
Method Article
In Vivo Modelagem do Morbid Genoma Humano utilizando Danio rerio
Neste Artigo
Resumo
Aqui, apresentamos uma abordagem sistemática para o desenvolvimento fisiologicamente relevante, sensível e específico In vivo Ensaios para interpretar variações na patologia humana. Manipulação genética transitória via microinjeção de WT e mutantes mRNA humano e morfolino (MO) oligonucleotídeos antisense aproveitar a rastreabilidade do desenvolvimento de embriões do peixe-zebra para rapidamente mutações ensaio patogênicos, especialmente, mas não exclusivamente, no contexto dos transtornos do desenvolvimento humano.
Resumo
Aqui, apresentamos métodos para o desenvolvimento de ensaios para consultar alterações nonsynonymous potencialmente clinicamente significativas usando em complementação vivo no peixe-zebra. Peixe-zebra (Danio rerio) é um sistema de animais útil devido à sua docilidade experimental; embriões são transparentes para permitir a visualização fácil, submetidos a um desenvolvimento rápido ex vivo, e pode ser manipulado geneticamente um Estes aspectos têm permitido avanços significativos na análise da embriogênese. processos moleculares e morfogenética de sinalização. Tomadas em conjunto, as vantagens desse modelo de vertebrados fazer zebrafish altamente receptivo para modelar os defeitos de desenvolvimento de doença pediátrica e, em alguns casos, distúrbios na idade adulta. Porque o genoma do peixe-zebra é altamente conservada com a dos seres humanos (~ 70% ortólogos), é possível para recapitular estados de doença humana em peixes-zebra. Isto é conseguido através da injecção da m humana mutanteRNA para induzir ganho dominante negativa ou de alelos de função, ou utilização de morfolino (MO), os oligonucleótidos anti-sentido para suprimir genes para imitar a perda de função de variantes. Através de complementação do fenótipo MO induzidas com ARNm humano tampado, a nossa abordagem permite a interpretação do efeito deletério de mutações em sequência da proteína humana, com base na capacidade de ARNm mutante para resgatar um mensurável, fenótipo fisiologicamente relevante. Modelando dos alelos humanos da doença ocorre através de microinjecção de embriões de peixes-zebra com MO e / ou ARNm humano no estádio de célula de 1-4, e fenotipagem até sete dias após a fertilização (dpf). Esta estratégia geral pode ser alargada a uma vasta gama de condições clínicas, como demonstrado no seguinte protocolo. Apresentamos nossos modelos estabelecidos para morfogenética de sinalização, integridade craniofacial, cardíaco, vascular, função renal, e esqueléticos fenótipos desordem muscular, bem como outros.
Introdução
A interpretação funcional da informação genética e atribuição do valor clínico preditivo de um genótipo representa um grande problema em genética médica e está se tornando cada vez mais pungente com a aceleração de viabilidade técnica e econômica do seqüenciamento do genoma. Por conseguinte, é necessário desenvolver e implementar novos paradigmas para testar a patogenicidade das variantes de significado desconhecido (USV) detectados em pacientes. Estes ensaios devem, então, ser exato, tempo e custo-eficiente, e do porto o potencial para catalisar uma transição para a utilidade clínica.
Enquanto o rato tem sido tradicionalmente a ferramenta de escolha no campo da modelagem de doenças humanas, zebrafish estão emergindo como um substituto científica e economicamente favorável. Ao contrário do rato, peixe-zebra biologia permite o acesso fácil e oportuno para todas as fases de desenvolvimento, auxiliados por claridade óptica dos embriões, que permite imagens em tempo real de desenvolvimento de patologias. 1 A relativamente recente geração de linhas de peixe-zebra mutante tem proporcionado testes adicionais e opções de modelagem, utilizada por muitos em estudos funcionais, mas esta tecnologia continua a ser limitado (revisado em 1,38). Não são apenas os mutantes genéticos com knock-ins de mutações específicas laboriosas para atingir, não são também acessíveis à análise de médio ou elevado rendimento para o ensaio de uma série de mutações num único gene. Importante, um único conjunto de testes pode proporcionar informação não só para o potencial patogénico de alelos, mas também para a direcção do efeito a nível celular (por exemplo, perda da função versus ganho de função), o qual é crítico para o modo de hereditariedade informando nas famílias, especialmente quando pequenos pedigrees humanos abrigam poucas informações sobre o modo de transmissão genética. Para posterior comparação dos usos do rato disponíveis e modelos de peixe-zebra, ver Tabela 1.
Observamos também que ore são limitações inerentes ao sistema modelo zebrafish. Embora D. rerio tem rápido desenvolvimento inicial de sistemas de órgãos, a maturidade sexual requer aproximadamente três meses. Devido a isso, distúrbios pré-natal e pediátrica de início são as mais susceptíveis a este modelo de expressão transitória. Enquanto ideal para a realização de telas de compostos grandes químicos, a utilização de mutantes genéticos não é viável para o teste sistemático de milhares de variantes nonsynonymous que contribuem para, e continuam a ser detectado em doenças pediátricas.
Os testes de complementação descrita aqui tirar proveito desta tratabilidade experimental, elevado grau de homologia, e preservação da função entre as proteínas humanas e peixe-zebra, em particular, para as moléculas necessárias para os processos de desenvolvimento conservadas. Figura 1 descreve a estratégia de identificação e testes para vários efeitos de alelos. A perda de função (LOF) e ensaios dominantes pode ser realizada. Para LOF, a experiência se inicia com a supressão do gene de interesse com um knockdown morfolino, e ensaiando para fenótipos que possam ser relevantes para o fenótipo clínico sob investigação. A supressão pode ser alcançada quer por bloqueio tradução visando um MO no ou perto do local de iniciação da tradução do locus de peixe-zebra (tradução bloqueador morfolino; tbMO), ou por interferência com splicing colocando um MO sobre uma junção de união, tipicamente induzindo ou inclusão de um intron ou exon aberrante pular (emenda bloqueio morpholino; SBMO).
Subsequentemente, o ARNm transcrito a partir do tampado ortólogo humano é introduzido e salvamento quantificável do fenótipo é medido. Uma vez que o ensaio é estabelecida, as mutações candidatas na mensagem humano pode ser introduzido e ensaiadas quanto à sua capacidade para resgatar o fenótipo MO induzida na mesma eficiência que WT ARNm humano. Por outro lado, para o candidato alelos dominantes, mRNA humano (mas não MO) é introduçãoed, com a expectativa de que o ARNm humano WT não irá afectar grandemente a anatomia e fisiologia do peixe-zebra, ao passo que a introdução de mutações de ensaio que tenham um efeito dominante vai induzir fenótipos análogos aos observados no estado clínico em humanos. Esta experiência pode ser refinado para dissecar ainda mais se o efeito dominante ocorre por um ganho de função (GOF), ou um mecanismo negativo dominante misturando WT e mutantes ARNm humano; para eventos GOF, a adição de ARNm humano WT deve ser irrelevante, ao passo que para os alelos dominantes-negativos, misturando de ARNm WT e mutantes devem alterar a gravidade do fenótipo induzido pela mensagem mutante. Em todos os casos, recomendamos que todas as combinações de injeções (MO com WT mRNA humano vs morpholino com mutante mRNA humano etc ser realizada, de preferência dentro da mesma ninhada de embriões (ver Figura 1) A interpretação é a seguinte.:
Para os testes de LOF:
- Se o knockdownproduz um fenótipo, que pode ser resgatada por equivalentemente ARNm mutante e WT, o alelo é provável benigna.
- Se o mutante de resgate do fenótipo knockdown é indistinguível da do próprio fenótipo knockdown, o alelo é um nulo funcional provável. O experimento não pode discriminar entre os verdadeiros valores nulos (sem proteína funcional) e os níveis de atividade da proteína ultrabaixo que não têm capacidade de resgate.
- Se o mutante de resgate do fenótipo knockdown é estatisticamente melhor do que o PB, mas pior do que a WT, o alelo é provável que um hypomorph como este resultado demonstra a perda parcial de função.
Para os testes dominantes:
- Se não houver um fenótipo de knockdown, mas a injecção de ARNm WT produz um fenótipo, um plano de emergência tem de ser utilizado se o experimento é prosseguir (ver abaixo).
- Se não houver um fenótipo de knockdown e injecção de ARNm WT produz um fenótipo, a experiência decorre, como de costume.
- Se injeçãodo mRNA mutante é equivalente ao de ARNm do tipo selvagem, o alelo podem ser benignos ou a perda de função, ou o ensaio pode ter falhado. Isso exige mais experimentação para discriminar entre essas opções.
- Se a injecção de ARNm mutante é indistinguível da MO knockdown, a função do produto do gene é provavelmente alterada de alguma forma. Para distinguir a mudança na função, uma titulação de ARNm mutante com o tipo selvagem de ARNm deve ser realizado.
- Se o resultado desta titulação é indistinguível do tipo selvagem ARNm sozinha, tem sido mostrado que o produto da proteína mutante utiliza a proteína do tipo selvagem como um substrato, tal como o seu efeito varia com a quantidade de titulação. Isto indica um fenótipo dominante negativo.
- Se o resultado desta titulação é indistinguível de ARNm mutante isoladamente, tem sido mostrado que o produto da proteína mutante não tem a mesma função que o de tipo selvagem, e, portanto, não é afectado pela quantidade de produto de tipo selvagem pré proteínaenviada. Isto demonstra que o alelo é provável ganho de função.
Plano de contingência:
- Se nenhum fenótipo apresenta a partir de MO knockdown, mas faz presente com mRNA WT, novas experiências podem ocorrer, apesar de enfatizar que esta situação não é ideal. ARNm humano WT deve ser titulada para minimizar o fenótipo, e também pode ser utilizado como um novo ponto de referência. Além disso coinjection do WT e mRNA humano mutante pode ser avaliado com base na capacidade de resgatar o mutante.
Protocolo
1. Bioinformática Análise
- Determinar se o gene humano de interesse tem um ortólogo peixe-zebra, e em caso afirmativo, quantas cópias. Recomendamos BLAST recíproco ( http://blast.ncbi.nlm.nih.gov/ ) da proteína humana contra o genoma do peixe-zebra, e subsequente BLAST o melhor zebrafish bater contra o genoma humano. Verdadeira orthologs será o melhor hit em cada instância.
- Determinar o tamanho da grelha de leitura aberta (ORF) do gene humano. Se mais de 6 kb, este modelo é intratável no momento por causa das limitações de alta qualidade transcrição in vitro de longo modelos.
- Obter ou gerar um construto contendo a ORF humana no pCS2 + vector espinha dorsal (ou um vector equivalente com um 5 'sítio SP6 de transcrição e de 3' do sinal de poliA).
- Projetar um MO para bloquear a emenda ou a tradução do gene zebrafish alvo. Se existir em múltiplas cópias no genoma do peixe-zebra, There são duas opções: a) projeto de MOs adicional, ou b) a identificação de um local de emenda totalmente preservada entre as cópias contra a qual um único MO pode ser eficaz. Algumas seqüências MO publicados estão disponíveis em www.zfin.org .
2. Análise da expressão no desenvolvimento de embriões Zebrafish
- Determinar se o ortólogo de peixe-zebra é expressa num contexto espácio-temporal relevante para a leitura fenotípica. Se não há dados disponíveis para a expressão do gene de interesse, realizar a transcrição reversa (RT)-PCR, utilizando ADNc de embriões de peixes-zebra inteiros ou hibridação in situ. (Ver 2,3).
3. Mutagênese sítio-dirigida
- Projeto iniciadores de mutagénese de 25-45 bases de comprimento, com a mutação desejada no centro. A temperatura de fusão do primário deve ser maior do que ou igual a 78 ° C. Projete uma cartilha mutagênese frente e verso para anelar a opostovertentes do plasmídeo.
- Obter iniciadores para a confirmação da sequência da ORF de pós-mutagénese, estes devem telha entre ~ 300 secções de pares de base para cobrir a totalidade da ORF.
- Montar a reacção de mutagénese, com uma polimerase de alta fidelidade e ciclo como se segue (1: 95 ° C 30 seg, 2: 95 ° C, 30 seg, 3: 55 ° C 1 min, 4: 68 ° C 6 min, 5: Ir a 2 de 18x, 6: 4 ° C para sempre, 7: end).
- Adicionar 1 ml de endonuclease de restrição Dpnl para eliminar por reacção barragem molde metilado; incubar a 37 ° C durante 2 horas.
- Transformar 2 ul de reacção de mutagénese em 20 ul de células competentes de acordo com protocolos padrão.
- Escolha 3-4 colônias e inocular 5 ml de meio LB contendo antibiótico apropriado. Agitar a 37 ° C durante a noite a 225 rpm.
- ADN Miniprep e determinar a concentração.
- Sequência do local de mutação para confirmar a presença da mutação de interesse.
- Seqüência inteira ORF para confirmar a integridade seqüência.
4. Transcrição in vitro de mRNA
- Usando um modelo de pCS2 + linearizado, gerar ARNm nivelada usando o mMessage mMachine SP6 kit (Ambion). Recomendamos o uso de metade das quantidades de componentes de reação.
- Purifica-se a amostra de ARN com LiCl precipitação ou fenol clorofórmio, tal como descrito nas instruções do kit.
- Determinar a concentração de ARNm utilizando a absorvência, a garantir a integridade do mRNA usando electroforese em gel, e armazenar a amostra em três ou mais partes alíquotas a -80 ° C até que esteja pronto para uso. Nós não recomendamos múltiplos ciclos de congelamento e descongelamento de alíquotas de mRNA.
5. Ensaio in vivo de variantes de perda de função
- Obter embriões de peixes-zebra a partir de acasalamentos zebrafish naturais, e mantê-las a 28 ° C em água de embriões em pratos de 6 ou 10 cm.
- Realizar uma curva de resposta à dose para avaliar a especificidade morfolino fenótipo MO MO eficácia e toxicidade. Injectar uma curva de pelo dosagemmenos três diferentes concentrações entre 1-10 ng MO em 50-100 (1-4 estágio de célula) embriões de peixe-zebra / lote. MOs eficientes devem dar origem a aumentos dose-dependentes da proporção de embriões afetados em um lote.
- Fenótipo embriões no estágio de desenvolvimento adequado com base na expressão do gene do peixe-zebra alvo de interesse e fase em que será observado um fenótipo relevante. Esta pode ser tanto quantitativa (como uma medida entre as estruturas anatómicas) ou qualitativas (com base em critérios fenotípicos padronizados). Para todas as injecções teve> 24 HPF: embriões devem ser tratados com PTU (0,003% de 1-fenil-2-tioureia em meios de embriões) a 24 hpf a redução máxima da formação de melanócitos.
- Para o bloqueio de tala-OMs, MO teste de eficiência através da extracção de ARN total de embriões a partir de lisados de todo, no ponto de tempo de pontuação fenotípica, gerar cDNA e realizar a RT-PCR do gene alvo, utilizando iniciadores que flanqueiam o local alvo MO.
- Para verificar a supressão efficiency de um todo tb-MO, a colheita de embriões e lisados de proteína de realizar imunotransferência para comparar os níveis de proteína versus controlo de destino. No entanto, esta abordagem não é passível de todos os genes-alvo porque não há reactividade cruzada limitada por muitos anticorpos comerciais para as proteínas de peixe-zebra. Dois métodos indirectos de exibir especificidade MO, incluem: a) demonstrar que existe um efeito dependente da dose de fenótipo, e b) mostra que a co-injecção de ARNm de tipo selvagem com TB-MO resgata o fenótipo de forma eficiente. Por essas razões, recomendamos um bloqueador de emenda quando possível porque a eficiência pode ser monitorado diretamente.
- Se for observado um fenótipo de avançar para o passo 5.7, se não for observado o fenótipo avançar para o passo 6.1.
- Para fenótipos qualitativos, selecione uma dose MO em que 50-75% dos embriões são afetados, pois fenótipos quantitativos, selecione uma dose MO em que a medida fenotípica é significativamente diferente do tipo selvagem (p <0,001). Injetar novos lotes de zebrafish embtabaco de enrolar (1-4 fase celular, n = 50-100/batch) com um cocktail contendo a dose "ensaio" de MO e uma curva de ARNm humano WT (variando 10-200 pg de ARNm de dosagem, essas doses assegurar superexpressão substancial acima a linha de base de um único transcrito, representando 0,25-0,5% do ARNm total de poli-A no embrião do peixe-zebra) 4.
- Realizar pontuação mascarado de lotes de injecção; escolher a dose mRNA WT com o resgate mais significativa em comparação MO sozinho para, esta é a dose "ensaio" de mRNA.
- Injetar novos lotes (1-4 estágio de célula; n = 50-100/batch) com a dose de ensaio de MO e da dose de ensaio de mRNA humano mutante. Fenótipo embriões no estágio apropriado e comparar os resultados para WT resgate mRNA humano usando um teste estatístico adequado (t-test ou qui-quadrado). Ver Figura 1 para os resultados e vá para a etapa 7. Injetar as doses de ensaio de WT e mRNA mutante sozinho para controlar os efeitos de toxicidade de mRNA.
6. Ensaio in vivo de variantes de DOMinant negativo ou ganho de Efeitos de função
- Se nenhuma perda de função fenótipo é observada (Passo 5,5) ou se o ARNm mutante dá origem ao fenótipo que não são significativamente diferentes MO isoladamente (Passo 5.7) a partir de, injecta-se uma curva de dosagem variando 10-200 pg de ARNm humano WT (recomendamos 25, 50 e 100 pg como um teste inicial) em lotes de embriões (1-4 estágio de célula; 50-100 embriões / lote).
- Na fase adequado (ver 5.3 acima), a conduta de pontuação fenotípica, e determinar a dose mais alta em que não há um número estatisticamente significativo de embriões mortos e / ou afectadas em comparação com os controlos não injectados. Esta é a dose "ensaio".
- Injetar a dose de ensaio de mRNA humano mutante (1-4 estágio de célula; 50-100 embriões / lote). Embriões fenótipo e comparar os resultados de pontuação a partir da dose de ensaio WT injeção de mRNA humano ou a concentração de MO ensaio. Veja a Figura 1 para os resultados.
- Se os resultados são indistinguíveis de MO, titular mRNA mutante humano com mRNA WTe comparar com WT humano e injeções de mRNA mutantes. Melhoria de fenótipos com mutantes mais WT lotes mRNA com injeção indica uma negativa dominante. Nenhuma melhoria indica um ganho de função.
7. Reproduzir nos resultados dos testes in vivo
- Repita a ensaios in vivo de um mínimo de três vezes.
8. Integrar Zebrafish em dados de patogenicidade in vivo com outras linhas de evidência
- Comparar os dados obtidos a partir de experimentos de patogenicidade peixe-zebra para: dados genéticos dentro de um pedigree (se aplicável), os dados de freqüência da população de controle, em estudos in vitro (ensaios baseados em células de estabilidade da proteína, localização celular, a produção de sinalização, ou atividade enzimática).
Resultados
Distúrbios recessivos e Pseudorecessive
Cílios são estruturas quase ubíquo no plano do corpo dos vertebrados, que desempenham funções de sinalização celular em múltiplos destinos celulares, incluindo a proliferação, a polaridade, diferenciação e manutenção de tecidos. Cinco disfunção desses organelos conduz a uma ampla gama de doenças genéticas humanas colectivamente como ciliopathies 6,7. Uma tal entidade clínica é a síndrome de Bardet-Biedl (BBS), uma doença pediátrica multissistêmica caracterizada por degeneração da retina, obesidade, hipogonadismo, polidactilia, e disfunção renal. 7 O desenvolvimento de ensaios in vivo de patogenicidade alelo era necessário para BBS porque a) é uma desordem geneticamente heterogênea causada principalmente por alterações nonsynonymous particulares que ocorreram em pelo menos 17 genes 7-10, e b) a herança oligogenic em> 25% das famílias BBS, onde a presença de alterações heterozigotos em um segundoGene BBS (além de mutações causais primárias recessivas) podem modular a penetrância clínica e expressividade. Normalmente, esses alelos terceiros são mudanças nonsynonymous heterozigotos de, a partir de um ponto de vista genético, potencial patogênico claro, necessitando, portanto, a interpretação exata do seu efeito biológico em função da proteína 11-13.
Para investigar o potencial de mutações que contribuem para a carga mutacional em BBS patogênico, inicialmente testado todas as alterações missense identificadas no BBS1 - BBS14. Nós e outros autores mostraram que a perda de proteínas do corpo basal dá origem a desregulação da polaridade celular planar (PCP; sinalização Wnt não canónicas). Manifestando-se como defeitos de extensão convergentes em embriões de peixe-zebra meados de somíticos 14 Utilizando esta leitura fenotípica fisiologicamente relevante, descobriram que a supressão de genes BBS resultou em encurtamento eixos corporais, somitos mais amplas e mais finas e largas, notochords torcida. 15 </ Sup> (Figura 2) MO-supressão induzida por defeitos produzidos a gastrulação, e co-injecção com mRNA humano resgatado significativamente (e reprodutivelmente), estes fenótipos como marcados por três diferentes métodos in vivo. Em primeiro lugar, os embriões foram marcados ao vivo de acordo com critérios fenotípicos qualitativos (Normal, Classe I e Classe II, para definições detalhadas das classes fenotípicas ver 15). Em seguida, durante a migração celular quantificada epiboly (uma fase de desenvolvimento precoce caracterizada pelo desbaste e espalhamento de camadas de células sobre a célula gema 16) pelo emprego de um marcador de fluoresceina para visualizar as células que migram. Finalmente, medimos comprimento do tronco somite em embriões de nove somito in situ cruzadas com um coquetel de krox20, PAX2 e riboprobes myoD, que foram plana montadas para análise morfológica.
Esta metodologia tem sido utilizada para testar em excesso de 500 alelos no espaço mutacional ciliar. Numaestudar sozinho, em ensaios in vivo de> 130 alelos produzida uma gama de contagens fenotípicos, como indicado pelo nosso protocolo (Figura 1) completo resgata foram classificados como benignos (não significativamente diferente de WT de salvamento), parcial resgata foram classificados como hypomorphs (melhorou significativamente a partir de MO, mas mais grave do que WT salvamento), a incapacidade de resgate foram classificados como nulos funcional (não significativamente diferente da MO), e fenótipos induzidas por ARNm mutante isoladamente em comparação com MO foram classificados como negativos dominantes.
Também se avaliou a sensibilidade e especificidade do ensaio de complementação in vivo em peixes-zebra. A especificidade foi confirmada por co-injecção de SNPs comuns (> 5% de frequência do alelo menor em populações saudáveis de controlo), os quais foram encontrados para dar fenótipos benignas em 14/17 testado (> 82%), e foi demonstrado que a sensibilidade a 98% como indicado de concordância entre os dados in vivo e argumentos genéticos suficientes to atribuir um alelo como causal em um BBS pedigree. 17 Além disso, os efeitos fenotípicos observados utilizando as três medidas Vivo (pontuação ao vivo, epiboly rastreamento e ISH morfometria) foram validados in vitro, utilizando estudos de localização de imunoeletrotransferência e celular. Enquanto a interpretação destes resultados requerido conhecimento prévio de pelo menos um mecanismo de patogênese da doença, este exemplo fornece evidências para comprovar a utilidade e robustez do nosso protocolo. Desde então, temos corroborado nossa na pontuação vivo com várias outras linhas de evidência experimental em um screening mutacional imparcial e estudo análise funcional do TTC21B, uma proteína de transporte intraflagellar retrógrada. 18
Distúrbios dominantes
Membro distrofias musculares cinturas (DMC) são uma classe autossômica de distrofia muscular, causando fraqueza muscular progride lenta nos quadris e ombros. Este geneticamente e mechanistically grupo heterogêneo de doenças é causada por ambas as mutações dominantes e recessivos em todo sarcolemal múltipla, sarcoméricos, citoplasmáticos e proteínas nucleares. Com base na apresentação de fenótipos clínicos e evidências do envolvimento muscular de ressonância magnética, foi investigada a causa de um LGMD dominante encontrada na Finlândia, EUA, e 19 famílias italianas. Sequenciação de candidatos posicionais dentro do locus mapeada revelou mutações no DNAJB6, um gene que codifica para um co-chaperonas de família HSP70 expressa como pelo menos duas isoformas de splice (nuclear e citoplasmática) em seres humanos. Para ganhar novos insights sobre a função DNAJB6 e sua relevância para LGMD, examinamos o seu papel na integridade muscular em zebrafish. RT-PCR de o peixe-zebra ortólogo (dnajb6b) detectou expressão tão cedo quanto a fase de cinco somito, que foi seguido por injecção de embriões com uma tala bloquear morfolino. Às 48 horas após a fertilização, a pontuação mascarado mostrou descolamento da lentafibras a partir de seus pontos de inserção. A especificidade deste fenótipo foi depois testado com um segundo MO não sobrepostos e resgatado subsequentemente com WT humana DNAJB6 ARNm.
Para consultar como a perda da função DNAJB6 conduz a defeitos de integridade muscular, introduzimos mutações missense encontrados em pacientes em transcritos humanos de ambas as isoformas e injectou-los em embriões de peixe-zebra. Embora a injecção de ARNm humano WT produziu um fenótipo sensível, estas alterações phenocopied a perda do efeito sobre a função dos mo quando projetada no citoplasma, mas não isoforma nuclear. Isto foi seguido por co-injecção de quantidades equimolares de ARNm mutante e WT, que apresentaram um aumento da gravidade do fenótipo muscular, sugerindo um efeito dominante. Para testar esta noção, mais injecções foram realizadas com a alteração proporções molares de ARNm mutante e WT. Consistente com a previsão, um excesso de ARNm mutante em comparação com o WT letalidade induzida em embriões, enquanto umn excesso de WT produziu um aumento progressivo de resgate. Isto foi seguido por experiências in vitro para determinar as propriedades oligimerization e possíveis interacções proteína. Estes mostraram que as mutações diminuir a actividade antiagregação de DNAJB6 citoplasmática e interferir com a rotação de ambos os mutantes e WT, bem como interagir com uma outra molécula, bag3, que também é relevante para o pathomechanism de DMC, desde LOF bag3 causa uma forma pediátrica distrofia muscular 20. Em seguida, perguntou se bag3 pode modular o fenótipo induzido por DNAJB6b mutante no peixe-zebra. Embora a injecção de WT bag3 sozinho produziu um fenótipo, co-injecção com DNAJB6 mutantes aumentou significativamente a severidade fenotípica, bag3 sugerindo que desempenha um papel na mediação da patogenicidade de tais mutantes 19.
| Transient Modelo Zebrafish | Mutant Zebrafish Linha | Camundongo transgênico Linha | |
| Idade de início do fenótipo humano sob investigação |
|
|
|
| Fenótipos possíveis |
|
|
|
| Tempo até à sua utilização (desde o nascimento) | 1-7 dias | > 3 meses | > 6 meses |
| Vazão | médio-alto | baixo | baixo |
| Vantagens |
|
|
|
Tabela 1. Comparação de modelos in vivo.

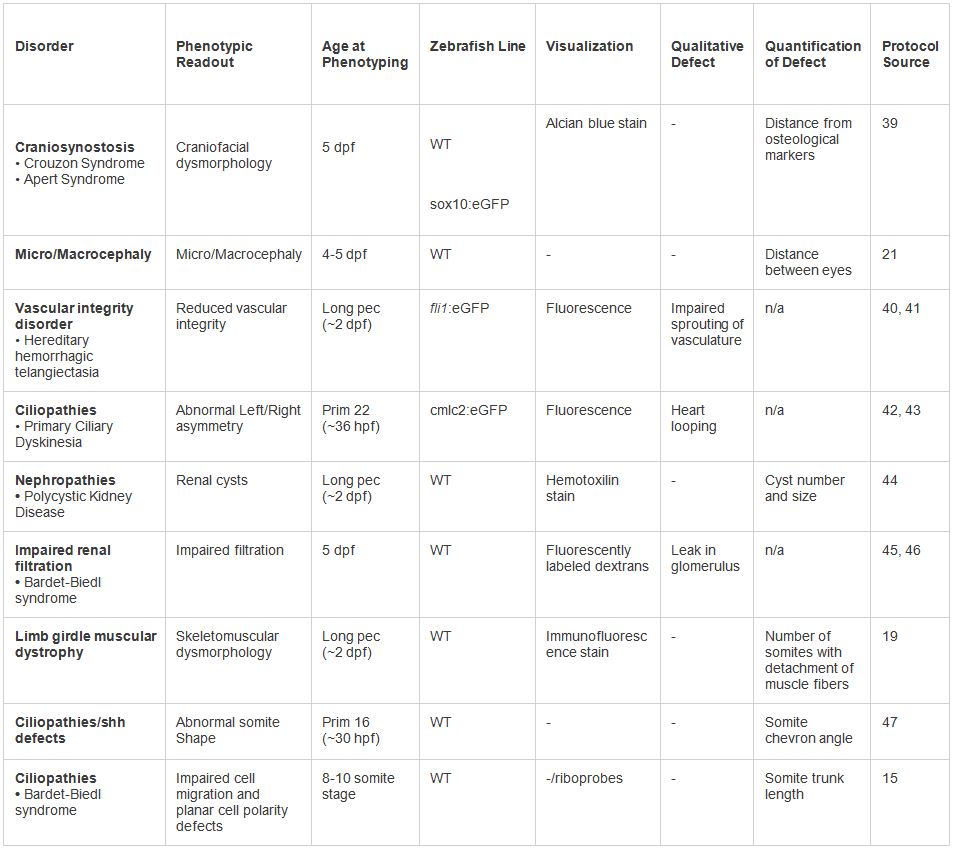
Tabela 2. Exemplos de modelagem em vivo de dysmorphologies humanos. Vários fenótipos testado sob o protocolo apresentado. Uma série de leituras fenotípicas e técnicas de visualização pode ser empregada com base no tipo de desordem. Clique aqui para ampliar a tabela.
{kind=link}

Figura 1. Nos testes funcionais in vivo de variantes nonsynonymous. Uma abordagem sistemática para o teste funcional de todosenguias de desconhecido ou a hipótese de significado. Knockdown do gene através morfolino microinjecção é seguida por uma série de (co) injecções de ambos WT e ARNm humano mutante. A análise estatística dos resultados fenotípicos informar a patogenicidade alelo ea função molecular. Resumidamente, para a perda de testes de funções: Se o knockdown produz um fenótipo, que pode ser resgatada por equivalentemente ARNm mutante e WT, o alelo é provável benigna (caixa verde). Se o resgate mutante do fenótipo knockdown é indistinguível do fenótipo knockdown, o alelo é um nulo funcional provável (caixa amarela). Se o mutante de resgate do fenótipo knockdown é estatisticamente melhor do que o PB, mas pior do que a WT, o alelo é provável que um hypomorph (caixa verde) Para os ensaios dominantes:. Se a injecção de ARNm mutante é equivalente à do tipo selvagem de ARNm , o alelo podem ser benignos ou a perda de função, ou o ensaio pode ter falhado (caixa verde). Se a injecção de ARNm mutante é indis tinguishable de MO knockdown, a função do produto do gene é provavelmente alterada de alguma forma. Para distinguir a mudança na função, dosear ARNm mutante com o tipo selvagem de mRNA. Se o resultado desta titulação é indistinguível do tipo selvagem ARNm sozinho, o produto da proteína mutante utiliza a proteína do tipo selvagem como um substrato, indicando, assim, um fenótipo negativo dominante (caixa azul). Se o resultado desta titulação é indistinguível de ARNm mutante sozinho, o produto da proteína mutante não tem a mesma função que o de tipo selvagem, e, assim, é provável que um ganho de função (caixa azul). Se apresenta um fenótipo a partir de MO knockdown, mas faz presente com ARNm WT, experimentação adicional pode ocorrer, WT ARNm humano deve ser titulada para minimizar o fenótipo, e também pode ser utilizado como um novo ponto de referência. Além disso coinjection do WT e mutantes mRNA humano pode ser avaliado com base na capacidade de resgate do mutante (caixa rosa).et = "_blank"> Clique aqui para ver a figura maior.

Figura 2. Avaliação quantitativa e qualitativa de MKS1 mutações detectadas em humanos. Defeitos de desenvolvimento em embriões mks1 morphant. Com base na gravidade, fenótipos foram classificadas em três grupos. Os exemplos de cada classe são mostrados (um), e a sua prevalência na sua coorte embrião (n = 100-160 embriões) foram tabulados (não mostrado). MO injetado embriões com Classe I fenótipos tinha grosseiramente morfologia normal, mas eram mais curtas, com tecido embrionário excessiva sobre a gema em relação ao controle embriões injetados na mesma idade somítica somites (8-9). Morphants Classe II foram diluídos, curto e tinha pouco desenvolvido estruturas de cabeça e cauda, e, adicionalmente, faltava definição somíticae simetria. Embriões Classe III foram severamente atrasado com pouco desenvolvidas e disforme somites, ondulado notochords e, normalmente, não sobreviveram após o estágio de 10 somite. Co-injecção de ARNm humano MKS1 resgatado cada um destes defeitos, que demonstra a especificidade dos fenótipos para mks1 supressão. Hibridação in situ de embriões na fase de 11 somito (± 1 somito) corado com krox20, PAX2 e ribossondas MyoD (b, c ). Os fenótipos foram quantificados através de medições a partir do primeiro para o último somito apreciável de cada embrião (setas), quantificado em c. Figura adaptado com permissão de 15.

Figura 3. Exemplos de modelagem em vivo de dismorfologia humano. (A) dismorfologia Craniofacial. MRNA controle injetado embrião (esquerda) e embrião injetado mutante (direita) coradas com Alcian blue em 5 dpf. Mutantes embriões mRNA injetados exibir nomeadamente as pequenas e disforme cabeça com uma desorganização geral do esqueleto craniofacial cartilaginoso incluindo abertas arcos branquiais e estruturas em falta ou mal formados. (B) Micro / macrocefalia. Controle uninjected embrião (esquerda) e kctd13 embrião MO-injetado (direita) a 5 dpf. Morphants exibir alargamento da cabeça, como se vê pelo espaço entre os olhos. 21 (c), a integridade vascular reduzida. Controle uninjected embrião (em cima) e eng embrião MO-injetado (em baixo) fotografada usando microscopia de fluorescência em 2 dpf em um FLI1: eGFP linha repórter transgênico. Morphants exibir prejudicada surgimento de vasos intersegmentares e outras estruturas vasculares. 41 (d) looping coração Altered. Hibridização in situ de uniembriões de tipo selvagem njected (à esquerda) mostra a expressão spaw na mesoderme lateral esquerdo, enquanto ccdc39 embriões mostraram morphant bilateral (direita), ou, na maioria dos casos, a expressão indetectável spaw (não mostrado). 43 (E) cistos nos rins. Embrião WT uninjected (em cima) e ift80 morphant (inferior). Morphants exibida grandes cistos renais (seta), edema de pericárdio (seta) e uma cauda enrolada. 44 (f) filtração glomerular reduzida. Visualização fluorescente de um embrião de controlo injectado (em cima) e ift80 morphant (inferior) 24 horas após a injecção de dextrano rodamina no coração. Dispersões de fluorescência ao longo do sistema vascular e é quase completamente evacuada pelos rins como pode ser visto a completa ausência de fluorescência do controlo. O morphant exibe persistente dextrano fluorescente, sugerindo diminuição da filtração glomerular. 46 (g), distrofia muscular. WT DNAJB6 mRNA embriões injetados (superior) mostram fibras musculares lentas normais abrangendo os somites normalmente entre myosepta adjacente conforme determinado pela imunomarcação com o anticorpo anti-miosina lenta. Mutant DNAJBb (inferior) mostrou parcial para completar desprendimento de fibras musculares de myosepta em um ou vários somites. 19 (h) ângulo somite. Magnified vista lateral ao vivo de controle uninjected (em cima) ou kif7 morphants (baixo) fotografada em 30 hpf. Morphants exibir somites anormalmente forma, atribuíveis ao ectópica Hedgehog sinalização no myotome zebrafish 47.
Todos os valores adaptado com permissão.
Discussão
Os métodos aqui descritos representam um protocolo geral aplicável para o ensaio de mudanças nonsynonymous associados com uma variedade de fenótipos de doenças genéticas humanas (Tabela 2, Figura 3). Nossas abordagens provaram ser úteis para avaliar o potencial impacto da variação fenótipos da doença, e para ajudar a dissecar mecanismos da doença (como a contribuição de mutações dominantes negativas para a Síndrome de Bardet-Biedl, uma doença autossômica recessiva, principalmente 17). Até à data, por meio do desenvolvimento da árvore de decisão apresentado, foram modelados a um custo razoável e tempo em excesso de 200 genes causalmente associada com desordens genéticas, a um excesso de 1000 alelos.
Apesar de não ser discutido em detalhe aqui, também têm demonstrado que estes métodos são apropriados para modelar outros tipos de lesões genéticas, como variantes do número de cópia (CNVs), assim como genética e interações. As análises de tais eventos são além do escopoDescrição da presente método, embora eles dependem fundamentalmente o mesmo princípio do teste sistemático de genes candidatos (incluindo os pares de genes injectados simultaneamente) para determinar a indução ou a exacerbação dos fenótipos clinicamente apropriados. Por exemplo, para elucidar quais dos 29 genes na 16p11.2 CNV pode ser relevante para o microcefalia observada observada nos pacientes com a duplicação de um segmento do genoma de 660 kb, mRNAs correspondentes a cada um dos 29 genes dentro do segmento da cabeça e foram injectados medidas de tamanho foram realizadas em dois dpf e 5 dpf, revelando uma grande contribuição de um único transcrito, KCTD13. 21 Além disso, temos usado este modelo para interações genéticas ensaio de lesões genômicas em pacientes com ambos síndrome de Bardet-Biedl e doença de Hirschsprung. 22 Por comparação de MO supressão dos genes causadores de duas identidades clínicos separadamente e simultaneamente, fomos capazes de identificar o fenótipo resultante como being de uma interacção sinérgica e não meramente aditivo gravidade.
Apesar de ter estabelecido alta sensibilidade (98%) e especificidade (> 82%) para as variantes que contribuem para ciliopathies 17, nós ainda não temos dados suficientes para determinar se estes são generalizáveis a todas as leituras fenotípicos em modelos de zebrafish. Para fazer isso, um grande número de alelos, previu geneticamente para ser benignos ou patogénicos, deve ser ensaiado, dentro de cada categoria fenotípica. Isto será particularmente importante para a implementação de tais ensaios em ambiente clínico, onde a interpretação funcional de VUSs pode informar o diagnóstico e tratamento somente se um entendimento robusto de falsos positivos e falsos negativos pode acompanhar a entrega de tais resultados aos médicos e pacientes. No entanto, estes métodos podem contribuir significativamente para uma melhor compreensão da paisagem de doença genética humana. Prevemos que esses modelos não só servir como uma fundadação para melhorar a interpretação da informação genética clínica, mas irá também ser empregues como modelos úteis para realizar telas terapêuticos. Os dados in vivo também pode ser comparado com in silico previsões computacionais a partir de fontes tais como PolyPhen 23, peneire 24, os SNPs & GO 25, ou MutPred 26 para mostrar concordância. Note-se que em um estudo anterior de previsão bancos de dados SNPs & GO e MutPred foram encontrados para ser mais preciso, com precisões atingindo apenas 0,82 e 0,81, respectivamente 27.
Embora tenhamos descrito a robustez destes métodos para um subconjunto de defeitos anatómicos pediátricos (Tabela 2, Figura 3), certos fenótipos são menos tratável por estes métodos. Algumas exceções, não obstante, existem três classes principais de distúrbios não passíveis de nosso protocolo. Desordens de início adulto (tais como a doença de Parkinson), representam um desafio para modelo num sistema embrionário. Lento progression fenótipos degenerativas (tais como demência fronto-temporal) pode necessitar de mais tempo do que a janela dpf sete da actividade MO para produzir um fenótipo. Outras tecnologias de genes tais como o knockdown de RNAi e siRNA estão disponíveis para interferir com ou degradam o gene alvo, mas foi mostrado que nenhum é tão específica, estável, não-tóxico, ou de longa duração como OMs 28, contribuindo assim para limitar o horizonte temporal para fenotipagem. Em terceiro lugar, algumas estruturas de vertebrados, tais como o pulmão de mamífero, não têm uma estrutura suficientemente ortólogos no embrião do peixe-zebra. Temos também um plano de contingência sugerido para investigação de casos em que WT injeção de mRNA humano leva a um fenótipo, embora nós advertimos esta é uma situação incomum e indesejável.
Alguns fenótipos da doença pode então exigir um maior grau de abstração e de sub-rogação. É possível que a função do gene divergiu suficientemente para enfraquecer a semelhança fenotípica entre modelo e true fenótipo, ou fisiologia do peixe-zebra que inerentemente complica os efeitos da doença induzida. Nesses casos, sugerimos posterior dissecação do fenótipo produzido antes da demissão. Nós produzimos alguns exemplos bem sucedidos em que os fenótipos problemáticas para este ensaio foram modelados em embriões de peixe-zebra. Por exemplo, as mutações em TCF8, um gene associado com a distrofia de Fuchs córnea (FCD), foram testadas usando o nosso protocolo usando defeitos gastrulação como uma leitura fenotípica substituto base nas funções conhecidas desta transcrição no início do desenvolvimento 29 Em outros casos, por exemplo. como a distrofia muscular em adultos causada por mutações em DNAJB6, fomos capazes de gerar fenótipos myofiber em embriões 5dpf apesar do fato de que os seres humanos são desprovidos de massa muscular apreciável patologia em seus primeiros três a quatro décadas de vida. 19
Além dos modelos aqui apresentados mutantes transientes, outros também tomaram vantage deste sistema transiente para modelar doenças humanas numa variedade de sistemas do corpo. Num exemplo, retinite pigmentosa foi modelada em peixes-zebra por o knockdown da RP2 gene, resultando em morte celular e diminuição da retina laminação da retina. Recuperação com ARNm humano de tipo selvagem resultou no desenvolvimento de todas as três camadas da laminação da retina, enquanto que quatro de cinco ARNm mutantes não conseguiram resgatar 30. Embora este modelo de um distúrbio sensorial humana baseia-se num fenótipo morfológico, também é possível ensaio resposta a estímulos tais como alarme sonoro ou inibição pré-pulso 47.
Recentemente, um modelo de peixe-zebra foi utilizada para investigar a patogênese da doença de Alzheimer através da proteína precursora de amilóide. 31 Os autores mostraram que o gene knockdown causado prejudicada crescimento axonal dos neurônios motores, que poderiam ser resgatados com mRNA humano. Esse modelo provou ser especialmente informativos, como modelos de mouse exibir apenas phenot sutilIPOS (único knockdown) ou letalidade pós-natal (double knockdown). A capacidade de avaliar os embriões de peixe-zebra in vivo ao longo do desenvolvimento ajudou a discernir o efeito patogénico de redução da proteína precursora amilóide, bem como a prova directa, desde que a proteína requer tanto um domínio extracelular e intracelular para um funcionamento adequado. Outros modelos notáveis incluem o de distrofias musculares adicionais 32, Diamante Blackfan anemia 33, síndrome Axenfeld-Reiger (desenvolvimento ocular e craniofacial) 34, doença inflamatória intestinal 35 (atividade antibacteriana), doença de Parkinson 36 (neurônio e perda de locomoção), e apreensão de 37 (hidrocefalia e hiperatividade).
O mais comum são as linhas mutantes de peixe-zebra que foram mostrados também recapitular um fenótipo da doença humana. Avaliado em 1,38, os modelos incluem leucemia, melanoma, cardiomiopatias dilatadas, a distrofia muscular de Duchenne,e muitos outros.
Divulgações
Os autores declaram que não têm interesses financeiros concorrentes.
Agradecimentos
Reconhecemos o apoio do Verão Research Fellowship a Duke University Dean (AN), American Heart Association (AHA) comunhão 11POST7160006 (CG), National Institutes of Health (NIH) concede R01-EY021872 do National Eye Institute (EED), R01HD04260 do Instituto Nacional de Saúde Infantil e Desenvolvimento (NK), R01DK072301 e R01DK075972 do Instituto Nacional de Diabetes e Doenças Digestivas renais (NK), e da União Europeia (Financiado pelo 7 º Programa-Quadro da UE sob GA nr 241955, projeto SYSCILIA;. EED, NK) NK é um Distinguished Jean e George W. Brumley Professor.
Materiais
| Name | Company | Catalog Number | Comments |
| Reagent | |||
| Phusion High-Fidelity DNA Polymerase | NEB | M0530S, M0530L | |
| DpnI restriction endonuclease | NEB | R0176L, R0176S | |
| Max Efficiency DH5α competent cells | Invitrogen | 18258-012 | |
| Big Dye Terminator | Applied Biosystems | 4337455 | |
| mMESSAGE mMACHINE Kit | Invitrogen | AM1340, AM1344, AM1348 | |
| Morpholino | Gene-Tools | n/a | |
| 1-phenyl-2-thiourea (PTU) | Sigma Aldrich | P7629 | Prepare as 0.003% PTU in embryo media |
| Paraformaldehyde (PFA) | Sigma Aldrich | P6148 | For embryos that must be fixed prior to phenotyping, prepare as 4% |
| Tricaine methane sulfonate | Western Chemical | N/A | For anesthetization and euthanasia |
| Equipment | |||
| PTC-225 Tetrad Thermal Cycler | BioRad | Any equivalent thermal cycler | |
| Nano Drop 2000 spectrophotometer | Thermo Scientific | ||
| SMZ 745T Stereomicroscope | Nikon | ||
| AZ100 Stereomicroscope | Nikon | ||
| DS Fi1 Digital Camera | Nikon | For color/fluorescent imaging | |
| DS QiMC Digital Camera | Nikon | For black/white imaging | |
| Advanced Resarch 3.2 Imaging Software | NIS- Elements | ||
Referências
- Lieschke, G. J., Currie, P. D. Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353-367 (2007).
- Nolan, T., Hands, R. E., Bustin, S. A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559-1582 (2006).
- Thisse, C., Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59-69 (2008).
- Detrich, H. W., Westerfield, M., Zon, L. I. Overview of the Zebrafish system. Methods Cell Biol. 59, 3-10 (1999).
- Gerdes, J. M., Davis, E. E., Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 137, 32-45 (2009).
- Hildebrandt, F., Benzing, T., Katsanis, N. Ciliopathies. N. Engl. J. Med. 364, 1533-1543 (2011).
- Zaghloul, N. A., Katsanis, N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 119, 428-437 (2009).
- Marion, V. Exome sequencing identifies mutations in LZTFL1, a BBSome and smoothened trafficking regulator, in a family with Bardet-Biedl syndrome with situs inversus and insertional polydactyly. J. Med. Genet. 49, 317-321 (2012).
- Otto, E. A. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat. Genet. 42, 840-850 (2010).
- Schaefer, E. Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur. J. Med. Genet. 54, 157-160 (2011).
- Katsanis, N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 13 Spec No 1, R65-R71 (2004).
- Badano, J. L. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 439, 326-330 (1038).
- Badano, J. L. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 12, 1651-1659 (2003).
- Gerdes, J. M. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 39, 1350-1360 (2007).
- Leitch, C. C. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 40, 443-448 (2008).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 203, 253-310 (1995).
- Zaghloul, N. A., et al. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 10602-10607 (2010).
- Davis, E. E. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189-196 (2011).
- Sarparanta, J., et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450-455 (2012).
- Selcen, D. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Annals of neurology. 65, 83-89 (2009).
- Golzio, C., et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 485, 363-367 (2012).
- de Pontual, L., et al. Epistasis between RET and BBS mutations modulates enteric innervation and causes syndromic Hirschsprung disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13921-13926 (2009).
- Adzhubei, I. A., et al. A method and server for predicting damaging missense mutations. Nature Methods. 7, 248-249 (2010).
- Kumar, P., Henikoff, S., Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073-1081 (2009).
- Calabrese, R., Capriotti, E., Fariselli, P., Martelli, P. L., Casadio, R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30, 1237-1244 (2009).
- Li, B., et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 25, 2744-2750 (2009).
- Thusberg, J., Olatubosun, A., Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 32, 358-368 (2011).
- Summerton, J. E. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 7, 651-660 (2007).
- Riazuddin, S. A., et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am. J. Hum. Genet. 86, 45-53 (2010).
- Shu, X., et al. Knockdown of the zebrafish ortholog of the retinitis pigmentosa 2 (RP2) gene results in retinal degeneration. Investigative Ophthalmology & Visual Science. 52, 2960-2966 (2011).
- Song, P., Pimplikar, S. W. Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PloS one. 7, e34209 (2012).
- Kawahara, G., Guyon, J. R., Nakamura, Y., Kunkel, L. M. Zebrafish models for human FKRP muscular dystrophies. Hum. Mol. Genet. 19, 623-633 (2010).
- Danilova, N., Sakamoto, K. M., Lin, S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 112, 5228-5237 (2008).
- Bohnsack, B. L., Kasprick, D. S., Kish, P. E., Goldman, D., Kahana, A. A zebrafish model of axenfeld-rieger syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Investigative Ophthalmology & Visual Science. 53, 7-22 (2012).
- Oehlers, S. H., et al. The inflammatory bowel disease (IBD) susceptibility genes NOD1 and NOD2 have conserved anti-bacterial roles in zebrafish. Disease Models & Mechanisms. 4, 832-841 (2011).
- Sheng, D., et al. Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS genetics. 6, e1000914 (2010).
- Teng, Y., et al. Loss of zebrafish lgi1b leads to hydrocephalus and sensitization to pentylenetetrazol induced seizure-like behavior. PloS one. 6, e24596 (2011).
- Santoriello, C., Zon, L. I. Hooked! Modeling human disease in zebrafish. J. Clin. Invest. 122, 2337-2343 (2012).
- Javidan, Y., Schilling, T. F. Development of cartilage and bone. Methods Cell Biol. 76, 415-436 (2004).
- Lawson, N. D., Weinstein, B. M. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev. Biol. 248, 307-318 (2002).
- Lee, N. Y. Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Molecular Biology of the Cell. 23, 2412-2423 (2012).
- Huang, C. J., Tu, C. T., Hsiao, C. D., Hsieh, F. J., Tsai, H. J. Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 228, 30-40 (2003).
- Merveille, A. C., et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 43, 72-78 (2011).
- Beales, P. L. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat. Genet. 39, 727-729 (2007).
- Drummond, I. A., Davidson, A. J. Zebrafish kidney development. Methods Cell Biol. 100, 233-260 (2010).
- Tobin, J. L., Beales, P. L. Restoration of renal function in zebrafish models of ciliopathies. Pediatr. Nephrol. 23, 2095-2099 (2008).
- Putoux, A. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat. Genet. 43, 601-606 (2011).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados