
Method Article
ADN-ARN simultánea extracción de los sedimentos costeros y cuantificación de 16S rRNA genes y las transcripciones de PCR en tiempo real
En este artículo
Resumen
A protocol to quantify bacterial 16S rRNA genes and transcripts from coastal sediments via real-time PCR is provided. The methodology includes the co-extraction of DNA and RNA; preparation of DNA-free RNA; and 16S rRNA gene and transcript quantification via RT-q-PCR, including standard curve construction.
Resumen
Tiempo real de reacción en cadena de polimerasa también conocida como PCR cuantitativa (q-PCR) es una herramienta ampliamente utilizada en la ecología microbiana de cuantificar la abundancia de genes de grupos taxonómicos y funcionales en muestras ambientales. Utilizado en combinación con una reacción de transcriptasa inversa (RT-q-PCR), también se puede emplear para cuantificar la transcripción de genes. q-PCR hace uso de la química de detección fluorescentes altamente sensibles que permiten la cuantificación de los amplicones de PCR durante la fase exponencial de la reacción. Por lo tanto, se evitan los sesgos asociados con PCR 'punto final' detectados en la fase de meseta de la reacción de PCR. Un protocolo para cuantificar bacterianas 16S rRNA genes y las transcripciones de los sedimentos costeros a través de tiempo real se proporciona PCR. En primer lugar, un método para la co-extracción de ADN y ARN a partir de sedimentos costeros, incluyendo los pasos adicionales requeridos para la preparación de ADN libre de ARN, se describe. En segundo lugar, una guía paso a paso para la cuantificación de los genes 16S rRNA y transcripts de los ácidos nucleicos extraídos a través de Q-PCR y RT-Q-PCR se describen. Esto incluye los detalles de la construcción de ADN y ARN estándar de curvas. se incluyen consideraciones clave para el uso de ensayos de RT-Q-PCR en ecología microbiana.
Introducción
Los microorganismos son la piedra angular de la función de los ecosistemas de conducción biosfera. La mayoría de los microorganismos permanecen uncultured 1. Por lo tanto, los enfoques basados moleculares son fundamentales para avanzar en nuestra comprensión de la diversidad y la función de los microorganismos en el medio ambiente. Central de estos enfoques es la extracción de ácidos nucleicos a partir de muestras ambientales y la posterior amplificación de los genes diana usando la reacción en cadena de la polimerasa (PCR).
El primer paso de la extracción de ADN / ARN objetivo para la lisis de las paredes celulares de la comunidad microbiana presente, eliminar las moléculas de ácido nucleico no deseadas (sustancias por ejemplo, orgánicos e inorgánicos) y retener de ADN / ARN en solución para análisis aguas abajo. Entre varias opciones disponibles en la literatura 2,3,4,5, incluyendo una gama de kits de extracción comercial, el método Griffiths 6 se emplea extensamente 7.8.9. Es rentable y particularly bien adaptado a los sedimentos ya que utiliza un paso de cordón vencer para lisar las células e incorpora medidas para minimizar la co-extracción de inhibidores de la PCR, tales como ácidos húmicos, mientras que se recupera el ADN y el ARN.
El segundo paso se utiliza la reacción en cadena de la polimerasa (PCR) para amplificar los genes diana, tales como el marcador taxonómico 16S rRNA, a partir de los ácidos nucleicos extraídos. Este enfoque ha sido y sigue para facilitar la exploración de la caja de negro no caracterizado microbiana 10,11. Sin embargo, los métodos de punto final basado en la PCR sufren de varias limitaciones que pueden sesgar la caracterización de comunidades microbianas 12. Para cuantificar con precisión la abundancia de genes / transcripción, PCR en tiempo real, también conocido como PCR cuantitativa (qPCR) debe ser utilizado. qPCR explota sistemas de colorantes fluorescentes reportero que rastrean la acumulación de amplificación después de cada ciclo de la PCR. Esto es significativo ya que significa que la cuantificación puede ocurrir durante la fase exponencial temprana, en vezque el punto final, la fase de la reacción de PCR, cuando el rendimiento del amplicón sigue siendo directamente proporcional a la abundancia inicial del gen diana.
Dos sistemas indicadores se utilizan comúnmente: una mancha de ácido nucleico de intercalación 13 y la 5 actividad '3' exonucleasa de la polimerasa de DNA 14. Desde el antiguo sistema informador se une indistintamente a todo el ADN de doble cadena, que puede dar lugar a una sobreestimación de la secuencia diana si se producen amplicones no específicos no deseados o dímeros de cebadores de productos. Con el fin de evitar esto, puede ser necesaria una amplia optimización de la amplificación. En este último sistema, la amplificación de la plantilla se realiza un seguimiento mediante una combinación de una actividad 5 'nucleasa de la polimerasa Taq que escinde un fluoróforo de una sonda interna. Esta característica aumenta la especificidad del ensayo debido a la utilización de una sonda fluorogénico que se une sólo a la sequenc específica de la diana complementariae entre el par de cebadores. Con las dos químicas de cuantificación se logra mediante la determinación del punto de cruce (Cp), donde la acumulación de amplicones de PCR, tal como se mide por un aumento en la fluorescencia, es significativamente por encima de la fluorescencia de fondo.
qPCR se ha utilizado ampliamente en la ecología microbiana para determinar la abundancia de genes en diferentes ambientes 15. Por otra parte, la transcripción inversa del ARN en ADNc se combina con qPCR y RT-qPCR para cuantificar la expresión génica. Por lo tanto, qPCR y RT-qPCR representan métodos rápidos y eficaces para la cuantificación de números de genes y / o de transcripción dentro de las muestras ambientales.
Los microorganismos en los sedimentos costeros en coche varios procesos de los ecosistemas, incluyendo la mineralización de la materia orgánica, la degradación de los contaminantes y el ciclo biogeoquímico de los macronutrientes como el nitrógeno 16,17,18. La comprensión exhaustiva de estas transformaciones requiere una amplia account de las poblaciones microbianas que contribuyen, incluyendo datos cuantitativos sobre la transcripción de genes y la abundancia. Aquí presentamos una serie de protocolos probado a fondo y simplificados y normalizados para la cuantificación de bacterias transcripción de genes y la abundancia de 16S rRNA en los sedimentos costeros. El protocolo describe la recogida de muestras, el ADN simultánea y la extracción de RNA, la preparación de ARN libre de ADN, control de calidad de ácidos nucleicos extraídos, generación de 16S rRNA-DNA y las normas -ARN y cuantificación de muestras ambientales. Se necesitan datos cuantitativos derivados de los métodos descritos aquí para arrojar luz sobre las comunidades microbianas que impulsan los ecosistemas costeros.
Protocolo
1. Extracción de ADN y ARN de los sedimentos marinos costeros
- Preparación de la ribonucleasa (ARNasa) y el material exento de espacio de trabajo
- Preparar agua destilada libre de ARNasa (dH 2 O) por tratamiento con 0,1% de pirocarbonato de dietilo (DEPC) y se incuba a 37 ° CO / N. Retire DEPC residual mediante tratamiento en autoclave de dH2O a 121 ° C durante 15 minutos. Uso tratada con DEPC H2O destilada para hacer todas las soluciones.
- Hornear el material de vidrio a 180 ° CO / N. Retire las tapas de plástico y llantas, en remojo en una solución de NaOH 2 M S / N, y enjuagarlos con DEPC tratada dH2O antes de su uso.
- Preparar una solución de tampón CTAB-fosfato según la Tabla 1.
- Preparar la solución de precipitación con PEG-NaCl según la Tabla 2.
- Limpiar todas las superficies de trabajo (es decir, banco, microcentrífuga, humo del capó) y micropipetas utilizando solución de RNasa descontaminar antes de comenzar. Use guantes durante todo el procedimiento.
- Utilice recipientes de plástico libre de ARNasa, junto con las puntas de filtro micropipeta para evitar la contaminación cruzada de la muestra.
- recogida de muestras del medio ambiente y de almacenamiento
- Recoger los sedimentos de la costa durante la marea baja usando un tubo Falcon de 50 ml estéril desde los 2,5 cm superiores. Recoger aproximadamente 15 a 20 g de sedimentos. Cierre con cuidado la tapa después de la recogida de muestras. El transporte inmediato al laboratorio en un refrigerador a 4 ° C para su procesamiento inmediato.
- Homogeneizar la muestra mediante la mezcla con una espátula estéril y alícuota 0,5 g de sedimento de peso en húmedo en un tubo de cordón de golpeo 2 ml. Alícuotas repeticiones adicionales, flash-congelación mediante la colocación en nitrógeno líquido. almacenar alícuotas a -80 ° C. Precaución: Use guantes y gafas de seguridad al manejar nitrógeno líquido.
Nota: Si el sitio campo no está situado cerca de un laboratorio, alícuota de 0,5 g de sedimento en 2 ml tubos de microcentrífuga estériles en el sitio en el campo. Flash-congelar los tubos al ponerlos inmediatamente en liquid de nitrógeno y el transporte al laboratorio. Almacene las muestras a -80 ° C hasta su procesamiento.
- Co-Extracción de ADN y ARN a partir de sedimentos
Nota: El siguiente protocolo es una versión modificada del método de Griffiths 6.- Añadir 500 l de tampón CTAB-fosfato y 500 fenol l: cloroformo: alcohol isoamílico (25: 24: 1) al tubo de lisis de talón 2 ml que contiene 0,5 g de sedimentos e invertir el tubo 5-10 veces para homogeneizar la muestra.
Precaución: Realice este paso en una campana de humos utilizando equipo protector adecuado (es decir, bata de laboratorio, guantes y gafas de seguridad) en todo momento. - Vortex a toda velocidad durante 2,5 minutos. Centrifugar a 16.000 xg durante 10 min a 4 ° C.
- En un humo del capó se extrajo la fase acuosa superior y traslado a un nuevo tubo de microcentrífuga de 1,5 ml estéril. Mantener las muestras en hielo, añadir 500 l de cloroformo enfriado con hielo: alcohol isoamílico (24: 1).
- Invertir varias veces hasta que una emulsión es visibLe. Centrifugar durante 10 min a 16.000 xg a 4 ° C.
- En un humo del capó se extrajo la fase acuosa superior y colocarlo en un nuevo tubo estéril de 1,5 ml. Precipitar los ácidos nucleicos mediante la adición de dos volúmenes de solución de 30% PEG- NaCl y mezclar bien.
- Incubar en hielo durante 2 hr o salir de muestras a 4 ° CO / N.
- Al final de las muestras de centrífuga de incubación a 16.000 xg durante 20 min a 4 ° C.
Nota: Un pellet puede ser visible en la parte inferior del tubo. - Retire con cuidado el sobrenadante utilizando una micropipeta, asegúrese de no perturbar el sedimento. Deja unos 10 l de solución de PEG en el tubo y añadir 1 ml de helado de etanol al 70%.
- Centrifugar durante 20 min a 16.000 xg a 4 ° C. Retire lentamente etanol con una micropipeta teniendo cuidado de no tocar el pellet.
- Se centrifuga durante 5 segundos y eliminar el etanol residual utilizando una micropipeta. Deja que el precipitado se seque al aire durante aproximadamente 5 minutos.
- Vuelva a suspender el precipitado en 50 l DEPC tratosH2O Examinar la calidad y el rendimiento de ADN / ARN mediante electroforesis en gel de agarosa correr 5 l en un gel de agarosa al 1% 19. Dividir los ácidos nucleicos en dos alícuotas, 15 l de ADN y 30 l de RNA.
- Almacenar el ADN a -80 ° C hasta que se necesite. Aislar el ARN como se describe en la siguiente sección.
- Añadir 500 l de tampón CTAB-fosfato y 500 fenol l: cloroformo: alcohol isoamílico (25: 24: 1) al tubo de lisis de talón 2 ml que contiene 0,5 g de sedimentos e invertir el tubo 5-10 veces para homogeneizar la muestra.
2. Preparación de ARN y la comprobación de la calidad del ADN libre de ARN
- La digestión de DNA
- Añadir 3 l de tampón de ADNasa I y 1,5 l de ADNasa I de 30 l de volumen de los ácidos nucleicos, se incuba a 37 ° C durante 30 minutos.
- Mezclar el reactivo de ADNasa Inactivación por agitación durante 30 segundos. Añadir 4,8 l de la solución a la muestra. Incubar a TA durante 5 min, de vez en cuando toque el fondo de los tubos para mezclar la solución.
- Centrifugar a 10.000 xg durante 90 segundos a temperatura ambiente, se transfiere el sobrenadante (ARN) en un nuevo tubo, teniendo cuidado de no transferir precipitado que puede inhibir reacciones posteriores.
- verificación de la calidad del ARN
- Añadir 2 l de ARN a 18 l de dH libre de RNAsa 2 O estéril para diluir 1:10.
- En las reacciones de PCR separadas, añadir 1 l de puro o dilución 1:10 de ARN a un tubo de PCR de 0,5 ml estéril. Añadir componentes de la reacción de PCR para amplificar el gen 16S rRNA según la Tabla 3. Incluir un (ej., ADN extraído de un cultivo bacteriano puro), un "PCR inhibidor de" control "positivo" (es decir, 1 l de ADN cultivo puro positiva y 1 l de ARN extraído limpio) y un control "negativo" de la estéril dH RNasa libre de 2 O.
- Ajuste el termociclador con los siguientes parámetros de amplificación: 95 ° C 5 min, (95 ° C 30 seg, 57 ° C 30 seg, 72 ° C 1 min) x 35 ciclos, 72 ° C 10 min.
- Ejecutar 10% del producto de PCR en un gel de agarosa al 1% a 85 V durante 40 min. Repetir el tratamiento con ADNasa I, si un producto de PCR se forma en la RN ambientalA muestras.
- Añadir 2 l de ARN a 18 l de dH libre de RNAsa 2 O estéril para diluir 1:10.
3. Generación de Primera cadena de ADNc a partir de ARN
- Añadir los reactivos como se indica en la Tabla 4 a una ARNasa / ADNasa libre de 0,2 ml tubo de PCR.
- Incubar la muestra a 65 ° C 5 min. Colocar en hielo durante al menos 1 min y luego se incuba a 25 ° C durante 10 min.
- Para el mismo tubo de añadir los reactivos listados en la Tabla 5.
- Incubar la muestra a 55 ° C durante 50 min. Inactivar la reacción a 72 ° C durante 10 min.
- ADNc almacenar a -20 ° C hasta su uso.
4. PCR cuantitativa
- Generación de patrones de ADN para qPCR
- Amplificar bacteriana secuencia 16S rRNA 20 (o cualquier otra secuencia de interés) a partir de ADN genómico extraído de un cultivo puro con los cebadores de PCR (q-1369F y 1492R 21).
- Se purifica el producto de PCR utilizando un kit comercial según las instrucciones del fabricante.
- Clonar la PCR purificadoproducto en un saliente 3 'T clonación sistema vector de acuerdo con las instrucciones del fabricante.
- Transformar el vector que contiene la PCR insertar en Escherichia coli JM109 células competentes de acuerdo con las instrucciones del fabricante.
- Plate 50 l de transformación Onto ampicilina (100 mg / ml), X-gal (20 mg / ml) y IPTG (0,5 mM) Luria Bertani placas de agar (triptona 10 g / L, extracto de levadura 5 g / L, NaCl 10 g / L, Agar 15 g / L). Incubar O / N a 37 ° C.
- Seleccione un solo transformante positivo blanco de la placa. Inocular el transformante positivo en 50 ml de caldo LB que contiene 100 mg / ml de ampicilina y se incuba O / N con agitación a 250 rpm a 37 ° C.
- A partir del cultivo / N O, aislar y purificar el plásmido utilizando un kit comercial según las instrucciones del fabricante.
- Linealizar el plásmido mediante un enzima de restricción apropiado que es capaz de cortar el plásmido una vez (por ejemplo, EcoRI). > Analizar la pureza del plásmido mediante la medición de la relación de absorción A 260 / A 280 19. Si la relación es inferior a 1,8, re-extraer el plásmido con fenol-cloroformo 19. Nota: DNA puro tiene una relación de 1,8.
- Determinar la concentración de ADN de plásmido mediante la absorbancia a 260 nm utilizando un espectrofotómetro 19.
Nota: Alternativamente, la precisión de la cuantificación puede ser mejorada mediante el uso de un colorante de unión a ADN fluorescente utilizado en conjunción con un fluorómetro. - Secuencia del plásmido de inserción para confirmar el tamaño del objetivo en las bases. Calcular la cantidad (es decir, porcentaje) de ADN del inserto en el plásmido (por ejemplo, en un vector de 3000 pb, un fragmento de ADN de 123 pb es 3,9% del ADN total) y se multiplica por la concentración obtenida en 4.1.10, para determinar la la concentración de ADN del objetivo (inserto).
- Utilice la siguiente ecuación para calcular las copias de la diana por l:
carga / 54067 / 54067eq1.jpg "/> - Calcular el objetivo Peso molecular (TMW) multiplicando el número de pares de bases del ADN diana por el peso molecular promedio de ADN de doble cadena (dsDNA, 660 Daltons por par de base).
- Diluir el ADN diana en un rango de 10 10 a 10 2 copias con 2 l de ADN y 18 l de dH estéril 2 O. Mezclar bien cada dilución y centrifugar brevemente antes de hacer la siguiente dilución.
- Generación de curva estándar de ARN para qPCR
- Desde el paso 4.1.5, pantalla un número de colonias de color blanco (aproximadamente 5) mediante PCR de colonias 19 usando el cebador inverso de destino (es decir, R1492) y el plásmido vector de cebador directo M13F.
Nota: Esta combinación de cebadores amplificará insertos clonados en la orientación sentido correcta con un sitio de promotor T7 aguas arriba. - Se purifica el producto de PCR utilizando un kit comercial siguiendo las instrucciones del fabricante. Cuantificar la PCR purificado pRODUCTO a 260 nm usando un espectrofotómetro.
- Añadir 200 ng de producto de PCR en un volumen final de 20 l con 7,5 mM de cada ribonucleótido, tampón de reacción 1 T7 y enzima polimerasa T7 1 para la reacción de transcripción in vitro.
- Incubar la reacción durante 4 horas a 37 ° C. Añadir 1 unidad de RNasa libre de DNasa I a la reacción, se incuba durante 15 minutos a 37 ° C para eliminar el molde de ADN.
- Recuperar el ARN transcrito in vitro por precipitación con etanol 19 y cuantificar el rendimiento de ARN usando un fluorímetro o mediante absorbancia a 260 nm.
- Calcular el número transcrito por l de ARN. Añadir el número de pares de bases en el ARN diana (por ejemplo, 123 bases) a la longitud del promotor de T7 (153 bases) y se multiplica por el peso molecular promedio de RNA, 340 Daltons por base.
- Añadir 500 ng de ARN objetivo cuantificado a una reacción de la transcriptasa inversa (RT) como se describe en la sección 3. En serie diluir el ADNc como se indica en 4.1.13 para su usocomo la curva estándar.
- Desde el paso 4.1.5, pantalla un número de colonias de color blanco (aproximadamente 5) mediante PCR de colonias 19 usando el cebador inverso de destino (es decir, R1492) y el plásmido vector de cebador directo M13F.
- Real-Time PCR
- ADN ambiental deshielo / cDNA muestras, estándares y reactivos qPCR en el hielo. Proteger la sonda fluorogénico de la luz.
- Diluir los estándares para la curva estándar como se indica en 4.1.14. Hacer diluciones 1:10 de ADN del medio ambiente / cDNA a partir ordenada a 10 -3 mediante la adición de 2 l de muestra a 18 l de dH 2 O. estéril
- Hacer una reacción mezcla maestra para el número total de reacciones más el 10% según la Tabla 6 (cebadores y la sonda) y la Tabla 7 (mezcla de reacción). Mezclar bien y centrifugar brevemente.
- Añadir 19 l de mezcla maestra y 1 l de plantilla (estándar, muestras del medio ambiente o el agua para el control negativo) a cada pocillo en una placa de 96 pocillos qPCR óptica. Realizar cada reacción (estándares, muestras ambientales y controles negativos) por triplicado.
- Cubrir la placa de 96 pocillos con una cubierta óptica q-PCR. Se centrifuga la placa brevemente. cargar elplaca en el bloque de calentamiento de la máquina qPCR y cierre la tapa.
- Abra el software gestor de qPCR. Haga clic en la pestaña "protocolo", haga clic en "Crear nuevo", añade las siguientes condiciones de amplificación: 3 min 95 ° C, (10 seg 95 ° C, 30 seg 60 ° C) x 40 ciclos. Ajustar el volumen de la muestra de 20 l, haga clic en "Aceptar" y guardar el protocolo.
- Haga clic en la pestaña "placa". En "Editar seleccionado" clic "seleccione fluoróforo", añade el colorante informe de sonda apropiada, por ejemplo, fluoresceína. Haga clic en Aceptar".
- Resaltar los pozos que contienen las normas, seleccione "estándar" en el menú "Tipo de muestra". Haga clic en "replicar serie" cambiar "replicar tamaño" a 3, haga clic en "aplicar". Haga clic en "serie de dilución" para añadir concentración inicial calculado para el estándar de 1, indicar el factor de dilución (10), seleccione "disminución" o "aumentar" en función de la orden se añadió la curva patróna la placa. Haga clic en "aplicar".
- Destaca también las posiciones que contienen las muestras desconocidas, seleccionar "desconocido" del "tipo de muestra" del menú desplegable. Haga clic en "replicar serie", y cambiar "replicar tamaño" a 3. Haga clic en "aplicar". Editar nombres de ejemplo en "nombre de la muestra". Repetir el procedimiento para No plantilla controles pozos (NTC).
- Haga clic en "Aceptar" en el cuadro de editor de plato y guardar el archivo. Haga clic en "Iniciar la ejecución".
- Al término de la carrera, la ventana de análisis de datos se abrirá. Haga clic en la pestaña de "cuantificación" para visualizar la curva estándar, descriptores estándar de la curva (Tabla 8) y gráficas de amplificación.
- Haga clic en la pestaña "datos de cuantificación" para los valores de Cp y la partida correspondiente cantidad de genes (SQ) de muestras desconocidas. tabla de exportación de software de hoja de cálculo, si es necesario.
- abundancias de genes se multiplican a partir de muestras desconocidas por el factor de dilución, la cantidad de sedimentos extracciado de los ácidos nucleicos y las de volumen total de se eluyeron en obtener copias de genes por gramo de peso húmedo del sedimento.
Nota: ADN eluyen de 0,5 g de sedimento en 50 l, diluir 1/10, añadir 1 l como molde para una reacción q-PCR; el cálculo de copias de genes g -1 sedimentos multiplicando: copias de genes l de ADN de dilución x volumen de elución del factor x 2 x.
Resultados
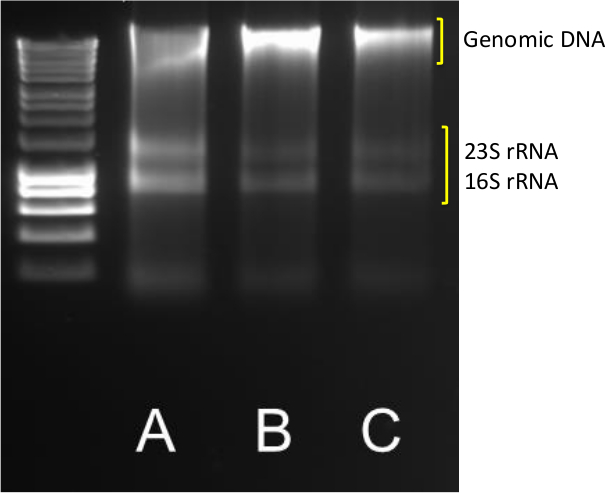
La extracción de ADN de buena calidad y el ARN de los sedimentos es el primer paso en la cuantificación de transcripción de genes y la abundancia. A exitosas rendimientos de extracción de grupos de ADN y ARN claras, como se indica en la Figura 1 para la muestra de CA, donde afilados bandas 23S y 16S rRNA son visibles, además de la banda de alto peso molecular de ADN genómico.

Figura 1. ADN / ARN de extracción. Los resultados típicos de la extracción de ADN / ARN a partir triplicado (ABC) 0,5 g sedimentos costeros. 5 l de ADN / ARN se ejecutan en un agarosa 1,4% a 85 V durante 40 min. Se utilizó un marcador molecular en el rango 10,037-200 pb. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Para preparar el ARN, una digestión del ADN co-extraído es obligatorio. Esto debe ir seguido de 16S rRNA punto final PCR del ARN para asegurar que el ADN se ha eliminado con éxito. Si el ADN se ha eliminado por completo sólo se observa una banda en el control positivo. Es importante utilizar tanto ordenada y una dilución 1:10 de ARN para asegurar inhibidores no impiden la formación de un producto de PCR. RNA ahora puede someterse a la reacción de la transcriptasa inversa para convertirlo en cDNA. Esto se puede realizar ya sea con cebadores específicos de genes o hexámeros aleatorios. Típicamente, la reacción se lleva a cabo en una máquina de PCR, para asegurar el perfil de temperatura óptima. Este cDNA se utilizó como molde en la reacción de qPCR posterior. ADN y ARN se cuantificaron para determinar la concentración de plantilla usado en cada reacción.
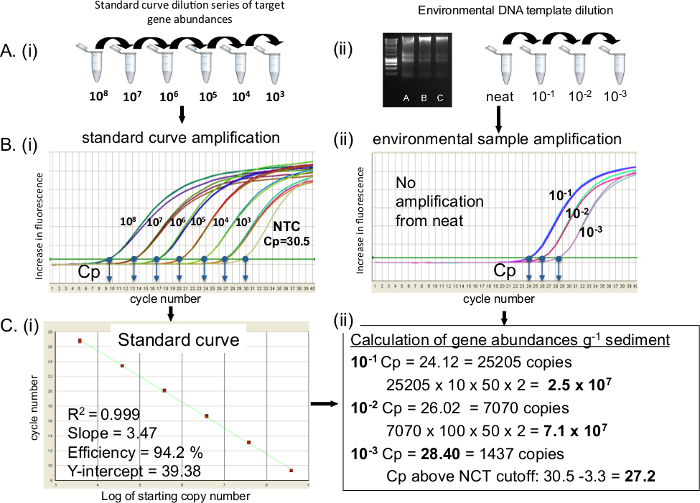
Un protocolo de reacción qPCR se perfila como objetivo ADN ARNr 16S. Por 16S rRNA transcripciones sustituir la norma cuRVE y plantilla con ADNc. Como muestras desconocidas se cuantifican en contra de la curva estándar, es imprescindible para asegurar que la curva estándar es de buena calidad. Figura 2 muestra la preparación de (A), las curvas de calibración y las diluciones de ADN del medio ambiente, (B), la amplificación de la curva estándar y muestras ambientales (C) conversión de la curva estándar a una regresión lineal y el cálculo del número de copias de genes. Cuando una serie de diluciones de 10 veces es correctamente preparado y amplifica la diferencia entre 3.3 Ciclo de cada dilución estándar que se ve (se tarda 3,3 ciclos para un incremento de diez veces en la plantilla en la eficiencia de amplificación 100%) (Figura 2Bi). Se desean curvas descriptores estándar, incluyendo el R 2 valores de 0,99 y eficiencia de la PCR en el intervalo de 90 a 110% (Figura 2C). Es importante reportar los valores Cp del control sin molde (NTC) si está presente. Si esto ocurre un corte de Cp para los estándares y las muestras desconocidas 3.3 ciclos ( es decir, un pliegue log) mayor que el valor Cp de la NTC se impone 20 (Figura 2Cii). Desconocido plantilla de ADN extraído de sedimento se cuantificó a partir de ordenado, 10 -1, 10 -2 y 10 -3 diluciones (B). La muestra pura no amplificó, los valores de Cp de 10 -1 a 10 -3 diluciones fueron 24.12, 26.02 y 28.40, respectivamente. El CNT Cp fue del 30,5, se impuso un punto de corte de 27,2 NCT. La conversión de la abundancia de genes de g-1 de peso húmedo del sedimento resultó en 2,5 x 10 7 y 7,1 x 10 7 10 -1 y 10 -2, respectivamente, para el intervalo de dilución. 10 -3 dilución Cp estaba por debajo del punto de corte NTC, y por lo tanto no se utilizó. En este caso, el 10 -2 fue seleccionada como la plantilla de dilución óptima.

Figura 2. 16S rRNA gen QPCR amplificación de ADN y las normas ambientales extraída de los sedimentos costeros. Preparación de (A) i) curva estándar de ADN y ii) diluciones de ADN del medio ambiente para la amplificación qPCR, (B) amplificación de qPCR de i) de la curva estándar de ADN y muestras ambientales ii), se muestran Cp para cada muestra. (C) i) de regresión lineal de la curva estándar con los descriptores de la curva estándar, ii) el cálculo de la abundancia de genes de muestras ambientales. NTC:. De Control Negativo Plantilla Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Reactivo | Cantidad |
| CTAB | 5 g |
| K 2 HPO 4 2 O x3H | 2,58 g |
| KH 2 PO 4 | 0,1 g |
| NaCl | 2,05 g |
| dH2O | hasta 100 ml |
Tabla 1. Preparación de tampón CTAB-fosfato.
| Reactivo | Cantidad |
| PEG 6000 | 30 g |
| NaCl | 9,35 g |
| dH2O | hasta 100 ml |
| Nota: añadir PEG 6000 lentamente bajo calentamiento moderado y la agitación de 50 ml de dH2O | |
Tabla 2. Preparación de la solución de precipitación con PEG-NaCl.
| Reactivo | Cantidad |
| ARN sin diluir / ARN diluido 1:10 | 1.0 l |
| 10x PCR Buffer | 5.0 l |
| dNTPs 10 mM | 1.0 l |
| cebador delantero 63F | 1.0 l |
| CAG GCC ATG TAA CAC GTC CAA | |
| cebador inverso 518R | 1.0 l |
| ATT ACC GCG TCG TCG GG | |
| Taq polimerasa | 0,4 l |
| DH libre de RNAsa 2 O | 40,6 l |
Tabla 3. calidad del RNA PCR master mix de verificación de ADN libre.
| Reactivo | Cantidad |
| ARN | 0,5-5 g |
| dNTPs 10 mM | 1 l |
| cebador inverso 10 M / M azar hexámeros 50 | 1 l |
| DEPC-H2O | Hasta 13 l |
Tabla 4. cDNA mezcla de reacción de síntesis A.
| Reactivo | Cantidad |
| Buffer 5x | 4 l |
| TDT | 1 l |
| inhibidor de ARNasa | 1 l |
| La transcriptasa inversa | 1 l |
Tabla 5. cDNA mezcla de reacción de síntesis B.
| Objetivo | nombre de imprimación | Secuencia | El recocido T (° C) | Referencia |
| bacterias 16S rRNA | Bact1369F | CGG TGA ATA CGT TCY CGG | 56 | Suzuki et al. 2000 19 |
| Prok1492R | GGW TAC CTT GTT ACG ACT T | |||
| TM1389F (sonda) | CTT GTA CAC CAC GCC CGT C |
| Reactivo | Cantidad |
| Mezcla 2x Maestro | 10 l |
| cebador directo (10 M) | 0,8 l |
| cebador inverso (10 M) | 0,8 l |
| Probe (10 M) | 0,4 l |
| Agua | 7,0 l |
| El volumen final | 19 l |
Tabla 7. tiempo real mezcla de reacción de PCR.
| descriptor | Significado |
| El coeficiente de correlación (R2) | Una medida de la linealidad de la curva estándar. Idealmente, debería ser R2 = 1. Valor de R 2 = 0,98-0,99 son aceptables. |
| Pendiente (α) | Una medida de la eficacia de la reacción. Idealmente, debería ser equivalente a -3,32. Valor en el rango entre -3,58 y -3,1. |
| Eficiencia (= 10 (-1 / pendiente) -1) | Si se trata de un 100%, las plantillas se duplica después de cada ciclo térmico durante la amplificación exponencial. Un buen rango de eficiencia es de entre 90 y 110%. |
| Y Intercepción (β) | El límite teórico de la detección de la reacción. Aunque no se utiliza para cuantificar la sensibilidad de reacción, es importante cuando se comparan diferentes curvas estándar del mismo objetivo. |
Tabla 8. QPCdescriptores de reacción R.
Discusión
La combinación de la extracción de ADN / ARN con qPCR proporciona un método rápido, preciso, relativamente rentable para la cuantificación sensible de la transcripción de genes y la abundancia de una serie de muestras ambientales, tales como sedimentos costeros.
La extracción de ácidos nucleicos inicial es el paso crítico para asegurar una visión representativa de la comunidad microbiana presente se logra 22. Una serie de limitaciones deben tenerse en cuenta para el protocolo de extracción: a) la consecución de la lisis celular total, b) co-extracción de compuestos inhibidores (por ejemplo, ácidos húmicos o polifenoles), c) la contaminación de ADN en la fracción de ARN, d) rápida degradación de los ácidos nucleicos extraídos cuando se almacena. Se deben tomar precauciones con el fin de evitar estas limitaciones. Por ejemplo, un cuidado particular tiene que ser tomada para asegurar que la extracción de ácidos nucleicos está optimizado para el tipo de muestra (por ejemplo, sedimentos, suelo, aguas residuales, etc.). Mejoras significativas en el rendimiento de ácido nucleico y la calidad, tanto para el ADN y el ARN se puede lograr mediante la realización de experimentos preliminares 23. Para minimizar el efecto de los compuestos inhibidores en aplicaciones basadas en PCR 24, ensayar una serie de diluciones a partir de ADN extraído y ARN. Para eludir la rápida degradación del ADN / ARN extraído de múltiples ciclos de congelación-descongelación y evitar la posible pérdida de información genética, almacenar múltiples alícuotas de pequeño volumen a -80 ° C.
Cuando se diseñan cuidadosamente, qPCR es un método robusto, altamente reproducible y sensible. En particular, los métodos para la amplificación y la construcción curva estándar descritos en este protocolo se pueden adaptar para cualquier objetivo gen de interés, incluyendo otros marcadores filogenéticos (es decir, 16S rRNA archaeal, 18S rRNA de hongos) o genes implicados en funciones importantes en el medio ambiente. Limitaciones conocidas en el uso de qPCR son: a) la generación de reproducible de alta calidad scurva stándar para la cuantificación absoluta, b) la elección de cebadores / sondas y optimización de las condiciones de ensayo Q-PCR, c) el uso de baja calidad / ácidos nucleicos cortados, d) la elección de la dilución de trabajo del ADN / ARN para evitar la inhibición . Por otra parte, hay que tener en cuenta que la técnica de PCR cuantitativa proporciona abundancia de genes / transcripción que no puede equiparar a la celda recuentos: Este es particularmente el caso cuando se dirigen los genes 16S y 18S rRNA, ya que los microorganismos tienen diferentes números de copias del gen ribosomal en su genoma 25.
curvas estándar de calidad pobres resultarán en la cuantificación inexacta del gen de interés. Es una buena práctica para almacenar una reserva de un alto nivel de concentración en pequeñas alícuotas a partir del cual se pueden hacer las curvas de calibración frescas. No guarde las curvas de calibración, siempre hacen diluciones frescas de un balance de la más alta concentración cada vez. Para la cuantificación exacta de las muestras ambientales asegurar el rango de concentraciones del StanDard curva se extiende por los valores esperados Cp de las muestras desconocidas. Al cuantificar las transcripciones, construir la curva estándar a partir de ARN no doble hebra de ADN. Cuando sea posible, la cuantificación de muestras que se van a comparar directamente debe ser completado dentro de un único ensayo para evitar la variación inter-ensayo 20. Esto no siempre es posible. Por lo tanto, para comparar la abundancia de genes generadas entre los ensayos, es aconsejable tener un azar sub-conjunto de muestras replicadas entre ensayos. Una amplia selección de conjuntos de cebadores y sondas están disponibles actualmente para qPCR focalización taxones microbianos y los grupos funcionales 15. Es necesario considerar cuidadosamente la hora de seleccionar estos para garantizar tanto la máxima cobertura y especificidad para el grupo objetivo. Si la eficacia de la reacción de un ensayo de qPCR no es satisfactoria, la solución de problemas comienza con la prueba de diferentes condiciones de ciclo térmico (como tiempo de recocido y / o la temperatura) y / o condiciones de reacción, por ejemplo, variando las concentraciones de cebador-sonda. Ona vez que se optimizan las condiciones de ensayo y de reacción Q-PCR, siempre llevar a cabo una prueba inicial de un intervalo de diluciones de ADN / ADNc para determinar la concentración de plantilla apropiado. Seleccione el rango de dilución resultante en el número de copias más alto como la dilución óptima plantilla para más ensayos.
En la actualidad, las tecnologías de secuenciación de próxima generación eficiente arrojan luz sobre la estructura de la comunidad microbiana y funciones en una plétora de ambientes 26,27,28. Sin embargo, estos conjuntos de datos se basan a menudo en el punto final bibliotecas amplificación de PCR y por lo tanto sólo proporcionan evaluaciones semi-cuantitativos de la abundancia de especies particulares. Por lo tanto, la capacidad de tiempo real técnica basada en PCR para apuntar marcadores taxonómicos específicos (de mayor dominio hasta el nivel de la tensión) permite la validación eficiente de los resultados obtenidos por secuenciación de próxima generación. Por otra parte, qPCR ha sido utilizado con éxito en combinación con otros métodos moleculares de la ecología microbiana tales como iso estableTope de sondeo (SIP) o microarrays filogenéticos / funcionales. Combinado con la primera herramienta, qPCR se puede utilizar para cuantificar la comunidad metabólicamente activo 29,30. Cuando se combina con el análisis de microarrays, qPCR ofrece una interpretación cuantitativa clave de las encuestas basadas en marcadores y genes funcionales filogenéticas de los entornos 31,32.
Por lo tanto, si se usa solo o en combinación con otras evaluaciones (a menudo basados en procesos) de la función del ecosistema, la PCR cuantitativa es una herramienta esencial para los ecologistas microbianos en la exploración de la relación entre las comunidades microbianas difícil de alcanzar y funciones de los ecosistemas.
Divulgaciones
The authors have nothing to disclose.
Agradecimientos
Esta publicación ha emanado de la investigación llevada a cabo con el apoyo financiero de Medio Ambiente del Consejo de Investigación Natural (NERC) bajo el número de concesión NERC NE / JO11959 / 1 y la Fundación Científica de Irlanda y la acción COFUND Marie-Curie subvención número 11 / GRIC / B2159awarded a CJS y el Consorcio de Investigación Académica del Este (ARC del Este).
Materiales
| Name | Company | Catalog Number | Comments |
| Diethylpyrocarbonate | Sigma | 40718 | Toxic, open under chemical hood |
| cetrimonium bromide (CTAB) | Sigma | 52365 | Irritant, open under chemical hood |
| potassium phosphate dibasic | Sigma | RES20765 | |
| potassium phosphate monobasic | Sigma | P9791 | |
| Phenol : Chloroform : Isoamylalcohol pH 8 | Sigma | P2069 | Equilibrate at pH 8 before using |
| Chloroform : Isoamylalcohol | Sigma | 25666 | |
| sodium chloride | VWR | 1.06404.0500 | |
| polyethylenglycol 6000 | VWR | 528877-100 | |
| Ethanol Molecular Grade | Sigma | E7148 | |
| Lysing Matrix E tubes | MP Biomedical | 116914050 | |
| Turbo DNAse | Ambion | AM1907 | |
| Taq polymerase | Sigma | D1806 | |
| dNTPs | Bioline | BIO-39028 | |
| SuperScript III | Life Technologies | 18080044 | |
| Rnase Inhibitor | Life Technologies | 10777019 | |
| RNAse/DNAse free 0.2 ml PCR tubes | Sarstedt | 72.985.002 | |
| SureCleanPlus | Bioline | BIO-37047 | |
| pGEM Easy T Vector | Promega | A1360 | |
| E. coli JM109 competent cells | Promega | L2005 | |
| Tryptone | Sigma | T7293 | |
| Yeast Extract | Sigma | 70161 | |
| Ampicillin | Sigma | 59349 | |
| X-Gal | Bioline | BIO-37035 | |
| IPTG | Bioline | BIO-37036 | |
| Agar | VWR | 20768.235 | |
| Plasmid Midi Kit | Qiagen | 12143 | |
| Qubit | Life Technologies | Q33216 | |
| Quant-IT DNA HS Assay | Life Technologies | Q-33120 | |
| Quant-IT RNA HS Assay | Life Technologies | Q32855 | |
| MEGAshortscript kit | Ambion | AM1354 | |
| TaqMan SensiFast Probe Lo-ROX kit | Bioline | BIO-84002 | |
| qPCR machine |
Referencias
- Epstein, S. S. The phenomenon of microbial uncultivability. Curr. Opin. Microbiol. 16 (5), 636-642 (2013).
- Zhou, J., Bruns, M. A., Tiedje, J. M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62, 316-322 (1996).
- Miller, D. N., Bryant, J. E., Madsen, E. L., Ghiorse, W. C. Evaluation and optimization of DNA extraction and purification procedures from soil and sediments samples. Appl. Environ. Microbiol. 65 (11), 4715-4724 (1999).
- Burgmann, H., Pesaro, M., Widmer, F., Zeyer, J. A strategy for optimizing quality and quantity of DNA extracted from soil. J. Microbiol. Methods. 45 (1), 7-20 (2001).
- Hurt, R. A., et al. Simultaneous recovery of RNA and DNA from soils and sediments. Appl. Environ. Microbiol. 67, 4495-4503 (2001).
- Griffiths, R. I., Whiteley, A. S., O'Donnell, A. G., Bailey, M. J. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl. Environ. Microbiol. 66 (12), 5488-5491 (2000).
- Martins, G., et al. Structure and activity of lacustrine sediment bacteria involved in nutrient and iron cycles. FEMS Microbiol. Ecol. 77 (3), 666-679 (2011).
- Kuffner, M., et al. Effects of season and experimental warming on the bacterial community in a temperate mountain forest soil assessed by 16S rRNA gene pyrosequencing. FEMS Microbiol. Ecol. 82 (3), 551-562 (2011).
- Tatti, E., et al. Influences of over winter conditions on denitrification and nitrous oxide-producing microorganism abundance and structure in an agricultural soil amended with different nitrogen sources. Agric. Ecosyst . Environ. 183, 47-59 (2014).
- Giovannoni, S. J., Britschgi, T. B., Moyer, C. L., Field, K. G. Genetic diversity in the Sargasso Sea bacterioplankton. Nature. 345, 60-62 (1990).
- Ward, D. W., Weller, R., Bateson, M. M. 16S rRNA sequences reveal numerous uncultured microorganisms a natural community. Nature. 345, 63-65 (1990).
- Suzuki, M. T., Giovannoni, S. J. Bias caused by template annealing in the amplification of mixture of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62, 625-630 (1996).
- Wittwer, C. T., Herrmann, M. G., Moss, A. A., Rasmussen, R. P. Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques. 22, 130-138 (1997).
- Holland, P. M., Abramson, R. D., Watson, R., Gelfand, D. H. Detection of Specific Polymerase Chain Reaction Product by Utilizing the 5' 3' Exonuclease Activity of Thermus aquaticus DNA Polymerase. PNAS. 88, 7276-7280 (1991).
- Smith, C. J., Osborn, A. M. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 67, 2-20 (2009).
- Park, S. J., Park, B. J., Rhee, S. K. Comparative analysis of archaeal 16S rRNA and amoA genes to estimate the abundance and diversity of ammonia-oxidizing archaea in marine sediments. Extremphiles. 12 (4), (2008).
- Urakawa, H., Yoshida, T., Nishimura, M., Ohwada, K. Characterization of depth-related population variation in microbial communities of a coastal marine sediment using 16S rDNA-based approaches and quinone profiling. Environ. Microbiol. 2, 542-554 (2008).
- Smith, C. J., Dong, L. F., Wilson, J., Stott, A., Osborn, A. M., Nedwell, D. B. Seasonal variation in denitrification and dissimilatory nitrate reduction to ammonia process rates and corresponding key functional genes along an estuarine nitrate gradient. Front. Microbiol. 6, 542(2015).
- Sambrook, J., Russell, D. W. Molecular Cloning: a laboratory manual. , Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. 205(2001).
- Smith, C. J., Nedwell, D. B., Dong, L. F., Osborn, A. M. Evaluation of Quantitative Polymerase Chain Reaction (Q-PCR) based approaches for determining gene copy and gene transcript numbers in environmental samples. Environ. Microbiol. 8 (5), 804-815 (2006).
- Suzuki, M. T., Taylor, L. T. DeLong E.F., Quantitative analysis of small-subunit rRNA genes in mixed microbial population via 5'-nuclease assays. Appl. Environ. Microbiol. 66, 4605-4614 (2000).
- Luna, G. M., Dell'Anno, A., Danovaro, R. DNA extraction procedure: a critical issue for bacterial diversity assessment in marine sediments. Environ. Microbiol. 8 (2), 308-320 (2005).
- Lever, M. A., Torti, A., Eickenbursch, P., Michaud, A. B., Šantl-Temkiv, T., Jørgensen, B. B. A modular method for the extraction of DNA and RNA, and the separation of DNA pools from diverse environmental sample types. Front. Microbiol. 6, 476(2015).
- Lloyd, K. G., MacGregor, B. J., Teske, A. Quantitative PCR methods for RNA and DNA in marine sediments: maximizing yield while overcoming inhibition. FEMS Microbol. Ecol. 72, 144-151 (2010).
- Klappenbach, J. A., Dumbar, J. M., Schmidt, T. M. rRNA Operon copy number reflects ecological strategies of bacteria. App. Environ. Microbiol. 66 (4), 1328-1333 (2000).
- Shi, Y., Tyson, G. W., Eppley, J. M., DeLong, E. F. Integrated metatrascriptomic and metagenomics analyses of stratified microbial assemblages in the open ocean. ISME J. 5, 999-1013 (2011).
- Howe, A. C., Jansson, J. K., Malfatti, S. A. Tringe S.G., Tiedje J.M., & Brown C.T. Tackling soil diversity with the assembly of large, complex metagenomes. PNAS. 111 (13), 4904-4909 (2014).
- Klaedtke, S., et al. Terroir is a key driver of seed-associated microbial assemblages. Environ. Microbiol. , (2015).
- Lueders, T., Wagner, B., Claus, P., Friedrich MW, Stable isotope probing of rRNA and DNA reveals a dynamic methylotroph community and trophic interactions with fungi and protozoa in oxic rice field soil). Environ. Microbiol. 6, 60-72 (2004).
- Kunapuli, U., Lueders, T., Meckenstock, R. U. The use of stable isotope probing to identify key iron-reducing microorganisms involved in anaerobic benzene degradation. ISME J. 1, 643-653 (2007).
- Bürgmann, H., et al. Transcriptional response of Silicibacter pomeroyi DSS-3 to dimethylsulfoniopropionate (DMSP). Environ. Microbiol. 9 (11), 2742-2755 (2007).
- He, J. Z., et al. Geochip: a comprehensive microarray for investigating biogeochemical ecological and environmental processes. ISME J. 1, 67-77 (2007).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados