200 pg de l'ARNm; ces doses assurer surexpression substantielle au-dessus la base d'une seule transcription, soit 0,25-0,5% de l'ARNm total polyA dans un embryon de poisson zèbre). 4
- Mener notation masqué des lots d'injection; choisir la dose de l'ARNm WT avec le sauvetage le plus important par rapport à MO seul, c'est la dose »de dosage" de l'ARNm.
- Injecter de nouveaux lots (stade de la cellule 1-4, n = 50-100/batch) avec la dose d'essai de MO et de la dose d'essai de l'ARNm humain mutant. embryons phénotypiques au stade approprié et comparer les résultats à WT sauvetage de l'ARNm humain à l'aide d'un test statistique approprié (t-test ou chi-carré). Voir la Figure 1 pour les résultats et passez à l'étape 7. Injecter les doses d'essai de WT et de l'ARNm mutant seul à contrôler les effets de la toxicité de l'ARNm.
6. Essai in vivo de la variante Dominante négative ou Gain des effets de fonction
- Si aucune perte de fonction phénotype est observé (étape 5.5) ou si l'ARNm mutant donne lieu à des phénotypes pas significativement différentes de MO seul (étape 5.7), injecter une courbe de dosage allant de 10 à 200 pg WT ARNm humain (nous recommandons 25, 50 , et 100 pg comme un test initial) en lots d'embryons (stade 1-4 cellulaire; 50-100 embryons / lot).
- Au stade approprié (voir 5.3 ci-dessus), effectuer score phénotypique, et de déterminer la dose la plus élevée à laquelle il n'y a pas un nombre statistiquement significatif d'embryons morts et / ou affectées par rapport aux contrôles non-injecté. Il s'agit de la dose "d'essai".
- Injecter la dose d'essai de l'ARNm humain mutant (stade 1-4 cellulaire; 50-100 embryons / lot). embryons phénotypiques et comparer les résultats à la notation de la dose d'essai de l'injection d'ARNm humain WT ou la concentration MO de dosage. Voir la Figure 1 pour les résultats.
- Si les résultats sont indiscernables de MO, titrer ARNm mutant humain avec l'ARNm WTet comparez-WT humaine et injections d'ARNm mutant. Amélioration des phénotypes avec des lots ARNm injectés mutantes ainsi WT indique un dominant négatif. Aucune amélioration indique un gain de fonction.
7. Reproduire des résultats des essais in vivo
- Répéter les essais in vivo d'un minimum de trois fois.
8. Intégrer poisson zèbre Données de pathogénicité in vivo avec autres sources de données
- Comparer les données de pathogénicité obtenus à partir d'expériences de poisson zèbre à: données génétiques au sein d'un pedigree (le cas échéant), les données de fréquence de la population de contrôle, des études in vitro (tests basés sur les cellules de la stabilité des protéines, la localisation cellulaire, sortie de signalisation ou l'activité enzymatique).
Résultats
Maladies récessives et Pseudorecessive
Les cils primaires sont des structures quasi-omniprésent sur le plan du corps des vertébrés qui jouent cellulaires rôles de signalisation dans plusieurs destins cellulaires, y compris la prolifération, la polarité, la différenciation et l'entretien des tissus. 5 dysfonctionnement de ces organites conduit à un large éventail de troubles génétiques humaines dénommés collectivement comme ciliopathies. 6,7 Une telle entité clinique est le syndrome de Bardet-Biedl (BBS), un trouble pédiatrique multisystémique caractérisée par une dégénérescence rétinienne, l'obésité, l'hypogonadisme, polydactylie, et la dysfonction rénale. 7 Le développement d'essais in vivo de la pathogénicité de l'allèle était nécessaire pour BBS parce que a) il s'agit d'un trouble génétiquement hétérogène causée par des changements non synonymes principalement privés survenant chez au moins 17 gènes 7-10, et b) l'héritage oligogénique dans> 25% des familles du BBS, dans lequel la présence de changements hétérozygotes dans un secondGène BBS (en plus de récessives mutations causales primaires) peut moduler la pénétrance clinique et expressivité. Typiquement, ces allèles tiers sont des changements non synonymes hétérozygotes, d'un point de vue génétique, le potentiel pathogène incertain, nécessitant une interprétation exacte de leur effet biologique sur la fonction des protéines 11-13.
Pour étudier le potentiel pathogène des mutations qui contribuent à charge mutationnelle dans BBS, nous avons d'abord testé tous les changements de faux-sens identifiées dans BBS1 - BBS14. Nous et d'autres ont montré que la perte de protéines basale du corps donne lieu à une dysrégulation de la polarité cellulaire planaire (PCP; signalisation Wnt non canonique). Manifestant comme des défauts d'extension convergentes dans des embryons de poisson zèbre mi-somitiques 14 Utilisation de cette lecture phénotypique physiologiquement pertinents, nous ont constaté que la suppression de gènes BBS entraîné axes raccourcissement du corps, somites plus larges et plus mince et large, notochords plié. 15 </ Sup> (Figure 2) suppression MO induite produite défauts de gastrulation, et co-injection avec de l'ARNm de l'homme sauvé de manière significative (et reproductible) ces phénotypes aussi marqué par trois différentes méthodes in vivo. D'abord, les embryons ont été marqués en direct selon des critères phénotypiques qualitatives (Normal, classes I et II, pour des définitions détaillées des classes phénotypiques voir 15). Ensuite, nous avons quantifié la migration cellulaire pendant épibolie (un stade précoce du développement caractérisé par l'amincissement et la propagation des couches de cellules sur la cellule jaune 16) en utilisant un traceur fluorescéine pour visualiser les cellules migrent. Enfin, nous avons mesuré la longueur du tronc somites chez les embryons de neuf somites in situ hybridées avec un cocktail de Krox20, PAX2 et ribosondes MYOD, qui ont été montés à plat pour l'analyse morphologique.
Cette méthodologie a été utilisée pour tester plus de 500 allèles dans l'espace mutationnel ciliaire. Dans unseule étude, les essais in vivo de> 130 allèles produit une gamme de scores phénotypiques, comme indiqué par notre protocole (Figure 1) complète sauvetages ont été classés comme bénins (non significativement différent de WT sauvetage), partielle sauvetages ont été classés comme hypomorphs (nettement améliorée de MO, mais plus sévère que WT sauvetage), l'insuffisance de sauvetage ont été classés comme non fonctionnelle (pas significativement différent de MO), et les phénotypes induits par l'ARNm mutant seuls comparativement à MO ont été classés comme négatifs dominants.
Nous avons également évalué la sensibilité et la spécificité du test in vivo de complémentation chez le poisson zèbre. La spécificité a été confirmée par co-injection SNP communs (> 5% la fréquence de l'allèle mineur dans les populations témoins en bonne santé), qui ont été trouvés à donner phénotypes bénins chez 14/17 testé (> 82%), et la sensibilité ont été présentés à 98% comme indiqué par concordance entre les données in vivo et des arguments génétiques suffisant to attribuer un allèle de causalité dans un BBS pedigree. 17 En outre, les effets phénotypiques observées en utilisant les trois mesures in vivo (score en direct, le suivi épibolie, et ISH morphométriques) ont été validés in vitro en utilisant des études de localisation immunoblot et cellulaire. Bien que l'interprétation de ces résultats Prérequis au moins un mécanisme de la pathogenèse de la maladie, cet exemple permet de preuve pour étayer l'utilité et la solidité de notre protocole. Depuis, nous avons corroboré notre meilleur marqueur in vivo avec plusieurs autres éléments de preuve expérimentale dans un criblage mutationnel impartiale et étude de l'analyse fonctionnelle des TTC21B, une protéine de transport intraflagellaire rétrograde. 18
Troubles dominantes
Dystrophies musculaires des ceintures (LGMD) sont une classe autosomique de la dystrophie musculaire, provoquant lente faiblesse musculaire progresse dans les hanches et les épaules. Cette génétiquement et moichanistically groupe hétérogène de maladies est causée par des mutations dominants et récessifs dans sarcolemmique multiple, sarcomériques, cytoplasmiques et les protéines nucléaires. Basé sur la présentation des phénotypes cliniques et des preuves de l'implication du muscle à partir de l'imagerie par résonance magnétique, nous avons enquêté sur la cause d'un LGMD dominante trouvé en finnois, États-Unis, et 19 familles italiennes. Le séquençage des candidats de position dans le locus mappé révélé des mutations dans DNAJB6, un gène codant pour une co-chaperon de la famille HSP70 exprimé en au moins deux isoformes d'épissage (nucléaire et cytoplasmique) chez les humains. Pour obtenir de nouvelles informations sur DNAJB6 fonction et sa pertinence pour LGMD, nous avons examiné son rôle dans l'intégrité du muscle chez le poisson zèbre. RT-PCR du poisson-zèbre orthologue (dnajb6b) détecté expression dès le stade de cinq somites, qui a été suivie par l'injection d'embryons avec une épissure bloquant morpholino. À 48 h après la fécondation, la notation masqué a montré détachement de la lenteurfibres à partir de leurs points d'insertion. La spécificité de ce phénotype a ensuite été testé avec un second MO non-cumul et secouru par la suite avec WT DNAJB6 ARNm humain.
Pour interroger comment la perte de DNAJB6 fonction conduit à des défauts dans l'intégrité musculaire, nous avons introduit des mutations faux-sens trouvées chez les patients dans les transcriptions de l'homme des deux isoformes et les ont injectées dans des embryons de poisson zèbre. Tandis que l'injection de l'ARNm humain WT produit aucun phénotype appréciable, ces changements phénocopies la perte des effets de fonction du MO lorsque conçu dans le cytoplasme, mais pas isoforme nucléaire. Elle a été suivie par co-injection de quantités équimolaires de l'ARNm mutant et WT, qui a montré une meilleure sévérité du phénotype musculaire, ce qui suggère un effet dominant. Pour tester cette idée, d'autres injections ont été réalisées avec la modification des rapports molaires de l'ARNm mutant et WT. Conformément à la prédiction, un excès d'ARNm mutant par rapport à létalité induite WT dans les embryons, tandis qu'unn plus de WT produit un sauvetage progressivement augmenté. Elle a été suivie par des expériences in vitro pour déterminer les propriétés de oligimerization et les interactions de protéines possibles. Ceux-ci ont montré que les mutations altèrent l'activité anti-agrégation de DNAJB6 cytoplasmique et interfèrent avec chiffre d'affaires des deux mutant et WT, mais aussi d'interagir avec une autre molécule, BAG3, qui est également pertinente pour le mécanisme pathogénique de LGMD, depuis LOF BAG3 provoque une forme pédiatrique de dystrophie musculaire 20. Nous avons ensuite demandé si BAG3 pourrait moduler le phénotype induit par DNAJB6b mutant chez le poisson zèbre. Tandis que l'injection de WT BAG3 seul produit aucun phénotype, co-injection avec DNAJB6 mutants augmenté de façon significative la sévérité phénotypique, ce qui suggère que BAG3 joue un rôle dans la médiation de la pathogénicité de ces mutants 19.
| Transient poisson zèbre modèle | Mutant poisson zèbre ligne | Lignée de souris transgénique | |
| Âge d'apparition du phénotype humain sous enquête |
|
|
|
| Phénotypes possibles |
|
|
|
| Temps jusqu'à l'utilisation (de la naissance) | 1-7 jours | > 3 mois | > 6 mois |
| Débit | moyen-élevé | faible | faible |
| Avantages |
|
|
|
Tableau 1. Comparaison des modèles in vivo.

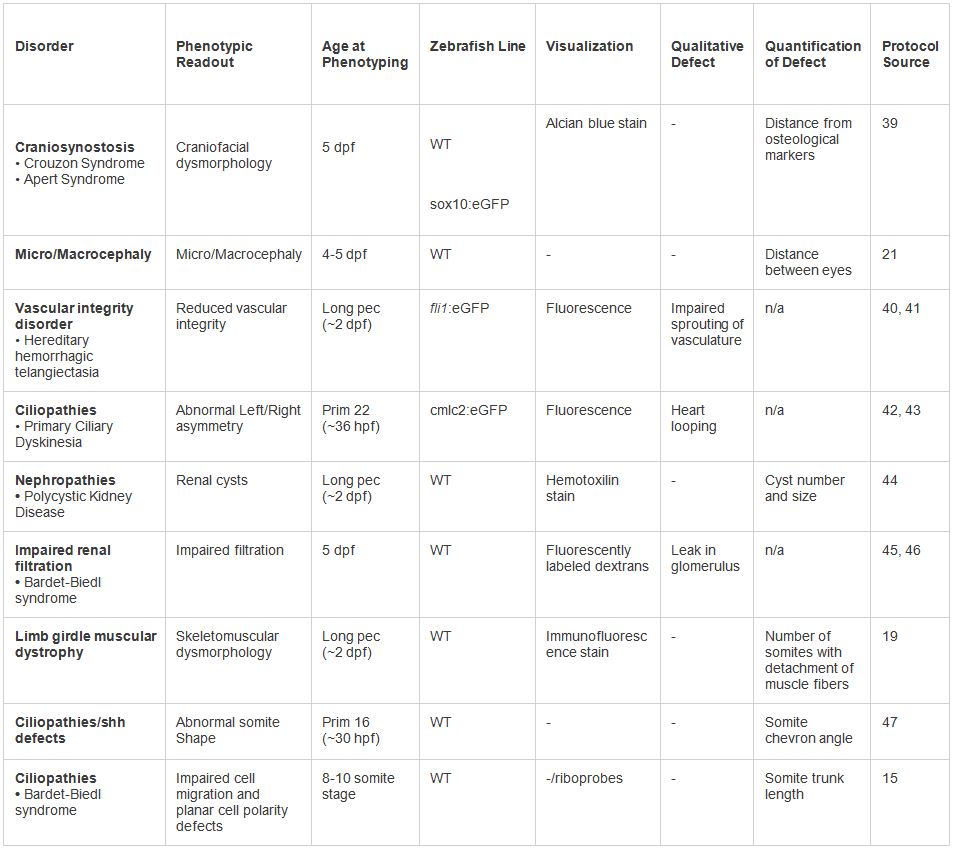
Tableau 2. Des exemples de la modélisation in vivo de dysmorphologies humaines. Divers phénotypes testés dans le cadre du protocole présenté. Une gamme de lectures phénotypiques et les techniques de visualisation peut être utilisée en fonction du type de trouble. Cliquez ici pour agrandir table.
{kind=link}

Figure 1. Dans les tests fonctionnels in vivo des variantes non synonymes. Une approche systématique à des tests fonctionnels de touseles de signification inconnue ou supposée. inactivation génique par l'intermédiaire d'un morpholino micro-injection est suivie d'une série de (co) injections de deux WT et ARNm humain mutant. Les analyses statistiques des résultats phénotypiques informer la pathogénicité des allèles et fonction moléculaire. En bref, la perte de tests de la fonction: Si l'effet de choc produit un phénotype qui peut être sauvé de façon équivalente par l'ARNm mutant et WT, l'allèle est probablement bénigne (boîte verte). Si le sauvetage mutant du phénotype knockdown est indiscernable de la phénotype knockdown, l'allèle est un nul fonctionnel probable (zone jaune). Si le sauvetage mutant du phénotype knockdown est statistiquement meilleure que la MO, mais pire que le WT, l'allèle est probablement un hypomorph (boîte verte) Pour les tests dominantes:. Si l'injection d'ARNm mutant est équivalente à celle de l'ARNm de type sauvage , l'allèle peut être soit bénigne ou perte de fonction ou le dosage peut avoir échoué (boîte verte). Si l'injection d'ARNm mutant est indis tinguishable de MO knockdown, la fonction du produit du gène est probablement altéré en quelque sorte. Pour discerner le changement de fonction, titrer ARNm mutant avec l'ARNm de type sauvage. Si le résultat de ce titrage est indiscernable de type sauvage ARNm seul, le produit de la protéine mutante utilise la protéine de type sauvage comme substrat, ce qui indique un phénotype dominant négatif (boîte bleue). Si le résultat de ce titrage est indiscernable à l'ARNm mutant seul, le produit de la protéine mutante n'a plus la même fonction que le type sauvage, et est donc probable un gain de fonction (boîte bleue). Si aucun phénotype présente de MO knockdown, mais il présente l'ARNm WT, une expérimentation plus poussée peut se produire, WT ARNm humain doit être adaptée afin de minimiser le phénotype, et peut également être utilisé comme un nouveau point de consigne. En outre co-injection d'ARNm humain WT et mutant peut être évaluée en fonction de la capacité de sauvetage du mutant (boîte rose).et = "_blank"> Cliquez ici pour agrandir la figure.

Figure 2. L'évaluation quantitative et qualitative des MKS1 mutations détectées chez les humains. Défauts de développement dans MKS1 embryons morphant. Sur la base de la gravité, les phénotypes ont été classés en trois groupes. Des exemples de chaque classe sont affichés (a), et leur prévalence au sein de leur cohorte d'embryons (n = 100-160 embryons) ont été compilés (non représenté). MO injecté embryons de classe I phénotypes avaient grossièrement morphologie normale mais étaient plus courtes, avec des tissus embryonnaires excessive sur le jaune par rapport à contrôler embryons injectés au même âge somites (8-9 somites). Morphants classe II ont été éclaircis, court et ont peu développé la tête et les structures de la queue, et en outre manqué définition somiteset de la symétrie. Embryons de classe III ont été sérieusement retardés avec somites peu développés et difforme, ondulé notochords, et généralement ne survivent pas dépassé le stade de 10 somites. Co-injection de MKS1 ARNm humain sauvé chacun de ces défauts, démontrant la spécificité des phénotypes à MKS1 suppression. L'hybridation in situ d'embryons au stade 11 somites (± 1 somite) colorées avec Krox20, PAX2 et ribosondes MYOD (b, c ). Phénotypes ont été quantifiés par des mesures de la première à la dernière somites appréciable de chaque embryon (flèches), quantifiés dans c. Figure adapté avec la permission de 15.

Figure 3. Des exemples de la modélisation in vivo de dysmorphie humaine. (A) une dysmorphie cranio-faciale. ARNm de contrôle injecté embryon (à gauche) et l'embryon injecté mutant (à droite) coloré au bleu Alcian à 5 dpf. Mutants embryons ARNm injectés présentent notamment les petites et difformes têtes avec une désorganisation générale du squelette cranio-facial cartilagineux dont écartés arcs branchiaux et manquantes ou structures malformées. (B) Micro / macrocéphalie. Contrôle non injectée embryon (à gauche) et kctd13 embryon MO-injecté (à droite) à 5 dpf. Morphants afficher l'élargissement de la tête, comme on le voit par l'espace entre les yeux. 21 (c) l'intégrité vasculaire réduit. Contrôle non injectée embryon (en haut) et eng embryon MO-injecté (en bas) imagées par microscopie de fluorescence à 2 DPF dans un FLI1: eGFP ligne de journaliste transgénique. Morphants présentent une déficience germination des navires intersegmentaire et autres structures vasculaires. 41 (d) boucle cardiaque altérée. L'hybridation in situ de l'UNIembryons de type sauvage njected (à gauche) montrent une expression de spaw dans le latéral gauche mésoderme, tandis que CCDC39 embryons morphant montré bilatérale (à droite) ou, dans la plupart des cas, l'expression de spaw indétectable (non représenté). 43 (e) des kystes rénaux. Embryon WT non injectée (en haut) et ift80 morphant (en bas). Morphants affichés gros kystes rénaux (flèche), œdème péricardique (pointe de flèche) et une queue enroulée. 44 (f) filtration glomérulaire réduit. Fluorescent visualisation d'un contrôle injecté embryon (en haut) et ift80 morphant (bas) 24 heures après l'injection de rhodamine dextran dans le coeur. Disperse fluorescence à travers le système vasculaire et est presque complètement évacué par les reins vu l'absence totale de fluorescence dans le contrôle. Le morphant affiche dextran fluorescent persistante, suggérant diminution de la filtration glomérulaire. 46 (g) La dystrophie musculaire. WT DNAJB6 mRNA embryons injectés (en haut) montrent myofibres lents ordinaires couvrant les somites normalement entre myosepta adjacent tel que déterminé par immunologique utilisant un anticorps anti-myosine lente. Mutant DNAJBb (en bas) montre partiel pour compléter détachement des fibres musculaires de myosepta dans un ou plusieurs somites. 19 (h) angle Somite. Magnifié vues latérales en direct de contrôle non injecté (en haut) ou kif7 morphants (en bas) imagées à 30 HPF. Morphants afficher somites forme anormale, attribuables à ectopique de signalisation Hedgehog dans le poisson-zèbre myotomes 47.
Tous les chiffres Adapté avec permission.
Discussion
Les méthodes décrites ici représentent un protocole général applicable à l'analyse des changements non synonymes associés à une variété de phénotypes de maladies génétiques humaines (tableau 2, figure 3). Nos approches se sont révélées utiles pour évaluer l'impact potentiel de variation sur les phénotypes de la maladie, et d'aider à disséquer les mécanismes des maladies (telles que la contribution des mutations dominantes négatives au syndrome de Bardet-Biedl, une maladie récessive autosomique principalement 17). À ce jour, grâce à l'élaboration de l'arbre de décision présenté, nous avons modélisé au coût et délai raisonnable au-delà de 200 gènes causalement liés à des troubles génétiques, à un excès d'allèles 1000.
Bien que n'étant pas discuté en détail ici, nous avons également montré que ces méthodes sont appropriées pour modéliser d'autres types de lésions génétiques, comme copie variation du nombre de copies (CNV), ainsi que et génétique des interactions. Les analyses de ces événements sont au-delà de la portée dela présente description de la méthode, même si elles reposent fondamentalement sur le même principe du test systématique des gènes candidats (y compris les paires de gènes injectés simultanément) afin de déterminer l'induction ou l'exacerbation des phénotypes cliniquement appropriées. Par exemple, pour élucider lequel des 29 gènes dans le 16p11.2 CNV pourrait être pertinent à la microcéphalie observée chez les patients ayant des duplications d'un segment génomique 660 kb, les ARNm correspondant à chacun des 29 gènes dans le secteur ont été injectés et la tête mesures de taille ont été réalisées à 2 DPF et 5 DPF, révélant une contribution majeure d'un seul transcrit, KCTD13. 21 En outre, nous avons utilisé ce modèle pour les interactions génétiques d'analyse de lésions génomiques chez les patients ayant à la fois le syndrome de Bardet-Biedl et la maladie de Hirschsprung. 22 En comparaison de MO suppression des gènes causaux des deux identités cliniques séparément et simultanément, nous avons pu identifier le phénotype résultant de being Une interaction synergique plutôt que de simplement la gravité de l'additif.
Malgré avoir établi une sensibilité élevée (98%) et la spécificité (> 82%) pour les variantes qui contribuent à ciliopathies 17, nous n'avons pas encore suffisamment de données pour déterminer si elles sont généralisables à tous les indicateurs phénotypiques dans les modèles de poisson-zèbre. Pour ce faire, un grand nombre d'allèles, prédit génétiquement pour être bénignes ou pathogènes, doivent être testés dans chaque catégorie phénotypique. Cela sera particulièrement important pour la mise en œuvre de ces tests en milieu clinique, dans laquelle l'interprétation fonctionnelle de VUSs peut informer diagnostic et le traitement seulement si une compréhension solide de faux positifs et de faux négatifs peut accompagner la livraison des résultats aux médecins et aux patients. Néanmoins, ces méthodes peuvent contribuer de manière significative à une meilleure compréhension du paysage de maladies génétiques humaines. Nous prévoyons que ces modèles ne seront pas seulement servir de fondationdation pour une meilleure interprétation de l'information génétique clinique, mais également être utilisé comme des modèles utiles pour mener écrans thérapeutiques. données in vivo peut également être comparé à in silico des prédictions informatiques provenant de sources telles que PolyPhen 23, tamiser 24 SNPs & GO 25, ou MutPred 26 pour montrer concordance. A noter que dans une étude précédente de prédiction bases de données SNP & GO et MutPred ont été jugées les plus précis, avec une précision atteignant seulement 0,82 et 0,81, respectivement 27.
Bien que nous ayons décrit la robustesse de ces méthodes pour un sous-ensemble de défauts anatomiques pédiatriques (tableau 2, figure 3), certains phénotypes sont moins dociles par ces méthodes. Certaines exceptions nonobstant, il ya trois grandes catégories de troubles ne se prêtent pas à notre protocole. Troubles à l'âge adulte (comme la maladie de Parkinson) représentent un défi pour modèle dans un système embryonnaire. Ralentissez progression phénotypes dégénératives (comme la démence fronto-temporale) peuvent nécessiter plus de temps que la fenêtre du DPF sept activité MO pour produire un phénotype. D'autres technologies de knockdown de gènes tels que l'ARNi et siRNA sont disponibles pour interférer ou dégrader le gène cible, mais il a été démontré qu'aucun n'est aussi précis, stable, non toxique, ou de longue durée comme OM 28, ce qui limite aussi le calendrier pour le phénotypage. Troisièmement, certaines structures vertébrés, tels que les poumons des mammifères, n'ont pas une structure suffisamment orthologue chez l'embryon de poisson zèbre. Nous avons également prévu un plan d'urgence proposé pour enquêter sur les cas dans lesquels l'injection d'ARNm humain WT conduit à un phénotype, mais nous mettons en garde s'agit d'une situation inhabituelle et indésirable.
Certains phénotypes de la maladie peuvent alors exiger un plus grand degré d'abstraction et de la maternité de substitution. Il est possible que la fonction du gène a divergé assez pour affaiblir la ressemblance phénotypique entre le modèle et true phénotype, ou que le poisson-zèbre physiologie complique intrinsèquement les effets de la maladie induite. Dans de tels cas, nous proposons en outre dissection du phénotype produite avant le licenciement. Nous avons réalisé quelques exemples réussis dans lequel phénotypes problématiques pour ce test ont été modélisés dans embryons de poissons zèbres. Par exemple, des mutations dans TCF8, un gène associé à Fuchs dystrophie cornéenne (FCD), ont été analysés en utilisant notre protocole en utilisant les défauts de gastrulation comme une lecture phénotypique de substitution basé sur les rôles connus de cette transcription dans le développement précoce. 29 Dans d'autres cas, comme comme la dystrophie musculaire de l'adulte causée par des mutations dans DNAJB6, nous avons pu générer des phénotypes de fibres musculaires dans les embryons 5dpf malgré le fait que les humains sont dépourvus de muscle appréciable pathologie dans leurs trois ou quatre premières années de la vie. 19
En plus des modèles mutants transitoires présentés ici, d'autres ont également pris Advantage de ce système transitoire pour modéliser les maladies humaines dans une variété de systèmes de l'organisme. Dans un exemple, la rétinite pigmentaire a été modélisée chez le poisson zèbre par le renversement de la RP2 du gène, entraînant la mort des cellules rétiniennes et une diminution de lamination rétine. Rescue avec l'ARNm humain de type sauvage a entraîné le développement de toutes les trois couches de lamination rétine, tandis que quatre sur cinq ARNm mutant n'a pas réussi à sauver. 30 Bien que ce modèle d'un trouble sensoriel humain est basé sur un phénotype morphologique, il est également possible d' réponse de dosage à des stimuli tels que le sursaut acoustique ou préimpulsion inhibition 47.
Récemment, un modèle de poisson zèbre a été utilisée pour étudier la pathogenèse de la maladie d'Alzheimer à travers la protéine précurseur de l'amyloïde. 31 Les auteurs ont montré que inactivation génique a causé une déficience croissance axonale des neurones moteurs, qui pourraient être sauvées avec un ARNm humain. Ce modèle s'est avéré particulièrement instructif, comme des modèles de souris affichent seulement phenot subtileYPES (knockdown simple) ou létalité post-natal (deux knockdown). La capacité d'évaluer les embryons de poisson zèbre in vivo au cours du développement a permis de discerner l'effet pathogène de la réduction de la protéine précurseur de l'amyloïde, ainsi que fourni des preuves directes que la protéine nécessite à la fois un domaine extracellulaire et intracellulaire pour le bon fonctionnement. Autres modèles notables comprennent que des dystrophies musculaires supplémentaires 32, Blackfan Diamond anémie 33, syndrome Axenfeld-Reiger (développement oculaire et craniofaciale) 34, la maladie inflammatoire de l'intestin 35 (activité antibactérienne), la maladie de Parkinson 36 (neurone et la perte de locomotion), et la saisie 37 (hydrocéphalie et l'hyperactivité).
Plus courantes sont les lignes de poisson zèbre mutant qui ont été montrés aussi de récapituler un phénotype de la maladie humaine. Revue en 1,38, les modèles comprennent la leucémie, le mélanome, cardiomyopathies dilatées, la dystrophie musculaire de Duchenne,et bien d'autres.
Déclarations de divulgation
Les auteurs déclarent qu'ils n'ont aucun intérêt financier concurrents.
Remerciements
Nous reconnaissons l'appui de Summer Research Fellowship d'un Duke University Dean (AN), l'American Heart Association (AHA) bourse 11POST7160006 (CG), National Institutes of Health (NIH) des subventions R01-EY021872 du National Eye Institute (EED), R01HD04260 de l' Institut national de la santé infantile et le développement (NK), R01DK072301 et R01DK075972 de l'Institut national de la Digestive du diabète et des troubles rénaux (NK), et l'Union européenne (Financé par l'UE 7 e PC sous AG nr 241955, projet SYSCILIA;. EED, NK) NK est un éminent Jean et George W. Brumley professeur.
matériels
| Name | Company | Catalog Number | Comments |
| Reagent | |||
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S, M0530L | |
| DpnI restriction endonuclease | NEB | R0176L, R0176S | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258-012 | |
| Big Dye Terminator | Applied Biosystems | 4337455 | |
| mMESSAGE mMACHINE Kit | Invitrogen | AM1340, AM1344, AM1348 | |
| Morpholino | Gene-Tools | n/a | |
| 1-phenyl-2-thiourea (PTU) | Sigma Aldrich | P7629 | Prepare as 0.003% PTU in embryo media |
| Paraformaldehyde (PFA) | Sigma Aldrich | P6148 | For embryos that must be fixed prior to phenotyping, prepare as 4% |
| Tricaine methane sulfonate | Western Chemical | N/A | For anesthetization and euthanasia |
| Equipment | |||
| PTC-225 Tetrad Thermal Cycler | BioRad | Any equivalent thermal cycler | |
| Nano Drop 2000 spectrophotometer | Thermo Scientific | ||
| SMZ 745T Stereomicroscope | Nikon | ||
| AZ100 Stereomicroscope | Nikon | ||
| DS Fi1 Digital Camera | Nikon | For color/fluorescent imaging | |
| DS QiMC Digital Camera | Nikon | For black/white imaging | |
| Advanced Resarch 3.2 Imaging Software | NIS- Elements | ||
Références
- Lieschke, G. J., Currie, P. D. Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353-367 (2007).
- Nolan, T., Hands, R. E., Bustin, S. A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559-1582 (2006).
- Thisse, C., Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69 (2008).
- Detrich, H. W., Westerfield, M., Zon, L. I. Overview of the Zebrafish system. Methods Cell Biol. 59, 3-10 (1999).
- Gerdes, J. M., Davis, E. E., Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 137, 32-45 (2009).
- Hildebrandt, F., Benzing, T., Katsanis, N. Ciliopathies. N. Engl. J. Med. 364, 1533-1543 (2011).
- Zaghloul, N. A., Katsanis, N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 119, 428-437 (2009).
- Marion, V. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet-Biedl syndrome with situs inversus and insertional polydactyly. J. Med. Genet. 49, 317-321 (2012).
- Otto, E. A. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 42, 840-850 (2010).
- Schaefer, E. Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur. J. Med. Genet. 54, 157-160 (2011).
- Katsanis, N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 13 Spec No 1, R65-R71 (2004).
- Badano, J. L. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 439, 326-330 (1038).
- Badano, J. L. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 12, 1651-1659 (2003).
- Gerdes, J. M. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 39, 1350-1360 (2007).
- Leitch, C. C. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 40, 443-448 (2008).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 203, 253-310 (1995).
- Zaghloul, N. A., et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 10602-10607 (2010).
- Davis, E. E. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189-196 (2011).
- Sarparanta, J., et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450-455 (2012).
- Selcen, D. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Annals of neurology. 65, 83-89 (2009).
- Golzio, C., et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 485, 363-367 (2012).
- de Pontual, L., et al. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13921-13926 (2009).
- Adzhubei, I. A., et al. A method and server for predicting damaging missense mutations. Nature Methods. 7, 248-249 (2010).
- Kumar, P., Henikoff, S., Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073-1081 (2009).
- Calabrese, R., Capriotti, E., Fariselli, P., Martelli, P. L., Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30, 1237-1244 (2009).
- Li, B., et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 25, 2744-2750 (2009).
- Thusberg, J., Olatubosun, A., Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 32, 358-368 (2011).
- Summerton, J. E. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 7, 651-660 (2007).
- Riazuddin, S. A., et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 86, 45-53 (2010).
- Shu, X., et al. Knockdown of the zebrafish ortholog of the retinitis pigmentosa 2 (RP2) gene results in retinal degeneration. Investigative Ophthalmology & Visual Science. 52, 2960-2966 (2011).
- Song, P., Pimplikar, S. W. Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PloS one. 7, e34209 (2012).
- Kawahara, G., Guyon, J. R., Nakamura, Y., Kunkel, L. M. Zebrafish models for human FKRP muscular dystrophies. Hum. Mol. Genet. 19, 623-633 (2010).
- Danilova, N., Sakamoto, K. M., Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 112, 5228-5237 (2008).
- Bohnsack, B. L., Kasprick, D. S., Kish, P. E., Goldman, D., Kahana, A. A zebrafish model of axenfeld-rieger syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Investigative Ophthalmology & Visual Science. 53, 7-22 (2012).
- Oehlers, S. H., et al. The inflammatory bowel disease (IBD) susceptibility genes NOD1 and NOD2 have conserved anti-bacterial roles in zebrafish. Disease Models & Mechanisms. 4, 832-841 (2011).
- Sheng, D., et al. Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS genetics. 6, e1000914 (2010).
- Teng, Y., et al. Loss of zebrafish lgi1b leads to hydrocephalus and sensitization to pentylenetetrazol induced seizure-like behavior. PloS one. 6, e24596 (2011).
- Santoriello, C., Zon, L. I. Hooked! Modeling human disease in zebrafish. J. Clin. Invest. 122, 2337-2343 (2012).
- Javidan, Y., Schilling, T. F. Development of cartilage and bone. Methods Cell Biol. 76, 415-436 (2004).
- Lawson, N. D., Weinstein, B. M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307-318 (2002).
- Lee, N. Y. Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Molecular Biology of the Cell. 23, 2412-2423 (2012).
- Huang, C. J., Tu, C. T., Hsiao, C. D., Hsieh, F. J., Tsai, H. J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 228, 30-40 (2003).
- Merveille, A. C., et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 43, 72-78 (2011).
- Beales, P. L. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 39, 727-729 (2007).
- Drummond, I. A., Davidson, A. J. Zebrafish kidney development. Methods Cell Biol. 100, 233-260 (2010).
- Tobin, J. L., Beales, P. L. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 23, 2095-2099 (2008).
- Putoux, A. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 43, 601-606 (2011).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.