
Method Article
Detección del número de copias alteraciones Uso de Secuenciación de los organismos unicelulares
En este artículo
Resumen
secuenciación de una sola célula es una herramienta cada vez más popular y accesible para hacer frente a los cambios genómicos en alta resolución. Proporcionamos un protocolo que utiliza la secuenciación de una sola célula para identificar alteraciones en el número de copias en las células individuales.
Resumen
La detección de los cambios genómicos en la resolución de una sola célula es importante para caracterizar la heterogeneidad genética y la evolución en los tejidos normales, el cáncer y las poblaciones microbianas. Los métodos tradicionales para evaluar la heterogeneidad genética se han visto limitados por la baja resolución, baja sensibilidad y / o especificidad baja. secuenciación de una sola célula se ha convertido en una herramienta poderosa para detectar la heterogeneidad genética con cuando se analiza adecuadamente alta resolución, alta sensibilidad y, de alta especificidad. Aquí proporcionamos un protocolo para el aislamiento, la amplificación del genoma completo, secuenciación y análisis de células individuales. Nuestro enfoque permite la identificación fiable de las variantes de número de copias megabase escala en células individuales. Sin embargo, los aspectos de este protocolo también se pueden aplicar para investigar otros tipos de alteraciones genéticas en las células individuales.
Introducción
Las alteraciones en el número de copias de ADN pueden variar en tamaño desde varios pares de bases (variantes de número de copia) (CNV) a la totalidad de los cromosomas (aneuploidía). Alteraciones del número de copias que afectan a grandes regiones del genoma puede tener consecuencias significativas fenotípicos mediante la alteración de la expresión de hasta miles de genes 1, 2. CNVs que están presentes en todas las células de una población se puede detectar por secuenciación granel o métodos de 3 a base de microarrays, 4. Sin embargo, las poblaciones también pueden ser genéticamente heterogénea, con CNVs existentes en un subconjunto de la población o incluso células individuales. La heterogeneidad genética es común en el cáncer, conduciendo la evolución del tumor, y también presente en los tejidos normales, con consecuencia desconocido 5, 6, 7, 8, 9 10.
Tradicionalmente, la heterogeneidad genética se evaluó ya sea por métodos citológicos o secuenciación granel. Citológicas enfoques, tales como hibridación in situ fluorescente (FISH), extensiones de cromosomas y cariotipo espectral (SKY), tienen la ventaja de identificar las alteraciones presentes en las células individuales, pero tienen altas tasas de error debido a los artefactos de la hibridación y la difusión 11. Estos enfoques también están limitados en su número de copias de la resolución de sólo revela los cambios que abarcan varias megabases. La secuenciación de ADN o microarrays de mayor, aunque superior en precisión y resolución, es menos sensible. Con el fin de detectar la heterogeneidad genética por los enfoques basados en la población, las variantes deben estar presentes en una fracción sustancial de las células en la población. La aparición de métodos para amplificar el ADN genómico a partir de células individuales ha permitido secuenciar el genoma de las células individuales. Seque una sola célulancing tiene las ventajas de alta resolución, alta sensibilidad, y, cuando se aplican los métodos de control de calidad apropiados, de alta precisión 12.
A continuación, describimos un método para detectar alteraciones del número de copias megabase escala en células individuales. Aislamos células individuales por microaspiraciones, amplificar el ADN genómico utilizando enlazador-adaptador de PCR, se preparan bibliotecas de secuenciación de próxima generación, y detectar las variantes de número de copias por tanto modelo oculto de Markov y segmentación binaria circular.
Protocolo
1. Aislamiento de células individuales
- Preparar el microaspirator
- Retire el extremo de plástico transparente del conjunto del tubo aspirador e inserte el extremo más estrecho en un extremo de un tubo de PVC de 1 pie de largo con 3/16 "de diámetro interior.
- Insertar la salida de un filtro de jeringa de 0,2 micras en el otro extremo del tubo de PVC 1 pie de largo con 3/6 "de diámetro interior.
- Inserte la entrada del filtro de jeringa de 0,2 micras en un extremo de un tubo de PVC de 6 pulgadas de largo con 5/16 "de diámetro interior.
- Opcional: Corte el tubo de PVC de 6 pulgadas de largo con 5/16 "de diámetro interno por la mitad e insertar un separador de agua en línea entre las dos mitades.
- Romper una pipeta serológica de 5 ml de plástico en la graduación de 1 ml e inserte el extremo roto en el extremo abierto del tubo de PVC con 5/16 "de diámetro interior.
- Insertar la salida de la pipeta serológica 5 ml de plástico en el extremo flexible de un conjunto de tubo de aspirador (donde el extremo de plástico transparentehabía sido eliminado).
- Para el almacenamiento, cubrir la boquilla rojo del conjunto del tubo aspirador con un tubo de plástico estéril.
- Es necesario realizar un tubo capilar de vidrio de un diámetro interior de 10 a 30 micras por lo que el centro del tubo cerca de una llama del quemador Bunsen y la aplicación de la tensión en cualquiera de los extremos del tubo. Romper el tubo estirado en el medio para crear dos agujas potenciales aspirador. Repita este paso con varios tubos, ya que sólo algunos tubos va a terminar con un diámetro interior que es apropiado para recoger células individuales.
- Para el almacenamiento, colocar dos tiras de plastilina en una placa de Petri de 15 cm y asegurar las agujas del aspirador a través de las tiras.
- recoger células
- Preparar una suspensión de células individuales según sea apropiado para el tipo de célula y el experimento. Por ejemplo, para preparar las células adherentes tales como líneas celulares de fibroblastos humanos, células de la cosecha de células tripsinización y transferir a un tubo cónico que contenía medio de comunicación adecuado.
- Antes, durante, o unaespués de la preparación de la suspensión de células individuales (dependiendo de cuánto tiempo se tarda en preparar la suspensión de una sola célula), la instalación de la campana para la amplificación del genoma completo.
- Rocíe la superficie de la campana, cajas de puntas de pipeta y puntas de pipeta con un 10% de lejía y limpiar con una toalla de papel. Repita este paso con un 70% de etanol.
- Añadir 8 l de agua desde el kit de amplificación de todo el genoma (WGA) a pocillos individuales de una placa de 96 pocillos PCR, un pocillo para cada celda a ser secuenciado. Cubrir la placa de 96 pocillos PCR con una tapa de una placa de cultivo de tejidos de 96 pocillos y el lugar en el hielo.
- Añadir 1.000 células a 10 ml de medio o solución salina tamponada con fosfato (PBS) en una placa de 15 cm Petri y colocar el plato en hielo para evitar que las células se adhieran al plato.
- Llevar las células en la placa de Petri y el pozo 96 de la placa de PCR con la tapa en hielo a un microscopio de luz con objetivo de 10X.
- Aumentar la apertura de la aguja de aspiración golpeando suavemente el extremo sacado de THe aguja de aspiración en una superficie dura tal que la punta se rompe. Inserte el extremo ancho de la aguja de aspiración en el extremo alejado de la microaspirator.
- Coloque la placa de Petri que contiene las células en la platina del microscopio. Coloque la boquilla del aspirador rojo en la boca. Utilice una mano para mover la aguja de aspiración y la otra mano para mover la placa de Petri que contiene las células. Identificar las células individuales a ser secuenciado.
- El uso de succión boca, dibujar una sola célula en la aguja de aspiración junto con ~ 1-2 l de los medios de comunicación o PBS. La transferencia de la célula en los 8 l de agua dentro de un solo pocillo de la placa de 96 pocillos PCR. Evitar la introducción de burbujas cuando se transfiere la célula.
- Repita el paso 1.2.7 hasta que se haya aislado el número deseado de células. Mantener la reacción PCR en hielo y cubierto con la tapa entre las células de la cosecha. Marcar los pozos que han recibido células.
- Al recoger células individuales, descongelar la única 10X tampón de lisis celular y la fragmentación de todo el gkit de amplificación enome. Después de recoger el número deseado de células, proceder inmediatamente a la amplificación del genoma completo.
2. Toda la amplificación del genoma
- Para evitar la contaminación durante la amplificación del genoma completo, añadir todos los reactivos dentro de una campana de cultivo de tejidos, utilizar puntas de pipeta con filtros y cambiar la punta de la pipeta en el medio pozos.
- Preparar una solución de lisis y la fragmentación de memoria temporal de trabajo desde el kit de amplificación de todo el genoma (WGA). Para cada conjunto de hasta 32 células, se combinan 32 l de 10x lisis de células individuales y el tampón de fragmentación y 2 l de solución de proteinasa K en un tubo de microcentrífuga. Vortex el tubo para mezclar las soluciones.
- Añadir 1 l de tampón de lisis y solución fragmentación de trabajo a cada pocillo y la pipeta hacia arriba y abajo para mezclar.
- Cubrir la placa y sellar todos los pocillos con una película de plástico. Centrifugar brevemente la placa en una mini placa de ruleta.
- ciclo térmico como follOWS: 50 ° C, 1 h y 99 ° C, 4 min.
- Se enfría la placa sobre hielo y centrifugar brevemente la placa en una mini placa de ruleta.
- Preparar una solución tampón de preparación de biblioteca de trabajo de todo el kit de amplificación del genoma. Para cada celda, se combinan 2 l de tampón de preparación de la biblioteca de células individuales 1x y 1 l de solución de estabilización biblioteca en un tubo de microcentrífuga. Preparar la solución de trabajo por varias células en el mismo tubo de microcentrífuga.
- Retire la película de plástico y añadir 3 l de solución de trabajo de tampón de preparación de la biblioteca a cada pocillo. Pipetear los contenidos del bien arriba y abajo para mezclar. Vuelva a colocar la lámina de plástico.
NOTA: La película de plástico puede ser reutilizado durante todo el proceso de amplificación del genoma hasta que los pozos empiezan a perforaciones en la película. Cuando esto ocurre, cambie a una nueva película de plástico. - Centrifugar brevemente la placa en un mini spinner placa y se incuba a 95 ° C durante 2 min.
- Se enfría la placa sobre hielo y brevementecentrifugar la placa en una mini placa de ruleta.
- Retire la película de plástico, añadir 1 l de enzima preparación biblioteca a cada pocillo, y la pipeta el contenido del bien arriba y abajo para mezclar. Mantenga la enzima preparación biblioteca en hielo o en un bloque frío a través de este paso. Vuelva a colocar la lámina de plástico.
- Centrifugar brevemente la placa en mini spinner placa y el ciclo térmico de la siguiente manera: 16 ° C, 20 min, 24 ° C, 20 min, 37 ° C, 20 min, 75 ° C, 5 min, y mantener a 4 ° C.
- Se enfría la placa sobre hielo y centrifugar brevemente la placa en una mini placa de ruleta.
- Preparar una mezcla de amplificación de trabajo de todo el kit de amplificación del genoma. Para cada celda, se combinan 48,5 l de agua, 7,5 l 10x amplificación de mezcla maestra y 5 l de ADN polimerasa WGA en un tubo de microcentrífuga. Preparar la mezcla de trabajo para varias células en el mismo tubo de microcentrífuga. Mantener la mezcla de trabajo en el hielo.
- Retire la película de plástico, añadir 61 l de trabajo mezcla de amplificaciónA cada pocillo, y contenido de la pipeta de bien arriba y abajo para mezclar. Mantener la mezcla de amplificación de trabajo en hielo o en un bloque frío a través de este paso. Vuelva a colocar la lámina de plástico.
- Centrifugar brevemente la placa en un mini spinner placa y el ciclo térmico de la siguiente manera: 95 ° C, 3 min, 25 ciclos de 94 ° C, 30 seg y 65 ° C, 5 min, y mantener a 4 ° C.
- Transferencia de 60 l de cada muestra a un tubo de microcentrífuga separado y se almacena a - 20 ° C para su uso en el paso 3.
- Añadir colorante de carga de ADN para el volumen restante de cada muestra (es decir, 3 l de ADN 6x carga de colorante a 15 l de muestra) y ejecute 5 l de cada reacción en un gel de agarosa al 1%.
NOTA: Las células que amplifican con éxito aparecerá como una mancha de 250 a 1000 pares de bases. Las muestras que no muestran una mancha, o sólo muestran una débil citología es poco probable que produzca datos de secuenciación útiles.
3. Secuenciación
- Purificar las muestras con perlas paramagnéticas.
- ThaLas muestras de ADN w en hielo. perlas paramagnéticas Vortex hasta que la solución es homogénea e incubar las perlas a temperatura ambiente durante al menos 30 minutos.
- Transferir 20 l de cada muestra de ADN en un tubo de microcentrífuga limpio. El resto de la muestra se puede almacenar a -20 ° C.
- Añadir 30 l (1,5x) de perlas paramagnéticas a cada muestra de ADN y agitar para mezclar. Se incuban las muestras a temperatura ambiente durante 10 min. Deje la solución madre de perlas a temperatura ambiente para su uso en el paso 3.4.
- Colocar los tubos en una tira tubo magnético durante 2 minutos, o hasta que los granos forman un sedimento y el sobrenadante es claro.
- Con una pipeta P200, eliminar la mayor cantidad del sobrenadante como sea posible sin molestar a los granos.
- Añadir 180 l de etanol al 80% a la mezcla de DNA-perla. Girar los tubos varias veces en relación con el imán para "aclarado" las perlas a través de la solución de etanol.
- Con una pipeta P200, eliminar la mayor cantidad de etanol como el lavado posible sin perturbar las perlas.
- Repita 3.1.6 y 3.1.7.
- Secar las cuentas por aproximadamente 10 minutos o hasta que ya no es visible etanol. Proceder al siguiente paso, cuando los granos aparecen grietas o caerse de la pared del tubo.
- Retire las muestras de la banda magnética. Añadir 40 l de 10 mM Tris pH 8.0 para eluir el ADN de las perlas y agitar las muestras para resuspender las perlas. Incubar durante 2 minutos a temperatura ambiente.
- Centrifugar brevemente las muestras para recoger el líquido en la parte inferior del tubo y se colocan los tubos en una tira magnética tubo durante al menos dos minutos. Transferir el eluyente en tubos de microcentrífuga limpios y sin molestar a los granos.
- la normalización de la muestra
- Cuantificar la concentración de cada muestra usando un espectrofotómetro. Las concentraciones deben variar de 10 a 30 ng / mL.
- Diluir cada muestra a 0,2 ng / l usando 10 mM Tris pH 8,0. Los siguientes pasos requerirán un mínimo de iNput de 5 l de esta dilución.
- la preparación de la biblioteca
- Preparar las bibliotecas de secuenciación siguiendo las instrucciones del kit de preparación de la biblioteca 13.
- la limpieza final
- Limpiar las muestras de acuerdo con la etapa 3.1. Para longitudes de lectura de 50 pb, 75 pb o 150 pb, utilizar una proporción de grano para degustar volumen de 1.5x, 1x, o 0,6x, respectivamente. Eluir con 15 l de 10 mM Tris pH 8,0.
- Ejecutar las muestras en un analizador de fragmentos para comprobar la distribución del tamaño de la biblioteca 14. La distribución del tamaño debe extenderse uniformemente desde 150 a la 900 pb.
- Cuantificar las muestras mediante qPCR
- El uso de un kit de cuantificación de la biblioteca, configure una reacción de PCR cuantitativa de acuerdo a las instrucciones del juego 15. Dejar un espacio en blanco la columna añadir las normas positivas (patrones de ADN del kit) y las normas negativo (agua u otra solución en blanco).
- ciclo térmico como folmínimos: 95 ° C, 5 min (velocidad de rampa de 4,8 ° C / s) y 35 ciclos de 95 ° C, 30 seg (velocidad de rampa de 4,8 ° C / s) y 60 ° C, 45 seg (velocidad de rampa de 2,5 ° C / seg).
- pooling
- Determinar cómo se necesitan muchos carriles en la celda de flujo y se dividen las muestras en grupos, con un grupo por cada carril.
- Para cada grupo de muestras, elegir la muestra con la concentración más baja sobre la base de los datos de qPCR de la etapa 3.5.2 y normalizar todas las muestras en ese grupo a que la concentración de uso de 10 mM Tris pH 8,0.
- Reunir las muestras en cada grupo.
- secuenciación
- Piscinas de carga en la máquina de secuenciación de próxima generación siguientes procedimientos estándar 16.
Análisis 4. Datos
NOTA: Se requiere un entorno basado en Unix para ejecutar los programas y scripts en esta sección. Instalar el software que se menciona en el protocolo después de su en guías de instala-. Todos los guiones se pueden encontrar en https://sourceforge.net/projects/singlecellseqcnv/.
- Recortar las lecturas de 40-nt usando fastx_trimmer partir de la versión 0.0.13 FASTX-Toolkit 17.
fastx_trimmer -Q33 -i example.fastq -l -o 40 example.trim.fastq - Alinear las lecturas con el genoma apropiado de referencia (mm.9 para el ratón, hg19 para el ser humano) usando BWA versión 0.6.1 con las opciones predeterminadas 18.
BWA ALN mm9.fa example.trim.fastq> example.sai
BWA Samse mm9.fa example.sai example.trim.fastq> example.sam - Retire las alineaciones de chrm y los cromosomas al azar, a continuación, ordenar e indexar el archivo resultante utilizando BAM SAMTools versión 0.1.19 19.
grep -v -w chrm example.sam | grep -v al azar> example.filtered.sam
samtools ver example.filtered.sam -uSh | samtools tipo - example.filtered
samtools índice example.filtered.bam - HMMcopy funcionar para detectar CNV= "Xref"> 20
- Utilice gcCounter en HMMcopy para generar un archivo de referencia GC porcentaje del genoma. Utilice la opción "-w 500000" para especificar el tamaño de la ventana. Utilizar la misma versión del archivo de referencia fasta utilizado en el paso 4.2, pero asegúrese de chrm y los cromosomas al azar se eliminan del archivo FASTA.
gcCounter -w 500000 mm9.fa> mm9_gc.wig - Utilice generateMap.pl en HMMcopy para generar un archivo de maniobra para mapeabilidad. Utilice la opción "-w 40" para especificar leer longitud. Utilizar el mismo archivo de referencia fasta utilizado en el paso 4.4.1.
generateMap.pl -b mm9.fa
generateMap.pl -w 40 -i -o mm9.fa mm9.fa mm9.bigwig - Utilice mapCounter en HMMcopy para generar un archivo de referencia mapeabilidad para el genoma. Utilice la opción "-w 500000" para especificar el tamaño de la ventana.
mapCounter -w 500000 mm9.bigwig> mm9_map.wig - Utilice readCounter en HMMcopy para generar un archivo de maniobra para cada archivo de BAM.
readCounter -w 500000 example.filtered.bam> input.wig - Modificar las rutas de acceso a los archivos de referencia en los guiones (R run_hmmcopy.mm9.r o run_hmmcopy.hg19.r), utilizar los archivos generados anteriormente en el apartado 4.4.1 (por ejemplo, mm9_gc.wig) y 4.4.3 (por ejemplo, mm9_map. peluca) para el GFile las variables y MFILE, que se refieren al archivo de archivo de referencia GC porcentaje y referencia mapeabilidad, respectivamente. A continuación, ejecute la secuencia de comandos proporcionada R "run_hmmcopy.mm9.r" o "run_hmmcopy.hg19.r".
R CMD run_hmmcopy.mm9.r LOTE
o
R CMD run_hmmcopy.hg19.r LOTE - Para el procesamiento por lotes de un conjunto de archivos BAM, BAM poner los archivos en una sola carpeta y ejecutar el guión previsto "HMMpipe.pl" en el paquete para llamar segmentos y calcular las puntuaciones de variabilidad (VS). Siga el formato:
Perl HMMpipe.pl Folder_to_BAMfiles mm.9
- Utilice gcCounter en HMMcopy para generar un archivo de referencia GC porcentaje del genoma. Utilice la opción "-w 500000" para especificar el tamaño de la ventana. Utilizar la misma versión del archivo de referencia fasta utilizado en el paso 4.2, pero asegúrese de chrm y los cromosomas al azar se eliminan del archivo FASTA.
- DNAcopy funcionar para detectar CNV 21.
- Utilice SAMtools versión 0.1.19 para extraer asignada de forma única lee de cada archivo de BAM.
samtools ver -h 0 -Fexample.filtered.bam x0004 | egrep -i "@ ^ | XT: A: T" | samtools ver -shū -> example.mapped.bam - Utilice MarkDuplicates en Picard versión 1.94 para marcar los duplicados de PCR en el archivo de BAM 22.
java-jar MarkDuplicates.jar ENTRADA SALIDA = example.mapped.bam = example.nondup.bam METRICS_FILE = example.dup REMOVE_DUPLICATES = true - Utilice coveragebed en bedtools versión 2.17.0 para contar mapeado lee en cada una de las ventanas dinámica 500kb mapeables predefinidos en el archivo de cama siempre "mm9.500k.dynamic.win.bed" o "hg19.500k.dynamic.win.bed" 23 .
coverageBed -abam example.nondup.bam -b mm9.500k.dynamic.win.bed Conteos | Ordenar -k1,1 -k2,2n> example.nondup.counts - Utilizar el script Perl proporcionado "normalizeGC.pl" para normalizar los recuentos de lectura basado en el archivo de referencia proporcionado contenido de GC "mm9.500k.dynamic.win.fa.gc.txt" o "hg19.500k.dynamic.win.fa. gc.txt ".
Perl normalizeGC.pl mm9.500k.dynamic.win.fa.gc.txt example.nondup.counts> example.bam.norm.counts - Utilice la secuencia de comandos R "dnacopy.r" proporcionado para llamar a los segmentos.
R CMD dnacopy.r LOTE
- Utilice SAMtools versión 0.1.19 para extraer asignada de forma única lee de cada archivo de BAM.
- identificar las VNC
- Excluir las células para las que el VS calculada en 4.4.6 es superior a 0,26.
- Filtrar los segmentos llamados por HMMcopy en 4.4.6 para incluir sólo aquellos para los que la ratio de la mediana de registro 2 es mayor que 0,4 (ganancia putativo) o menos de -0.35 (pérdida putativo).
- Filtrar los segmentos llamados por DNAcopy en 4.5.5 para incluir sólo aquellos para los que significa el segmento es mayor que 1,32 o menos de 0,6.
- La superposición de los segmentos de 4.6.2 y 4.6.3, llamando CNV sólo en las regiones donde tanto HMMcopy y DNAcopy identifican una ganancia o pérdida putativo putativo.
Resultados
El aspirador montado debería ser similar a la de la Figura 1A. La aguja debe establecerse de tal manera que es suficientemente ancha para acomodar una sola célula, pero no tan ancho que un gran volumen se redacta con la célula individual. De aspiración de las células individuales es más fácil cuando hay entre uno y cinco células en un campo de 10 veces (Figura 1B).
Si la amplificación del genoma entero tiene éxito, la muestra aparece como una mancha en un gel de agarosa (Figura 2, carriles 1, 2, 4, 5, 6, y 7). Un frotis débil o ausente indica una reacción de amplificación fallado y la muestra no debe ser secuenciado (Figura 2, carriles 3 y 8).
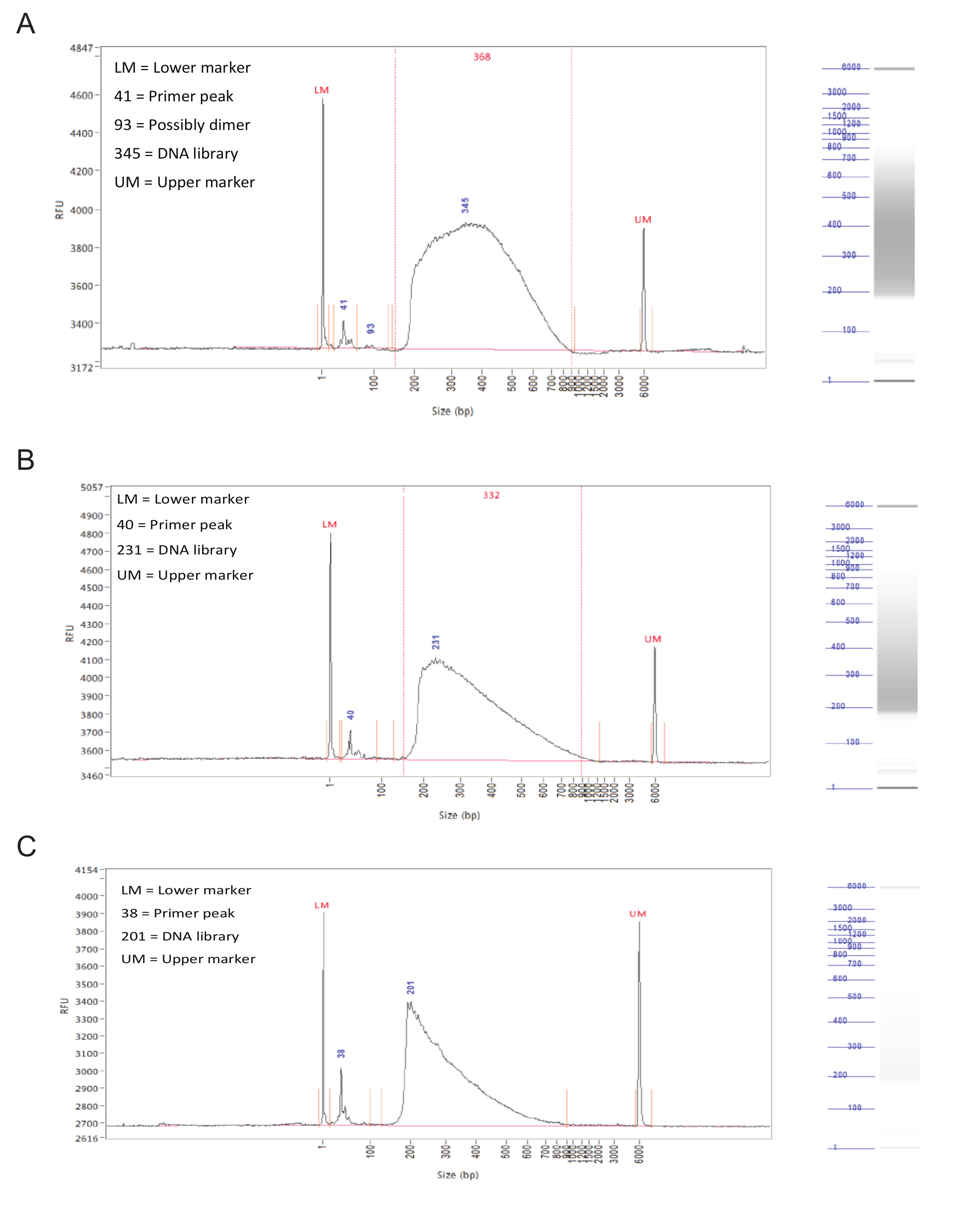
Después de la preparación de la biblioteca, la distribución de tamaño de los fragmentos de las muestras debe ser evaluada por electroforesis capilar en un analizador de fragmentos.Con éxito bibliotecas preparadas tendrán una distribución bastante uniforme de tamaños de los fragmentos 150 a 900 pb (Figura 3, A, B). Preparación biblioteca Error dará lugar a una distribución de tamaño de fragmento asimétricos y tales bibliotecas no debe ser secuenciado (Figura 3C).
El procesamiento de los datos de secuenciación a través del modelo oculto de Markov (HMMcopy) y la segmentación binaria circular (DNAcopy) analizará el genoma de cada célula en segmentos de número de copias estimado. Estos segmentos pueden ser filtrados para identificar aquellos con un número de copias estimado consistente con la ganancia o pérdida en una sola célula (Tabla 1). Estos segmentos filtrados desde HMMcopy y DNAcopy entonces se debe solapar para identificar alta confianza CNV.

Figura 1. Aislamiento de células individuales. ( A) Un ensamblado microaspirator. (B) Un campo 10X muestra disociada células (flechas) y la aguja microaspirator (esquina inferior derecha). De aspiración de las células individuales es más fácil cuando hay entre uno y cinco células en un campo de 10X. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2. amplificación del genoma entero. electroforesis en gel de agarosa de 5 l de los productos de amplificación de todo el genoma. Las muestras que amplifican con éxito aparecerá frotis tan brillante de 100 pb a 1 kb (carriles 1, 2, 4, 5, 6 y 7) y puede ser secuenciado. Las muestras que no se amplifican con éxito producirá manchas débiles o sin citología (carriles 3 y 8) y no debe ser secuenciado.large.jpg "target =" _ blank "> Haga clic aquí para ver una versión más grande de esta figura.

Figura 3. Preparación de la biblioteca. Los resultados representativos de un analizador de fragmentos. Los gráficos muestran el tamaño del fragmento (en pb) en el eje X y unidades de fluorescencia relativa (RFU) en el eje Y. A la derecha de cada gráfica es un carril de gel simulado. (A) Resultados para una muestra ideal, con una distribución uniforme entre 150 y 900 pb y sin picos afilados o sesgo hacia un lado. Esta muestra es aceptable para la secuenciación. (B) Los resultados de una muestra bien, con una distribución de tamaño sesgados hacia tamaños de los fragmentos inferiores. Si bien esto no es óptima, la muestra todavía puede ser secuenciado. (C) Resultados de una muestra fracasado, en los que predominan los pequeños tamaños de los fragmentos. Esto es probablemente causado por la incubación prolongada durante el paso de tagmentationde la preparación de la biblioteca. Esta muestra no debe ser secuenciado. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| HMMcopy | |||||||
| Muestra | Segmento | Chr | comienzo | Fin | Estado | Mediana | |
| D15-4998 | 23 | CHR8 | 144500001 | 146500000 | 6 | 0.5008794 | |
| D15-4998 | 29 | chr10 | 67000001 | 134500000 | 6 | 0.4031945 | |
| D15-4998 | 52 | dohr19 | 1 | 20.000.000 | 6 | 0.4616884 | |
| D15-4998 | 57 | chry | 1 | 59500000 | 2 | -1.506532 | |
| DNAcopy | |||||||
| Muestra | Chrom | Inicio bin | fin bin | comienzo | Fin | Seg media | med seg |
| D15-4998 | chr10 | 88 | 197 | 62612945 | 129971511 | 1.4688 | 0,157 |
| D15-4998 | chr19 | 0 | 31 | 0 | 28416392 | 1.4141 | 0.1674 |
| D15-4998 | CHRX | 77 | 126 | 51659160 | 95343369 | 1.3548 | 0.1874 |
| D15-4998 | chry | 0 | 14 | 0 | 23805358 | -2,7004 | 0.3591 |
Tabla 1. Análisis de los datos. segmentos filtrados generados por HMMcopy y DNAcopy de una sola célula. La superposición de estos dos resultados revela una ganancia en el cromosoma 10 de 67 a 130 Mb, así como una ganancia en el cromosoma 19 de 0 a 20 Mb. Hemos encontrado que las ganancias en la parte proximal del cromosoma 19 son un artefacto de secuenciación sola célula 12.
Discusión
Tradicionalmente, la identificación de las VNC y aneuploidía en el nivel de células individuales necesarios métodos citológicos como el pescado y el cielo. Ahora, la secuenciación de una sola célula se ha convertido en un enfoque alternativo para tales preguntas. secuenciación de una sola célula tiene ventajas sobre el pescado y SKY, ya que es a la vez el genoma de ancho y de alta resolución. Por otra parte, cuando se aplican métodos de control de calidad apropiados, secuenciación sola célula puede proporcionar una evaluación más fiable de la CNV y aneuploidía, ya que no es susceptible a la hibridación y la difusión de artefactos inherentes a la pesca y el cielo. Sin embargo, muchas de las recientes aplicaciones de secuenciación de células individuales no han sido documentados por evaluación exhaustiva de la sensibilidad y especificidad de los métodos y análisis. De hecho, algunos de los métodos analíticos utilizados por otros estudios están asociados con altas frecuencias (> 50%) de los falsos positivos CNV llama 12. El enfoque que describimos ha sido probado rigurosamente el uso de células de knCNV propia carga con el fin de determinar las tasas de descubrimiento de verdaderos y falsos y optimizar la sensibilidad y especificidad de la detección CNV 12. Usando el control de calidad y enfoques analíticos descritos en este protocolo, aproximadamente el 20% de las ganancias de 5 Mb, el 75% de las pérdidas de 5 Mb, y todas las VNC superior a 10 Mb puede ser detectado. A pesar de la determinación de la tasa de falso descubrimiento de la secuenciación de una sola célula es difícil, hemos estimado que sea inferior al 25%. Este protocolo se puede aplicar a las células de una variedad de fuentes, los guiones se pueden modificar para ajustar la resolución de la detección de CNV, y el protocolo se pueden adaptar para identificar otros tipos de alteraciones genómicas.
Hay una variedad de medios de disociar tejidos frescos en células individuales, y muchas publicaciones describen procedimientos optimizados para tejidos específicos, tales como la piel 24 y el cerebro 25. Preferimos para aislar células individuales por microaspiración ya que permite fo r evaluación visual de cada celda a ser secuenciado. Sin embargo, también es posible aislar las células individuales por clasificación de células activadas por fluorescencia (FACS) 26 y los dispositivos de microfluidos 27. Si el aislamiento de células individuales y la amplificación del genoma completo se realiza de forma manual, es razonable para aislar y amplificar hasta cuarenta celdas en una sola sesión. Con el fin de obtener datos de secuenciación sola célula de alta calidad, es crucial que la amplificación de los genomas de células individuales es uniforme y completa. Encontramos que la calidad de las células individuales aisladas, así como la eficiencia de la lisis y la amplificación tiene un impacto significativo en la calidad de los datos de secuenciación. Como tal, las células se deben cosechar de su entorno nativo justo antes del aislamiento y la amplificación de todo el genoma debe comenzar inmediatamente después de que se aislaron células. Además, la etapa de lisis y la fragmentación se debe seguir exactamente como se describe en el 2.3 a 2.6 pasos.
"> Los algoritmos se pueden ajustar para cambiar la resolución de la detección de CNV, con efectos sobre la sensibilidad y especificidad 12 opuestas. También es posible ajustar los umbrales para detectar aneuploidía de todo el cromosoma en el contexto de tetraploidía 11. Sin embargo, nos encontramos con que nuestro enfoque se limita a la detección de las VNC superior a 5 Mb, como el ruido introducido durante la amplificación del genoma completo complica la detección de variantes más pequeñas 12. las futuras mejoras en los métodos de amplificación de todo el genoma en última instancia, deben mejorar la resolución de la detección de la CNV mediante la secuenciación de una sola célula.Secuenciación de células individuales permite la investigación no sólo de alteraciones de número de copias, sino también variaciones de nucleótidos individuales 28, 29 y la variación estructural 30. El protocolo para el aislamiento de células individuales se puede aplicar para responder a estos otros Questions. Sin embargo, la elección del método de amplificación de todo el genoma depende de la aplicación específica. El método descrito en este protocolo, que se basa en la reacción en cadena de la polimerasa, es el más adecuado para la detección de alteraciones de número de copias ya que está asociada con niveles más bajos de sesgo de amplificación 32. Para la investigación de otros tipos de alteraciones genómicas, tales como polimorfismos de nucleótido único, se cree que otros métodos de amplificación de todo el genoma a ser más adecuado 31, 32.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Agradecemos a Stuart Levine para comentarios sobre este manuscrito. Este trabajo fue apoyado por los Institutos Nacionales de Salud de subvención GM056800 y el Fondo de Investigación del Cáncer Curt mármol Kathy y Angelika Amon y en parte por el Instituto Koch programa de subsidios para P30-CA14051. Angelika Amon es también un investigador del Instituto Médico Howard Hughes y la Fundación para la Investigación Biomédica Glenn. Kak es apoyado por la Beca de Formación NIGMS T32GM007753.
Materiales
| Name | Company | Catalog Number | Comments |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177 | |
| PVC tubing (ID 3/16", OD 5/16", ~1 foot) | VWR | 89068-500 | |
| PVC tubing (ID 5/16", OD 7/16", ~6 inches) | VWR | 89068-508 | |
| Acrodisc Syringe Filter with HT Tuffryn Membrane (diameter 25 mm, pore size 0.2 µm) | VWR | 28144-040 | |
| Serological pipette (5 mL) | BioExpress | P-2837-5 | Any plastic 5 mL pipette should suffice |
| In-Line Water Trap for Oxygen Use | Amazon.com | 700220210813 | |
| Capillary Melting Point Tubes (ID 0.8-1.1 mm, 100 mm length) | VWR | 34502-99 | |
| Modeling clay | VWR | 470156-850 | |
| Petri dish (150 mm diameter) | VWR | 25384-326 | Surface should be non-adherent to facilitate aspiration of single cells |
| Hard-Shell Full-Height 96-Well Semi-Skirted PCR Plates | Bio-Rad | HSS9601 | Any 96-well PCR plate should suffice |
| 96-well tissue culture plate | VWR | 62406-081 | Any 96-well tissue culture plate should suffice |
| GenomePlex Single Cell Whole Genome Amplification Kit | Sigma | WGA4 | |
| Microseal 'A' Film | Bio-Rad | MSA5001 | |
| Mini plate spinner | Thomas Scientific | 1225Z37 | |
| Thermal cycler | Bio-Rad | 1861096 | Any thermal cycler should suffice so long as it can accommodate a 96-well PCR plate |
| Agencourt Ampure Beads | Beckman Coulter | A63880 | |
| Dynamag-2 Magnet | Thermo Fisher Scientific | 12321D | Any similar magnetic tube strip should suffice |
| Nextera XT DNA Library Preparation Kit | Illumina | FC-131-1096 | |
| Nextera XT Index Kit | Illumina | FC-121-1012 | |
| Complete kit (optimized for Roche LightCycler 480) | Kapa Biosystems | KK4845 | This kit is optimized for the Roche LightCycler 480 real-time PCR machine. If using a different machine, substitute a kit optimized for your machine. |
Referencias
- Kahlem, P. Transcript Level Alterations Reflect Gene Dosage Effects Across Multiple Tissues in a Mouse Model of Down Syndrome. Genome Res. 14 (7), 1258-1267 (2004).
- Torres, E. M. Effects of Aneuploidy on Cellular Physiology and Cell Division in Haploid Yeast. Science. 317 (5840), 916-924 (2007).
- Itsara, A. Population Analysis of Large Copy Number Variants and Hotspots of Human Genetic Disease. Am. J. Human Gen. 84 (2), 148-161 (2009).
- Sudmant, P. H. Global diversity, population stratification, and selection of human copy-number variation. Science. 349 (6253), aab3761 (2015).
- Navin, N. Inferring tumor progression from genomic heterogeneity. Genome Res. 20 (1), 68-80 (2010).
- Torres, L., Ribeiro, F. R., Pandis, N., Andersen, J. A., Heim, S., Teixeira, M. R. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast cancer res and treat. 102 (2), 143-155 (2007).
- Yates, L. R. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 21 (7), 751-759 (2015).
- Forsberg, L. A. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Human Gen. 90 (2), 217-228 (2012).
- Laurie, C. C. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Gen. 44 (6), 642-650 (2012).
- Jacobs, K. B. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Gen. 44 (6), 651-658 (2012).
- Knouse, K. A., Wu, J., Whittaker, C. A., Amon, A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. 111 (37), 13409-13414 (2014).
- Knouse, K. A., Wu, J., Amon, A. Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. 26 (3), 376-384 (2016).
- . KAPA Library Quantification Kit Available from: https://www.kapabiosystems.com/product-applications/products/next-generation-sequencing-2/library-quantification/ (2016)
- . DNAcopy Available from: https://bioconductor.org/packages/release/bioc/html/DNAcopy.html (2016)
- Lichti, U., Anders, J., Yuspa, S. H. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat. Protoc. 3 (5), 799-810 (2008).
- Brewer, G. J., Torricelli, J. R. Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2 (6), 1490-1498 (2007).
- Navin, N. Tumour evolution inferred by single-cell sequencing. Nature. 472 (7341), 90-94 (2011).
- Szulwach, K. E., Chen, P. Single-Cell Genetic Analysis Using Automated Microfluidics to Resolve Somatic Mosaicism. PLoS ONE. 10 (8), e0135007 (2015).
- Zong, C., Lu, S., Chapman, A. R., Xie, X. S. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science. 338 (6114), 1622-1626 (2012).
- Wang, Y. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 512 (7513), 155-160 (2014).
- Zhang, C. Z. Chromothripsis from DNA damage in micronuclei. Nature. 522 (7555), 179-184 (2015).
- Macaulay, I. C., Voet, T. Single Cell Genomics: Advances and Future Perspectives. PLoS Genetics. 10 (1), e1004126 (2014).
- Gawad, C., Koh, W., Quake, S. R. Single-cell genome sequencing: current state of the science. Nat. Rev. Gen. , 1-14 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados