
Method Article
In Vivo Modellazione del genoma umano Morbid utilizzando Danio rerio
In questo articolo
Riepilogo
Qui vi presentiamo un approccio sistematico per lo sviluppo fisiologicamente rilevanti, sensibile e specifico In vivo Saggi per interpretare variazione nella patologia umana. Manipolazione genetica transitoria tramite microiniezione di WT e mutanti mRNA umano e morpholino (MO) oligonucleotidi antisenso sfruttare la trattabilità del zebrafish embrione in via di sviluppo a rapida mutazioni patogene del test, soprattutto, ma non esclusivamente, nel contesto di disturbi dello sviluppo umano.
Abstract
Qui vi presentiamo i metodi per lo sviluppo di test per interrogare i cambiamenti non sinonime di potenziale significato clinico utilizzando in complementazione vivo in zebrafish. Zebrafish (Danio rerio) sono un sistema animale utile per la loro trattabilità sperimentale, gli embrioni sono trasparenti per permettere la visualizzazione facile, sottoporsi a sviluppo rapido ex vivo, e possono essere manipolati geneticamente 1 Questi aspetti hanno permesso significativi progressi nell'analisi di embriogenesi,. processi molecolari e morfogenetica segnalazione. Nel loro insieme, i vantaggi di questo modello vertebrato zebrafish rendono altamente suscettibili di modellazione dei difetti dello sviluppo nella malattia pediatrica, e in alcuni casi, disturbi ad insorgenza nell'età adulta. Poiché il genoma zebrafish è altamente conservata con quella degli esseri umani (~ 70% ortologhi), è possibile ricapitolare stati di malattia umani in zebrafish. Questo si realizza sia attraverso l'iniezione di m mutante umanaRNA per indurre dominante negativo o aumento di alleli funzionali, o l'utilizzo di morpholino (MO) oligonucleotidi antisenso per sopprimere i geni per imitare la perdita di varianti di funzione. Attraverso la complementazione dei fenotipi MO indotte con capped mRNA umano, il nostro approccio permette l'interpretazione degli effetti deleteri delle mutazioni sulla sequenza della proteina umana basata sulla capacità di mRNA mutante per salvare un misurabile, fisiologicamente rilevanti fenotipo. Modellazione degli alleli malattia umani avviene tramite microiniezione di embrioni di zebrafish con MO e / o mRNA umano allo stadio delle cellule 1-4, e la fenotipizzazione fino a sette giorni dopo la fecondazione (DPF). Questa strategia generale può essere esteso ad una vasta gamma di fenotipi di malattia, come dimostrato nel seguente protocollo. Vi presentiamo i nostri modelli stabiliti per morfogenetica segnalazione, craniofacciale cardiaco, l'integrità, vascolare, la funzione renale, e scheletrici fenotipi disturbo muscolare, così come gli altri.
Introduzione
L'interpretazione funzionale delle informazioni genetiche e di assegnazione del valore clinico predittivo di un genotipo rappresenta un grave problema in genetica medica e sta diventando sempre più pregnante con l'accelerazione di fattibilità tecnica ed economica di tutto il genoma sequenziamento. Pertanto, è necessario sviluppare e implementare nuovi paradigmi per testare la patogenicità di varianti di significato sconosciuto (VUS) rilevate nei pazienti. Questi test devono quindi essere precisi, nel tempo ed economicamente efficiente, e porto il potenziale per catalizzare una transizione di utilità clinica.
Mentre il mouse è sempre stato lo strumento di scelta nel campo della modellazione malattia umana, zebrafish stanno emergendo come un surrogato scientificamente ed economicamente favorevole. A differenza del topo, biologia zebrafish permette l'accesso facile e tempestivo a tutte le fasi di sviluppo, aiutati dalla chiarezza ottica di embrioni che consente l'imaging in tempo reale di sviluppo patologie. 1 La relativamente recente generazione di linee mutanti di zebrafish ha fornito ulteriori test e opzioni di modellazione, impiegato da molti in studi funzionali, ma questa tecnologia continua ad essere limitato (recensione a 1,38). Non solo i mutanti genetici con knock-in di mutazioni specifiche faticosi da raggiungere, anche questi non sono suscettibili di un'analisi di medio o high-throughput per la sperimentazione di una serie di mutazioni in un singolo gene. Importante, un'unica suite di test può fornire informazioni non solo per il potenziale patogeno di alleli, ma anche per la direzione dell'effetto a livello cellulare (ad es perdita di funzione vs guadagno di funzione), che è critica per la modalità di ereditarietà informare nelle famiglie, specialmente quando i piccoli alberi genealogici umani nutrono scarse informazioni sulle modalità di trasmissione genetica. Per ulteriore confronto degli usi di modelli disponibili zebrafish mouse e, vedi Tabella 1.
Notiamo anche che lari sono limitazioni intrinseche al sistema modello di zebrafish. Sebbene D. rerio ha un rapido sviluppo iniziale dei sistemi di organi, la maturità sessuale richiede circa tre mesi. A causa di questo, disturbi prenatale e pediatrica insorgenza sono i più adatti a questo modello di espressione transiente. Mentre ideale per condurre grandi schermi composti chimici, l'uso di mutanti genetici non è fattibile per il test sistematico di migliaia di varianti nonsynonymous che contribuiscono, e continuano ad essere rilevato in disturbi pediatrici.
I test di complementazione qui descritti approfittare di questa trattabilità sperimentale, alto grado di omologia, e la conservazione della funzione tra proteine umane e zebrafish, in modo particolare per le molecole necessarie per i processi di sviluppo conservati. Figura 1 illustra la sperimentazione e la strategia di identificazione per i vari effetti allele. Sia perdita di funzione (OL) e saggi dominanti possono essere eseguite. Per LOF, l'esperimento ha inizio con la soppressione del gene di interesse con un atterramento morpholino, e del test per i fenotipi che potrebbero essere rilevanti per il fenotipo clinico in esame. Soppressione può essere ottenuto sia bloccando traduzione di mira una MO o in prossimità del sito di inizio della traduzione del pesce zebra locus (traduzione bloccante morpholino; tbMO) o interferendo con splicing mettendo un MO su un nodo di giunzione, tipicamente inducendo o inserimento di un introne o exon skipping aberrante (giuntura blocco morpholino; SBMO).
Successivamente, mRNA ricoperto dalla trascrizione umano orthologous è introdotto e soccorso quantificabile del fenotipo è misurata. Una volta che il test è stabilita, mutazioni candidati nel messaggio umano possono essere introdotti ed analizzati per la loro capacità di salvare il fenotipo MO indotta presso la stessa efficienza di WT mRNA umano. Al contrario, per il candidato alleli dominanti, mRNA umano (ma non MO) è introndr con l'aspettativa che WT mRNA umano non sarà grossolanamente influenzare zebrafish anatomia e fisiologia, che, l'introduzione di mutazioni di prova che hanno un effetto dominante sarà indurre fenotipi analoghi a quelli osservati nella condizione clinica umana. Questo esperimento può essere a grana fine di sezionare ulteriormente se l'effetto dominante si verifica da un guadagno di funzione (GOF) o un meccanismo dominante negativo dal taglio di WT e mutante mRNA umano, per gli eventi GOF, aggiunta di WT mRNA umano dovrebbe essere irrilevante, mentre per gli alleli dominanti negative, la miscelazione di mRNA WT e mutanti dovrebbe alterare la gravità del fenotipo indotto dal messaggio mutante. In tutti i casi, si consiglia di tutte le combinazioni di iniezioni (MO con WT mRNA umano vs morpholino con mRNA umano mutante ecc essere effettuata, preferibilmente entro la stessa frizione di embrioni (vedi Figura 1) L'interpretazione è la seguente.:
Per le prove di LOF:
- Se il colpoproduce un fenotipo che può essere salvato in modo equivalente da mRNA mutante e WT, l'allele è probabile benigna.
- Se il salvataggio del fenotipo mutante knockdown è indistinguibile dal fenotipo knockdown stessa, l'allele nullo funzionale è un probabile. L'esperimento non può discriminare tra i veri valori nulli (nessuna proteina funzionale) ei livelli di attività della proteina ultrabasse che non hanno la capacità di salvataggio.
- Se il paracadute mutante del fenotipo knockdown è statisticamente migliore del MO, ma peggio del WT, l'allele è probabile una hypomorph quanto questo risultato dimostra parziale perdita di funzioni.
Per i test dominanti:
- Se non vi è alcun fenotipo knockdown, ma l'iniezione di mRNA WT produce un fenotipo, un piano di emergenza deve essere utilizzato se l'esperimento è procedere (vedere sotto).
- Se non vi è alcun fenotipo knockdown e l'iniezione di mRNA WT produce alcun fenotipo, l'esperimento procede come al solito.
- Se l'iniezionedi mRNA mutante è equivalente a quello di tipo selvaggio mRNA, l'allele può essere benigni o perdita di funzione, oppure il dosaggio può essere guasto. Ciò richiede ulteriori sperimentazioni di discriminare tra queste opzioni.
- Se l'iniezione di mRNA mutante è indistinguibile da MO knockdown, la funzione del prodotto del gene è probabile alterato in qualche modo. Di discernere il cambiamento di funzione, una titolazione di mRNA mutante con wild-type mRNA deve essere effettuata.
- Se il risultato di questa titolazione è indistinguibile da solo tipo selvaggio mRNA, è stato dimostrato che il prodotto proteina mutante usa la proteina wild-type come substrato, come il suo effetto varia con la quantità di titolazione. Questo indica un fenotipo negativo dominante.
- Se il risultato di questa titolazione è indistinguibile da sola mutante mRNA, è stato dimostrato che il prodotto proteina mutante non ha più la stessa funzione del tipo selvatico, e quindi non è influenzata dalla quantità di proteina wild-type prodotto preinviato. Ciò dimostra che l'allele è probabile guadagno di funzione.
Piano di emergenza:
- Se nessun fenotipo presenta da MO atterramento, ma lo fa presente con mRNA WT, ulteriori sperimentazioni può avvenire, anche se si sottolinea che questa situazione non è ideale. WT mRNA umano deve essere titolata per minimizzare il fenotipo, e può essere utilizzato anche come un nuovo set point. Ulteriore coiniezione di WT e mutanti mRNA umano può essere valutata in base alla capacità salvataggio del mutante.
Protocollo
1. Analisi Bioinformatica
- Determinare se il gene umano di interesse ha un ortologo zebrafish, e in tal caso, il numero di copie. Si consiglia di BLAST reciproca ( http://blast.ncbi.nlm.nih.gov/ ) della proteina umana contro il genoma di zebrafish, e la successiva BLAST delle migliori zebrafish colpire contro il genoma umano. Vero ortologhi sarà il miglior colpo in ogni istanza.
- Determinare le dimensioni della cornice di lettura aperta (ORF) del gene umano. Se più di 6 kb, questo modello è intrattabile attualmente a causa di limitazioni di alta qualità trascrizione in vitro di modelli lunghi.
- Ottenere o generare un costrutto contenente la ORF umano nel pCS2 + vettore backbone (o vettore equivalente con un 5 'SP6 sito trascrizione e 3' del segnale polyA).
- Progettare un MO per bloccare splicing o la traduzione del gene zebrafish mirato. Se esistono più copie nel genoma zebrafish there sono due opzioni: a) progettazione di ulteriore MO, oppure b) individuazione di un sito di splicing interamente conservata tra le copie contro le quali un singolo MO possono essere efficaci. Alcune sequenze MO pubblicate sono disponibili presso www.zfin.org .
2. Analisi di espressione in via di sviluppo Zebrafish Embryo
- Determinare se il ortologo zebrafish è espressa in un contesto spazio-temporale rilevante per la lettura fenotipica. Se sono disponibili per il gene di interesse sono dati espressione, condurre trascrizione inversa (RT)-PCR utilizzando cDNA da intere zebrafish embrioni o ibridazione in situ. (Vedi 2,3).
3. Mutagenesi sito-diretta
- Progettazione primer mutagenesi 25-45 basi di lunghezza, con la mutazione desiderata al centro. La temperatura di fusione del primer dovrà essere maggiore o uguale a 78 ° C. Progettare un primer mutagenesi avanti e retromarcia per temprare al contrariofilamenti del plasmide.
- Primer per ottenere la conferma della sequenza del ORF post-mutagenesi, effetti che devono piastrelle attraverso ~ 300 sezioni di paia di basi per coprire l'intera ORF.
- Assemblare la reazione di mutagenesi con una polimerasi ad alta fedeltà e ciclo come segue (1: 95 ° C 30 sec, 2: 95 ° C 30 sec, 3: 55 ° C 1 min, 4: 68 ° C 6 min, 5: Go a 2 18x, 6: 4 ° C per sempre, 7: fine).
- Aggiungere 1 ml DpnI endonucleasi di restrizione per reazione per rimuovere diga template metilata; incubare a 37 ° C per 2 h.
- Trasformare 2 microlitri di reazione di mutagenesi in 20 microlitri cellule competenti secondo protocolli standard.
- Scegli 3-4 colonie e inoculare 5 ml di mezzo LB contenente antibiotico appropriato. Agitare a 37 ° C durante la notte a 225 rpm.
- Miniprep DNA e determinare la concentrazione.
- Sequenza il sito di mutazione per confermare la presenza della mutazione di interesse.
- Sequenza intera ORF per confermare integrità della sequenza.
4. In vitro Trascrizione mRNA
- Utilizzando un modello + pCS2 linearizzato, generare mRNA capped utilizzando il mMessage mMachine SP6 kit (Ambion). Si consiglia di utilizzare la metà delle quantità dei componenti di reazione.
- Purificare il campione di RNA con precipitazioni LiCl o fenolo cloroformio come descritto nelle istruzioni del kit.
- Determinare la concentrazione di mRNA utilizzando assorbanza, assicurare l'integrità dell'mRNA mediante elettroforesi su gel, e conservare il campione in tre o più aliquote a -80 ° C fino al momento dell'uso. Si consiglia di non più cicli di gelo-disgelo di aliquote di mRNA.
5. Saggio in vivo di perdita Variante di Funzione
- Ottenere embrioni di zebrafish di accoppiamenti zebrafish naturali, e di mantenerli a 28 ° C in acqua embrione in 6 o 10 piatti cm.
- Condurre una curva dose-risposta morpholino per valutare fenotipo specificità, MO efficacia e la tossicità MO. Iniettare una curva dose di aalmeno tre diverse concentrazioni tra 1-10 ng MO in 50-100 (1-4 stadio cella) zebrafish embrioni / batch. MO efficienti dovrebbero dar luogo a un aumento dose-dipendente della percentuale di embrioni affetti in un batch.
- Embrioni fenotipo adeguato stadio di sviluppo basate su espressione genica mirata zebrafish di interesse e fase in cui sarà osservato un fenotipo rilevante. Questo può essere sia quantitativa (come una misura tra strutture anatomiche) o qualitativa (basata su criteri fenotipici standardizzati). Per tutte le iniezioni segnati> 24 HPF: gli embrioni devono essere trattati con PTU (0,003% 1-fenil-2-tiourea in mezzi di embrioni) a 24 HPF al massimo la riduzione della formazione di melanociti.
- Per splice-blocking MOS, prova MO efficienza estraendo l'RNA totale da tutta embrione lisati al punto di scoring fenotipica tempo, generare cDNA e condurre RT-PCR del gene bersaglio utilizzando primer fiancheggianti il sito di destinazione MO.
- Per verificare l'effic soppressioneiency di un tb-MO, raccolto interi embrioni lisati proteici e condurre immunoblotting per confrontare i livelli della proteina mirata contro controllo. Tuttavia, questo approccio non è suscettibile di tutti i geni bersaglio perché vi è limitata reattività crociata per molti anticorpi commerciali alle proteine zebrafish. Due metodi indiretti di mostrare MO specificità, comprendono: a) dimostrare che esiste un effetto dose-dipendente sul fenotipo, e b) che mostra che la co-iniezione di mRNA tipo selvatico con tb-MO salva il fenotipo in modo efficiente. Per queste ragioni, si consiglia un bloccante dei giunzione quando possibile, perché l'efficienza può essere monitorato direttamente.
- Se si osserva un fenotipo procedere al punto 5.7, se non si osserva alcun fenotipo passare al punto 6.1.
- Per fenotipi qualitativi, selezionare una dose MO in cui 50-75% degli embrioni sono affetti; per fenotipi quantitativi, selezionare una dose MO in cui la misura fenotipico è significativamente diversa da wild-type (p <0.001). Iniettare nuovi lotti di zebrafish embarrotolare (1-4 stadio cella; n = 50-100/batch) con un cocktail contenente la dose "dosaggio" di MO e una curva dose di mRNA WT umana (che vanno 10-200 pg mRNA; queste dosi assicurano sovraespressione sostanziale sopra la linea di base di ogni singolo verbale, che rappresenta 0,25-0,5% del totale polyA mRNA in un embrione di pesce zebra). 4
- Condurre punteggio mascherato di lotti di iniezione; scegliere la dose mRNA WT con il più significativo di soccorso in confronto al solo MO, questa è la dose "test" di mRNA.
- Iniettare nuovi lotti (1-4 stadio cella n = 50-100/batch) con la dose test di MO e la dose test di mutante mRNA umano. Embrioni fenotipo nella fase appropriata e confronta i risultati a WT salvataggio mRNA umano utilizzando un test statistico appropriato (t-test o di chi-quadrato). Vedere la Figura 1 per i risultati e passare al punto 7. Iniettare le dosi del saggio di WT e mRNA mutante da solo per controllare gli effetti di tossicità mRNA.
6. Saggio in vivo della Variante Dominant negativo o Guadagno di Funzione Effetti
- Se non si osservano perdita di funzione fenotipo (Passo 5.5) o se mRNA mutante dà luogo a fenotipi non significativamente diversi da sola MO (Passo 5.7), iniettare una curva dose vanno 10-200 pg WT mRNA umano (si consiglia 25, 50 , e 100 pg come prova iniziale) in partite di embrioni (1-4 cellule di stadio; 50-100 embrioni / partita).
- Nella fase appropriata (vedere 5.3 sopra), condurre scoring fenotipica, e di determinare la dose massima a cui non vi è un numero statisticamente significativo di embrioni morti e / o colpiti in confronto ai controlli uninjected. Questo è il dosaggio "dosaggio".
- Iniettare la dose test di mutante mRNA umano (1-4 stadio cella; 50-100 embrioni / partita). Embrioni fenotipo e confronta i risultati al gol dalla dose test di WT iniezione mRNA umano o la concentrazione di saggio MO. Vedere la Figura 1 per i risultati.
- Se i risultati sono indistinguibili da MO, titolare mRNA mutante umano con mRNA WTe confrontare WT umana e iniezioni di mRNA mutanti. Miglioramento dei fenotipi con lotti mRNA a iniezione mutanti più WT indica un dominante negativo. Nessun miglioramento indica un guadagno di funzione.
7. Riprodurre in vivo test Risultati
- Ripetere il test in vivo un minimo di tre volte.
8. Integrare Zebrafish in dati patogenicità in vivo con altre linee di evidenza
- Confrontare i dati ottenuti da esperimenti di patogenicità zebrafish a: dati genetici all'interno di un pedigree (se applicabile), i dati di frequenza nella popolazione di controllo, studi in vitro (saggi cellulari di stabilità proteica, localizzazione cellulare, la produzione di segnalazione, o l'attività enzimatica).
Risultati
Malattie recessive e Pseudorecessive
Ciglia primarie sono strutture quasi onnipresenti sul piano del corpo dei vertebrati, che svolgono un ruolo di segnalazione cellulare in molteplici destini delle cellule, tra cui la proliferazione, la polarità, la differenziazione e la manutenzione dei tessuti. 5 erettile di questi organelli porta ad una vasta gamma di malattie genetiche umane di cui collettivamente ciliopathies. 6,7 Un tale entità clinica è la sindrome di Bardet-Biedl (BBS), una malattia pediatrica multisistemica caratterizzata da degenerazione della retina, l'obesità, ipogonadismo, polidattilia, e disfunzione renale. 7 Lo sviluppo di test in vivo di allele patogenicità era necessaria per BBS perché a) è un disordine geneticamente eterogenea causata da cambiamenti soprattutto non sinonime privati che si verificano in almeno 17 geni 7-10 e b) eredità oligogenic in> 25% delle famiglie BBS, in cui la presenza di cambiamenti eterozigoti in un secondoBBS gene (in aggiunta a recessive mutazioni causali primari) può modulare penetranza clinica e di espressività. In genere, queste terze alleli sono sinonime cambiamenti eterozigoti, da un punto di vista genetico, potenziale patogeno chiaro necessitando perciò di corretta interpretazione del loro effetto biologico sulla funzione della proteina. 11-13
Per studiare il potenziale patogenetico delle mutazioni che contribuiscono al carico mutazionale in BBS, inizialmente abbiamo testato tutte le modifiche missenso individuate nel BBS1 - BBS14. Sia noi che altri hanno dimostrato che la perdita di proteine del corpo basale dà luogo a una disregolazione di polarità planare delle cellule (PCP; non canonici di segnalazione Wnt). Che si manifesta come convergenti difetti di estensione in embrioni di zebrafish metà somitic 14 Usando questo fisiologicamente rilevanti lettura fenotipica, abbiamo hanno trovato che la soppressione dei geni BBS provocato assi accorciati corpo, somiti più ampie e più sottile e largo, notochords piegato. 15 </ Sup> (Figura 2) soppressione MO indotta prodotta difetti gastrulazione, e co-iniezione con mRNA umano salvato significativamente (e riproducibile) questi fenotipi come segnati da tre diversi metodi in vivo. In primo luogo, gli embrioni sono stati segnati dal vivo secondo criteri fenotipici qualitativi (Normale, di classe I e di classe II, per le definizioni dettagliate di classi fenotipiche vedere 15). Successivamente, abbiamo quantificato la migrazione delle cellule durante epiboly (uno stadio di sviluppo iniziale caratterizzato dalla assottigliamento e la diffusione di strati di cellule sopra la cella tuorlo 16) impiegando un fluoresceina tracciante per visualizzare le cellule che migrano. Infine, abbiamo misurato la lunghezza del tronco somite in embrioni di nove somite in situ ibridato con un cocktail di Krox20, PAX2 e ribosonde MyoD, che sono stati piatto montato per l'analisi morfologica.
Questa metodologia è stata utilizzata per testare in eccesso di 500 alleli nello spazio mutazionale ciliare. In unastudiare da solo, la sperimentazione in vivo di> 130 alleli prodotto una gamma di punteggi fenotipiche, come indicato dal nostro protocollo (Figura 1) completo salvataggi sono stati classificati come benigni (non significativamente diverso da WT salvataggio), parziale salvataggi sono stati classificati come hypomorphs (migliorato significativamente da MO, ma più grave di WT di salvataggio), il mancato soccorso sono stati classificati come nullo funzionale (non significativamente diverso da MO), e fenotipi indotti da mutante mRNA da solo rispetto a MO sono stati classificati come dominanti negativi.
Abbiamo anche valutato la sensibilità e la specificità del test in complementazione vivo in zebrafish. La specificità è stata confermata mediante co-iniezione di SNPs comuni (> 5% minore frequenza allele in popolazioni di controllo sani); questi sono stati trovati a dare fenotipi benigne in 14/17 testate (> 82%), e la sensibilità è risultata pari al 98%, come indicato di concordanza tra i dati in vivo e argomenti genetici sufficiente to attribuire un allele come causale in una BBS pedigree. 17 Inoltre, gli effetti fenotipici osservati utilizzando le tre misure in vivo (punteggio dal vivo, epiboly monitoraggio e ISH morfometria) sono stati validati in vitro utilizzando gli studi di localizzazione immunoblotting e cellulari. Mentre l'interpretazione di questi risultati richiede la conoscenza preventiva di almeno un meccanismo di patogenesi della malattia, questo esempio fornisce la prova per dimostrare l'utilità e la robustezza del nostro protocollo. Da allora abbiamo confermato la nostra in vivo marcature con molteplici altre linee di prove sperimentali in uno screening mutazionale imparziale e funzionale studio di analisi di TTC21B, una proteina di trasporto intraflagellare retrogrado 18.
Disturbi dominanti
Arto cintura distrofie muscolari (LGMD) sono una classe autosomica di distrofia muscolare, causando debolezza muscolare lenta progressione nei fianchi e le spalle. Questo geneticamente e mechanistically gruppo eterogeneo di malattie è causata da due mutazioni dominanti e recessivi in tutta sarcolemmal multipla, sarcomeriche, citoplasmatici e proteine nucleari. Sulla base della presentazione di fenotipi clinici e le prove del coinvolgimento muscolare da risonanza magnetica, abbiamo studiato la causa di una LGMD dominante trovato in Finlandia, Stati Uniti, e le famiglie italiane 19. Sequenziamento di candidati posizionali all'interno del locus mappato rivelato mutazioni in DNAJB6, un gene codificante una co-chaperone della famiglia HSP70 espresso in almeno due isoforme di splicing (nucleari e citoplasmatiche) negli esseri umani. Per ottenere ulteriori approfondimenti DNAJB6 funzione e la sua rilevanza per LGMD, abbiamo esaminato il suo ruolo nella integrità muscolare in zebrafish. RT-PCR del pesce zebra ortologo (dnajb6b) rilevata espressione già nella fase cinque somite, che è stata seguita da iniezione di embrioni con una giunzione-blocking morpholino. A 48 ore dopo la fecondazione, scoring mascherato ha mostrato distacco lentofibre dai loro punti di inserimento. La specificità di questo fenotipo è stato poi testato con un secondo non sovrapponibile MO salvato e successivamente con WT umana DNAJB6 mRNA.
Per eseguire una query come la perdita di DNAJB6 funzione porta a difetti di integrità muscolare, abbiamo introdotto mutazioni missense trovati in pazienti in trascrizioni umane di entrambe le isoforme e iniettato in embrioni di zebrafish. Mentre l'iniezione di mRNA umano WT prodotto alcun fenotipo apprezzabile, questi cambiamenti phenocopied la perdita di effetti funzionali del MO quando ingegnerizzato nel citoplasmatica, ma isoforma non nucleare. Questo è stato seguito da coiniezione di quantità equimolari di mRNA mutante e WT, che ha mostrato una maggiore gravità del fenotipo muscolare, suggerendo un effetto dominante. Per verificare questa ipotesi, ulteriori iniezioni sono state effettuate con alterazione rapporti molari mutante mRNA e WT. Coerentemente con la previsione, un eccesso di mRNA mutante rispetto WT letalità indotta negli embrioni, mentre unan eccesso di WT ha prodotto un progressivo aumento di soccorso. Questo è stato seguito da esperimenti in vitro per determinare le proprietà oligimerization e possibili interazioni proteina. Questi hanno mostrato che le mutazioni alterano l'attività antiaggregante del DNAJB6 citoplasmatica e interferiscono con la rotazione di entrambi mutante e WT, così come interagire con un'altra molecola, BAG3, che è anche rilevante per il meccanismo patogenetico di LGMD, in quanto LOF BAG3 causa una forma pediatrica di distrofia muscolare 20. Abbiamo poi chiesto se BAG3 potrebbe modulare il fenotipo indotto dal DNAJB6b mutante in zebrafish. Mentre l'iniezione di WT BAG3 solo prodotto alcun fenotipo, coiniezione con DNAJB6 mutanti aumentato significativamente la severità fenotipica, suggerendo che BAG3 gioca un ruolo nel mediare la patogenicità di tali mutanti 19.
| Transient modello Zebrafish | Mutant Zebrafish Linea | Topo transgenico Linea | |
| Età di insorgenza del fenotipo umano sotto inchiesta |
|
|
|
| Fenotipi possibili |
|
|
|
| Tempo fino al momento dell'uso (dalla nascita) | 1-7 giorni | > 3 mesi | > 6 mesi |
| Throughput | medio-alta | basso | basso |
| Vantaggi |
|
|
|
Tabella 1. Confronto di modelli in vivo.

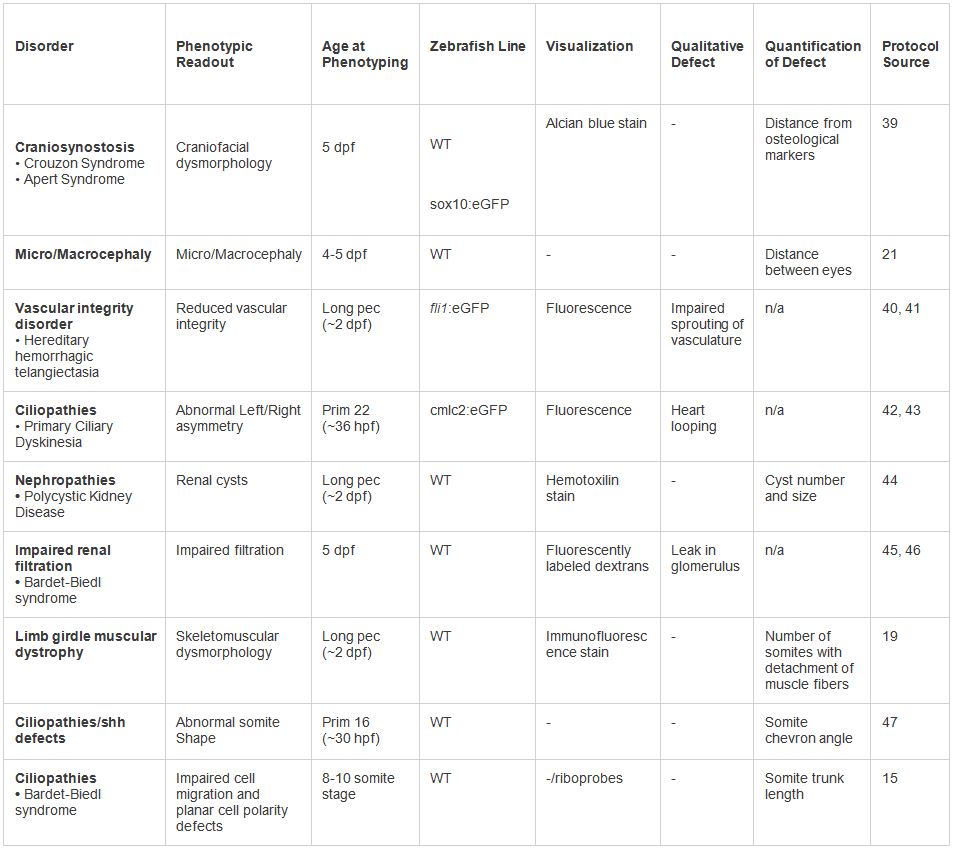
Tabella 2. Esempi di modellazione in vivo di dysmorphologies umani. Vari fenotipi testati sotto il protocollo presentato. Una serie di letture fenotipici e tecniche di visualizzazione possono essere impiegati in base al tipo di disturbo. Clicca qui per vedere tabella più grande.
{kind=link}

Figura 1. Nel test funzionali in vivo di varianti non sinonime. Un approccio sistematico alla verifica funzionale di tuttiEles di significato sconosciuto o ipotizzato. Gene knockdown via morfolino microiniezione è seguito da una serie di (co) iniezioni di entrambi WT e mutante mRNA umano. Le analisi statistiche dei risultati fenotipici informare la patogenicità allele e la funzione molecolare. In breve, per la perdita di test di funzionalità: se il colpo produce un fenotipo che può essere salvato in modo equivalente da mRNA mutante e WT, l'allele è probabile benigna (scatola verde). Se il salvataggio mutante del fenotipo knockdown è indistinguibile dal fenotipo knockdown, l'allele è un nullo funzionale probabile (scatola gialla). Se il salvataggio del fenotipo mutante knockdown è statisticamente migliore del MO, ma peggio del WT, l'allele è probabile una hypomorph (scatola verde) Per i test dominanti:. Se l'iniezione di mRNA mutante è equivalente a quello di tipo selvaggio mRNA , l'allele può essere benigni o perdita di funzione, o il test potrebbe non essere riuscito (scatola verde). Se l'iniezione di mRNA mutante è Indis distinguibile da MO knockdown, la funzione del prodotto del gene è probabile alterato in qualche modo. Per discernere il cambiamento di funzione, titolare mRNA mutante con wild-type mRNA. Se il risultato di questa titolazione è indistinguibile da solo tipo selvaggio mRNA, il prodotto proteina mutante usa la proteina wild-type come substrato, indicando quindi un fenotipo negativo dominante (scatola blu). Se il risultato di questa titolazione è indistinguibile da sola mutante mRNA, il prodotto proteina mutante non ha più la stessa funzione del tipo selvatico, e quindi è probabile un guadagno di funzione (scatola blu). Se nessun fenotipo presenta da MO knockdown, ma fa presente con mRNA WT, può verificarsi ulteriori sperimentazioni, WT mRNA umano deve essere titolata per minimizzare il fenotipo, e può essere utilizzato anche come un nuovo set point. Ulteriore coiniezione di WT e mutanti mRNA umano può essere valutata in base alla capacità salvataggio del mutante (scatola rosa).et = "_blank"> Clicca qui per ingrandire la figura.

Figura 2. Valutazione quantitativa e qualitativa delle MKS1 mutazioni rilevate nell'uomo. Difetti dello sviluppo in MKS1 embrioni morphant. In base alla gravità, fenotipi sono stati classificati in tre gruppi. Esempi di ogni classe sono indicati (a), e loro prevalenza nel loro coorte dell'embrione (n = 100-160 embrioni) è stato tabulati (non mostrato). MO iniettato embrioni con classe I fenotipi avevano grossolanamente morfologia normale, ma erano più brevi, con un eccesso di tessuto embrionale sul tuorlo rispetto al controllo embrioni iniettati alla stessa età somitic (8-9 somiti). Morphants Classe II sono stati assottigliati, corti e avevano poco sviluppata la testa e le strutture di coda, e inoltre mancava definizione somitice la simmetria. Embrioni di classe III sono stati forti ritardi con somiti poco sviluppate e deforme, ondulato notochords, e in genere non sopravvivere oltre la fase 10-somite. Co-iniezione di umano MKS1 mRNA salvato ognuno di questi difetti, dimostrando la specificità dei fenotipi di MKS1 soppressione. Ibridazione in situ di embrioni allo stadio 11-somite (± 1 somite) colorati con Krox20, PAX2 e ribosonde MyoD (b, c ). Fenotipi sono stati quantificati da misurazioni dal primo all'ultimo apprezzabile somite di ogni embrione (frecce), quantificato in c. Figura adattato con il permesso di 15.

Figura 3. Esempi di modellazione in vivo di dismorfismi umana. (A), dismorfismi cranio-facciale. MRNA di controllo iniettato embrione (a sinistra) e mutante embrioni iniettati (a destra) colorato con blu alcian a 5 dpf. Mutanti mRNA a iniezione embrioni mostrano in particolare le piccole e deformi teste con una disorganizzazione generale dello scheletro cranio-facciale cartilagineo tra divaricate archi branchiali e mancanti o strutture malformate. (B) micro / macrocefalia. Controllo uninjected embrione (a sinistra) e kctd13 embrione MO-iniettato (a destra) a 5 dpf. Morphants visualizzano ampliamento della testa, come visto dallo spazio tra gli occhi. 21 (c) integrità vascolare ridotto. Controllo uninjected embrione (in alto) e eng embrione MO-iniettato (in basso) ripreso con un microscopio a fluorescenza a 2 dpf in un FLI1: eGFP linea giornalista transgenico. Morphants visualizzare compromessa germinazione dei vasi intersegmentale e altre strutture vascolari. 41 (d) Altered looping cuore. L'ibridazione in situ di uninjected embrioni wild-type (sinistra) mostrano espressione spaw nel mesoderma piastra laterale sinistra, mentre ccdc39 embrioni morphant mostravano bilaterale (destra) o, in molti casi, espressione spaw non rilevabile (non mostrato). 43 (e) cisti renali. Uninjected embrione WT (in alto) e ift80 morphant (in basso). Morphants visualizzati grandi cisti renali (freccia), edema pericardico (punta di freccia) e una coda arricciata. 44 (f) Riduzione di filtrazione glomerulare. Visualizzazione fluorescente di un controllo iniettato embrione (in alto) e ift80 morphant (inferiore) 24 ore dopo l'iniezione di rodamina destrano nel cuore. Disperde fluorescenza in tutto il sistema vascolare e viene quasi completamente evacuati dai reni come visto la completa assenza di fluorescenza nel controllo. Il morphant mostra persistente fluorescenti destrano, suggerendo ridotta filtrazione glomerulare. 46 (g) distrofia muscolare. WT DNAJB6 mRNA embrioni iniettati (in alto) mostrano normali miofibre lenti che coprono i somiti normalmente tra myosepta adiacenti, come determinato dal immunoistochimica utilizzando anticorpi anti-miosina lenta. Mutant DNAJBb (in basso) ha mostrato parziale a completa distacco di miofibre da myosepta in una o più somiti. 19 (h) Angolo somite. Ingrandita viste laterali live di controllo uninjected (in alto) o kif7 morphants (in basso) impressi a 30 HPF. Morphants visualizzare somiti forma anomala, attribuibili a ectopica di segnalazione Hedgehog nel myotome zebrafish 47.
Tutte le cifre adattate con il permesso.
Discussione
I metodi qui descritti rappresentano un protocollo generale applicabile al dosaggio dei cambiamenti nonsynonymous associate con una varietà di fenotipi malattia genetica umana (Tabella 2, Figura 3). I nostri approcci si sono dimostrati utili per valutare il potenziale impatto di variazione su fenotipi di malattia, e per contribuire a sviscerare i meccanismi della malattia (come il contributo delle mutazioni dominanti negative per la sindrome di Bardet-Biedl, un disordine autosomico recessivo soprattutto 17). Ad oggi, attraverso lo sviluppo dell'albero decisionale presentato, abbiamo modellato al costo e di tempo ragionevole al di sopra di 200 geni causalmente connessi con malattie genetiche, ad un eccesso di 1.000 alleli.
Anche se non è discusso in dettaglio qui, abbiamo anche dimostrato che questi metodi sono appropriati per modellare altri tipi di lesioni genetiche, come il numero di copie (CNV varianti) e di genetica e interazioni. Le analisi di questi eventi sono al di là del campo di applicazione delmetodo della presente descrizione, pur fondamentalmente basano sullo stesso principio di test sistematici di geni candidati (incluse coppie di geni iniettati contemporaneamente) per determinare induzione o esacerbazione di fenotipi clinicamente appropriate. Ad esempio, per chiarire che dei 29 geni nel 16p11.2 CNV potrebbe essere rilevante per l'osservato microcefalia osservata nei pazienti con duplicazioni di 660 kb segmento genomico, mRNA corrispondente a ciascuno dei 29 geni all'interno del segmento sono stati iniettati e la testa misurazione delle dimensioni sono stati condotti a 2 dpf e 5 dpf, rivelando un importante contributo di un singolo trascritto, KCTD13 21. Inoltre, abbiamo utilizzato questo modello per analisi interazioni genetiche di lesioni genomiche in pazienti con entrambi sindrome di Bardet-Biedl e malattia di Hirschsprung. 22 Al confronto di MO soppressione dei geni causali delle due identità clinici separatamente e contemporaneamente, siamo stati in grado di identificare il fenotipo risultante da being una interazione sinergica piuttosto che semplicemente additivo gravità.
Pur avendo stabilito alta sensibilità (98%) e specificità (> 82%) per le varianti che contribuiscono al ciliopathies 17, noi non abbiamo ancora dati sufficienti per determinare se questi sono generalizzabili a tutte le letture fenotipiche nei modelli di zebrafish. Per fare ciò, un gran numero di alleli, predisse geneticamente per essere benigni o patogeni, deve essere testato all'interno di ciascuna categoria fenotipica. Ciò sarà particolarmente importante per l'attuazione di tali dosaggi nel setting clinico, in cui interpretazione funzionale VUSs può informare la diagnosi e la gestione solo se una conoscenza solida di falsi positivi e falsi negativi può accompagnare la consegna di tali risultati ai medici e pazienti. Tuttavia, questi metodi possono contribuire significativamente verso una migliore comprensione del paesaggio di malattia genetica umana. Prevediamo che questi modelli non serviranno solo come fonzione per una migliore interpretazione delle informazioni genetiche cliniche, ma anche essere impiegato come modelli utili per condurre schermate terapeutiche. I dati in vivo può anche essere paragonato al silico predizioni computazionali da fonti come PolyPhen 23, setacciare 24 SNPs & GO 25, o MutPred 26 per mostrare concordanza. Si noti che in un precedente studio di previsione database SNP & GO e MutPred sono risultati essere il più preciso, con precisioni di solo 0,82 e 0,81 raggiungendo, rispettivamente 27.
Anche se abbiamo delineato la robustezza di questi metodi per un sottoinsieme di difetti anatomici pediatrici (Tabella 2, Figura 3), certi fenotipi sono meno trattabili con questi metodi. Alcune eccezioni nonostante, ci sono tre classi principali di disturbi non riconducibili al nostro protocollo. Disturbi ad insorgenza nell'età adulta (come il morbo di Parkinson), rappresentano una sfida al modello in un sistema embrionale. Lento progression fenotipi degenerative (come la demenza frontotemporale) possono richiedere più tempo di quanto la finestra dpf sette delle attività MO per produrre un fenotipo. Altre tecnologie knockdown gene come RNAi e siRNA sono a disposizione per interferire o degradare il gene bersaglio, ma è stato dimostrato che nessuno è così specifica, stabile, non tossico, o di lunga durata, come MOs 28, quindi anche limitando il periodo di tempo per fenotipizzazione. Terzo, alcune strutture vertebrati, come il polmone di mammifero, non hanno una struttura sufficientemente ortologhi nell'embrione zebrafish. Abbiamo anche fornito un piano di emergenza suggerito per le indagini di quei casi in cui WT iniezione di mRNA umano porta ad un fenotipo, anche se avvertiamo questa è una situazione insolita e indesiderabile.
Alcuni fenotipi malattia possono quindi richiedere un maggior grado di astrazione e la maternità surrogata. E 'possibile che la funzione del gene è discostato sufficientemente per indebolire la somiglianza fenotipica tra modello e true fenotipo, o che zebrafish fisiologia complica inerentemente gli effetti della malattia indotta. In questi casi, si consiglia ulteriore dissezione del fenotipo prodotto prima licenziamento. Abbiamo prodotto alcuni esempi di successo in cui fenotipi problematiche per questo saggio sono stati modellati in embrioni di zebrafish. Per esempio, mutazioni in TCF8 un gene associato Fuchs distrofia corneale (FCD) sono stati analizzati utilizzando il nostro protocollo utilizzando difetti gastrulazione come visualizzatore fenotipica surrogato base ai ruoli noti di questo trascritto nel primo sviluppo. 29 In altri casi, come ad come adulto-inizio distrofia muscolare causata da mutazioni in DNAJB6, siamo stati in grado di generare fenotipi miofibre in embrioni 5dpf nonostante il fatto che gli esseri umani sono privi di patologia muscolare apprezzabile nei loro primi tre o quattro decenni di vita 19.
Oltre ai modelli mutanti transitori presentati qui, altri hanno preso advantage di questo sistema transitorio per modellare malattia umana in una varietà di sistemi del corpo. In un esempio, retinite pigmentosa stato modellato in zebrafish dal knockdown del gene RP2, con conseguente morte delle cellule retiniche e diminuita laminazione retinica. Salvataggio con mRNA wild-type umana ha portato allo sviluppo di tutti e tre strati di laminazione retinica, mentre quattro su cinque mRNA mutanti riuscito a salvare. 30 Anche tale modello di un disturbo sensoriale umano è basato su un fenotipo morfologico, è anche possibile risposta saggio a stimoli come stimoli acustici o prepulse inibizione 47.
Recentemente, un modello di zebrafish è stato utilizzato per studiare la malattia di Alzheimer attraverso la patogenesi proteina precursore dell'amiloide. 31 Gli autori hanno dimostrato che knockdown gene causato alterata crescita degli assoni dei neuroni motori, che potrebbero essere salvati con mRNA umano. Questo modello ha dimostrato di essere particolarmente informativo, come i modelli del mouse visualizzano solo phenot sottileipi (singolo atterramento) o letalità postnatale (doppio atterramento). La capacità di valutare gli embrioni di zebrafish in vivo durante tutto lo sviluppo ha aiutato a discernere l'effetto patogeno di ridotta proteina precursore dell'amiloide, così come previsto prova diretta che la proteina richiede sia un dominio extracellulare e intracellulare per il corretto funzionamento. Altri modelli importanti sono quella di altre distrofie muscolari 32, Diamante Blackfan 33, Axenfeld-Reiger sindrome (sviluppo oculare e cranio-facciale) 34, la malattia infiammatoria intestinale 35 (attività antibatterica), malattia di Parkinson 36 (neurone e la perdita di locomozione), e il sequestro 37 (idrocefalo e iperattività).
Più comuni sono le linee di zebrafish mutanti che hanno dimostrato anche di ricapitolare un fenotipo malattia umana. Inviato a 1,38, i modelli includono la leucemia, il melanoma, cardiomiopatie dilatate, la distrofia muscolare di Duchenne,e molti altri.
Divulgazioni
Gli autori dichiarano di non avere interessi finanziari in competizione.
Riconoscimenti
Noi riconosciamo il sostegno di una della Duke University Dean Estate Research Fellowship (AN), American Heart Association (AHA) fellowship 11POST7160006 (CG), National Institutes of Health (NIH) sovvenzioni R01-EY021872 dal National Eye Institute (EED), R01HD04260 dal Istituto Nazionale della Salute dei bambini e lo sviluppo (NK), R01DK072301 e R01DK075972 dal National Institute of Diabetes Digestiva e Malattie renali (NK), e l'Unione europea (finanziato da UE 7 ° PQ sotto GA nr 241955, progetto SYSCILIA,. EED, NK) NK è un Distinguished Jean e George W. Brumley prof.
Materiali
| Name | Company | Catalog Number | Comments |
| Reagent | |||
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S, M0530L | |
| DpnI restriction endonuclease | NEB | R0176L, R0176S | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258-012 | |
| Big Dye Terminator | Applied Biosystems | 4337455 | |
| mMESSAGE mMACHINE Kit | Invitrogen | AM1340, AM1344, AM1348 | |
| Morpholino | Gene-Tools | n/a | |
| 1-phenyl-2-thiourea (PTU) | Sigma Aldrich | P7629 | Prepare as 0.003% PTU in embryo media |
| Paraformaldehyde (PFA) | Sigma Aldrich | P6148 | For embryos that must be fixed prior to phenotyping, prepare as 4% |
| Tricaine methane sulfonate | Western Chemical | N/A | For anesthetization and euthanasia |
| Equipment | |||
| PTC-225 Tetrad Thermal Cycler | BioRad | Any equivalent thermal cycler | |
| Nano Drop 2000 spectrophotometer | Thermo Scientific | ||
| SMZ 745T Stereomicroscope | Nikon | ||
| AZ100 Stereomicroscope | Nikon | ||
| DS Fi1 Digital Camera | Nikon | For color/fluorescent imaging | |
| DS QiMC Digital Camera | Nikon | For black/white imaging | |
| Advanced Resarch 3.2 Imaging Software | NIS- Elements | ||
Riferimenti
- Lieschke, G. J., Currie, P. D. Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353-367 (2007).
- Nolan, T., Hands, R. E., Bustin, S. A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559-1582 (2006).
- Thisse, C., Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69 (2008).
- Detrich, H. W., Westerfield, M. 3rd, Zon, L. I. Overview of the Zebrafish system. Methods Cell Biol. 59, 3-10 (1999).
- Gerdes, J. M., Davis, E. E., Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 137, 32-45 (2009).
- Hildebrandt, F., Benzing, T., Katsanis, N. Ciliopathies. N. Engl. J. Med. 364, 1533-1543 (2011).
- Zaghloul, N. A., Katsanis, N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 119, 428-437 (2009).
- Marion, V. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet-Biedl syndrome with situs inversus and insertional polydactyly. J. Med. Genet. 49, 317-321 (2012).
- Otto, E. A. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 42, 840-850 (2010).
- Schaefer, E. Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur. J. Med. Genet. 54, 157-160 (2011).
- Katsanis, N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 13 Spec No 1, R65-R71 (2004).
- Badano, J. L. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 439, 326-330 (1038).
- Badano, J. L. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 12, 1651-1659 (2003).
- Gerdes, J. M. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 39, 1350-1360 (2007).
- Leitch, C. C. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 40, 443-448 (2008).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 203, 253-310 (1995).
- Zaghloul, N. A., et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 10602-10607 (2010).
- Davis, E. E. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189-196 (2011).
- Sarparanta, J., et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450-455 (2012).
- Selcen, D. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Annals of neurology. 65, 83-89 (2009).
- Golzio, C., et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 485, 363-367 (2012).
- de Pontual, L., et al. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13921-13926 (2009).
- Adzhubei, I. A., et al. A method and server for predicting damaging missense mutations. Nature Methods. 7, 248-249 (2010).
- Kumar, P., Henikoff, S., Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073-1081 (2009).
- Calabrese, R., Capriotti, E., Fariselli, P., Martelli, P. L., Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30, 1237-1244 (2009).
- Li, B., et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 25, 2744-2750 (2009).
- Thusberg, J., Olatubosun, A., Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 32, 358-368 (2011).
- Summerton, J. E. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 7, 651-660 (2007).
- Riazuddin, S. A., et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 86, 45-53 (2010).
- Shu, X., et al. Knockdown of the zebrafish ortholog of the retinitis pigmentosa 2 (RP2) gene results in retinal degeneration. Investigative Ophthalmology & Visual Science. 52, 2960-2966 (2011).
- Song, P., Pimplikar, S. W. Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PloS one. 7, e34209(2012).
- Kawahara, G., Guyon, J. R., Nakamura, Y., Kunkel, L. M. Zebrafish models for human FKRP muscular dystrophies. Hum. Mol. Genet. 19, 623-633 (2010).
- Danilova, N., Sakamoto, K. M., Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 112, 5228-5237 (2008).
- Bohnsack, B. L., Kasprick, D. S., Kish, P. E., Goldman, D., Kahana, A. A zebrafish model of axenfeld-rieger syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Investigative Ophthalmology & Visual Science. 53, 7-22 (2012).
- Oehlers, S. H., et al. The inflammatory bowel disease (IBD) susceptibility genes NOD1 and NOD2 have conserved anti-bacterial roles in zebrafish. Disease Models & Mechanisms. 4, 832-841 (2011).
- Sheng, D., et al. Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS genetics. 6, e1000914(2010).
- Teng, Y., et al. Loss of zebrafish lgi1b leads to hydrocephalus and sensitization to pentylenetetrazol induced seizure-like behavior. PloS one. 6, e24596(2011).
- Santoriello, C., Zon, L. I. Hooked! Modeling human disease in zebrafish. J. Clin. Invest. 122, 2337-2343 (2012).
- Javidan, Y., Schilling, T. F. Development of cartilage and bone. Methods Cell Biol. 76, 415-436 (2004).
- Lawson, N. D., Weinstein, B. M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307-318 (2002).
- Lee, N. Y. Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Molecular Biology of the Cell. 23, 2412-2423 (2012).
- Huang, C. J., Tu, C. T., Hsiao, C. D., Hsieh, F. J., Tsai, H. J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 228, 30-40 (2003).
- Merveille, A. C., et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 43, 72-78 (2011).
- Beales, P. L. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 39, 727-729 (2007).
- Drummond, I. A., Davidson, A. J. Zebrafish kidney development. Methods Cell Biol. 100, 233-260 (2010).
- Tobin, J. L., Beales, P. L. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 23, 2095-2099 (2008).
- Putoux, A. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 43, 601-606 (2011).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati