
Method Article
ARN-ADN Extraction simultanée de sédiments côtiers et Quantification de 16S ARNr et Transcriptions par PCR en temps réel
Dans cet article
Résumé
A protocol to quantify bacterial 16S rRNA genes and transcripts from coastal sediments via real-time PCR is provided. The methodology includes the co-extraction of DNA and RNA; preparation of DNA-free RNA; and 16S rRNA gene and transcript quantification via RT-q-PCR, including standard curve construction.
Résumé
Temps réel Réaction de polymérisation en chaîne aussi connu comme la PCR quantitative (q-PCR) est un outil largement utilisé dans l'écologie microbienne pour quantifier les abondances de gènes des groupes taxonomiques et fonctionnels dans les échantillons environnementaux. Utilisé en combinaison avec une réaction de transcriptase inverse (RT-PCR quantitative), elle peut également être utilisée pour quantifier la transcription de gènes. q-PCR utilise des chimies de détection par fluorescence très sensibles qui permettent la quantification des amplicons PCR pendant la phase exponentielle de la réaction. Par conséquent, les préjugés associés à 'point final' PCR détectés dans la phase de plateau de la réaction PCR sont évités. Un protocole pour quantifier les gènes bactériens ARNr 16S et transcriptions de sédiments côtiers par PCR en temps réel est fourni. Tout d'abord, un procédé de co-extraction de l'ADN et de l'ARN à partir des sédiments côtiers, comprenant les étapes supplémentaires nécessaires à la préparation de l'ARN exempt d'ADN, est décrit. Deuxièmement, un guide étape par étape pour la quantification des gènes ARNr 16S et transcripts à partir des acides nucléiques extraits par Q-PCR et RT-PCR quantitative est décrite. Cela comprend les détails de la construction des courbes d'ADN standard et de l'ARN. Considérations clés pour l'utilisation de tests RT-q-PCR en écologie microbienne sont inclus.
Introduction
Microorganismes sont la pierre angulaire de la fonction biosphère de l'écosystème de conduite. La majorité des micro - organismes restent incultes 1. Par conséquent, les approches fondées moléculaires sont fondamentales pour faire progresser notre compréhension de la diversité et de la fonction de micro-organismes dans l'environnement. Au centre de ces approches est l'extraction d'acides nucléiques à partir d'échantillons environnementaux et l'amplification subséquente des gènes cibles en utilisant la réaction en chaîne par polymérase (PCR).
La première étape d'extraction d' ADN / ARN vise à lyser les parois cellulaires de la communauté microbienne présente, éliminer les molécules indésirables non acides nucléiques (substances par exemple, organiques et inorganiques) et de conserver l' ADN / ARN en solution pour une analyse ultérieure en aval. Parmi plusieurs options disponibles dans la littérature 2,3,4,5, y compris une gamme de kits d'extraction commerciale, la méthode Griffiths 6 est largement utilisé 7,8,9. Il est rentable et particularly bien adapté aux sédiments car il utilise une étape de bourrelet battant pour lyser les cellules et intègre les étapes consistant à réduire au minimum la co-extraction des inhibiteurs de la PCR, tels que les acides humiques, tout en récupérant simultanément l'ADN et l'ARN.
La seconde étape utilise la réaction en chaîne par polymérase (PCR) pour amplifier des gènes cibles, tels que le marqueur taxonomique ARNr 16S, à partir des acides nucléiques extraits. Cette approche a été et continue à faciliter l'exploration de la non caractérisée microbienne boîte noire 10,11. Cependant, les méthodes de point final basé sur la PCR souffrent de diverses limitations qui peuvent biaiser la caractérisation des communautés microbiennes 12. Pour quantifier précisément les abondances gène / de transcription, PCR en temps réel, également connu sous le nom de PCR quantitative (qPCR) doit être utilisé. qPCR exploite des systèmes de colorants rapporteurs fluorescents qui permettent de suivre l'accumulation d'amplicons après chaque cycle de la PCR. Ceci est important car cela signifie que la quantification peut se produire pendant la phase exponentielle précoce, plutôtque le point final, la phase de la réaction PCR, lorsque le rendement de l'amplicon est toujours directement proportionnelle à l'abondance initiale du gène cible.
Deux systèmes rapporteurs sont couramment utilisés: un colorant intercalant d'acide nucléique 13 et de l'activité 5 '3' exonucléase de l'ADN polymerase 14. Depuis l'ancien système rapporteur se lie indistinctement à tous les ADN double brin, il peut conduire à une surestimation de la séquence cible si amplicons non spécifiques indésirables ou des dimères d'amorces par produits se produisent. Pour contourner cela, une vaste optimisation de l'amplification peut être nécessaire. Dans ce dernier système, modèle amplification est suivie à l' aide d' une combinaison d'une activité 5 'nuclease de la Taq polymérase qui clive un fluorophore à partir d' une sonde interne. Cette caractéristique accroît la spécificité de l'essai en raison de l'utilisation d'une sonde fluorogène qui se lie uniquement à la sequenc spécifique à la cible complémentairee entre la paire d'amorce. Avec les deux chimies la quantification est réalisée en déterminant le point de passage (Cp) où l'accumulation d'amplicons de PCR, telle que mesurée par une augmentation de la fluorescence est significativement supérieure à la fluorescence de fond.
qPCR a été largement utilisé en écologie microbienne afin de déterminer les abondances de gènes dans différents environnements 15. En outre, la transcription inverse de l'ARN en ADNc est combiné avec qPCR et RT-qPCR pour quantifier l'expression génique. Par conséquent, la RT-PCR quantitative et qPCR représentent des procédés rapides et efficaces pour la quantification du nombre de gènes et / ou de transcription au sein des échantillons environnementaux.
Microorganismes dans les sédiments côtiers conduisent divers processus écosystémiques, y compris la minéralisation de la matière organique, la dégradation des polluants et le cycle biogéochimique des macronutriments tels que 16,17,18 d'azote. La compréhension exhaustive de ces transformations nécessite une accoun complètet des populations microbiennes qui contribuent, y compris des données quantitatives sur gènes et de transcription abondances. Ici, nous introduisons une série de soigneusement testés, des protocoles simplifiés et normalisés pour la quantification des gènes et de transcription abondances ARNr 16S bactérienne dans les sédiments côtiers. Le protocole décrit le prélèvement des échantillons, l'ADN simultanée et l'extraction de l'ARN, préparation d'ARN sans ADN, le contrôle de la qualité des acides nucléiques extraits, génération de ARNr 16S-ADN et les normes de -ARN et la quantification des échantillons environnementaux. Les données quantitatives tirées des méthodes décrites ici sont nécessaires pour faire la lumière sur les communautés microbiennes de conduite des écosystèmes côtiers.
Protocole
1. ADN et ARN Extraction de sédiments marins côtiers
- Préparation de la ribonucléase (RNAse) matériau -free et espace de travail
- Préparer de l' eau distillée sans RNase (dH 2 O) par traitement avec 0,1% de pyrocarbonate de diéthyle (DEPC) et incuber à 37 ° CO / N. Retirer DEPC résiduelle par autoclavage dH 2 O à 121 ° C pendant 15 min. Utilisez DEPC traité dH 2 O pour faire toutes les solutions.
- Cuire au four toute la verrerie à 180 ° CO / N. Retirer les couvercles et les jantes en plastique, les tremper dans une solution de 2 M NaOH O / N, et les rincer à l' DEPC traité dH 2 O avant utilisation.
- Préparer une solution tampon CTAB phosphate selon le tableau 1.
- Préparer la solution de précipitation PEG-NaCl selon le tableau 2.
- Nettoyez toutes les surfaces de travail (c. -à- banc, microcentrifugeuse, la hotte aspirante) et micropipettes en utilisant une solution RNAse décontaminer avant de commencer. Porter des gants pendant toute la procédure.
- Utilisez plasticware sans RNAse ainsi que des conseils de filtre micropipette pour éviter la contamination croisée échantillon.
- la collecte d'échantillons de l'environnement et de stockage
- Collecter les sédiments côtiers à marée basse en utilisant un 50 ml tube Falcon stérile de 2,5 cm supérieurs. Collecter environ 15-20 g de sédiments. Bien refermer le couvercle après le prélèvement de l'échantillon. Transport immédiatement au laboratoire dans une glacière à 4 ° C pour un traitement immédiat.
- Homogénéiser l'échantillon en mélangeant avec une spatule stérile et aliquote de 0,5 g de sédiment de poids humide dans un tube à cordon de battement 2 ml. Aliquotes répétitions supplémentaires, flash-gel en plaçant dans l'azote liquide. aliquotes Conserver à -80 ° C. Attention: Porter des gants et des lunettes de sécurité lors de la manipulation de l'azote liquide.
Note: Si le site de terrain ne se trouve pas à proximité d'un laboratoire, aliquote de 0,5 g de sédiments dans 2 ml microtubes stériles sur le site de terrain. Flash geler les tubes en les mettant immédiatement en liazote quid et le transport au laboratoire. Conserver les échantillons à -80 ° C jusqu'à la transformation.
- Co-extraction de l'ADN et de l'ARN à partir des sédiments
Remarque: Le protocole suivant est une version modifiée de la méthode Griffiths 6.- Ajouter 500 pi de tampon CTAB-phosphate et 500 pi de phénol: chloroforme: alcool isoamylique (25: 24: 1) dans le tube de perles lyser 2 ml contenant 0,5 g de sédiments et d'inverser le tube 5-10 fois pour homogénéiser l'échantillon.
Attention: Effectuez cette étape dans un hottes portant un équipement de protection approprié (c. -à , manteau de laboratoire, des gants et des lunettes de sécurité) en tout temps. - Vortex à pleine vitesse pendant 2,5 minutes. Centrifugeuse à 16.000 xg pendant 10 min à 4 ° C.
- Dans un hottes extraire la couche aqueuse supérieure et transférer à un nouveau 1,5 ml microtube stérile. Garder les échantillons sur la glace, ajouter 500 pi de glace froide chloroforme: alcool isoamylique (24: 1).
- Inversez plusieurs fois jusqu'à ce qu'une émulsion est Visible. Centrifugeuse pendant 10 min à 16 000 xg à 4 ° C.
- Dans un hottes extraire la couche aqueuse supérieure et le placer dans un nouveau 1,5 ml tube stérile. Précipiter les acides nucléiques en ajoutant deux volumes de solution à 30% PEG- NaCl et bien mélanger.
- Incuber sur la glace pendant 2 heures ou à laisser des échantillons à 4 ° CO / N.
- A la fin de centrifuger les échantillons d'incubation à 16 000 xg pendant 20 min à 4 ° C.
Remarque: Une pastille peut être visible au fond du tube. - Retirez délicatement le surnageant à l'aide d'une micropipette, assurez-vous de ne pas perturber le culot. Laisser environ 10 pi de solution PEG dans le tube et ajouter 1 ml de glace-froid à 70% d'éthanol.
- Centrifugeuse pendant 20 min à 16 000 xg à 4 ° C. Retirer lentement l'éthanol avec une micropipette en prenant soin de ne pas toucher le culot.
- Centrifuger pendant 5 sec et éliminer l'éthanol résiduel en utilisant une micropipette. Laissez le culot sécher à l'air pendant environ 5 min.
- Remettre en suspension le culot dans 50 ul traitée au DEPCH 2 O. Examiner la qualité et le rendement de l' ADN / ARN par électrophorèse sur gel d' agarose en retrait de 5 pi sur un gel d'agarose à 1% 19. Divisez les acides nucléiques en deux aliquotes, 15 pi de l'ADN et 30 pi pour l'ARN.
- Stocker l'ADN à -80 ° C jusqu'à leur utilisation. Isoler ARN tel que décrit dans la section suivante.
- Ajouter 500 pi de tampon CTAB-phosphate et 500 pi de phénol: chloroforme: alcool isoamylique (25: 24: 1) dans le tube de perles lyser 2 ml contenant 0,5 g de sédiments et d'inverser le tube 5-10 fois pour homogénéiser l'échantillon.
2. Préparation de l'ARN et de Contrôle de la qualité de l'ARN sans ADN
- Digestion de l'ADN
- Ajouter 3 pi de tampon de DNAse I et de 1,5 pi de DNase I à 30 ul volume d'acides nucléiques, incuber à 37 ° C pendant 30 min.
- Mélanger le réactif ADNase Inactivation par tourbillonnement pendant 30 s. Ajouter 4,8 ul de la solution à l'échantillon. Incuber à température ambiante pendant 5 min, de temps en temps appuyez sur le fond des tubes pour mélanger la solution.
- Centrifugeuse à 10.000 xg pendant 90 secondes à la température ambiante, transférer le surnageant (ARN) dans un nouveau tube, en prenant soin de ne pas transférer précipité qui peut inhiber les réactions en aval.
- Contrôle de la qualité de l'ARN
- Ajouter 2 ul d'ARN à 18 pi de stériles dH sans RNAse-2 O pour diluer 1:10.
- Dans les réactions PCR séparées, ajouter 1 pl de dilution pur ou 1:10 de l'ARN à 0,5 ml tube stérile PCR. Ajouter PCR composants de réaction pour amplifier le gène ARNr 16S selon le tableau 3. Inclure un (par exemple., L' ADN extrait d'une culture bactérienne pure), un «PCR inhibiteur" contrôle "positif" (ie, 1 pl de l' ADN de culture pure positive et 1 pi d'ARN extrait pur) et un contrôle «négatif» de dH sans RNAse stérile 2 O.
- Régler le thermocycleur avec les paramètres d'amplification suivantes: 95 ° C 5 min, 95 ° C (30 s, 57 ° C 30 sec, 72 ° C 1 min) x 35 cycles, 72 ° C 10 min.
- Exécuter 10% du produit de PCR sur un gel d'agarose à 1% à 85 V pendant 40 min. Répéter le traitement à la DNase I, si un produit PCR est formé dans l'environnement RNA échantillons.
- Ajouter 2 ul d'ARN à 18 pi de stériles dH sans RNAse-2 O pour diluer 1:10.
3. Génération de premier brin d'ADNc à partir d'ARN
- Ajouter les réactifs selon le tableau 4 à un RNAse / DNAse 0,2 ml de tube PCR.
- Incuber l'échantillon à 65 ° C 5 min. Placer sur de la glace pendant au moins 1 min, puis on incube à 25 ° C pendant 10 min.
- Dans le même tube ajouter les réactifs listés dans le Tableau 5.
- Incuber l'échantillon à 55 ° C pendant 50 min. Inactiver la réaction à 72 ° C pendant 10 min.
- ADNc de magasin à -20 ° C jusqu'à utilisation.
4. PCR quantitative
- Génération de normes d'ADN pour qPCR
- Amplifier la séquence d' ARNr 16S bactérienne 20 (ou toute autre séquence d'intérêt) à partir de l' ADN génomique extrait d' une culture pure avec des amorces de PCR (q-1369F et 1492R 21).
- On purifie le produit PCR en utilisant un kit commercial selon les instructions du fabricant.
- Clone du PCR purifiéproduit en un débord 3 'T clonage système de vecteur selon les instructions du fabricant.
- Transformer le vecteur contenant le PCR insérer dans Escherichia coli cellules compétentes JM109 selon les instructions du fabricant.
- Plaque 50 pi de transformation ONTO ampicilline (100 pg / ml), X-gal (20 pg / ml) et IPTG (0,5 mM) de Luria Bertani plaques de gélose (tryptone 10 g / L, extrait de levure 5 g / L, NaCl 10 g / l, agar 15 g / L). Incuber O / N à 37 ° C.
- Sélectionnez un seul transformant blanc positif de la plaque. Inoculer le transformant positif dans 50 ml de bouillon LB contenant 100 ug / ml d'ampicilline et incuber O / N agitation à 250 tours par minute à 37 ° C.
- De l'O / N culture, d'isoler et de purifier le plasmide en utilisant un kit commercial selon les instructions du fabricant.
- Linéariser le plasmide en utilisant une enzyme de restriction appropriée qui est capable de couper le plasmide une fois (par exemple Eco RI). > Analyse de la pureté du plasmide en mesurant le rapport d'absorption A260 / 280 19. Si le ratio est inférieur à 1,8, re-extraire le plasmide avec du phénol-chloroforme 19. Remarque: L'ADN pur a un rapport de 1,8.
- Déterminer la concentration de l' ADN plasmidique par l' absorbance à 260 nm en utilisant un spectrophotomètre 19.
Remarque: En variante, la précision de la quantification peut être améliorée par l'utilisation d'un colorant de liaison à l'ADN fluorescent utilisé en conjonction avec un fluoromètre. - Séquence du plasmide insérer pour confirmer la taille de la cible dans des bases. Calculer la quantité (c. -à- pourcentage) d'ADN inséré dans le plasmide (par exemple dans un vecteur de 3000 pb, un fragment d'ADN de 123 pb est de 3,9% de l' ADN total) et multiplier par la concentration obtenue dans l' 04/01/10, pour déterminer la la concentration d'ADN de la cible (insert).
- Utilisez l'équation suivante pour calculer les copies de cible par ul:
charge / 54067 / 54067eq1.jpg "/> - Calculer le poids cible moléculaire (TMW) en multipliant le nombre de paires de bases de l'ADN cible par le poids moléculaire moyen de l'ADN double brin (dsDNA, 660 Daltons par paire de base).
- Diluer l'ADN cible dans une plage de 10 10 à 10 2 copies en utilisant 2 pi d'ADN et 18 pi de dH 2 O stérile Mélanger chaque puits de dilution et centrifuger brièvement avant de prendre la prochaine dilution.
- Génération de la courbe standard d'ARN pour qPCR
- De l' étape 4.1.5, écran un certain nombre de colonies blanches (environ 5) par PCR sur colonie 19 utilisant l'amorce inverse cible (ie, R1492) et le plasmide vecteur avant l' amorce M13F.
Remarque: Cette combinaison d'amorces va amplifier les inserts clonés dans l'orientation sens correct avec un site promoteur T7 en amont. - Purifier le produit PCR en utilisant un kit commercial en suivant les instructions du fabricant. Quantifier le p PCR purifiéroduit à 260 nm en utilisant un spectrophotomètre.
- Ajouter 200 ng de produit de PCR dans un volume final de 20 ul avec 7,5 mM de chaque ribonucleotide, un tampon de réaction 1 de T7 et une polymerase de T7 pour l' enzyme in vitro réaction de transcription.
- Incuber la réaction pendant 4 heures à 37 ° C. Ajouter 1 unité de RNase DNase I à la réaction, incuber pendant 15 min à 37 ° C pour éliminer la matrice d'ADN.
- Récupérer l'ARN transcrit in vitro par précipitation à l' éthanol 19 et de quantifier les rendements d'ARN en utilisant un fluorimètre ou par absorbance à 260 nm.
- Calculer le nombre de transcription par ul d'ARN. Ajouter le nombre de paires de bases dans l'ARN cible (par exemple, 123 bases) de la longueur du promoteur T7 (153 bases) et multiplier par le poids moléculaire moyen de l' ARN, 340 Daltons par base.
- Ajouter 500 ng d'ARN cible quantifiée à une réaction de la transcriptase inverse (RT), comme indiqué dans la section 3. En série dilué l'ADNc tel que décrit dans 4.1.13 pour une utilisationcomme la courbe standard.
- De l' étape 4.1.5, écran un certain nombre de colonies blanches (environ 5) par PCR sur colonie 19 utilisant l'amorce inverse cible (ie, R1492) et le plasmide vecteur avant l' amorce M13F.
- PCR en temps réel
- ADN environnemental Thaw / échantillons d'ADNc, des normes et des réactifs qPCR sur la glace. Protéger la sonde fluorogène de la lumière.
- Diluer les normes pour la courbe standard comme indiqué dans 4.1.14. Faire des dilutions 1:10 de l'ADN de l' environnement / ADNc de pur à 10 -3 en ajoutant 2 ul d'échantillon à 18 pi de dH stérile 2 O.
- Faire une réaction maître de mélange pour le nombre total de réactions plus 10% selon le tableau 6 (amorces et la sonde) et le tableau 7 (mélange réactionnel). Bien mélanger et centrifuger brièvement.
- Ajouter 19 pi de mélange maître et 1 pl de modèle (standard, l'échantillon de l'environnement ou de l'eau pour le contrôle négatif) à chaque puits dans une plaque à 96 puits optique qPCR. Effectuez chaque réaction (normes, des échantillons environnementaux et des contrôles négatifs) en triple.
- Couvrir la plaque de 96 puits avec un couvercle optique q-PCR. Centrifuger brièvement la plaque. Chargez lela plaque dans le bloc de chauffage de la machine qPCR et fermer le couvercle.
- Ouvrez le logiciel de gestion qPCR. Cliquez sur l'onglet "protocole", cliquez sur "créer un nouveau", ajouter les conditions d'amplification suivantes: 3 min 95 ° C, (10 sec 95 ° C, 30 sec 60 ° C) x 40 cycles. Régler le volume de l'échantillon à 20 pi, cliquez sur "OK" et enregistrez le protocole.
- Cliquez sur l'onglet "Plate". Sous "Modifier la sélection" cliquez sur "sélectionner fluorophore", ajouter le rapport colorant pour la sonde appropriée, par exemple, la fluorescéine. Cliquez sur "OK".
- Mettez en surbrillance les puits contenant les normes, sélectionnez "standard" dans le menu "de type d'échantillon". Cliquez sur "répliquer série" pour changer "répliquer taille" à 3, cliquez sur "appliquer". Cliquez sur "série de dilution" pour ajouter la concentration de départ calculée pour la norme 1, indiquer le facteur de dilution (10), sélectionnez «en baisse» ou «augmenter», selon l'ordre a été ajouté la courbe standardà la plaque. Cliquez sur "Appliquer".
- Mettez en surbrillance et positions contenant les échantillons inconnus, sélectionnez «inconnu» du «type d'échantillon" dans le menu déroulant. Cliquez sur "répliquer série" et changer "répliquer taille" à 3. Cliquez sur "appliquer". Modifier les noms d'échantillons sous "nom de l'échantillon". Répéter la procédure pour les contrôles sans matrice (NTC) puits.
- Cliquez sur "OK" sur la boîte de l'éditeur de plaque et enregistrer le fichier. Cliquez sur "start run".
- À l'issue de la course, la fenêtre d'analyse de données ouvrira. Cliquez sur l'onglet "quantification" pour afficher la courbe standard, les descripteurs standards de la courbe (tableau 8) et des parcelles d'amplification.
- Cliquez sur l'onglet "Données de quantification" pour les valeurs de Cp et correspondant à partir du gène quantité (SQ) d'échantillons inconnus. table Exporter vers un logiciel tableur si nécessaire.
- abondances géniques Multiply à partir d'échantillons inconnus par le facteur de dilution, la quantité de sédiments extraccié et les acides nucléiques volume total ont été élues pour obtenir des copies de gènes par gramme de sédiments de poids humide.
Note: Éluer l'ADN de 0,5 g de sédiments dans 50 ul, diluer 1/10, ajouter 1 pl comme matrice pour une réaction q-PCR; calculer copies du gène g -1 sédiments en multipliant: copies du gène pi de l' ADN x dilution du volume x facteur d'élution x 2.
Résultats
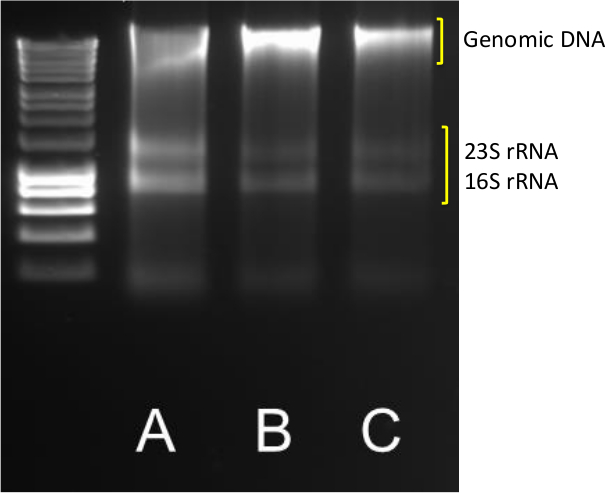
L'extraction des ADN de bonne qualité et de l'ARN à partir des sédiments est la première étape dans la quantification de gènes et de transcription abondances. Un succès des rendements d'extraction des bandes d'ADN et d' ARN claires comme indiqué sur la Figure 1 pour l' échantillon AC, où des bandes 23S et 16S ARNr vives sont visibles en plus de la masse moléculaire génomique bande haute de l' ADN.

Figure 1. ADN / extraction de l' ARN. Les résultats typiques de l' extraction d' ADN / ARN de triplicate (ABC) 0,5 g de sédiments côtiers. 5 pi d'ADN / ARN a été exécuté sur un agarose de 1,4% à 85 V pendant 40 min. Un marqueur moléculaire dans l'intervalle 10,037-200 pb a été utilisé. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Pour préparer l'ARN, une digestion de l'ADN co-extraite est obligatoire. Ceci doit être suivi d'ARNr 16S point final par PCR de l'ARN pour s'assurer que l'ADN a été supprimé avec succès. Si l'ADN a été complètement retirée seulement une bande dans le contrôle positif est observé. Il est important d'utiliser à la fois pur et une dilution 1:10 de l'ARN pour assurer les inhibiteurs ne sont pas empêcher la formation d'un produit de PCR. L'ARN peut maintenant subir la réaction de la transcriptase inverse pour la convertir en ADNc. Ceci peut être réalisé soit avec des amorces spécifiques d'un gène ou d'hexamères aléatoires. Typiquement, la réaction est effectuée dans une machine de PCR, afin d'assurer un profil de température optimal. Cet ADNc est utilisé comme matrice dans la réaction subséquente qPCR. ADN et l'ARN est quantifié pour déterminer la concentration de matrice utilisée dans chaque réaction.
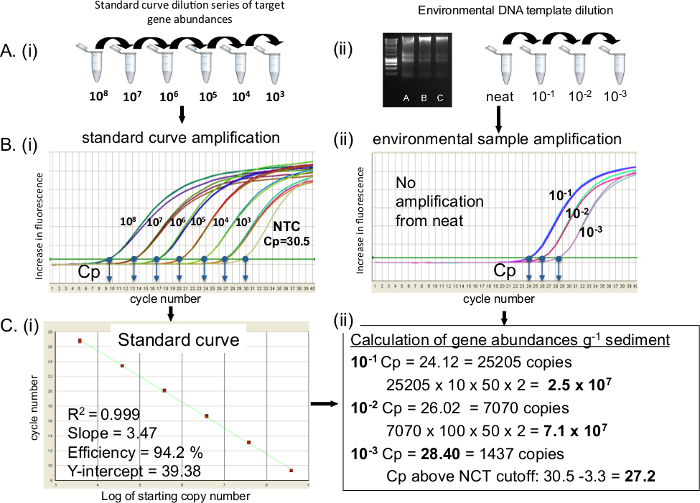
Un protocole de réaction qPCR est décrit cible ARNr 16S ADN. Pour 16S ARNr transcriptions remplacent la norme curve et la matrice avec de l'ADNc. Que des échantillons inconnus sont quantifiés sur la courbe d' étalonnage, il est impératif de veiller à ce que la courbe d' étalonnage est de bonne qualité. La figure 2 montre la préparation de (A), des courbes d' étalonnage et les dilutions d'ADN de l' environnement, (B), l' amplification de la courbe standard et des échantillons environnementaux (C) la conversion de la courbe standard à une régression linéaire et le calcul du nombre de copies du gène. Quand une gamme de dilution de 10 fois est correctement préparé et amplifié une différence de 3,3 entre chaque cycle de dilution standard est vu (il faut 3,3 cycles pour une augmentation de dix fois dans le modèle à 100% d' efficacité d'amplification) (Figure 2Bi). Courbes standard descripteurs, y compris les valeurs de R 2 de 0,99 et une efficacité de PCR dans l'intervalle de 90 à 110% (figure 2C) sont souhaitées. Il est important de signaler les valeurs de Cp du pas de contrôle de gabarit (NTC) si elle est présente. Si cela se produit une coupure de Cp des normes et des échantillons inconnus 3.3 cycles ( ie, un pli du journal) supérieure à la valeur de Cp du CNT est imposée 20 (Figure 2Cii). Modèle inconnu à partir d' ADN extrait a été quantifié à partir de sédiments propres, 10 -1, 10 -2 et 10 -3 dilutions (B). L'échantillon pur n'a pas amplifier, les valeurs de Cp de 10 -1 à 10 -3 dilutions étaient 24.12, 26.02 et 28.40 respectivement. Le CNT Cp était de 30,5, une coupure NCT de 27,2 a été imposée. Convertir les abondances de gène à g -1 sédiment de poids humide a donné lieu à 2,5 x 10 7 et 7,1 x 10 7 à 10 -1 et 10 -2 respectivement pour la gamme de dilution. 10 -3 dilution Cp était inférieur au seuil de NTC, et n'a donc pas utilisé. Dans ce cas , le 10 -2 a été choisi comme matrice dilution optimale.

Figure 2. gène ARNr 16S qPCR amplification des normes et de l' ADN extrait de l' environnement de sédiments côtiers. Préparation de (A) i) courbe standard de l' ADN et ii) des dilutions d'ADN pour l' environnement qPCR amplification, (B) qPCR amplification de i) la courbe standard de l' ADN et ii) des échantillons environnementaux, Cp pour chaque échantillon sont présentés. (C) i) de régression linéaire de la courbe standard avec des descripteurs de courbe standards; ii) le calcul des abondances de gènes à partir d' échantillons environnementaux. NTC:. Template Control Négatif S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
| Réactif | Montant |
| CTAB | 5 g |
| K 2 HPO 4 X3H 2 O | 2,58 g |
| KH 2 PO 4 | 0,1 g |
| NaCl | 2,05 g |
| dH2Û | à 100 ml |
Tableau 1. Préparation de tampon CTAB-phosphate.
| Réactif | Montant |
| PEG 6000 | 30 g |
| NaCl | 9,35 g |
| dH2Û | à 100 ml |
| Note: ajouter PEG 6000 lentement sous chauffage et l' agitation de 50 ml de dH 2 O. modérée | |
Tableau 2. Préparation de la solution de précipitation PEG-NaCl.
| Réactif | Montant |
| ARN non dilué / 01h10 ARN dilué | 1,0 ul |
| Tampon PCR 10x | 5,0 ul |
| dNTP 10 mM | 1,0 ul |
| 63F amorce sens | 1,0 ul |
| CAG GCC TAA CAC ATG CAA GTC | |
| 518R amorce inverse | 1,0 ul |
| ATT ACC GCG GCT GCT GG | |
| la polymérase Taq | 0,4 pi |
| DH 2 O libre RNAse | 40,6 pi |
Tableau 3. qualité de l' ARN maître contrôle de PCR mélange sans ADN.
| Réactif | Montant |
| ARN | 0,5-5 pg |
| dNTP 10 mM | 1 ul |
| Amorce antisens 10 uM / Aléatoire hexamères 50 uM | 1 ul |
| DEPC-H 2 O | Jusqu'à 13 ul |
Tableau 4. ADNc réaction de synthèse mélange A.
| Réactif | Montant |
| Buffer 5x | 4 ul |
| TNT | 1 ul |
| RNase Inhibitor | 1 ul |
| transcriptase inverse | 1 ul |
Tableau 5. ADNc de la réaction de synthèse de mélange B.
| Cible | nom Primer | Séquence | Recuit T (° C) | Référence |
| bactéries ARNr 16S | Bact1369F | CGG TGA ATA CGT TCY CGG | 56 | Suzuki et al. , 2000 19 |
| Prok1492R | GGW TAC CTT GTT ACG ACT T | |||
| TM1389F (sonde) | CTT GTA CAC ACC GCC CGT C |
| Réactif | Montant |
| 2x Master Mix | 10 ul |
| amorce Forward (10 uM) | 0,8 pi |
| Amorce antisens (10 uM) | 0,8 pi |
| Sonde (10 pM) | 0,4 pi |
| Eau | 7,0 pi |
| Le volume final | 19 pi |
Tableau 7. Temps réel réaction PCR mélange.
| Descriptor | Importance |
| Le coefficient de corrélation (R 2) | Une mesure de la linéarité de la courbe standard. Idéalement , il devrait être R 2 = 1. Valeur de R 2 = 0,98-0,99 sont acceptables. |
| Slope (α) | Une mesure de l'efficacité de la réaction. Idéalement, il devrait être équivalent à -3,32. Valeur dans la plage comprise entre -3,58 et -3,1. |
| Efficacité (= 10 (-1 / pente -1)) | Si elle est de 100%, les modèles double après chaque cycle thermique pendant l'amplification exponentielle. Une bonne gamme d'efficacité se situe entre 90 et 110%. |
| Y Intercept (β) | La limite théorique de la détection de la réaction. Bien que non utilisé pour quantifier la sensibilité de la réaction, il est important lorsque l'on compare les différentes courbes d'étalonnage de la même cible. |
Tableau 8. qPCR descripteurs de réaction.
Discussion
La combinaison de l'ADN d'extraction / d'ARN avec qPCR fournit une méthode rapide, précise, relativement rentable pour la quantification sensible du gène et de transcription abondances à partir d'une gamme d'échantillons environnementaux, tels que les sédiments côtiers.
L'extraction d'acide nucléique initiale est l'étape critique pour assurer une vue représentative de la communauté microbienne présente est atteint 22. Un certain nombre de limites doivent être pris en considération pour le protocole d'extraction: a) la réalisation de la lyse totale des cellules, b) la co-extraction des composés inhibiteurs (par exemple, des acides humiques ou polyphénoliques), c) l' ADN contaminant dans la fraction d'ARN, d) dégradation rapide des acides nucléiques extraits lorsqu'ils sont stockés. Des précautions doivent être prises afin de contourner ces limitations. Par exemple, un soin particulier doit être pris pour assurer que l'extraction des acides nucléiques est optimisée pour le type d'échantillon (par exemple, les sédiments, le sol, les eaux usées, etc.). Des améliorations significatives dans le rendement d'acide nucléique et de la qualité à la fois l' ADN et l' ARN peuvent être obtenus en réalisant des expériences préliminaires 23. Afin de minimiser l'effet des composés inhibiteurs sur les applications basées sur la PCR 24, tester une gamme de dilutions de l'ADN extrait et de l' ARN. Pour contourner la dégradation rapide de l'ADN / ARN extrait à partir de plusieurs cycles de gel-dégel et d'éviter la perte possible de l'information génétique, enregistrer plusieurs petites portions de volume à -80 ° C.
Lorsqu'ils sont conçus avec soin, qPCR est une méthode robuste, hautement reproductible et sensible. Notamment, les méthodes d'amplification et de construction de courbe standard décrites dans ce protocole peuvent être adaptés à toute cible de gène d'intérêt, y compris d' autres marqueurs phylogénétiques (ie, archées ARNr 16S, 18S fongiques ARNr) ou des gènes impliqués dans des fonctions importantes dans l'environnement. Limitations connues dans l'utilisation de qPCR sont: a) la génération de reproductibles de haute qualité scourbe de tandard pour la quantification absolue, b) le choix des amorces / sondes et l'optimisation des conditions d'essai q-PCR, c) l'utilisation de faible qualité / acides nucléiques cisaillées, d) le choix de la dilution de travail de l'ADN / ARN pour éviter l'inhibition . En outre, il doit être considéré que la technique qPCR fournit abondances gène / transcription qui peut ne pas assimiler à la cellule compte: ceci est particulièrement le cas lorsque les gènes 16S et 18S sont ciblés, comme les micro-organismes ont différents nombres de copies du gène ribosomal dans leur génome 25.
courbes standard de mauvaise qualité se traduira par une quantification inexacte du gène d'intérêt. Il est de bonne pratique pour stocker un stock de normes de concentration élevées en petites portions à partir de laquelle les courbes standard fraîches peuvent être faites. Ne pas stocker des courbes standard, toujours faire des dilutions fraîches à partir d'un stock de la plus forte concentration à chaque fois. Pour la quantification précise des échantillons environnementaux assurer la gamme de concentrations de l'stancourbe de dard couvre les valeurs Cp attendues des échantillons inconnus. Pour quantifier la transcription, la construction de la courbe standard à partir d'ARN pas l'ADN double brin. Lorsque cela est possible, la quantification des échantillons d'être directement comparés doit être achevée dans un seul essai pour éviter inter-essai variation 20. Cela peut ne pas être toujours possible. Par conséquent, pour comparer les abondances de gènes générées entre les dosages, il est conseillé d'avoir un sous-ensemble aléatoire d'échantillons répliqués entre les dosages. Une large sélection de amorce et de sonde ensembles sont actuellement disponibles pour qPCR ciblage taxa microbienne et des groupes fonctionnels 15. Une attention particulière est nécessaire lors de la sélection de ces pour assurer à la fois la couverture maximale et une spécificité pour le groupe cible. Si le rendement de la réaction d'un qPCR est pas satisfaisante, le dépannage commence par tester différentes conditions de cyclage thermique (tel que le temps de recuit et / ou de la température) et / ou les conditions de réaction, par exemple, des concentrations variant amorce-sonde. Once du dosage et les conditions de réaction q-PCR sont optimisés, toujours effectuer un premier test d'une série de dilutions d'ADN / ADNc pour déterminer la concentration de modèle approprié. Sélectionnez la plage de dilution résultant du nombre de copies plus élevé que la dilution du modèle optimal pour d'autres essais.
Actuellement, les technologies de séquençage de prochaine génération mettent efficacement la lumière sur la structure et les fonctions de la communauté microbienne dans une multitude d'environnements 26,27,28. Cependant, ces ensembles de données sont souvent basées sur point final PCR bibliothèques amplicon et fournissent donc que des évaluations semi-quantitatives de l'abondance des taxons particuliers. Par conséquent, la capacité de la technique à base de PCR en temps réel pour cibler les marqueurs taxinomiques spécifiques (de domaine supérieur jusqu'au niveau de déformation) permet de valider l'efficacité des résultats obtenus par séquençage de la prochaine génération. En outre, qPCR a été utilisé avec succès en combinaison avec d'autres méthodes moléculaires de l'écologie microbienne tels que iso stabletope sondage (SIP) ou microarrays phylogénétiques / fonctionnels. Combiné avec l'ancien outil, qPCR peut être utilisée pour quantifier la communauté métaboliquement active 29,30. Lorsqu'il est combiné avec l' analyse des microréseaux, qPCR fournit une interprétation quantitative clé des enquêtes basées sur des marqueurs et gènes fonctionnels phylogénétiques des environnements 31,32.
Par conséquent, qu'il soit utilisé seul ou en combinaison avec d'autres (souvent basées sur des processus) des évaluations de la fonction de l'écosystème, la PCR quantitative est un outil essentiel pour les écologistes microbiens dans l'exploration du lien insaisissable entre les communautés microbiennes et les fonctions des écosystèmes.
Déclarations de divulgation
The authors have nothing to disclose.
Remerciements
Cette publication a émané de la recherche menée avec le soutien financier du Natural Environment Research Council (NERC) sous le numéro de subvention NERC NE / JO11959 / 1 et de la Science Foundation Ireland & Marie-Curie d'action COFUND sous le numéro Grant 11 / GSS / B2159awarded à CJS et l'Academic Research Consortium de l'Est (ARC Est).
matériels
| Name | Company | Catalog Number | Comments |
| Diethylpyrocarbonate | Sigma | 40718 | Toxic, open under chemical hood |
| cetrimonium bromide (CTAB) | Sigma | 52365 | Irritant, open under chemical hood |
| potassium phosphate dibasic | Sigma | RES20765 | |
| potassium phosphate monobasic | Sigma | P9791 | |
| Phenol : Chloroform : Isoamylalcohol pH 8 | Sigma | P2069 | Equilibrate at pH 8 before using |
| Chloroform : Isoamylalcohol | Sigma | 25666 | |
| sodium chloride | VWR | 1.06404.0500 | |
| polyethylenglycol 6000 | VWR | 528877-100 | |
| Ethanol Molecular Grade | Sigma | E7148 | |
| Lysing Matrix E tubes | MP Biomedical | 116914050 | |
| Turbo DNAse | Ambion | AM1907 | |
| Taq polymerase | Sigma | D1806 | |
| dNTPs | Bioline | BIO-39028 | |
| SuperScript III | Life Technologies | 18080044 | |
| Rnase Inhibitor | Life Technologies | 10777019 | |
| RNAse/DNAse free 0.2 ml PCR tubes | Sarstedt | 72.985.002 | |
| SureCleanPlus | Bioline | BIO-37047 | |
| pGEM Easy T Vector | Promega | A1360 | |
| E. coli JM109 competent cells | Promega | L2005 | |
| Tryptone | Sigma | T7293 | |
| Yeast Extract | Sigma | 70161 | |
| Ampicillin | Sigma | 59349 | |
| X-Gal | Bioline | BIO-37035 | |
| IPTG | Bioline | BIO-37036 | |
| Agar | VWR | 20768.235 | |
| Plasmid Midi Kit | Qiagen | 12143 | |
| Qubit | Life Technologies | Q33216 | |
| Quant-IT DNA HS Assay | Life Technologies | Q-33120 | |
| Quant-IT RNA HS Assay | Life Technologies | Q32855 | |
| MEGAshortscript kit | Ambion | AM1354 | |
| TaqMan SensiFast Probe Lo-ROX kit | Bioline | BIO-84002 | |
| qPCR machine |
Références
- Epstein, S. S. The phenomenon of microbial uncultivability. Curr. Opin. Microbiol. 16 (5), 636-642 (2013).
- Zhou, J., Bruns, M. A., Tiedje, J. M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62, 316-322 (1996).
- Miller, D. N., Bryant, J. E., Madsen, E. L., Ghiorse, W. C. Evaluation and optimization of DNA extraction and purification procedures from soil and sediments samples. Appl. Environ. Microbiol. 65 (11), 4715-4724 (1999).
- Burgmann, H., Pesaro, M., Widmer, F., Zeyer, J. A strategy for optimizing quality and quantity of DNA extracted from soil. J. Microbiol. Methods. 45 (1), 7-20 (2001).
- Hurt, R. A., et al. Simultaneous recovery of RNA and DNA from soils and sediments. Appl. Environ. Microbiol. 67, 4495-4503 (2001).
- Griffiths, R. I., Whiteley, A. S., O'Donnell, A. G., Bailey, M. J. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl. Environ. Microbiol. 66 (12), 5488-5491 (2000).
- Martins, G., et al. Structure and activity of lacustrine sediment bacteria involved in nutrient and iron cycles. FEMS Microbiol. Ecol. 77 (3), 666-679 (2011).
- Kuffner, M., et al. Effects of season and experimental warming on the bacterial community in a temperate mountain forest soil assessed by 16S rRNA gene pyrosequencing. FEMS Microbiol. Ecol. 82 (3), 551-562 (2011).
- Tatti, E., et al. Influences of over winter conditions on denitrification and nitrous oxide-producing microorganism abundance and structure in an agricultural soil amended with different nitrogen sources. Agric. Ecosyst . Environ. 183, 47-59 (2014).
- Giovannoni, S. J., Britschgi, T. B., Moyer, C. L., Field, K. G. Genetic diversity in the Sargasso Sea bacterioplankton. Nature. 345, 60-62 (1990).
- Ward, D. W., Weller, R., Bateson, M. M. 16S rRNA sequences reveal numerous uncultured microorganisms a natural community. Nature. 345, 63-65 (1990).
- Suzuki, M. T., Giovannoni, S. J. Bias caused by template annealing in the amplification of mixture of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 62, 625-630 (1996).
- Wittwer, C. T., Herrmann, M. G., Moss, A. A., Rasmussen, R. P. Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques. 22, 130-138 (1997).
- Holland, P. M., Abramson, R. D., Watson, R., Gelfand, D. H. Detection of Specific Polymerase Chain Reaction Product by Utilizing the 5' 3' Exonuclease Activity of Thermus aquaticus DNA Polymerase. PNAS. 88, 7276-7280 (1991).
- Smith, C. J., Osborn, A. M. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 67, 2-20 (2009).
- Park, S. J., Park, B. J., Rhee, S. K. Comparative analysis of archaeal 16S rRNA and amoA genes to estimate the abundance and diversity of ammonia-oxidizing archaea in marine sediments. Extremphiles. 12 (4), (2008).
- Urakawa, H., Yoshida, T., Nishimura, M., Ohwada, K. Characterization of depth-related population variation in microbial communities of a coastal marine sediment using 16S rDNA-based approaches and quinone profiling. Environ. Microbiol. 2, 542-554 (2008).
- Smith, C. J., Dong, L. F., Wilson, J., Stott, A., Osborn, A. M., Nedwell, D. B. Seasonal variation in denitrification and dissimilatory nitrate reduction to ammonia process rates and corresponding key functional genes along an estuarine nitrate gradient. Front. Microbiol. 6, 542(2015).
- Sambrook, J., Russell, D. W. Molecular Cloning: a laboratory manual. , Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. 205(2001).
- Smith, C. J., Nedwell, D. B., Dong, L. F., Osborn, A. M. Evaluation of Quantitative Polymerase Chain Reaction (Q-PCR) based approaches for determining gene copy and gene transcript numbers in environmental samples. Environ. Microbiol. 8 (5), 804-815 (2006).
- Suzuki, M. T., Taylor, L. T. DeLong E.F., Quantitative analysis of small-subunit rRNA genes in mixed microbial population via 5'-nuclease assays. Appl. Environ. Microbiol. 66, 4605-4614 (2000).
- Luna, G. M., Dell'Anno, A., Danovaro, R. DNA extraction procedure: a critical issue for bacterial diversity assessment in marine sediments. Environ. Microbiol. 8 (2), 308-320 (2005).
- Lever, M. A., Torti, A., Eickenbursch, P., Michaud, A. B., Šantl-Temkiv, T., Jørgensen, B. B. A modular method for the extraction of DNA and RNA, and the separation of DNA pools from diverse environmental sample types. Front. Microbiol. 6, 476(2015).
- Lloyd, K. G., MacGregor, B. J., Teske, A. Quantitative PCR methods for RNA and DNA in marine sediments: maximizing yield while overcoming inhibition. FEMS Microbol. Ecol. 72, 144-151 (2010).
- Klappenbach, J. A., Dumbar, J. M., Schmidt, T. M. rRNA Operon copy number reflects ecological strategies of bacteria. App. Environ. Microbiol. 66 (4), 1328-1333 (2000).
- Shi, Y., Tyson, G. W., Eppley, J. M., DeLong, E. F. Integrated metatrascriptomic and metagenomics analyses of stratified microbial assemblages in the open ocean. ISME J. 5, 999-1013 (2011).
- Howe, A. C., Jansson, J. K., Malfatti, S. A. Tringe S.G., Tiedje J.M., & Brown C.T. Tackling soil diversity with the assembly of large, complex metagenomes. PNAS. 111 (13), 4904-4909 (2014).
- Klaedtke, S., et al. Terroir is a key driver of seed-associated microbial assemblages. Environ. Microbiol. , (2015).
- Lueders, T., Wagner, B., Claus, P., Friedrich MW, Stable isotope probing of rRNA and DNA reveals a dynamic methylotroph community and trophic interactions with fungi and protozoa in oxic rice field soil). Environ. Microbiol. 6, 60-72 (2004).
- Kunapuli, U., Lueders, T., Meckenstock, R. U. The use of stable isotope probing to identify key iron-reducing microorganisms involved in anaerobic benzene degradation. ISME J. 1, 643-653 (2007).
- Bürgmann, H., et al. Transcriptional response of Silicibacter pomeroyi DSS-3 to dimethylsulfoniopropionate (DMSP). Environ. Microbiol. 9 (11), 2742-2755 (2007).
- He, J. Z., et al. Geochip: a comprehensive microarray for investigating biogeochemical ecological and environmental processes. ISME J. 1, 67-77 (2007).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.