
Method Article
Der Nachweis von Copy Number Änderungen Single Cell-Sequenzierung
In diesem Artikel
Zusammenfassung
Einzelne Zell Sequenzierung ist ein immer beliebter und zugängliches Werkzeug für genomische Veränderungen bei hoher Auflösung Adressierung. Wir bieten ein Protokoll, das einzelne Zelle Sequenzierung verwendet Kopienzahl Veränderungen in einzelnen Zellen zu identifizieren.
Zusammenfassung
Nachweis von genomischen Veränderungen an Einzelzellauflösung ist wichtig für die genetische Heterogenität und Evolution in normalen Geweben, Krebserkrankungen und mikrobiellen Populationen zu charakterisieren. Traditionelle Methoden für genetische Heterogenität der Beurteilung wurden durch eine geringe Auflösung, geringe Empfindlichkeit und / oder geringe Spezifität begrenzt. Einzelne Zell-Sequenzierung zum Nachweis genetischer Heterogenität mit hoher Auflösung, hohe Empfindlichkeit als ein mächtiges Werkzeug entstanden und, wenn sie in geeigneter analysiert, eine hohe Spezifität. Hier stellen wir ein Protokoll für die Isolierung, Amplifikation des gesamten Genoms, Sequenzierung und Analyse von Einzelzellen. Unser Ansatz ermöglicht die sichere Identifizierung von Megabasen-Skala Kopienzahl-Varianten in einzelnen Zellen. Jedoch können Aspekte dieses Protokoll auch zur Untersuchung anderer Arten von genetischen Veränderungen in Einzelzellen angewendet werden.
Einleitung
Veränderungen in der DNA-Kopienzahl kann in der Größe von mehreren Basenpaaren reichen (copy number variants) (CNVs) auf ganzer Chromosomen (Aneuploidie). Die Kopienzahl Veränderungen große Bereiche des Genoms beeinflussen können erhebliche phänotypische Folgen haben durch den Ausdruck von bis zu Tausenden von Genen verändern 1, 2. CNV , die in allen Zellen der Population vorhanden sind , können durch Substanz- Sequenzierung oder Mikroarray-basierten Verfahren 3, 4 erfaßt werden. Jedoch Populationen können auch genetisch heterogen sein, wobei CNVs in einer Teilmenge der Population vorhandenen oder sogar einzelne Zellen. Genetische Heterogenität in Krebs gemeinsamen, Tumorentwicklung Fahren und derzeit auch in normalen Geweben mit unbekannter Folge 5, 6, 7, 8, 9 10.
Traditionell wurde die genetische Heterogenität entweder durch zytologische Ansätze oder Bulk-Sequenzierung beurteilt. Zytologischen Ansätze wie Fluoreszenz - in - situ - Hybridisierung (FISH), Chromosom - Spreads, und die spektrale Karyotypisierung (SKY), haben den Vorteil der Identifizierung von Veränderungen in einzelnen Zellen , sondern weisen eine hohe Fehlerraten aufgrund von Hybridisierungsartefakten und 11 ausbreitet. Diese Ansätze sind begrenzt, auch in ihrer Auflösung nur für Kopienzahl enthüllt Änderungen über mehrere Megabasen. Die Sequenzierung oder Microarrays von Bulk-DNA, obwohl eine höhere Genauigkeit und Auflösung, ist weniger empfindlich. Um genetische Heterogenität von populationsbasierte Ansätze zu erkennen, müssen die Varianten in einem wesentlichen Anteil der Zellen in der Population vorhanden sein. Die Entstehung von Methoden, um die genomische DNA aus einzelnen Zellen zu verstärken, wurde es möglich, das Genom von einzelnen Zellen zu sequenzieren. Einzelzelle sequeber bouncing hat die Vorteile einer hohen Auflösung, hohe Empfindlichkeit, und bei geeigneter Qualitätskontrollverfahren angewendet werden, die hohe Genauigkeit 12.
Hier beschreiben wir ein Verfahren zum Nachweis von Megabasen-Skala Kopienzahl Veränderungen in einzelnen Zellen. Wir isolieren einzelne Zellen durch microaspiration, verstärken genomischen DNA-Linker-Adapter unter Verwendung von PCR vorbereiten Bibliotheken für die Sequenzierung der nächsten Generation, und erkennen Kopienzahl durch versteckte Varianten sowohl Markov-Modell und Kreis binäre Segmentierung.
Protokoll
1. Isolierung von Einzelzellen
- Bereiten Sie die microaspirator
- Entfernen Sie die durchsichtige Plastik Ende aus dem Ansaugrohr Kassette und legen Sie das schmale Ende in ein Ende einer 1 Meter langen PVC-Schlauch mit 3/16 "Innendurchmesser.
- Setzen Sie den Ausgang eines 0,2 & mgr; m Spritzenfilter in das andere Ende des 1 Meter langen PVC-Schläuche mit 3/6 "Innendurchmesser.
- Setzen Sie den Einlass der 0,2 & mgr; m Spritzenfilter in ein Ende eines 6-Zoll-langen PVC-Schlauch mit 5/16 "Innendurchmesser.
- Optional: Schneiden Sie die 6-Zoll-langen PVC-Schlauch mit 5/16 "Innendurchmesser in der Mitte und legen Sie Wasserfalle zwischen den beiden Hälften einer in-line.
- Break a 5 ml Kunststoff serologischen Pipette in der 1 ml Graduierung und setzen Sie das gebrochene Ende in das offene Ende des PVC-Schlauch mit 5/16 "Innendurchmesser.
- Setzen Sie den Ausgang des 5 ml Kunststoff serologischen Pipette in das flexible Ende einer Saugvorrichtung Rohranordnung (wo die durchsichtige Kunststoff Endeentfernt worden war).
- Für die Lagerung, decken Sie die rote Mundstück der Saugvorrichtung Rohranordnung mit einem sterilen Kunststoffrohr.
- Zeichne eine Glaskapillare mit einem Innendurchmesser von 10-30 um, indem die Mitte des Rohres in der Nähe einer Bunsenbrennerflamme zu bringen und die Spannung an beiden Enden des Rohres aufgebracht wird. Brechen Sie das gezogene Rohr in der Mitte zwei potentielle Absauger Nadeln zu schaffen. Wiederholen Sie diesen Schritt mit mehreren Rohren als nur einige Rohre mit einem Innendurchmesser am Ende wird, die für die Kommissionierung einzelner Zellen geeignet ist.
- Für die Lagerung, legen zwei Streifen aus Knetmasse in einer 15 cm Petrischale und sichern Sie die Saugvorrichtung Nadeln über den Streifen.
- Pick-Zellen
- Bereiten Sie eine Einzelzellsuspension als geeignet für den Zelltyp und Experiment. Um beispielsweise adhärente Zellen herzustellen, wie beispielsweise humanen Fibroblastenzelllinien, Ernte die Zellen durch Trypsinisierung und Transfer Zellen einem konischen Rohr entsprechenden Medien enthält.
- Vor, während oderN ach der Herstellung der Einzelzellsuspension (je nachdem, wie lange es dauert, die Einzelzellsuspension herzustellen), Aufbau der Haube für Amplifikation des gesamten Genoms.
- Spray bis die Oberfläche der Haube, Pipettenspitze Boxen und Pipettenspitzen mit 10% Bleichmittel und wischen Sie mit einem Papiertuch. Wiederholen Sie diesen Schritt mit 70% Ethanol.
- Aus 8 ul Wasser aus der Amplifikation des gesamten Genoms (WGA) Satz an einzelne Vertiefungen einer 96-Well-PCR-Platte, eine Vertiefung für jede Zelle sequenziert werden. Decken Sie die 96-Well-PCR-Platte mit einem Deckel aus einer 96-Well-Gewebekulturplatte und auf Eis.
- Hinzufügen 1000 Zellen auf 10 ml Medium oder Phosphat-gepufferter Salzlösung (PBS) in einer 15 cm Petrischale und die Schale auf Eis um die Zellen zu verhindern, dass an der Schale anhaften.
- Bringen Sie die Zellen in der Petrischale und die 96-Well-PCR-Platte mit Deckel auf Eis zu einem Lichtmikroskop mit 10X-Objektiv.
- Erhöhen Sie die Öffnung der Nadel Absauger durch sanft die herausgezogen Ende th tippene Aspirator Nadel auf einer harten Oberfläche, so dass die Spitze abbricht. Legen Sie das breite Ende der Saugvorrichtung Nadel in das klare Ende des microaspirator.
- Legen Sie die Petrischale mit Zellen auf dem Mikroskoptisch. Setzen Sie den roten Mundstück der Saugvorrichtung in den Mund. Mit einer Hand die Saugvorrichtung Nadel und die andere Hand zu bewegen, um die Petrischale mit Zellen zu bewegen. Identifizieren Sie einzelne Zellen sequenziert werden.
- Mit Mund abgesaugt, ziehen eine einzelne Zelle in die Nadel Absauger zusammen mit ~ 1-2 ul Medium oder PBS. Übertragen Sie die Zelle in die 8 ul Wasser in einem einzigen Vertiefung der 96-Well-PCR-Platte. Vermeiden Sie Blasen einzuführen, wenn die Zelle zu übertragen.
- Wiederholen Sie Schritt 1.2.7 bis die gewünschte Anzahl von Zellen isoliert wurden. Halten Sie die PCR-Platte auf Eis und bedeckt mit Deckel zwischen Kommissionierung Zellen. Markieren Sie die Vertiefungen, die Zellen erhalten haben.
- Während einzelne Zellen Kommissionierung, tauen die 10X einzelne Zell-Lyse und Fragmentierung Puffer aus der ganzen genome Amplifikationskit. Nachdem die gewünschte Anzahl von Zellen Kommissionierung, gehen Sie sofort zu Amplifikation des gesamten Genoms.
2. Whole Genome Amplification
- Um zu verhindern Kontamination während der Amplifikation des gesamten Genoms, fügen Sie alle Reagenzien in einer Gewebekultur Kapuze, verwenden Pipettenspitzen mit Filter und die Pipettenspitze wechseln zwischen Brunnen.
- Bereiten Sie eine Arbeits Lyse und Fragmentierung Pufferlösung aus der Amplifikation des gesamten Genoms (WGA) Kit. Für jeden Satz von bis zu 32 Zellen kombinieren 32 ul 10fach Einzel Zelllyse und Fragmentierungspuffer und 2 & mgr; l Proteinase K-Lösung in einem Reaktionsgefäß. Vortex die Röhre, die Lösungen zu mischen.
- 1 & mgr; l der Arbeits Lyse und Fragmentierung Pufferlösung in jede Vertiefung und Pipette nach oben und unten zu mischen.
- Decken Sie die Platte und Dichtung alle Vertiefungen mit einer Kunststoff-Folie. Zentrifuge kurz die Platte in einem Mini-Platte Spinner.
- Thermal-Zyklus als FollOWS: 50 ° C, 1 h und 99 ° C, 4 min.
- Kühlen Sie die Platte auf Eis und kurz zentrifugiert werden, die Platte in einem Mini-Platte Spinner.
- Bereiten Sie eine Arbeitsbibliothek Vorbereitung Pufferlösung aus der Amplifikation des gesamten Genoms Kit. Für jede Zelle kombinieren 2 ul 1x Einzelzellbibliothek Präparationspuffer und 1 ul Bibliothek Stabilisierungslösung in einem Reaktionsgefäß. Herstellung der Arbeitslösung für mehrere Zellen in dem gleichen Reaktionsgefäß.
- Entfernen Sie die Kunststoff-Folie und fügen Sie 3 ul der Arbeitsbibliothek Zubereitung Pufferlösung in jede Vertiefung. Pipette den Inhalt der gut nach oben und unten zu mischen. Setzen Sie die Kunststoff-Folie.
HINWEIS: Die Kunststofffolie kann während des gesamten Genoms Amplifikationsverfahren wiederverwendet werden, bis die Vertiefungen zu Bohrungen in dem Film zu starten. Wenn dies der Fall ist, wechseln Sie zu einer neuen Kunststoff-Folie. - Zentrifuge kurz die Platte in einem Mini-Platte Spinner und Inkubation bei 95 ° C für 2 min.
- Kühlen Sie die Platte auf Eis und kurzZentrifuge, die Platte in einem Mini-Platte Spinner.
- Entfernen Sie die Kunststoff-Folie, fügen 1 ul Bibliothek Enzympräparat in jede Vertiefung und pipettiert den Inhalt der gut nach oben und unten zu mischen. Halten Sie die Bibliothek Enzympräparat, auf Eis oder in einem kalten Block in diesem Schritt. Setzen Sie die Kunststoff-Folie.
- Zentrifuge kurz die Platte in Mini-Platte Spinner und Wärmezyklus wie folgt: 16 ° C, 20 min, 24 ° C, 20 min, 37 ° C, 20 min, 75 ° C, 5 min, und halten bei 4 ° C.
- Kühlen Sie die Platte auf Eis und kurz zentrifugiert werden, die Platte in einem Mini-Platte Spinner.
- Bereiten Sie eine Arbeits Verstärkung Mischung aus der Amplifikation des gesamten Genoms Kit. Für jede Zelle kombinieren 48,5 ul Wasser, 7,5 ul 10x-Amplifizierung Mastermix und 5 & mgr; l WGA-DNA-Polymerase in einem Reaktionsgefäß. Bereiten Sie die Arbeitsmischung für mehrere Zellen in dem gleichen Reaktionsgefäß. Halten Sie die Arbeitsmischung auf Eis.
- Entfernen Sie die Kunststoff-Folie, fügen Sie 61 ul Arbeits Verstärkung mixin jede Vertiefung und Pipette Inhalt gut nach oben und unten zu mischen. Halten Sie die Arbeits Verstärkung Mischung auf Eis oder in einem kalten Block in diesem Schritt. Setzen Sie die Kunststoff-Folie.
- Zentrifuge kurz die Platte in einem Mini-Platte Spinner und Wärmezyklus wie folgt: 95 ° C, 3 min, 25 Zyklen von 94 ° C, 30 sec und 65 ° C, 5 min, und halten bei 4 ° C.
- Transfer 60 ul jeder Probe auf einem separaten Mikrozentrifugenröhrchen und lagern bei - 20 ° C zur Verwendung in Schritt 3.
- Hinzufügen DNA loading dye zu dem verbleibenden Volumen jeder Probe (dh 3 ul 6x DNA loading dye bis 15 & mgr; l der Probe) und führen 5 & mgr; l von jeder Reaktion auf einem 1% Agarosegel.
Hinweis: Zellen, die erfolgreich verstärkt werden von 250 bis 1000 bp als Abstrich erscheinen. Die Proben, die zeigen keinen Abstrich zeigen oder nur ein schwaches Abstrich sind unwahrscheinlich nützliche Sequenzdaten zu erhalten.
3. Sequenzierung
- Reinige Proben mit paramagnetischen Kügelchen.
- Thaw DNA-Proben auf Eis. Vortex paramagnetischen Kügelchen, bis die Lösung homogen und die Kügelchen bei Raumtemperatur für mindestens 30 min inkubiert.
- Übertragen Sie 20 ul jeder DNA-Probe in ein sauberes Reaktionsgefäß. Der Rest der Probe kann bei -20 ° C gelagert werden.
- In 30 ul (1,5x) von paramagnetischen Perlen zu jeder DNA-Probe und Wirbel zu mischen. Inkubieren der Proben bei Raumtemperatur für 10 min. Lassen Sie die Stammlösung von Kügelchen bei Raumtemperatur zur Verwendung in Schritt 3.4.
- Die Röhrchen auf einem Magnetstreifen Rohr für 2 min, oder bis die Kügelchen ein Pellet bilden, und der Überstand klar ist.
- Verwendung eines P200-Pipette zu entfernen, so viel von dem Überstand wie möglich, ohne die Perlen stören.
- Hinzufügen 180 & mgr; l 80% Ethanol zu der DNA-Bead-Mischung. Drehen Sie die Rohre mehrmals relativ zum Magneten, um die Perlen durch die Ethanollösung "spülen".
- Mit Hilfe einer Pipette P200, entfernen Sie so viel von der Ethanolwasch wie mögBLE ohne die Perlen zu stören.
- Wiederholen 3.1.6 und 3.1.7.
- Luft trocknen die Kügelchen für ca. 10 min oder bis kein Ethanol mehr sichtbar ist. Fahren Sie mit dem nächsten Schritt, wenn die Perlen geknackt erscheinen oder fallen die Wand des Rohres ab.
- Entfernen Sie die Proben aus dem Magnetstreifen. Werden 40 ul 10 mM Tris pH 8,0, die DNA von den Kügelchen eluiert und die Proben vortexen die Perlen zu resuspendieren. Inkubieren für 2 min bei Raumtemperatur.
- kurz zentrifugieren, die Proben der Flüssigkeit am Boden des Röhrchens zu sammeln, und die Röhrchen auf einem Magnetstreifen Rohr Platz für mindestens zwei Minuten. Übertragen Sie das Eluens in saubere Mikrozentrifugenröhrchen ohne die Perlen zu stören.
- Beispiel Normalisierung
- Quantifizieren der Konzentration jeder Probe mit einem Spektralphotometer. Die Konzentrationen sollten 10 bis 30 ng / & mgr; l liegen.
- Verdünnte jede Probe auf 0,2 ng / & mgr; l unter Verwendung von 10 mM Tris pH 8,0. In den folgenden Schritten wird ein Minimum i erfordernnput von 5 & mgr; l dieser Verdünnung.
- Bibliothek Vorbereitung
- Bereiten Sie Sequenzierungsbibliotheken indem Sie die Anweisungen der Bibliothek Vorbereitung Kit 13.
- Endgültige Bereinigung
- Reinigen Sie die Proben gemäß Schritt 3.1. Für Leselängen von 50 bp, 75 bp oder 150 bp, ein Verhältnis von Perle verwenden Volumen von 1.5x zu probieren, 1x oder 0.6x sind. Eluieren mit 15 ul 10 mM Tris pH 8,0.
- Führen Sie die Proben auf einem Fragment Analysator die Größenverteilung der Bibliothek 14 zu überprüfen. Die Größenverteilung sollte gleichmäßig 150-900 bp verteilt werden.
- Quantifizieren Proben unter Verwendung von qPCR
- Mit Hilfe einer Bibliothek Quantifizierung Kit, Set 15 eine qPCR Reaktion gemäß dem Kit - Anweisungen auf. Lassen Sie eine Spalte leer hinzuzufügen positive Standards (DNA-Standards aus dem Kit) und negativen Standards (Wasser oder andere leere Lösung).
- Wärmezyklus wie folvor: 95 ° C, 5 min (Aufheizrate von 4,8 ° C / sec) und 35 Zyklen von 95 ° C, 30 sec (Aufheizrate von 4,8 ° C / sec) und 60 ° C, 45 sec (Rampenrate von 2,5 ° C / sec).
- Pooling
- Ermitteln Sie, wie viele Fahrspuren auf der Fließzelle benötigt werden, und teilen Sie die Proben in Gruppen, wobei eine Gruppe pro Spur.
- Für jede Gruppe von Proben, wählen Sie die Probe mit der niedrigsten Konzentration auf der Grundlage der qPCR Daten aus Schritt 3.5.2 und normalisieren alle Proben in dieser Gruppe auf dieser Konzentration unter Verwendung von 10 mM Tris pH 8,0.
- Pool, die Proben in jeder Gruppe zusammen.
- Die Sequenzierung
- Last Pools auf der nächsten Generation Sequenzierung Maschine nach Standardverfahren 16.
4. Datenanalyse
HINWEIS: Ein Unix-basierten Umgebung ist erforderlich, um die Programme und Skripte in diesem Abschnitt auszuführen. Installieren Sie die Software in dem Protokoll nach ihrer in den genannten stallation Führungen. Alle Skripte können in https://sourceforge.net/projects/singlecellseqcnv/ finden.
- Schneiden Sie das liest bis 40-nt mit fastx_trimmer von FastX-Toolkit Version 0.0.13 17.
fastx_trimmer -Q33 -i example.fastq -l 40 -o example.trim.fastq - Richten Sie die liest in den entsprechenden Referenzgenom (mm9 für Maus, hg19 für Menschen) unter Verwendung von BWA - Version 0.6.1 mit den Standardoptionen 18.
BWA aln mm9.fa example.trim.fastq> example.sai
BWA Samse mm9.fa example.sai example.trim.fastq> example.sam - Entfernen Ausrichtungen chrm und zufällige Chromosomen, dann sortieren und Index die resultierende BAM - Datei mit samtools Version 0.1.19 19.
grep -v -w chrm example.sam | grep -v zufällig> example.filtered.sam
samtools sehen -uSh example.filtered.sam | samtools Art - example.filtered
samtools Index example.filtered.bam - Führen Sie HMMcopy zu erkennen CNVs= "Xref"> 20
- Verwenden Sie gcCounter in HMMcopy einen GC-Prozentsatz Referenzdatei für das Genom zu erzeugen. Verwenden Sie Option "-w 500000" Fenstergröße angeben. Verwenden Sie die gleiche Version der fasta Referenzdatei in Schritt 4.2, aber stellen Sie sicher, dass chrm und zufällige Chromosomen aus der fasta Datei entfernt.
gcCounter -w 500000 mm9.fa> mm9_gc.wig - Verwenden Sie generateMap.pl in HMMcopy eine wackeln Datei für mappability zu erzeugen. Verwenden Sie Option "-w 40" angeben Länge lesen. Verwenden Sie die gleiche fasta Referenzdatei verwendet in Schritt 4.4.1.
generateMap.pl -b mm9.fa
generateMap.pl -w 40 -i mm9.fa mm9.fa -o mm9.bigwig - Verwenden Sie mapCounter in HMMcopy eine mappability Referenzdatei für das Genom zu erzeugen. Verwenden Sie Option "-w 500000" Fenstergröße angeben.
mapCounter -w 500000 mm9.bigwig> mm9_map.wig - Verwenden Sie Read in HMMcopy eine wackeln-Datei für jede BAM-Datei zu erzeugen.
Read -w 500000 example.filtered.bam> input.wig - Ändern Sie die Pfade zu den Referenzdateien in den R - Skripten (run_hmmcopy.mm9.r oder run_hmmcopy.hg19.r), verwenden Sie die erzeugten Dateien oben in 4.4.1 (zB mm9_gc.wig) und 4.4.3 (zB mm9_map. Perücke) für die Variablen gfile und mfile, die am GC Prozentsatz Referenzdatei und mappability Referenzdatei beziehen sich auf. Führen Sie dann die bereitgestellte R-Skript "run_hmmcopy.mm9.r" oder "run_hmmcopy.hg19.r".
R CMD BATCH run_hmmcopy.mm9.r
oder
R CMD BATCH run_hmmcopy.hg19.r - Für die Stapelverarbeitung von einer Reihe von BAM-Dateien, legen Sie die BAM-Dateien in einem einzigen Ordner und führen Sie das Script über "HMMpipe.pl" in der Packungssegmente zu nennen und Variabilität Scores berechnen (VS). Folgen Sie dem Format:
perl HMMpipe.pl Folder_to_BAMfiles mm9
- Verwenden Sie gcCounter in HMMcopy einen GC-Prozentsatz Referenzdatei für das Genom zu erzeugen. Verwenden Sie Option "-w 500000" Fenstergröße angeben. Verwenden Sie die gleiche Version der fasta Referenzdatei in Schritt 4.2, aber stellen Sie sicher, dass chrm und zufällige Chromosomen aus der fasta Datei entfernt.
- Führen Sie DNAcopy auf CNVs 21 erkennen.
- Verwenden Sie samtools Version 0.1.19 zu extrahieren eindeutig abgebildet liest aus jeder BAM-Datei.
samtools sehen -h -F 0x0004 example.filtered.bam | egrep -i "^ @ | XT: A: U" | samtools -shū Ansicht -> example.mapped.bam - Verwenden Sie MarkDuplicates in Picard Version 1.94 auf PCR - Duplikate in der BAM markieren 22 einreichen.
java -jar MarkDuplicates.jar INPUT = example.mapped.bam OUTPUT = example.nondup.bam METRICS_FILE = example.dup REMOVE_DUPLICATES = true - Verwenden Sie coveragebed in bedtools 2.17.0 Version zu zählen in jedem der vordefinierten dynamischen 500kb mappable Fenster in der mitgelieferten BED Datei "mm9.500k.dynamic.win.bed" oder "hg19.500k.dynamic.win.bed" abgebildet liest 23 .
coverageBed -abam example.nondup.bam -b mm9.500k.dynamic.win.bed -counts | Art -k1,1 -k2,2n> example.nondup.counts - Verwenden Sie das mitgelieferte Perl-Skript "normalizeGC.pl", um die Lesezählungen normalisieren auf Grundlage des bereitgestellten GC-Gehalt Referenzdatei "mm9.500k.dynamic.win.fa.gc.txt" oder "hg19.500k.dynamic.win.fa. gc.txt ".
perl normalizeGC.pl mm9.500k.dynamic.win.fa.gc.txt example.nondup.counts> example.bam.norm.counts - Verwenden Sie das mitgelieferte R Skript "dnacopy.r" die Segmente zu nennen.
R CMD BATCH dnacopy.r
- Verwenden Sie samtools Version 0.1.19 zu extrahieren eindeutig abgebildet liest aus jeder BAM-Datei.
- Identifizieren CNVs
- Zellen ausschließen, für die die VS berechnet in 4.4.6 0,26 übersteigt.
- Filter , die Segmente genannt durch HMMcopy in 4.4.6 , um nur die , für die der Medianwert log 2 -Verhältnis größer als 0,4 (putative gain) oder weniger als -0,35 (putative Verlust).
- Filtern Sie die von DNAcopy genannt Segmente in 4.5.5, um nur diejenigen, für die das Segment bedeuten größer als 1,32 oder weniger als 0,6.
- Overlap die Segmente aus 4.6.2 und 4.6.3, ruft CNVs nur in Regionen, in denen beide HMMcopy und DNAcopy ein mutmaßliches Gewinn oder vermeintlichen Verlust identifizieren.
Ergebnisse
Die zusammengebaute Aspirator sollte ähnlich dem in Figur 1A angezeigt. Die Nadel sollte so gezogen werden, dass er ausreichend breit ist, um eine einzelne Zelle aufnehmen, aber nicht so breit, dass ein großes Volumen mit der einzelnen Zelle erstellt. Einzelzellen Ansaugrauchmelder ist am einfachsten , wenn es zwischen einem und fünf Zellen in einem 10fach - Feld (1B).
Wenn Amplifikation des gesamten Genoms erfolgreich ist, erscheint die Probe als Abstrich auf einem Agarosegel (2, Spuren 1, 2, 4, 5, 6 und 7). Eine schwache oder fehlende Abstrich zeigt eine ausgefallene Amplifikationsreaktion und die Probe nicht sequenziert werden soll (Abbildung 2, Bahnen 3 und 8).
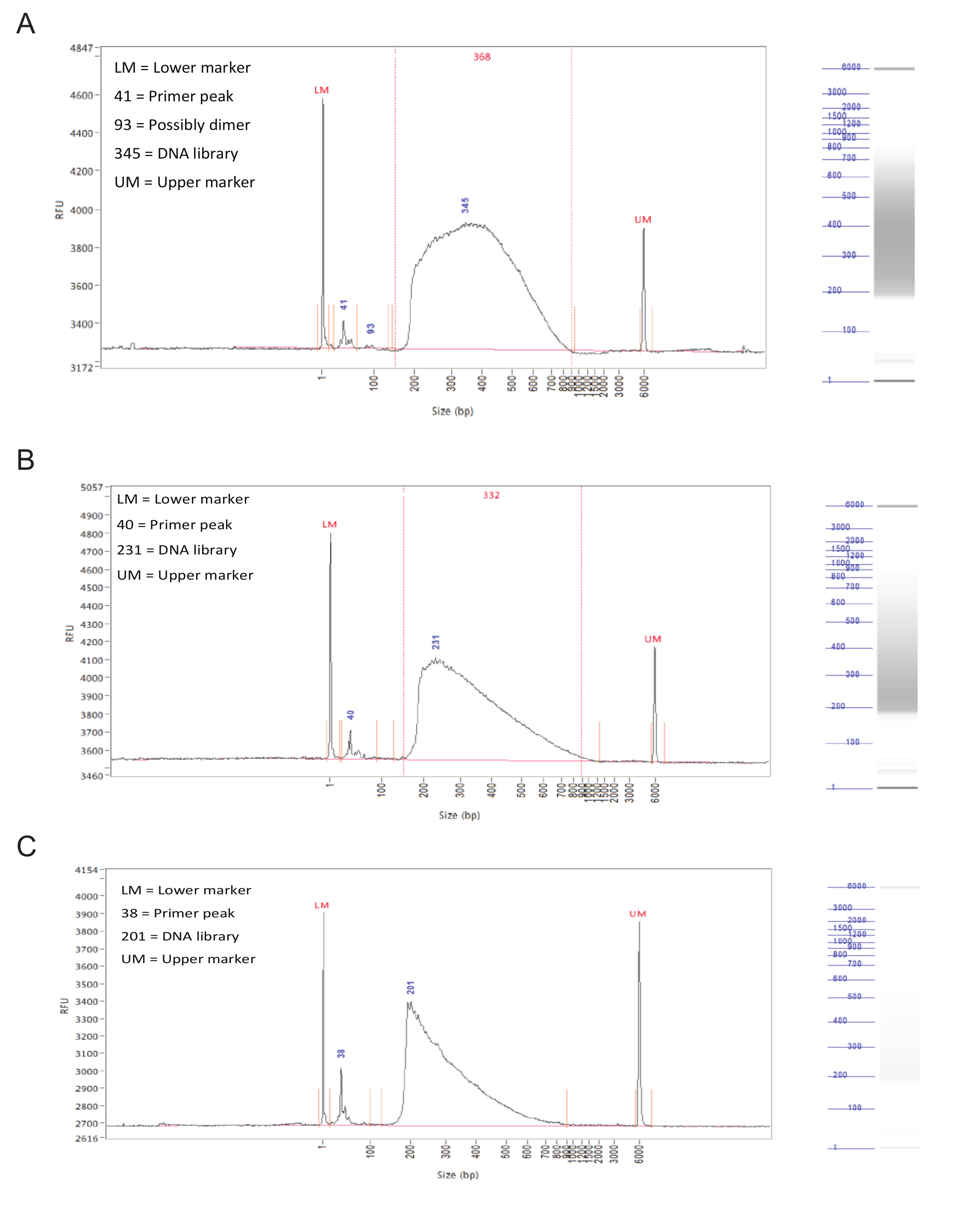
Folgende Bibliothek Vorbereitung, die Bruchgrößenverteilung der Proben sollte mittels Kapillarelektrophorese auf einem Fragment Analysators beurteilt.Erfolgreich vorbereiteten Bibliotheken eine ziemlich gleichmäßige Verteilung der Fragmentgrößen von 150 bis 900 bp haben (Abbildung 3, A, B). Fehlgeschlagen Bibliothek Vorbereitung wird in einer schrägen Bruchgrößenverteilung führen und solche Bibliotheken sollten nicht sequenziert werden (Abbildung 3C).
Sequenzierung verarbeitet Daten über den Hidden-Markov-Modell (HMMcopy) und Kreis binäre Segmentierung (DNAcopy) das Genom jeder Zelle in Segmente der geschätzten Kopienzahl analysieren. Diese Segmente können dann gefiltert werden , die mit einer geschätzten Kopienzahl im Einklang mit Gewinn oder Verlust in einer einzelnen Zelle (Tabelle 1) zu identifizieren. Diese gefilterten Segmente aus HMMcopy und DNAcopy sollte dann überlappt werden hohe Vertrauen CNVs zu identifizieren.

Abbildung 1. Einzelzellisolierung. ( A) Gebaute microaspirator. (B) A 10X Feld zeigt dissoziierten Zellen (Pfeile) und microaspirator Nadel (untere rechte Ecke). Einzelzellen Ansaugrauchmelder ist am einfachsten, wenn es zwischen einem und fünf Zellen in 10-facher Bereich sind. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
{kind=link}

Abbildung 2. Amplifikation des gesamten Genoms. Agarose-Gelelektrophorese von 5 ul whole genome Amplifikationsprodukte. Proben, die erfolgreich amplifiziert werden als helle Ausstrichen von 100 bp bis 1 kb (Spuren 1, 2, 4, 5, 6 und 7) erscheinen und sequenziert werden können. Die Proben, die nicht erfolgreich verstärken sie werden schwach schmiert oder keinen Abstrich (Bahnen 3 und 8) produzieren und nicht sequenziert werden soll.large.jpg "target =" _ blank "> Bitte hier klicken, um eine größere Version dieser Figur zu sehen.

Abbildung 3. Bibliothek Vorbereitung. Repräsentative Ergebnisse aus einem Fragment Analysator. Die Diagramme zeigen Fragmentgröße (in bp) auf der X-Achse und der relativen Fluoreszenzeinheiten (RFU) auf der Y-Achse. Auf der rechten Seite jedes Graphen ist eine simulierte Gel Spur. (A) Ergebnisse für eine ideale Probe, mit einer gleichmäßigen Verteilung zwischen 150 und 900 bp und ohne scharfe Spitzen oder Neigung zu einer Seite. Diese Probe ist akzeptabel für die Sequenzierung. (B) Die Ergebnisse einer in Ordnung Probe, mit einer Größenverteilung verzerrt zu niedrigeren Fragmentgrößen. Dies ist zwar nicht optimal ist, kann die Probe noch sequenziert werden. (C) Ergebnisse von einem ausgefallenen Probe, mit überwiegend kleinen Fragmentgrößen. Dies wird wahrscheinlich bei längerer Inkubation während des tagmentation Schritt verursachtder Bibliothek Vorbereitung. Diese Probe sollte nicht sequenziert werden. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
{kind=link}
| HMMcopy | |||||||
| Sample | Segment | Chr | Anfang | Ende | Bundesland | Median | |
| D15-4998 | 23 | CHR 8 | 144500001 | 146500000 | 6 | 0.5008794 | |
| D15-4998 | 29 | chr10 | 67000001 | 134500000 | 6 | 0.4031945 | |
| D15-4998 | 52 | chr19 | 1 | 20000000 | 6 | 0.4616884 | |
| D15-4998 | 57 | Chry | 1 | 59500000 | 2 | -1,506532 | |
| DNAcopy | |||||||
| Sample | chrom | Starten bin | End bin | Anfang | Ende | seg Mittelwert | seg med |
| D15-4998 | chr10 | 88 | 197 | 62612945 | 129971511 | 1,4688 | 0,157 |
| D15-4998 | chr19 | 0 | 31 | 0 | 28416392 | 1,4141 | 0,1674 |
| D15-4998 | CHRX | 77 | 126 | 51659160 | 95343369 | 1,3548 | 0,1874 |
| D15-4998 | Chry | 0 | 14 | 0 | 23805358 | -2,7004 | 0,3591 |
Tabelle 1. Datenanalyse. Gefiltert Segmente erzeugt durch HMMcopy und DNAcopy aus einer einzigen Zelle. diese beiden Ergebnisse Overlapping zeigt einen Gewinn auf dem Chromosom 10 67-130 Mb sowie einen Gewinn auf dem Chromosom 19 von 0 bis 20 Mb. Wir haben festgestellt , dass sich die Gewinne auf dem proximalen Abschnitt des Chromosoms 19 sind ein Artefakt der einzelnen Zelle Sequenzierung 12.
Diskussion
Traditionell Identifizierung CNVs und Aneuploidie auf der Ebene einzelner Zellen zytologischen Methoden erforderlich wie FISH und SKY. Nun einzelne Zelle Sequenzierung als Alternative für solche Fragen entstanden. Einzelne Zell Sequenzierung hat Vorteile gegenüber FISH und SKY, da sie sowohl genomweite und hohe Auflösung. Wenn darüber hinaus geeignete Qualitätskontrollverfahren angewendet werden, können einzelne Zelle Sequenzierung eine zuverlässigere Beurteilung von CNVs bieten und Aneuploidie, da sie nicht anfällig für die Hybridisierung und Verbreitung Artefakte inhärenten FISH und SKY ist. Allerdings haben viele der jüngsten Anwendungen der einzelnen Zelle Sequenzierung nicht durch gründliche Bewertung der Sensitivität und Spezifität der Methoden und Analysen belegt. Tatsächlich sind einige der analytischen Ansätze in anderen Studien verwendet , die mit hohen Frequenzen (> 50%) von falsch positiven CNV 12 nennt. Der Ansatz, den wir beschreiben wurde unter Verwendung von Zellen von kn rigoros getesteteigene CNV Belastung um 12 wahr und falsch Entdeckung Raten und die Optimierung der Sensitivität und Spezifität der CNV Erkennung zu bestimmen. Verwendung der Qualitätskontrolle und Analyse in diesem Protokoll beschriebenen Ansätze, rund 20% der 5 Mb Gewinne, 75% 5 Mb Verluste und alle CNVs über 10 Mb detektiert werden. Obwohl die Bestimmung der falschen Entdeckung Rate der einzelnen Zelle Sequenzierung ist schwierig, wir haben es geschätzt als 25% weniger. Dieses Protokoll kann zu Zellen von einer Vielzahl von Quellen verwendet werden, können die Scripts modifiziert werden, um die Auflösung von CNV Detektion einzustellen, und das Protokoll angepasst werden kann, andere Arten von genomischen Veränderungen zu identifizieren.
Es gibt eine Vielzahl von Mitteln von frischem Gewebe in einzelne Zellen dissoziiert, und viele Veröffentlichungen beschreiben Verfahren für spezifische Gewebe optimiert, wie Haut 24 und das Gehirn 25. Wir bevorzugen einzelne Zellen durch microaspiration zu isolieren, wie es erlaubt fo r visuelle Beurteilung jeder Zelle sequenziert werden. Es ist jedoch auch möglich , einzelne Zellen durch fluoreszenzaktivierte Zellsortierung (FACS) 26 und Mikrofluidik - Vorrichtungen 27 zu isolieren. Wenn einzelne Zellisolierung und Amplifikation des gesamten Genoms von Hand durchgeführt wird, ist es sinnvoll, zu isolieren und zu vierzig Zellen in einer einzigen Sitzung verstärken werden. Um eine hohe Qualität einzelne Zelle Sequenzierungsdaten zu erhalten, ist es von entscheidender Bedeutung, dass die Amplifikation von Einzelzellgenome einheitlich und vollständig sind. Wir finden, dass die Qualität der einzelnen Zellen isoliert sowie die Effizienz der Lyse und Verstärkung auf die Qualität der Sequenzierungsdaten einen wesentlichen Einfluss hat. Als solche sollten die Zellen aus ihrer natürlichen Umgebung nur vor der Isolierung und Amplifikation des gesamten Genoms beginnen sollte sofort geerntet werden, nachdem die Zellen isoliert werden. Darüber hinaus sollte die Lyse und Fragmentierungsschritt genau befolgt werden, wie in den Schritten 2.3-2.6 beschrieben.
"> Die Algorithmen können eingestellt werden , um die Auflösung von CNV Erkennung zu verändern, mit 12 Auswirkungen auf die Empfindlichkeit und Spezifität gegenüber. Es ist auch möglich , die Schwellenwerte einzustellen Ganz Chromosom Aneuploidie in der Einstellung des Tetraploidie 11 zu erkennen. Allerdings finden wir , dass unser Ansatz ist begrenzt CNVs größer als 5 Mb zu erfassen, als Rauschen während der Amplifikation des gesamten Genoms erschwert die Erkennung von kleineren Varianten 12. Zukünftige Verbesserungen in Amplifikation des gesamten Genoms Ansätze eingeführt wird, sollte die Auflösung der CNV - Detektion einzelnen Zelle Sequenzierung letztlich verbessern.Einzelne Zell Sequenzierung ermöglicht die Untersuchung nicht nur Kopienzahl Veränderungen , sondern auch Einzel - Nukleotid - Variationen 28, 29 und Strukturvariation 30. Unser Protokoll für einzelne Zellisolierung können diese anderen que zu beantworten angewendet werdenstions. Allerdings hängt die Wahl des gesamten Genoms Amplifikationsverfahren von der spezifischen Anwendung. Das Verfahren in diesem Protokoll beschrieben, die auf der Polymerasekettenreaktion basiert, eignet sich am besten zum Erfassen Änderungen Kopienzahl , weil es mit einem niedrigeren Verstärkungs Vorspannung 32 verbunden ist. Für andere Arten von genomischen Veränderungen zu untersuchen, wie beispielsweise Einzelnukleotid - Polymorphismen, andere Verfahren zur Amplifikation des gesamten Genoms sind vermutlich besser geeignet 31, 32 zu sein.
Offenlegungen
Die Autoren haben nichts zu offenbaren.
Danksagungen
Wir danken Stuart Levine für Kommentare zu diesem Manuskript. Diese Arbeit wurde von den National Institutes of Health Grants GM056800 und Kathy und Curt Marble Cancer Research Fund an Angelika Amon und zum Teil durch die Koch-Institut Förderpreis P30-CA14051 unterstützt. Angelika Amon ist auch ein Ermittler des Howard Hughes Medical Institute und der Glenn-Stiftung für biomedizinische Forschung. KAK wird durch die NIGMS Ausbildungsbeihilfe T32GM007753 unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177 | |
| PVC tubing (ID 3/16", OD 5/16", ~1 foot) | VWR | 89068-500 | |
| PVC tubing (ID 5/16", OD 7/16", ~6 inches) | VWR | 89068-508 | |
| Acrodisc Syringe Filter with HT Tuffryn Membrane (diameter 25 mm, pore size 0.2 µm) | VWR | 28144-040 | |
| Serological pipette (5 mL) | BioExpress | P-2837-5 | Any plastic 5 mL pipette should suffice |
| In-Line Water Trap for Oxygen Use | Amazon.com | 700220210813 | |
| Capillary Melting Point Tubes (ID 0.8-1.1 mm, 100 mm length) | VWR | 34502-99 | |
| Modeling clay | VWR | 470156-850 | |
| Petri dish (150 mm diameter) | VWR | 25384-326 | Surface should be non-adherent to facilitate aspiration of single cells |
| Hard-Shell Full-Height 96-Well Semi-Skirted PCR Plates | Bio-Rad | HSS9601 | Any 96-well PCR plate should suffice |
| 96-well tissue culture plate | VWR | 62406-081 | Any 96-well tissue culture plate should suffice |
| GenomePlex Single Cell Whole Genome Amplification Kit | Sigma | WGA4 | |
| Microseal 'A' Film | Bio-Rad | MSA5001 | |
| Mini plate spinner | Thomas Scientific | 1225Z37 | |
| Thermal cycler | Bio-Rad | 1861096 | Any thermal cycler should suffice so long as it can accommodate a 96-well PCR plate |
| Agencourt Ampure Beads | Beckman Coulter | A63880 | |
| Dynamag-2 Magnet | Thermo Fisher Scientific | 12321D | Any similar magnetic tube strip should suffice |
| Nextera XT DNA Library Preparation Kit | Illumina | FC-131-1096 | |
| Nextera XT Index Kit | Illumina | FC-121-1012 | |
| Complete kit (optimized for Roche LightCycler 480) | Kapa Biosystems | KK4845 | This kit is optimized for the Roche LightCycler 480 real-time PCR machine. If using a different machine, substitute a kit optimized for your machine. |
Referenzen
- Kahlem, P. Transcript Level Alterations Reflect Gene Dosage Effects Across Multiple Tissues in a Mouse Model of Down Syndrome. Genome Res. 14 (7), 1258-1267 (2004).
- Torres, E. M. Effects of Aneuploidy on Cellular Physiology and Cell Division in Haploid Yeast. Science. 317 (5840), 916-924 (2007).
- Itsara, A. Population Analysis of Large Copy Number Variants and Hotspots of Human Genetic Disease. Am. J. Human Gen. 84 (2), 148-161 (2009).
- Sudmant, P. H. Global diversity, population stratification, and selection of human copy-number variation. Science. 349 (6253), aab3761 (2015).
- Navin, N. Inferring tumor progression from genomic heterogeneity. Genome Res. 20 (1), 68-80 (2010).
- Torres, L., Ribeiro, F. R., Pandis, N., Andersen, J. A., Heim, S., Teixeira, M. R. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast cancer res and treat. 102 (2), 143-155 (2007).
- Yates, L. R. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 21 (7), 751-759 (2015).
- Forsberg, L. A. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Human Gen. 90 (2), 217-228 (2012).
- Laurie, C. C. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Gen. 44 (6), 642-650 (2012).
- Jacobs, K. B. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Gen. 44 (6), 651-658 (2012).
- Knouse, K. A., Wu, J., Whittaker, C. A., Amon, A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. 111 (37), 13409-13414 (2014).
- Knouse, K. A., Wu, J., Amon, A. Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. 26 (3), 376-384 (2016).
- . KAPA Library Quantification Kit Available from: https://www.kapabiosystems.com/product-applications/products/next-generation-sequencing-2/library-quantification/ (2016)
- . DNAcopy Available from: https://bioconductor.org/packages/release/bioc/html/DNAcopy.html (2016)
- Lichti, U., Anders, J., Yuspa, S. H. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat. Protoc. 3 (5), 799-810 (2008).
- Brewer, G. J., Torricelli, J. R. Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2 (6), 1490-1498 (2007).
- Navin, N. Tumour evolution inferred by single-cell sequencing. Nature. 472 (7341), 90-94 (2011).
- Szulwach, K. E., Chen, P. Single-Cell Genetic Analysis Using Automated Microfluidics to Resolve Somatic Mosaicism. PLoS ONE. 10 (8), e0135007 (2015).
- Zong, C., Lu, S., Chapman, A. R., Xie, X. S. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science. 338 (6114), 1622-1626 (2012).
- Wang, Y. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 512 (7513), 155-160 (2014).
- Zhang, C. Z. Chromothripsis from DNA damage in micronuclei. Nature. 522 (7555), 179-184 (2015).
- Macaulay, I. C., Voet, T. Single Cell Genomics: Advances and Future Perspectives. PLoS Genetics. 10 (1), e1004126 (2014).
- Gawad, C., Koh, W., Quake, S. R. Single-cell genome sequencing: current state of the science. Nat. Rev. Gen. , 1-14 (2016).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten