
Method Article
Simultaneous DNA-RNA Extraction from Coastal Sediments and Quantification of 16S rRNA Genes and Transcripts by Real-time PCR
In This Article
Summary
A protocol to quantify bacterial 16S rRNA genes and transcripts from coastal sediments via real-time PCR is provided. The methodology includes the co-extraction of DNA and RNA; preparation of DNA-free RNA; and 16S rRNA gene and transcript quantification via RT-q-PCR, including standard curve construction.
Abstract
Real Time Polymerase Chain Reaction also known as quantitative PCR (q-PCR) is a widely used tool in microbial ecology to quantify gene abundances of taxonomic and functional groups in environmental samples. Used in combination with a reverse transcriptase reaction (RT-q-PCR), it can also be employed to quantify gene transcripts. q-PCR makes use of highly sensitive fluorescent detection chemistries that allow quantification of PCR amplicons during the exponential phase of the reaction. Therefore, the biases associated with 'end-point' PCR detected in the plateau phase of the PCR reaction are avoided. A protocol to quantify bacterial 16S rRNA genes and transcripts from coastal sediments via real-time PCR is provided. First, a method for the co-extraction of DNA and RNA from coastal sediments, including the additional steps required for the preparation of DNA-free RNA, is outlined. Second, a step-by-step guide for the quantification of 16S rRNA genes and transcripts from the extracted nucleic acids via q-PCR and RT-q-PCR is outlined. This includes details for the construction of DNA and RNA standard curves. Key considerations for the use of RT-q-PCR assays in microbial ecology are included.
Introduction
Microorganisms are the corner-stone of the biosphere driving ecosystem function. The majority of microorganisms remain uncultured1. Therefore molecular based approaches are fundamental to advance our understanding of the diversity and function of microorganisms in the environment. Central to these approaches is the extraction of nucleic acids from environmental samples and the subsequent amplification of target genes using the Polymerase Chain Reaction (PCR).
The first step of DNA/RNA extraction aims to lyse the cell walls of the microbial community present, remove undesired non nucleic-acid molecules (e.g., organic and inorganic substances) and retain DNA/RNA in solution for further downstream analysis. Among several options available in the literature2,3,4,5, including a range of commercial extraction kits, the Griffiths method6 is widely employed7,8,9. It is cost-effective and particularly well suited to sediments as it uses a bead-beating step to lyse cells and incorporates steps to minimize the co-extraction of PCR inhibitors, such as humic acids, whilst simultaneously recovering DNA and RNA.
The second step uses the polymerase chain reaction (PCR) to amplify target genes, such as the 16S rRNA taxonomic marker, from the extracted nucleic acids. This approach has and continues to facilitate the exploration of the uncharacterized microbial black box10,11. However, end-point PCR-based methods suffer from various limitations that can bias the characterization of microbial communities12. To accurately quantify gene/transcript abundances, real-time PCR, also known as quantitative PCR (qPCR) must be used. qPCR exploits fluorescent reporter dye systems that track amplicon accumulation after every cycle of the PCR. This is significant as it means that quantification can occur during the early exponential phase, rather than the end-point, phase of the PCR reaction, when the amplicon yield is still directly proportional to the initial abundance of the target gene.
Two reporter systems are commonly used: an intercalating nucleic acid stain13 and the 5' 3' exonuclease activity of the DNA polymerase14. Since the former reporter system binds indistinctively to all double-stranded DNA, it may lead to an overestimation of the target sequence if unwanted non-specific amplicons or primer dimers by products occur. In order to circumvent this, extensive optimization of amplification may be required. In the latter system, template amplification is tracked using a combination of a 5' nuclease activity of Taq polymerase that cleaves a fluorophore from an internal probe. This feature increases the specificity of the assay due to the utilization of a fluorogenic probe that binds only to the complementary target-specific sequence between the primer pair. With both chemistries quantification is achieved by determining the crossing-point (Cp) where the accumulation of PCR amplicons, as measured by an increase in fluorescence, is significantly above background fluorescence.
qPCR has been extensively used in microbial ecology to determine gene abundances in different environments15. Moreover, reverse transcription of RNA to cDNA is combined with qPCR and RT-qPCR to quantify gene expression. Hence, qPCR and RT-qPCR represent fast, effective methods for the quantification of gene and/or transcript numbers within environmental samples.
Microorganisms in coastal sediments drive various ecosystem processes, including the mineralization of organic matter, the degradation of pollutants and the biogeochemical cycling of macronutrients such as nitrogen16,17,18. The exhaustive understanding of these transformations requires a comprehensive account of the contributing microbial populations, including quantitative data on gene and transcript abundances. Here we introduce a series of thoroughly tested, streamlined and standardized protocols for the quantification of bacterial 16S rRNA gene and transcript abundances in coastal sediments. The protocol outlines sample collection, simultaneous DNA and RNA extraction, DNA-free RNA preparation, quality checking of extracted nucleic acids, generation of 16S rRNA-DNA and -RNA standards and quantification of environmental samples. Quantitative data derived from the methods described here are needed to shed light on microbial communities driving coastal ecosystems.
Protocol
1. DNA & RNA Extraction from Marine Coastal Sediments
- Preparation of ribonuclease (RNAse)-free material and workspace
- Prepare RNAse-free distilled water (dH2O) by treating with 0.1% diethylpyrocarbonate (DEPC) and incubate at 37 °C O/N. Remove residual DEPC by autoclaving dH2O at 121 °C for 15 min. Use DEPC treated dH2O to make all solutions.
- Bake all glassware at 180 °C O/N. Remove plastic lids and rims, soak them in a solution of 2 M NaOH O/N, and rinse them with DEPC treated dH2O before use.
- Prepare a CTAB-Phosphate buffer solution as per Table 1.
- Prepare the PEG-NaCl precipitation solution as per Table 2.
- Clean all working surfaces (i.e., bench, microcentrifuge, fume-hood) and micropipettes using RNAse-decontaminating solution before starting. Wear gloves during the entire procedure.
- Use RNAse-free plasticware along with micropipette filter tips to avoid sample cross-contamination.
- Environmental sample collection and storage
- Collect coastal sediment during low tide using a sterile 50 ml falcon tube from the top 2.5 cm. Collect approximately 15-20 g of sediments. Carefully close the lid after sample collection. Transport immediately to the lab in a cooler at 4 °C for immediate processing.
- Homogenize the sample by mixing with a sterile spatula and aliquot 0.5 g of wet weight sediment into a 2 ml bead beating tube. Aliquot additional replicates, flash-freeze by placing in liquid nitrogen. Store aliquots at -80 °C. Caution: Wear protective gloves and safety goggles when handling liquid nitrogen.
Note: If the field site is not located close to a laboratory, aliquot 0.5 g of sediment into 2 ml sterile microcentrifuge tubes at the field site. Flash-freeze the tubes by putting them immediately into liquid nitrogen and transport to the laboratory. Store samples at -80 °C until processing.
- Co-Extraction of DNA and RNA from sediment
Note: The following protocol is a modified version of the Griffiths method6.- Add 500 µl CTAB-Phosphate buffer and 500 µl phenol:chloroform:isoamyl alcohol (25:24:1) to the 2 ml bead-lysing tube containing 0.5 g of sediment and invert the tube 5-10 times to homogenize the sample.
Caution: Perform this step in a fume-hood wearing appropriate protective gear (i.e., laboratory coat, gloves and safety goggles) at all times. - Vortex at full speed for 2.5 min. Centrifuge at 16,000 x g for 10 min at 4 °C.
- In a fume-hood extract the top aqueous layer and transfer to a new 1.5 ml sterile microcentrifuge tube. Keeping the samples on ice, add 500 µl of ice-cold chloroform:isoamyl alcohol (24:1).
- Invert several times until an emulsion is visible. Centrifuge for 10 min at 16,000 x g at 4 °C.
- In a fume-hood extract the top aqueous layer and place it in a new 1.5 ml sterile tube. Precipitate the nucleic acids by adding two volumes of 30% PEG- NaCl solution and mix well.
- Incubate on ice for 2 hr or leave samples at 4 °C O/N.
- At the end of incubation centrifuge samples at 16,000 x g for 20 min at 4 °C.
Note: A pellet may be visible at the bottom of the tube. - Gently remove the supernatant using a micropipette, make sure not to disturb the pellet. Leave around 10 µl of PEG solution in the tube and add 1 ml of ice-cold 70% ethanol.
- Centrifuge for 20 min at 16,000 x g at 4 °C. Slowly remove ethanol with a micropipette taking care not to touch the pellet.
- Centrifuge for 5 sec and remove residual ethanol using a micropipette. Leave the pellet to air dry for approximately 5 min.
- Re-suspend the pellet in 50 µl DEPC-treated H2O. Examine the quality and yield of DNA/RNA by agarose gel electrophoresis running 5 µl on a 1% agarose gel19. Divide the nucleic acids into two aliquots, 15 µl for DNA and 30 µl for RNA.
- Store the DNA at -80 °C until needed. Isolate RNA as outlined in the next section.
- Add 500 µl CTAB-Phosphate buffer and 500 µl phenol:chloroform:isoamyl alcohol (25:24:1) to the 2 ml bead-lysing tube containing 0.5 g of sediment and invert the tube 5-10 times to homogenize the sample.
2. Preparation of RNA and Quality Check of DNA-free RNA
- Digestion of DNA
- Add 3 µl of DNAse I Buffer and 1.5 µl of DNAse I to 30 µl volume of nucleic acids, incubate at 37 °C for 30 min.
- Mix the DNAse Inactivation reagent by vortexing for 30 sec. Add 4.8 µl of the solution to the sample. Incubate at RT for 5 min, occasionally tap the bottom of the tubes to mix the solution.
- Centrifuge at 10,000 x g for 90 sec at RT, transfer the supernatant (RNA) into a new tube, taking care not to transfer precipitate which can inhibit downstream reactions.
- RNA quality check
- Add 2 µl of RNA to 18 µl of sterile RNAse-free dH2O to dilute 1:10.
- In separate PCR reactions, add 1 µl of neat or 1:10 dilution of RNA to a sterile 0.5 ml PCR tube. Add PCR reaction components to amplify the 16S rRNA gene as per Table 3. Include a "positive" (e.g., DNA extracted from a pure bacterial culture), a "PCR inhibitor" control (i.e., 1 µl of positive pure culture DNA and 1 µl of neat extracted RNA) and a "negative" control of sterile RNAse-free dH2O.
- Set the thermocycler with the following amplification parameters: 95 °C 5 min, (95 °C 30 sec, 57 °C 30 sec, 72 °C 1 min) x 35 cycles, 72 °C 10 min.
- Run 10% of the PCR product on a 1% agarose gel at 85 V for 40 min. Repeat the DNAse I treatment if a PCR product is formed in the environmental RNA samples.
- Add 2 µl of RNA to 18 µl of sterile RNAse-free dH2O to dilute 1:10.
3. Generation of First Strand cDNA from RNA
- Add the reagents as per Table 4 to a RNAse/DNAse free 0.2 ml PCR tube.
- Incubate the sample at 65 °C 5 min. Place on ice for at least 1 min and then incubate at 25 °C for 10 min.
- To the same tube add the reagents listed in Table 5.
- Incubate the sample at 55 °C for 50 min. Inactivate the reaction at 72 °C for 10 min.
- Store cDNA at -20 °C until use.
4. Quantitative PCR
- Generation of DNA standards for qPCR
- Amplify bacterial 16S rRNA sequence20 (or any other sequence of interest) from genomic DNA extracted from pure culture with q-PCR primers (1369F & 1492R21).
- Purify the PCR product using a commercial kit according to manufacturer's instructions.
- Clone the purified PCR product into a 3'-T overhang cloning vector system according to manufacturer's instructions.
- Transform the vector containing the PCR insert into Escherichia coli JM109 competent cells according to manufacturer's instructions.
- Plate 50 µl of transformation onto ampicillin (100 µg/ml), X-gal (20 µg/ml) and IPTG (0.5 mM) Luria Bertani agar plates (Tryptone 10 g/L, Yeast Extract 5 g/L, NaCl 10 g/L, Agar 15 g/L). Incubate O/N at 37 °C.
- Select a single positive white transformant from the plate. Inoculate the positive transformant in 50 ml LB broth containing 100 µg/ml ampicillin and incubate O/N shaking at 250 rpm at 37 °C.
- From the O/N culture, isolate and purify the plasmid using a commercial kit according to manufacturer's instructions.
- Linearize the plasmid using an appropriate restriction enzyme that is capable of cutting the plasmid once (e.g., EcoRI).
- Analyze the purity of the plasmid by measuring the absorption ratio A260/A28019. If the ratio is below 1.8, re-extract the plasmid with phenol-chloroform19. Note: Pure DNA has a 1.8 ratio.
- Determine the concentration of plasmid DNA by absorbance at 260 nm using a spectrophotometer19.
Note: Alternatively, the accuracy of quantification can be improved by use of a fluorescent DNA-binding dye used in conjunction with a fluorometer. - Sequence the plasmid insert to confirm the size of the target in bases. Calculate the amount (i.e., percentage) of insert DNA in the plasmid (e.g., in a 3,000 bp vector, a 123 bp DNA fragment is 3.9% of total DNA) and multiply it by the concentration obtained in 4.1.10, to determine the DNA concentration of the target (insert).
- Use the following equation to calculate the copies of target per µl:

- Calculate the Target Molecular Weight (TMW) by multiplying the number of base pairs of the target DNA by the average molecular weight of double strand DNA (dsDNA, 660 Daltons per base pair).
- Dilute the target DNA in a range from 1010 to 102 copies using 2 µl of DNA and 18 µl of sterile dH2O. Mix each dilution well and spin down briefly before making the next dilution.
- Generation of RNA standard curve for qPCR
- From step 4.1.5, screen a number of white colonies (approximately 5) by colony PCR19 using the target reverse primer (i.e., R1492) and the forward vector plasmid primer M13F.
Note: This combination of primers will amplify inserts cloned in the correct sense orientation with a T7 promoter site upstream. - Purify the PCR product using a commercial kit following manufacturer's instruction. Quantify the purified PCR product at 260 nm using a spectrophotometer.
- Add 200 ng of PCR product in a final volume of 20 µl with 7.5 mM of each ribonucleotide, 1 T7 reaction buffer and 1 T7 polymerase enzyme for the in vitro transcription reaction.
- Incubate the reaction for 4 hr at 37 °C. Add 1 unit of RNase-free DNase I to the reaction, incubate for 15 min at 37 °C to remove DNA template.
- Recover the in vitro transcribed RNA by ethanol precipitation19 and quantify RNA yields using a fluorimeter or by absorbance at 260 nm.
- Calculate transcript number per µl of RNA. Add the number of base pairs in the target RNA (e.g., 123 bases) to the length of the T7 promoter (153 bases) and multiply by the average molecular weight of RNA, 340 Daltons per base.
- Add 500 ng of quantified target RNA to a reverse transcriptase (RT) reaction as outlined in section 3. Serially dilute the cDNA as outlined in 4.1.13 for use as the standard curve.
- From step 4.1.5, screen a number of white colonies (approximately 5) by colony PCR19 using the target reverse primer (i.e., R1492) and the forward vector plasmid primer M13F.
- Real-Time PCR
- Thaw environmental DNA/cDNA samples, standards and qPCR reagents on ice. Protect the fluorogenic probe from light.
- Dilute the standards for the standard curve as outlined in 4.1.14. Make 1:10 dilutions of the environmental DNA/cDNA from neat to 10-3 by adding 2 µl of sample to 18 µl of sterile dH2O.
- Make a master mix reaction for the total number of reactions plus 10% as per Table 6 (primers and probe) and Table 7 (reaction mix). Mix well and briefly centrifuge.
- Add 19 µl of master mix and 1 µl of template (standard, environmental sample or water for negative control) to each well in a 96-well optical qPCR plate. Perform each reaction (standards, environmental samples and negative controls) in triplicate.
- Cover the 96-well plate with a q-PCR optical cover. Centrifuge the plate briefly. Load the plate into the heating block of the qPCR machine and close the lid.
- Open the qPCR manager software. Click the "protocol tab", click "create new", add the following amplification conditions: 3 min 95 °C, (10 sec 95 °C, 30 sec 60 °C) x 40 cycles. Set sample volume to 20 µl, click "OK" and save the protocol.
- Click the "Plate" tab. Under "edit selected" click "select fluorophore", add the report dye for appropriate probe, e.g., fluorescein. Click "OK".
- Highlight wells containing the standards, select "standard" from the "sample type" menu. Click "replicate series" to change "replicate size" to 3, click "apply". Click "dilution series" to add calculated starting concentration for standard 1, indicate the dilution factor (10), select "decreasing" or "increasing" depending on the order the standard curve was added to the plate. Click "apply".
- Highlight well positions containing the unknown samples, select "unknown" from the "sample type" drop down menu. Click "replicate series", and change "replicate size" to 3. Click "apply". Edit sample names under "sample name". Repeat procedure for No Template Controls (NTC) wells.
- Click "OK" on the plate editor box and save file. Click "start run".
- On completion of the run, the data analysis window will open. Click the "quantification tab" to display the standard curve, standard curve descriptors (Table 8) and amplification plots.
- Click on the "quantification data" tab for Cp values and corresponding starting quantity of gene (SQ) of unknown samples. Export table to spreadsheet software if required.
- Multiply gene abundances from unknown samples by the dilution factor, the quantity of sediment extracted from and the total volume nucleic acids were eluted in to obtain gene copies per gram of wet weight sediment.
Note: Elute DNA from 0.5 g of sediment in 50 µl, dilute 1/10, add 1 µl as template to a q-PCR reaction; calculate gene copies g-1 sediment by multiplying: gene copies µl of DNA x dilution factor x elution volume x 2.
Results
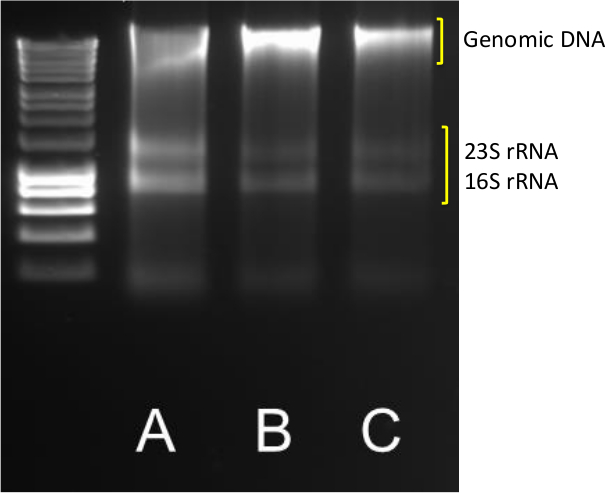
The extraction of good quality DNA and RNA from sediments is the first step in quantifying gene and transcript abundances. A successful extraction yields clear DNA and RNA bands as indicated in Figure 1 for sample A-C, where sharp 23S and 16S rRNA bands are visible in addition to the high molecular weight genomic DNA band.

Figure 1. DNA/RNA extraction. Typical results from DNA/RNA extraction from triplicate (A-B-C) 0.5 g coastal sediments. 5 µl of DNA/RNA was run on a 1.4% agarose at 85 V for 40 min. A molecular marker in the 10,037-200 bp range was used. Please click here to view a larger version of this figure.
{kind=link}
To prepare RNA, a digestion of the co-extracted DNA is mandatory. This must be followed by 16S rRNA end-point PCR of the RNA to ensure that the DNA has been successfully removed. If the DNA has been completely removed only a band in the positive control is observed. It is important to use both neat and a 1:10 dilution of RNA to ensure inhibitors are not preventing the formation of a PCR product. RNA can now undergo the reverse transcriptase reaction to convert it to cDNA. This can be performed with either gene-specific primers or random hexamers. Typically, the reaction is carried out in a PCR machine, to ensure optimal temperature profile. This cDNA is used as template in the subsequent qPCR reaction. DNA and RNA is quantified to determine the template concentration used in each reaction.
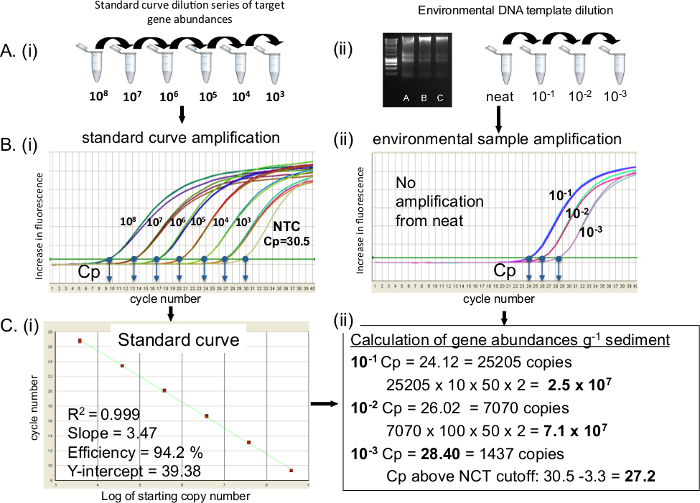
A qPCR reaction protocol is outlined targeting 16S rRNA DNA. For 16S rRNA transcripts substitute the standard curve and template with cDNA. As unknown samples are quantified against the standard curve, it is imperative to ensure that the standard curve is of good quality. Figure 2 shows the preparation of (A), standard curves and environmental DNA dilutions, (B), amplification of the standard curve and environmental samples (C) conversion of standard curve to a linear regression and calculation of gene copy numbers. When a 10-fold dilution range is correctly prepared and amplified a 3.3 cycle difference between each standard dilution is seen (it takes 3.3 cycles for a ten-fold increase in template at 100% amplification efficiency) (Figure 2Bi). Standard curves descriptors, including the R2 values of 0.99 and PCR efficiencies within the range of 90 to 110% (Figure 2C) are desired. It is important to report Cp values from the no template control (NTC) if present. If this occurs a Cp cutoff for standards and unknown samples 3.3 cycles (i.e., a log fold) higher than the Cp value of the NTC is imposed20 (Figure 2Cii). Unknown template extracted from sediment DNA was quantified from neat, 10-1, 10-2 and 10-3 dilutions (B). The neat sample did not amplify, the Cp values of 10-1 to 10-3 dilutions were 24.12, 26.02 and 28.40 respectively. The NTC Cp was 30.5, an NCT cutoff of 27.2 was imposed. Converting gene abundances to g-1 wet weight sediment resulted in 2.5 x 107 and 7.1 x 107 for 10-1 and 10-2 respectively for the dilution range. 10-3 dilution Cp was below the NTC cutoff, and was therefore not used. In this case the 10-2 was selected as the optimal template dilution.

Figure 2. 16S rRNA gene qPCR amplification of standards and environmental DNA extracted from coastal sediments. Preparation of (A) i) DNA standard curve and ii) environmental DNA dilutions for qPCR amplification, (B) qPCR amplification of i) DNA standard curve and ii) environmental samples, Cp for each sample are shown. (C) i) Linear regression of standard curve with standard curve descriptors; ii) calculation of gene abundances from environmental samples. NTC: Negative Template Control. Please click here to view a larger version of this figure.
{kind=link}
| Reagent | Amount |
| CTAB | 5 g |
| K2HPO4x3H2O | 2.58 g |
| KH2PO4 | 0.1 g |
| NaCl | 2.05 g |
| dH2O | up to 100 ml |
Table 1. Preparation of CTAB-phosphate buffer.
| Reagent | Amount |
| PEG 6000 | 30 g |
| NaCl | 9.35 g |
| dH2O | up to 100 ml |
| Note: add PEG 6000 slowly under moderate heating and stirring of 50 ml of dH2O. | |
Table 2. Preparation of PEG-NaCl precipitation solution.
| Reagent | Amount |
| Undiluted RNA / 1:10 diluted RNA | 1.0 µl |
| 10x PCR Buffer | 5.0 µl |
| dNTPs 10 mM | 1.0 µl |
| 63F forward primer | 1.0 µl |
| CAG GCC TAA CAC ATG CAA GTC | |
| 518R reverse primer | 1.0 µl |
| ATT ACC GCG GCT GCT GG | |
| Taq polymerase | 0.4 µl |
| RNAse-free dH2O | 40.6 µl |
Table 3. DNA-free RNA quality check PCR master mix.
| Reagent | Amount |
| RNA | 0.5-5 μg |
| dNTPs 10 mM | 1 µl |
| Reverse primer 10 μM/ Random Hexamers 50 µM | 1 µl |
| DEPC-H2O | Up to 13 µl |
Table 4. cDNA synthesis reaction mix A.
| Reagent | Amount |
| Buffer 5x | 4 µl |
| DTT | 1 µl |
| RNAse Inhibitor | 1 µl |
| Reverse Transcriptase | 1 µl |
Table 5. cDNA synthesis reaction mix B.
| Target | Primer name | Sequence | Annealing T (°C) | Reference |
| 16S rRNA bacteria | Bact1369F | CGG TGA ATA CGT TCY CGG | 56 | Suzuki et al. 2000 19 |
| Prok1492R | GGW TAC CTT GTT ACG ACT T | |||
| TM1389F (probe) | CTT GTA CAC ACC GCC CGT C |
Table 6. Primer and probes for 16S rRNA q-PCR assay.
| Reagent | Amount |
| 2x Master Mix | 10 μl |
| Forward primer (10 μM) | 0.8 μl |
| Reverse primer (10 μM) | 0.8 μl |
| Probe (10 μM) | 0.4 μl |
| Water | 7.0 μl |
| Final volume | 19 μl |
Table 7. Real time PCR reaction mix.
| Descriptor | Significance |
| Correlation coefficient (R2) | A measure of the linearity of the standard curve. Ideally it should be R2=1. Value of R2=0.98-0.99 are acceptable. |
| Slope (α) | A measure of reaction efficiency. Ideally it should be equivalent to -3.32. Value in the range between -3.58 and -3.1. |
| Efficiency (=10(-1/slope)-1) | If it is 100%, the templates doubles after each thermal cycle during exponential amplification. A good efficiency range is between 90 and 110%. |
| Y Intercept (β) | The theoretical limit of detection of the reaction. Although not used to quantify reaction sensitivity, it is important when comparing different standard curves of the same target. |
Table 8. qPCR reaction descriptors.
Discussion
The combination of DNA/RNA extraction with qPCR provides a fast, accurate, relatively cost-effective method for the sensitive quantification of gene and transcript abundances from a range of environmental samples, such as coastal sediments.
The initial nucleic acid extraction is the critical step to ensure a representative view of the microbial community present is achieved22. A number of limitations need to be considered for the extraction protocol: a) the achievement of total cell lysis, b) co-extraction of inhibitory compounds (e.g., humic acids or polyphenolics), c) contaminating DNA in the RNA fraction, d) rapid degradation of extracted nucleic acids when stored. Precautions must be taken in order to circumvent these limitations. For example, particular care has to be taken to ensure that the extraction of nucleic acids is optimized for the sample type (e.g., sediments, soil, wastewater, etc.). Significant improvements in nucleic acid yield and quality for both DNA and RNA can be achieved by carrying out preliminary experiments23. To minimize the effect of inhibitory compounds on PCR-based applications24, test a range of dilutions from the extracted DNA and RNA. To circumvent the rapid degradation of the extracted DNA/RNA from multiple freeze-thaw cycles and avoid possible loss of genetic information, store multiple small volume aliquots at -80 °C.
When carefully designed, qPCR is a robust, highly reproducible and sensitive method. Notably, the methods for amplification and standard curve construction outlined in this protocol can be adapted for any gene target of interest, including other phylogenetic markers (i.e., archaeal 16S rRNA, fungal 18S rRNA) or genes involved in important functions in the environment. Known limitations in the use of qPCR are: a) the generation of reproducible high-quality standard curve for absolute quantification, b) the choice of primer/probes and optimization of q-PCR assay conditions, c) the use of low quality/sheared nucleic acids, d) the choice of the working dilution of DNA/RNA to avoid inhibition. Moreover, it has to be considered that qPCR technique provides gene/transcript abundances which may not equate to cell counts: this is particularly the case when 16S and 18S rRNA genes are targeted, as microorganisms have different copy numbers of the ribosomal gene in their genome25.
Poor quality standard curves will result in inaccurate quantification of the gene of interest. It is good practice to store a stock of high concentration standards in small aliquots from which fresh standard curves can be made. Do not store standard curves, always make fresh dilutions from a stock of the highest concentration each time. For accurate quantification of environmental samples ensure the range of concentrations of the standard curve spans the expected Cp values of the unknown samples. When quantifying transcripts, construct the standard curve from RNA not double stranded DNA. When possible, quantification of samples to be directly compared should be completed within a single assay to avoid inter-assay variation20. This may not always be possible. Therefore, to compare gene abundances generated between assays it is advisable to have a random sub-set of samples replicated between assays. A wide selection of primer and probe sets are currently available for qPCR targeting microbial taxa and functional groups15. Careful consideration is needed when selecting these to ensure both maximal coverage and specificity for the target group. If the reaction efficiency of a qPCR assay is not satisfactory, troubleshooting starts with testing different thermal cycling conditions (as annealing time and/or temperature) and/or reaction conditions, e.g., varying primer-probe concentrations. Once the q-PCR assay and reaction conditions are optimized, always conduct an initial test of a range of DNA/cDNA dilutions to determine the appropriate template concentration. Select the dilution range resulting in the highest copy number as the optimal template dilution for further assays.
Currently, next-generation sequencing technologies efficiently shed light on microbial community structure and functions in a plethora of environments26,27,28. However, these datasets are often based on end-point PCR amplicon libraries and therefore provide only semi-quantitative assessments of the abundance of particular taxa. Hence, the capability of real time PCR-based technique to target specific taxonomic markers (from higher domain down to strain level) allows efficient validation of the results obtained by next-generation sequencing. Moreover, qPCR has been successfully used in combination with other microbial ecology molecular methods such as stable isotope probing (SIP) or phylogenetic/functional microarrays. Combined with the former tool, qPCR can be used to quantify the metabolically active community29,30. When combined with microarray analysis, qPCR provides key quantitative interpretation of phylogenetic marker-based and functional gene surveys of environments31,32.
Therefore, whether used alone or in combination with other (often process-based) assessments of ecosystem function, quantitative PCR is an essential tool for microbial ecologists in the exploration of the elusive link between microbial communities and ecosystem functions.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This publication has emanated from research conducted with the financial support of the Natural Environment Research Council (NERC) under grant number NERC NE/JO11959/1 and Science Foundation Ireland & the Marie-Curie Action COFUND under Grant Number 11/SIRG/B2159awarded to CJS and the Eastern Academic Research Consortium (Eastern ARC).
Materials
| Name | Company | Catalog Number | Comments |
| Diethylpyrocarbonate | Sigma | 40718 | Toxic, open under chemical hood |
| cetrimonium bromide (CTAB) | Sigma | 52365 | Irritant, open under chemical hood |
| potassium phosphate dibasic | Sigma | RES20765 | |
| potassium phosphate monobasic | Sigma | P9791 | |
| Phenol : Chloroform : Isoamylalcohol pH 8 | Sigma | P2069 | Equilibrate at pH 8 before using |
| Chloroform : Isoamylalcohol | Sigma | 25666 | |
| sodium chloride | VWR | 1.06404.0500 | |
| polyethylenglycol 6000 | VWR | 528877-100 | |
| Ethanol Molecular Grade | Sigma | E7148 | |
| Lysing Matrix E tubes | MP Biomedical | 116914050 | |
| Turbo DNAse | Ambion | AM1907 | |
| Taq polymerase | Sigma | D1806 | |
| dNTPs | Bioline | BIO-39028 | |
| SuperScript III | Life Technologies | 18080044 | |
| Rnase Inhibitor | Life Technologies | 10777019 | |
| RNAse/DNAse free 0.2 ml PCR tubes | Sarstedt | 72.985.002 | |
| SureCleanPlus | Bioline | BIO-37047 | |
| pGEM Easy T Vector | Promega | A1360 | |
| E. coli JM109 competent cells | Promega | L2005 | |
| Tryptone | Sigma | T7293 | |
| Yeast Extract | Sigma | 70161 | |
| Ampicillin | Sigma | 59349 | |
| X-Gal | Bioline | BIO-37035 | |
| IPTG | Bioline | BIO-37036 | |
| Agar | VWR | 20768.235 | |
| Plasmid Midi Kit | Qiagen | 12143 | |
| Qubit | Life Technologies | Q33216 | |
| Quant-IT DNA HS Assay | Life Technologies | Q-33120 | |
| Quant-IT RNA HS Assay | Life Technologies | Q32855 | |
| MEGAshortscript kit | Ambion | AM1354 | |
| TaqMan SensiFast Probe Lo-ROX kit | Bioline | BIO-84002 | |
| qPCR machine |
References
- Epstein, S. S. The phenomenon of microbial uncultivability. Curr. Opin. Microbiol. 16 (5), 636-642 (2013).
- Zhou, J., Bruns, M. A., Tiedje, J. M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62, 316-322 (1996).
- Miller, D. N., Bryant, J. E., Madsen, E. L., Ghiorse, W. C. Evaluation and optimization of DNA extraction and purification procedures from soil and sediments samples. Appl. Environ. Microbiol. 65 (11), 4715-4724 (1999).
- Burgmann, H., Pesaro, M., Widmer, F., Zeyer, J. A strategy for optimizing quality and quantity of DNA extracted from soil. J. Microbiol. Methods. 45 (1), 7-20 (2001).
- Hurt, R. A., et al. Simultaneous recovery of RNA and DNA from soils and sediments. Appl. Environ. Microbiol. 67, 4495-4503 (2001).
- Griffiths, R. I., Whiteley, A. S., O'Donnell, A. G., Bailey, M. J. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl. Environ. Microbiol. 66 (12), 5488-5491 (2000).
- Martins, G., et al. Structure and activity of lacustrine sediment bacteria involved in nutrient and iron cycles. FEMS Microbiol. Ecol. 77 (3), 666-679 (2011).
- Kuffner, M., et al. Effects of season and experimental warming on the bacterial community in a temperate mountain forest soil assessed by 16S rRNA gene pyrosequencing. FEMS Microbiol. Ecol. 82 (3), 551-562 (2011).
- Tatti, E., et al. Influences of over winter conditions on denitrification and nitrous oxide-producing microorganism abundance and structure in an agricultural soil amended with different nitrogen sources. Agric. Ecosyst . Environ. 183, 47-59 (2014).
- Giovannoni, S. J., Britschgi, T. B., Moyer, C. L., Field, K. G. Genetic diversity in the Sargasso Sea bacterioplankton. Nature. 345, 60-62 (1990).
- Ward, D. W., Weller, R., Bateson, M. M. 16S rRNA sequences reveal numerous uncultured microorganisms a natural community. Nature. 345, 63-65 (1990).
- Suzuki, M. T., Giovannoni, S. J. Bias caused by template annealing in the amplification of mixture of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62, 625-630 (1996).
- Wittwer, C. T., Herrmann, M. G., Moss, A. A., Rasmussen, R. P. Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques. 22, 130-138 (1997).
- Holland, P. M., Abramson, R. D., Watson, R., Gelfand, D. H. Detection of Specific Polymerase Chain Reaction Product by Utilizing the 5' 3' Exonuclease Activity of Thermus aquaticus DNA Polymerase. PNAS. 88, 7276-7280 (1991).
- Smith, C. J., Osborn, A. M. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 67, 2-20 (2009).
- Park, S. J., Park, B. J., Rhee, S. K. Comparative analysis of archaeal 16S rRNA and amoA genes to estimate the abundance and diversity of ammonia-oxidizing archaea in marine sediments. Extremphiles. 12 (4), (2008).
- Urakawa, H., Yoshida, T., Nishimura, M., Ohwada, K. Characterization of depth-related population variation in microbial communities of a coastal marine sediment using 16S rDNA-based approaches and quinone profiling. Environ. Microbiol. 2, 542-554 (2008).
- Smith, C. J., Dong, L. F., Wilson, J., Stott, A., Osborn, A. M., Nedwell, D. B. Seasonal variation in denitrification and dissimilatory nitrate reduction to ammonia process rates and corresponding key functional genes along an estuarine nitrate gradient. Front. Microbiol. 6, 542(2015).
- Sambrook, J., Russell, D. W. Molecular Cloning: a laboratory manual. , Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. 205(2001).
- Smith, C. J., Nedwell, D. B., Dong, L. F., Osborn, A. M. Evaluation of Quantitative Polymerase Chain Reaction (Q-PCR) based approaches for determining gene copy and gene transcript numbers in environmental samples. Environ. Microbiol. 8 (5), 804-815 (2006).
- Suzuki, M. T., Taylor, L. T. DeLong E.F., Quantitative analysis of small-subunit rRNA genes in mixed microbial population via 5'-nuclease assays. Appl. Environ. Microbiol. 66, 4605-4614 (2000).
- Luna, G. M., Dell'Anno, A., Danovaro, R. DNA extraction procedure: a critical issue for bacterial diversity assessment in marine sediments. Environ. Microbiol. 8 (2), 308-320 (2005).
- Lever, M. A., Torti, A., Eickenbursch, P., Michaud, A. B., Šantl-Temkiv, T., Jørgensen, B. B. A modular method for the extraction of DNA and RNA, and the separation of DNA pools from diverse environmental sample types. Front. Microbiol. 6, 476(2015).
- Lloyd, K. G., MacGregor, B. J., Teske, A. Quantitative PCR methods for RNA and DNA in marine sediments: maximizing yield while overcoming inhibition. FEMS Microbol. Ecol. 72, 144-151 (2010).
- Klappenbach, J. A., Dumbar, J. M., Schmidt, T. M. rRNA Operon copy number reflects ecological strategies of bacteria. App. Environ. Microbiol. 66 (4), 1328-1333 (2000).
- Shi, Y., Tyson, G. W., Eppley, J. M., DeLong, E. F. Integrated metatrascriptomic and metagenomics analyses of stratified microbial assemblages in the open ocean. ISME J. 5, 999-1013 (2011).
- Howe, A. C., Jansson, J. K., Malfatti, S. A. Tringe S.G., Tiedje J.M., & Brown C.T. Tackling soil diversity with the assembly of large, complex metagenomes. PNAS. 111 (13), 4904-4909 (2014).
- Klaedtke, S., et al. Terroir is a key driver of seed-associated microbial assemblages. Environ. Microbiol. , (2015).
- Lueders, T., Wagner, B., Claus, P., Friedrich MW, Stable isotope probing of rRNA and DNA reveals a dynamic methylotroph community and trophic interactions with fungi and protozoa in oxic rice field soil). Environ. Microbiol. 6, 60-72 (2004).
- Kunapuli, U., Lueders, T., Meckenstock, R. U. The use of stable isotope probing to identify key iron-reducing microorganisms involved in anaerobic benzene degradation. ISME J. 1, 643-653 (2007).
- Bürgmann, H., et al. Transcriptional response of Silicibacter pomeroyi DSS-3 to dimethylsulfoniopropionate (DMSP). Environ. Microbiol. 9 (11), 2742-2755 (2007).
- He, J. Z., et al. Geochip: a comprehensive microarray for investigating biogeochemical ecological and environmental processes. ISME J. 1, 67-77 (2007).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved