
Method Article
Détection de Copy Number Transformations Utilisation unique cellule de séquençage
Dans cet article
Résumé
séquençage cellulaire unique est un outil de plus en plus populaire et accessible pour aborder les changements génomiques à haute résolution. Nous fournissons un protocole qui utilise le séquençage d'une cellule unique pour identifier des altérations du nombre de copies dans des cellules individuelles.
Résumé
Détection des changements génomiques à la résolution de la cellule unique est important pour la caractérisation de l'hétérogénéité génétique et l'évolution dans les tissus normaux, les cancers et les populations microbiennes. Les méthodes classiques d'évaluation de l'hétérogénéité génétique a été limitée par la faible résolution, la faible sensibilité et / ou une faible spécificité. séquençage cellulaire unique a émergé comme un outil puissant pour détecter l'hétérogénéité génétique avec une haute résolution, une sensibilité élevée et, lorsqu'elles sont analysées de manière appropriée, une spécificité élevée. Ici, nous fournissons un protocole pour l'isolement, l'amplification du génome complet, le séquençage et l'analyse des cellules individuelles. Notre approche permet l'identification fiable des variantes du nombre de copies megabase échelle dans des cellules individuelles. Cependant, certains aspects de ce protocole peuvent également être appliquées pour enquêter sur d'autres types d'altérations génétiques dans des cellules individuelles.
Introduction
Les modifications du nombre de copies d'ADN peuvent varier en taille de plusieurs paires de bases (nombre de copies variantes) (CNV) à des chromosomes entiers (aneuploïdie). Modifications du nombre de copies qui affectent les grandes régions du génome peuvent avoir des conséquences importantes phénotypiques en modifiant l'expression de jusqu'à des milliers de gènes 1, 2. CNV qui sont présents dans toutes les cellules d'une population peut être détectée par un séquençage en masse ou des méthodes basées sur microréseaux 3, 4. Cependant, les populations peuvent également être génétiquement hétérogène, avec CNV existant dans un sous-ensemble de la population ou même des cellules individuelles. Hétérogénéité génétique est fréquente dans le cancer, entraînant l' évolution de la tumeur et également présent dans les tissus normaux, avec des conséquences inconnues 5, 6, 7, 8, 9 , 10.
Traditionnellement, l'hétérogénéité génétique a été évaluée soit par des approches cytologiques ou séquençage en vrac. Approches cytologiques, telles que l' hybridation fluorescente in situ (FISH), les écarts de chromosomes, et caryotype spectral (SKY) dans, ont l'avantage d'identifier les altérations présentes dans les cellules individuelles , mais ont des taux d'erreur élevés dus à des artefacts d'hybridation et de propagation 11. Ces approches sont également limitées dans leur nombre de copies résolution uniquement révélant des changements sur plusieurs mégabases. Séquençage ou microarrays d'ADN en vrac, bien plus élevé dans la précision et la résolution, est moins sensible. Afin de détecter l'hétérogénéité génétique par des approches basées sur la population, les variantes doivent être présents dans une fraction substantielle des cellules dans la population. L'émergence des procédés pour amplifier l'ADN génomique à partir de cellules individuelles a permis de séquencer le génome des cellules individuelles. Seule cellule sequencing présente les avantages de haute résolution, haute sensibilité, et, lorsque les méthodes de contrôle de la qualité appropriées sont appliquées, de haute précision 12.
Ici, nous décrivons un procédé de détection mégabases échelle du nombre de copies des altérations dans des cellules individuelles. Nous isolons des cellules individuelles par microaspiration, amplifier l'ADN génomique en utilisant linker-adaptateur PCR, préparer des banques pour le séquençage de prochaine génération, et de détecter le nombre de copies des variantes à la fois par le modèle de Markov caché et la segmentation circulaire binaire.
Protocole
1. Isoler cellules isolées
- Préparer le microaspirator
- Retirez l'extrémité plastique transparent de l'ensemble de tube d'aspiration et insérer l'extrémité étroite à une extrémité d'un tube de PVC 1 pieds de long avec 3/16 "de diamètre intérieur.
- Insérer la sortie d'un filtre à seringue de 0,2 um dans l'autre extrémité de la tubulure en PVC 1 avec les pieds de long 6/3 "de diamètre intérieur.
- Insérer l'entrée du filtre à seringue de 0,2 um dans une extrémité d'un tuyau en PVC de 6 pouces de long avec 5/16 "de diamètre intérieur.
- Facultatif: Couper le tube de PVC 6 pouces de long avec 5/16 "de diamètre intérieur en deux et insérer un piège à eau en ligne entre les deux moitiés.
- Pause d'une pipette sérologique 5 ml en plastique à la graduation 1 ml et insérez l'extrémité cassée dans l'extrémité ouverte du tube en PVC avec 5/16 "de diamètre intérieur.
- Insérer la sortie de la pipette sérologique de 5 ml en matière plastique souple à l'extrémité d'un ensemble de tube d'aspiration (où l'extrémité en plastique transparentont été supprimés).
- Pour le stockage, recouvrir l'embout buccal rouge de l'ensemble de tube d'aspiration avec un tube en plastique stérile.
- Tirer un tube capillaire en verre à un diamètre intérieur de 10 à 30 um en portant le milieu du tube à proximité d'une flamme du bec Bunsen et appliquer une tension sur les deux extrémités du tube. Briser le tube étiré au milieu pour créer deux aiguilles d'aspiration potentiels. Répétez cette étape avec plusieurs tubes comme seuls quelques tubes finiront avec un diamètre intérieur qui est approprié pour la cueillette des cellules individuelles.
- Pour le stockage, placer deux bandes de pâte à modeler dans un plat de 15 cm de Pétri et fixer les aiguilles d'aspiration à travers les bandes.
- Choisissez cellules
- Préparer une suspension cellulaire unique comme il convient pour le type de cellule et d'expérimentation. Par exemple, pour préparer des cellules adhérentes telles que des lignées humaines de fibroblastes de cellules, les cellules de la récolte par des cellules trypsinisation et de transfert à un tube conique contenant les médias appropriés.
- Avant, pendant ouprès avoir préparation de la suspension unique de cellules (selon le temps qu'il faut pour préparer la suspension cellulaire unique), la configuration de la hotte pour l'amplification du génome entier.
- Vaporiser en bas de la surface de la hotte, les boîtes de pointes de pipettes et embouts de pipette avec 10% de l'eau de Javel et essuyer avec une serviette en papier. Répétez cette étape avec 70% d'éthanol.
- Ajouter 8 pi d'eau du kit d'amplification du génome entier (WGA) à des puits individuels d'une plaque de 96 puits PCR, un puits pour chaque cellule à séquencer. Couvrir le 96 puits plaque PCR avec un couvercle à partir d'une plaque de culture de tissus à 96 puits et placer sur la glace.
- Ajouter 1000 cellules à 10 ml de milieu ou tampon phosphate salin (PBS) dans une boîte de 15 cm de Pétri et placez le plat sur la glace pour empêcher les cellules d'adhérer à l'antenne.
- Apportez les cellules dans la boîte de Pétri et le 96-puits plaque PCR avec couvercle sur la glace à un microscope optique avec objectif 10X.
- Augmenter l'ouverture de l'aiguille d'aspiration en tapotant doucement l'extrémité tirée hors du ièmee aiguille d'aspiration sur une surface dure telle que la pointe se détache. Insérez l'extrémité large de l'aiguille d'aspiration dans l'extrémité claire de la microaspirator.
- Placez la boîte de Pétri contenant des cellules sur la platine du microscope. Placer l'embout buccal rouge de l'aspirateur dans la bouche. Utilisez une main pour déplacer l'aiguille d'aspiration et l'autre main pour déplacer la boîte de Pétri contenant des cellules. Identifier des cellules individuelles à séquencer.
- Utilisation de la bouche d'aspiration, dessiner une seule cellule dans l'aiguille d'aspiration ainsi que ~ 1-2 pi de médias ou de PBS. Transférer la cellule dans les 8 pi d'eau à l'intérieur d'un seul puits de la plaque de 96 puits PCR. Évitez d'introduire des bulles lors du transfert de la cellule.
- Répétez l'étape 1.2.7 jusqu'à ce que le nombre désiré de cellules ont été isolées. Gardez la plaque PCR sur la glace et recouvert de couvercle entre les cellules de cueillette. Marquez les puits qui ont reçu des cellules.
- Tout en ramassant des cellules individuelles, décongeler le seul 10X lyse des cellules et un tampon de fragmentation de l'ensemble du gkit d'amplification enome. Après avoir choisi le nombre désiré de cellules, procéder immédiatement à l'amplification du génome entier.
2. Whole Genome Amplification
- Pour éviter la contamination lors de l'amplification du génome entier, ajouter tous les réactifs dans une culture de tissu hotte, utiliser des embouts de pipette avec des filtres, et changer la pointe de la pipette entre les puits.
- Préparer une solution de travail d'un tampon de lyse et la fragmentation de la trousse d'amplification du génome entier (WGA). Pour chaque ensemble de 32 cellules, mélanger 32 pi de 10x lyse cellulaire unique et un tampon de fragmentation et 2 pl de solution de proteinase K dans un tube à centrifuger. Vortex le tube pour mélanger les solutions.
- Ajouter 1 pl de lyse et une solution tampon de fragmentation de travail à chaque puits et la pipette de haut en bas pour mélanger.
- Couvrir la plaque et sceller tous les puits avec un film plastique. Centrifuger brièvement la plaque dans une mini-plaque spinner.
- Cycle thermique comme follOWS: 50 ° C, 1 h et 99 ° C, 4 min.
- Refroidir la plaque sur la glace et centrifuger brièvement la plaque dans une mini-plaque spinner.
- Préparer une solution de tampon de préparation bibliothèque de travail de l'ensemble du kit d'amplification du génome. Pour chaque cellule, mélanger 2 pl de tampon 1x préparation de bibliothèque de cellules simples et 1 pl de solution de stabilisation bibliothèque dans un tube de microcentrifugation. Préparer la solution de travail à plusieurs cellules dans le même tube de microcentrifugation.
- Retirez le film plastique et ajouter 3 pi de solution de travail de tampon de préparation bibliothèque pour chaque puits. Pipeter le contenu du bien haut et en bas pour mélanger. Remplacer le film plastique.
NOTE: Le film plastique peut être réutilisé pendant tout le processus d'amplification du génome jusqu'à ce que les puits commencent à percer des trous dans le film. Lorsque cela se produit, passer à un nouveau film plastique. - Centrifuger brièvement la plaque dans une mini-plaque spinner et incuber à 95 ° C pendant 2 min.
- Refroidir la plaque sur la glace et brièvementcentrifuger la plaque dans une mini-plaque spinner.
- Retirez le film plastique, ajouter 1 pl bibliothèque enzyme de préparation à chaque puits, et la pipette le contenu du puits de haut en bas pour mélanger. Gardez l'enzyme de préparation bibliothèque sur la glace ou dans un bloc froid tout au long de cette étape. Remplacer le film plastique.
- Centrifuger brièvement la plaque en mini spinner de plaque et cycle thermique comme suit: 16 ° C, 20 min, 24 ° C, 20 min, 37 ° C, 20 min, 75 ° C, 5 min, et maintenir à 4 ° C.
- Refroidir la plaque sur la glace et centrifuger brièvement la plaque dans une mini-plaque spinner.
- Préparer une amplification mélange de travail de l'ensemble du kit d'amplification du génome. Pour chaque cellule, mélanger 48,5 ul d'eau, 7,5 ul 10x amplification mélange maître, et 5 ul d'ADN polymérase WGA dans un tube à centrifuger. Préparer le mélange travaillant pour plusieurs cellules dans le même tube de microcentrifugeuse. Gardez le mélange de travail sur la glace.
- Retirez le film plastique, ajouter 61 pi de travail amplification mixà chaque puits, et le contenu de la pipette de bien haut et en bas pour mélanger. Gardez l'amplification mélange de travail sur la glace ou dans un bloc froid tout au long de cette étape. Remplacer le film plastique.
- Centrifuger brièvement la plaque dans une mini-plaque spinner et cycle thermique comme suit: 95 ° C, 3 min, 25 cycles de 94 ° C, 30 sec et 65 ° C, 5 min, et maintenir à 4 ° C.
- Transfert de 60 pi de chaque échantillon à un tube de microcentrifugeuse séparé et stocker à - 20 ° C pour une utilisation à l'étape 3.
- Ajouter l' ADN du chargement de colorant sur le volume restant de chaque échantillon ( par exemple 3 pi de colorant de charge 6x ADN à 15 ul de l' échantillon) et exécuter 5 ul de chaque réaction sur un gel d'agarose à 1%.
NOTE: Les cellules qui ont amplifié avec succès apparaîtra comme un frottis de 250 à 1000 pb. Les échantillons qui ne présentent pas un frottis ou seulement montrent un léger frottis sont peu susceptibles de produire des données de séquençage utiles.
3. séquençage
- Purifier échantillons avec des billes paramagnétiques.
- ThaLes échantillons d'ADN w sur la glace. des billes paramagnétiques vortex jusqu'à ce que la solution est homogène et incuber les billes à la température ambiante pendant au moins 30 min.
- Transférer 20 ul de chaque échantillon d'ADN dans un tube propre. Le reste de l'échantillon peut être stocké à -20 ° C.
- Ajouter 30 ul (1.5x) des billes paramagnétiques à chaque échantillon d'ADN et de vortex pour mélanger. Incuber les échantillons à température ambiante pendant 10 min. Laissez la solution mère de perles à la température ambiante pour une utilisation à l'étape 3.4.
- Placer les tubes sur une bande de tube magnétique pendant 2 minutes, ou jusqu'à ce que les billes forment un culot et le surnageant est clair.
- En utilisant une pipette P200, enlever autant de surnageant que possible sans perturber les perles.
- Ajouter 180 ul de 80% d'éthanol au mélange ADN-perle. Faire tourner les tubes plusieurs fois par rapport à l'aimant de "rinçage" des perles à travers la solution d'éthanol.
- En utilisant une pipette P200, enlever autant de lavage à l'éthanol comme possible sans déranger les perles.
- Répétez 3.1.6 et 3.1.7.
- Air sécher les billes pendant environ 10 minutes ou jusqu'à ce que plus l'éthanol est visible. Passez à l'étape suivante lorsque les perles apparaissent fissurés ou tomber de la paroi du tube.
- Retirer les échantillons de la bande magnétique. Ajouter 40 pl de 10 mM de Tris, pH 8,0, pour éluer l'ADN à partir des billes et vortexer les échantillons pour remettre en suspension les billes. Incuber pendant 2 minutes à la température ambiante.
- centrifuger brièvement les échantillons pour collecter le liquide au fond du tube et placer les tubes sur une bande magnétique du tube pendant au moins deux minutes. Transférer l'éluant dans des tubes à centrifuger propres sans déranger les perles.
- normalisation Sample
- Quantifier la concentration de chaque échantillon à l'aide d'un spectrophotomètre. Les concentrations devraient se situer entre 10 et 30 ng / pl.
- Diluer chaque échantillon à 0,2 ng / pl en utilisant 10 mM de Tris pH 8,0. Les étapes suivantes nécessiteront un i minimumnput de 5 pi de cette dilution.
- préparation Bibliothèque
- Préparer les bibliothèques de séquençage en suivant les instructions du kit de préparation de la bibliothèque 13.
- nettoyage final
- Nettoyer les échantillons selon l'étape 3.1. Pour des longueurs de lecture de 50 pb, 75 pb ou 150 pb, utiliser un rapport de bourrelet à volume de 1,5x, 1x ou 0.6x échantillon, respectivement. Éluer avec 15 ul de 10 mM de Tris pH 8,0.
- Exécutez les échantillons sur un analyseur de fragment pour vérifier la distribution de la taille de la bibliothèque 14. La distribution de la taille doit être répartie uniformément 150-900 pb.
- Quantifier les échantillons en utilisant qPCR
- L' utilisation d' un kit de quantification bibliothèque, mettre en place une réaction qPCR selon les instructions du kit 15. Laisser une ébauche de colonne pour ajouter des normes positives (normes d'ADN du kit) et des normes négatives (eau ou autre solution à blanc).
- Cycle thermique Assembas: 95 ° C, 5 min (vitesse de rampe de 4,8 ° C / s) et 35 cycles de 95 ° C, 30 sec (vitesse de rampe de 4,8 ° C / sec) et 60 ° C, 45 sec (taux de 2,5 rampe ° C / sec).
- mise en commun
- Déterminer le nombre de voies sur la cuve à circulation sont nécessaires et divisent les échantillons en groupes, avec un groupe par voie.
- Pour chaque groupe d'échantillons, pour choisir l'échantillon avec la concentration la plus faible sur la base des données provenant de l'étape qPCR 3.5.2 et normaliser tous les échantillons dans ce groupe pour que la concentration en utilisant 10 mM de Tris pH 8,0.
- Réunir les échantillons dans chaque groupe ensemble.
- séquençage
- Pools de charge sur la prochaine génération de machines de séquençage suivantes procédures standard 16.
Analyse 4. Données
NOTE: Un environnement basé sur Unix est requis pour exécuter les programmes et les scripts de cette section. Installez le logiciel mentionné dans le protocole suivant leur en guides d'ins-. Tous les scripts peuvent être trouvés à https://sourceforge.net/projects/singlecellseqcnv/.
- Coupez le lit à 40 nt en utilisant fastx_trimmer de la version FASTX-Toolkit 0.0.13 17.
fastx_trimmer -Q33 -i example.fastq -l 40 -o example.trim.fastq - Alignez le lit au génome approprié de référence (mm9 pour la souris, hg19 pour l' homme) en utilisant la version 0.6.1 BWA avec les options par défaut 18.
bwa aln mm9.fa example.trim.fastq> example.sai
bwa Samse mm9.fa example.sai example.trim.fastq> example.sam - Retirer les alignements de CHRM et chromosomes aléatoires, puis trier et indexer le fichier BAM résultant en utilisant la version 0.1.19 samtools 19.
grep -v -w CHRM example.sam | aléatoire> example.filtered.sam -v grep
samtools voir example.filtered.sam -uSh | samtools tri - example.filtered
samtools index example.filtered.bam - Exécutez HMMcopy pour détecter CNV= "Xref"> 20
- Utilisez gcCounter dans HMMcopy pour générer un fichier de référence en pourcentage de GC pour le génome. Utilisez l'option "-w 500000" pour spécifier la taille de la fenêtre. Utilisez la même version du fichier de référence fasta utilisé dans l'étape 4.2, mais assurez-vous CHRM et les chromosomes aléatoires sont retirés du fichier FASTA.
gcCounter -w 500000 mm9.fa> mm9_gc.wig - Utilisez generateMap.pl dans HMMcopy pour générer un fichier de manœuvre pour cartographiabilité. Utilisez l'option "-w 40" pour spécifier la longueur lue. Utilisez le même fichier de référence fasta utilisé dans l'étape 4.4.1.
generateMap.pl -b mm9.fa
generateMap.pl -w 40 -i mm9.fa mm9.fa -o mm9.bigwig - Utilisez mapCounter dans HMMcopy pour générer un fichier de référence de cartographiabilité pour le génome. Utilisez l'option "-w 500000" pour spécifier la taille de la fenêtre.
mapCounter -w 500000 mm9.bigwig> mm9_map.wig - Utilisez ReadCounter dans HMMcopy pour générer un fichier de manoeuvre pour chaque fichier BAM.
ReadCounter -w 500000 example.filtered.bam> input.wig - Modifiez les chemins d'accès aux fichiers de référence dans les scripts de R (run_hmmcopy.mm9.r ou run_hmmcopy.hg19.r), utiliser les fichiers générés ci - dessus en 4.4.1 (par exemple, mm9_gc.wig) et 4.4.3 (par exemple, mm9_map. perruque) pour les variables gfile et MFILE, qui se réfèrent au fichier de fichier de référence en pourcentage de GC et la référence de cartographiabilité, respectivement. Ensuite, exécutez le script R fourni "de run_hmmcopy.mm9.r" ou "run_hmmcopy.hg19.r".
R CMD BATCH run_hmmcopy.mm9.r
ou
R CMD BATCH run_hmmcopy.hg19.r - Pour le traitement d'un ensemble de fichiers BAM par lots, mettre les fichiers BAM dans un seul dossier et exécutez le script fourni "HMMpipe.pl" dans le paquet pour appeler des segments et calculer les scores de variabilité (VS). Suivez le format:
perl HMMpipe.pl Folder_to_BAMfiles mm9
- Utilisez gcCounter dans HMMcopy pour générer un fichier de référence en pourcentage de GC pour le génome. Utilisez l'option "-w 500000" pour spécifier la taille de la fenêtre. Utilisez la même version du fichier de référence fasta utilisé dans l'étape 4.2, mais assurez-vous CHRM et les chromosomes aléatoires sont retirés du fichier FASTA.
- Exécutez DNAcopy pour détecter CNV 21.
- Utilisez samtools la version 0.1.19 pour extraire mappé unique lit de chaque fichier BAM.
samtools voir -h -F 0x0004 example.filtered.bam | egrep -i "^ @ | XT: A: U" | samtools voir -SHU -> example.mapped.bam - Utilisez MarkDuplicates dans Picard Version 1.94 pour marquer les doublons de PCR dans le BAM fichier 22.
java -jar MarkDuplicates.jar INPUT = example.mapped.bam OUTPUT = example.nondup.bam METRICS_FILE = example.dup REMOVE_DUPLICATES = true - Utilisez coveragebed dans bedtools la version 2.17.0 de compter mappé lit dans chacune des fenêtres de cartographiables de 500kb dynamique prédéfinis dans le fichier BED fourni "de mm9.500k.dynamic.win.bed" ou "hg19.500k.dynamic.win.bed" 23 .
coverageBed -abam example.nondup.bam -b mm9.500k.dynamic.win.bed -counts | Tri -k1,1 -k2,2n> example.nondup.counts - Utilisez le script Perl fourni "normalizeGC.pl" pour normaliser les chiffres de lecture sur la base du fichier de référence fourni de contenu GC "mm9.500k.dynamic.win.fa.gc.txt" ou "hg19.500k.dynamic.win.fa. gc.txt ".
perl normalizeGC.pl mm9.500k.dynamic.win.fa.gc.txt example.nondup.counts> example.bam.norm.counts - Utilisez le R script "dnacopy.r" fourni pour appeler les segments.
R CMD BATCH dnacopy.r
- Utilisez samtools la version 0.1.19 pour extraire mappé unique lit de chaque fichier BAM.
- Identifier CNV
- Exclure les cellules pour lesquelles le VS calculé 4.4.6 dépasse 0,26.
- Filtrer les segments appelés par HMMcopy en 4.4.6 pour inclure uniquement ceux pour lesquels le ratio médian log 2 est supérieur à 0,4 (gain putative) ou inférieure à -0,35 (perte putative).
- Filtrer les segments appelés par DNAcopy à 4.5.5 pour inclure seulement ceux pour lesquels le segment moyenne est supérieure à 1,32 ou inférieur à 0,6.
- Superposer les segments de 4.6.2 et 4.6.3, appelant CNV seulement dans les régions où les deux HMMcopy et DNAcopy identifient un gain ou une perte putative putatif.
Résultats
L'aspirateur assemblé doit ressembler à celle de la figure 1A. L'aiguille doit être établie de telle sorte qu'elle est suffisamment large pour accueillir une seule cellule, mais pas si grand qu'un volume est établi avec la cellule unique. Aspiration des cellules individuelles est plus facile quand il y a entre un et cinq cellules dans un champ 10X (figure 1B).
Si l' amplification du génome entier est réussie, l'échantillon apparaîtra comme un frottis sur un gel d' agarose (figure 2, lignes 1, 2, 4, 5, 6 et 7). Un frottis faible ou absent indique une réaction d'amplification a échoué et l'échantillon ne doit pas être séquencée (figure 2, pistes 3 et 8).
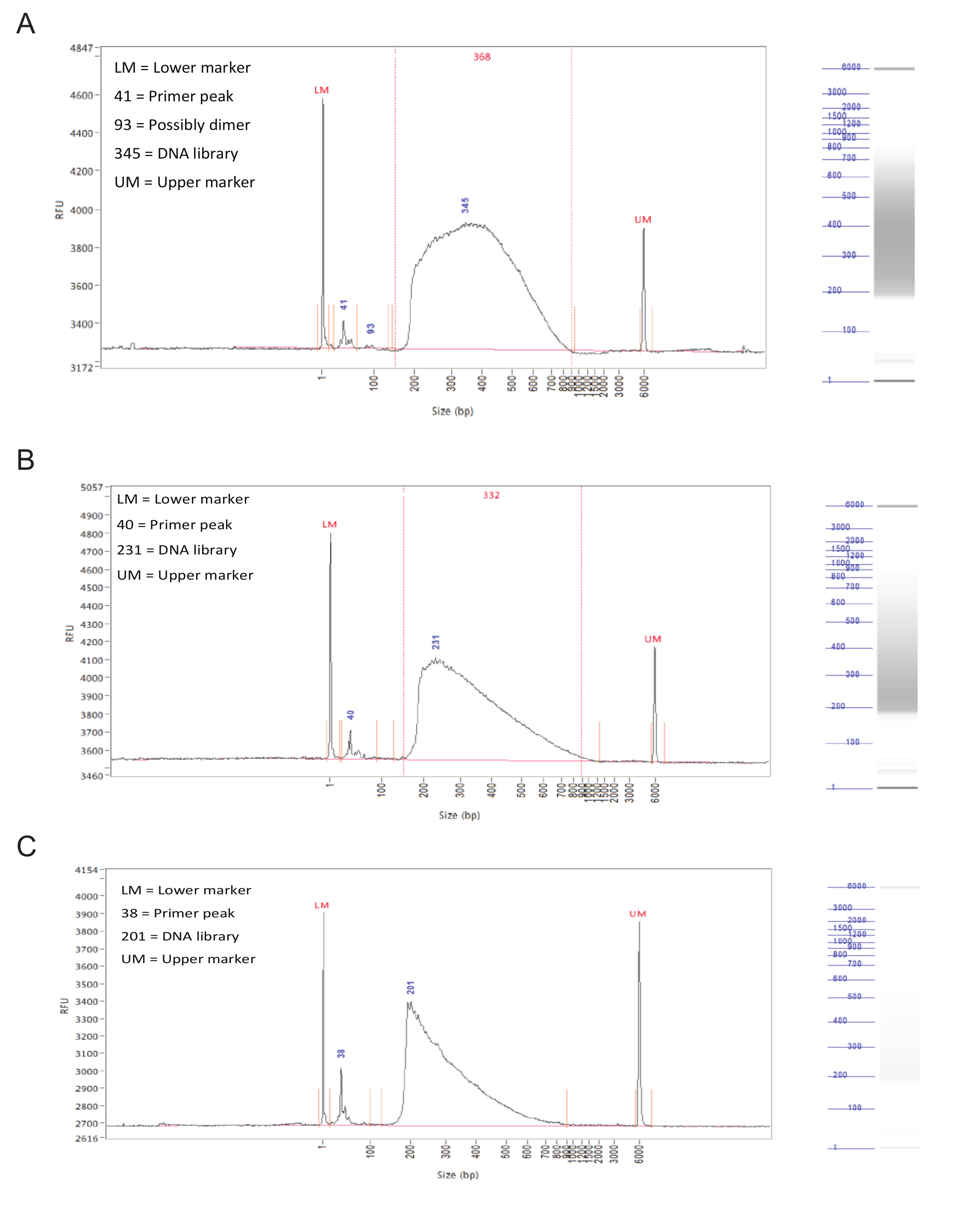
Après la préparation de bibliothèque, la distribution de la taille des fragments des échantillons doit être évalué par électrophorèse capillaire sur un analyseur de fragment.Avec succès les bibliothèques préparées ont une distribution plutôt même des tailles de fragment de 150 à 900 pb (figure 3, A, B). Préparation de la bibliothèque Échec se traduira par une distribution asymétrique de la taille des fragments et ces bibliothèques ne doit pas être séquencés (figure 3C).
Le traitement des données de séquençage au moyen du modèle de Markov caché (HMMcopy) et segmentation binaire circulaire (DNAcopy) va analyser le génome de chaque cellule en segments d'un nombre de copies estimé. Ces segments peuvent ensuite être filtrés pour identifier ceux avec un nombre de copies estimé compatible avec le gain ou la perte d'une seule cellule (tableau 1). Ces segments filtrés de HMMcopy et DNAcopy devraient ensuite être superposées pour identifier haute confiance CNV.

Figure 1. isolement cellulaire unique. ( A) Une assemblée microaspirator. (B) champ A 10X montrant dissocié les cellules (flèches) et aiguille microaspirator (coin inférieur droit). Aspiration des cellules individuelles est plus facile quand il y a entre un et cinq cellules dans un champ 10X. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 2. génome entier amplification. Agarose électrophorèse sur gel de 5 pi de produits entiers d'amplification du génome. Les échantillons qui amplifient avec succès apparaîtra comme frottis lumineux de 100 pb à 1 kb (pistes 1, 2, 4, 5, 6 et 7) et peut être séquencée. Les échantillons qui n'amplifient pas correctement produira des frottis faibles ou pas de frottis (voies 3 et 8) et ne doit pas être séquencés.large.jpg "target =" _ blank "> S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.

Figure 3. Préparation Bibliothèque. Les résultats représentatifs d'un analyseur de fragment. Les graphiques montrent la taille des fragments (en pb) sur l'axe X et les unités de fluorescence relative (RFU) sur l'axe des ordonnées. A droite de chaque graphique est une voie de gel simulé. (A) Résultats pour un échantillon idéal, avec une répartition uniforme entre 150 et 900 pb et sans pics ou polarisation vers un côté coupants. Cet échantillon est acceptable pour le séquençage. (B) Les résultats d'une échantillon correct, avec une distribution de taille biaisés vers des tailles de fragments inférieurs. Bien que ce soit pas optimal, l'échantillon peut encore être séquencée. (C) Résultats d'un échantillon a échoué, avec prédominance de petites tailles de fragments. Ceci est probablement dû à une incubation prolongée au cours de l'étape de tagmentationpréparation de bibliothèque. Cet échantillon ne doit pas être séquencé. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
| HMMcopy | |||||||
| Échantillon | Segment | Chr | Début | Fin | Etat | Médian | |
| D15-4998 | 23 | CHR8 | 144500001 | 146500000 | 6 | 0.5008794 | |
| D15-4998 | 29 | chr10 | 67000001 | 134500000 | 6 | 0.4031945 | |
| D15-4998 | 52 | chr19 | 1 | 20000000 | 6 | 0.4616884 | |
| D15-4998 | 57 | Chry | 1 | 59500000 | 2 | -1,506532 | |
| DNAcopy | |||||||
| Échantillon | Chrom | Démarrer bin | Fin bin | Début | Fin | moyenne Seg | med Seg |
| D15-4998 | chr10 | 88 | 197 | 62612945 | 129971511 | 1.4688 | 0,157 |
| D15-4998 | chr19 | 0 | 31 | 0 | 28416392 | 1,4141 | 0,1674 |
| D15-4998 | CHRX | 77 | 126 | 51659160 | 95343369 | 1.3548 | 0,1874 |
| D15-4998 | Chry | 0 | 14 | 0 | 23805358 | -2,7004 | 0,3591 |
Tableau 1. Analyse des données. segments Filtré générés par HMMcopy et DNAcopy à partir d'une seule cellule. Chevauchement ces deux résultats révèle un gain sur le chromosome 10 de 67 à 130 Mb ainsi qu'un gain sur le chromosome 19 de 0 à 20 Mb. Nous avons constaté que les gains sur la partie proximale du chromosome 19 sont un artefact de séquençage d'une seule cellule 12.
Discussion
Traditionnellement, l'identification CNV et aneuploïdie au niveau des cellules individuelles requises méthodes cytologiques telles que FISH et SKY. Maintenant, le séquençage de la cellule unique a émergé comme une approche alternative pour ces questions. séquençage cellulaire unique a des avantages sur FISH et SKY car il est à la fois l'ensemble du génome et de haute résolution. En outre, lorsque les méthodes de contrôle de la qualité appropriées sont appliquées, le séquençage de la cellule unique peut fournir une évaluation plus fiable du CNV et aneuploïdie comme il est pas sensible aux artefacts d'hybridation et de propagation inhérents à FISH et SKY. Cependant, la plupart des applications récentes de séquençage de la cellule unique ne sont pas étayées par une évaluation approfondie de la sensibilité et la spécificité des méthodes et des analyses. En effet, certaines des approches analytiques utilisées par d' autres études sont associés à des fréquences élevées (> 50%) des faux positifs CNV appelle 12. L'approche que nous décrivons a été rigoureusement testé en utilisant des cellules de knpropre fardeau CNV afin de déterminer les taux de découverte vrais et faux et d' optimiser la sensibilité et la spécificité de la détection CNV 12. Utilisation du contrôle de la qualité et des approches analytiques décrites dans ce protocole, environ 20% des 5 gains Mb, 75% de 5 défaites Mb, et tout CNV dépassant 10 Mb peut être détectée. Bien que la détermination du taux de fausse découverte du séquençage d'une seule cellule est difficile, nous avons estimé qu'il soit inférieur à 25%. Ce protocole peut être appliqué à des cellules d'une variété de sources, les scripts peuvent être modifiées pour ajuster la résolution de la détection NVC, ainsi que le protocole peut être adapté pour identifier d'autres types d'altérations génomiques.
Il existe une variété de moyens de dissociation de tissus frais dans des cellules individuelles, et de nombreuses publications décrivent les procédures optimisées pour des tissus spécifiques, tels que la peau et le cerveau 24 25. Nous préférons pour isoler des cellules uniques par microaspiration car elle permet fo r évaluation visuelle de chaque cellule à séquencer. Cependant, il est également possible d'isoler des cellules uniques par fluorescence activée par tri cellulaire (FACS) 26 et 27 des dispositifs microfluidiques. Si l'isolement d'une cellule unique et l'amplification du génome entier est effectuée manuellement, il est raisonnable d'isoler et d'amplifier jusqu'à quarante cellules en une seule séance. Afin d'obtenir des données de séquençage d'une cellule unique de haute qualité, il est crucial que l'amplification des génomes de cellules uniques est uniforme et complète. Nous constatons que la qualité des cellules individuelles isolées, ainsi que l'efficacité de la lyse et l'amplification a un impact significatif sur la qualité des données de séquençage. Par conséquent, les cellules doivent être récoltées à partir de leur environnement naturel, juste avant l'isolement et l'amplification du génome entier devrait commencer immédiatement après que les cellules sont isolées. En outre, l'étape de lyse et de la fragmentation doit être suivie exactement comme décrit dans les étapes 2.3-2.6.
"> Les algorithmes peuvent être ajustés pour changer la résolution de la détection CNV, avec des effets sur la sensibilité et la spécificité 12 opposées. Il est également possible d'ajuster les seuils pour détecter l' ensemble du chromosome aneuploïdie dans le cadre de tétraploïdie 11. Cependant, nous constatons que notre approche est limitée à la détection CNV supérieure à 5 Mb, comme le bruit introduit lors de l' amplification du génome entier complique la détection de petites variantes 12. les améliorations futures dans les approches d'amplification du génome entier devraient finalement augmenter la résolution de la détection CNV utilisant le séquençage d'une seule cellule.Séquençage cellulaire unique permet une enquête non seulement des modifications du nombre de copies , mais aussi des variations de nucléotides simples 28, 29 et variation structurelle 30. Notre protocole d'isolement d'une cellule unique peut être appliqué pour répondre à ces autres Questions. Cependant, le choix de la méthode d'amplification du génome entier dépend de l'application spécifique. La méthode décrite dans ce protocole, qui est basé sur la réaction en chaîne de la polymérase, est le mieux adapté pour détecter le nombre de copies des modifications car elle est associée à des niveaux inférieurs de polarisation d'amplification 32. Pour étudier d' autres types d'altérations génomiques, tels que des polymorphismes de nucléotides simples, d' autres procédés d'amplification du génome entier sont censés être plus appropriés 31, 32.
Déclarations de divulgation
Les auteurs n'ont rien à dévoiler.
Remerciements
Nous remercions Stuart Levine pour les commentaires sur ce manuscrit. Ce travail a été soutenu par les National Institutes of Health Grant GM056800 et Kathy et Curt Marble Cancer Research Fund à Angelika Amon et en partie par l'Institut Koch de soutien Grant P30-CA14051. Angelika Amon est aussi un enquêteur du Howard Hughes Medical Institute et la Fondation Glenn pour la recherche biomédicale. KAK est soutenu par le NIGMS Training Grant T32GM007753.
matériels
| Name | Company | Catalog Number | Comments |
| Aspirator tube assemblies for calibrated microcapillary pipettes | Sigma | A5177 | |
| PVC tubing (ID 3/16", OD 5/16", ~1 foot) | VWR | 89068-500 | |
| PVC tubing (ID 5/16", OD 7/16", ~6 inches) | VWR | 89068-508 | |
| Acrodisc Syringe Filter with HT Tuffryn Membrane (diameter 25 mm, pore size 0.2 µm) | VWR | 28144-040 | |
| Serological pipette (5 mL) | BioExpress | P-2837-5 | Any plastic 5 mL pipette should suffice |
| In-Line Water Trap for Oxygen Use | Amazon.com | 700220210813 | |
| Capillary Melting Point Tubes (ID 0.8-1.1 mm, 100 mm length) | VWR | 34502-99 | |
| Modeling clay | VWR | 470156-850 | |
| Petri dish (150 mm diameter) | VWR | 25384-326 | Surface should be non-adherent to facilitate aspiration of single cells |
| Hard-Shell Full-Height 96-Well Semi-Skirted PCR Plates | Bio-Rad | HSS9601 | Any 96-well PCR plate should suffice |
| 96-well tissue culture plate | VWR | 62406-081 | Any 96-well tissue culture plate should suffice |
| GenomePlex Single Cell Whole Genome Amplification Kit | Sigma | WGA4 | |
| Microseal 'A' Film | Bio-Rad | MSA5001 | |
| Mini plate spinner | Thomas Scientific | 1225Z37 | |
| Thermal cycler | Bio-Rad | 1861096 | Any thermal cycler should suffice so long as it can accommodate a 96-well PCR plate |
| Agencourt Ampure Beads | Beckman Coulter | A63880 | |
| Dynamag-2 Magnet | Thermo Fisher Scientific | 12321D | Any similar magnetic tube strip should suffice |
| Nextera XT DNA Library Preparation Kit | Illumina | FC-131-1096 | |
| Nextera XT Index Kit | Illumina | FC-121-1012 | |
| Complete kit (optimized for Roche LightCycler 480) | Kapa Biosystems | KK4845 | This kit is optimized for the Roche LightCycler 480 real-time PCR machine. If using a different machine, substitute a kit optimized for your machine. |
Références
- Kahlem, P. Transcript Level Alterations Reflect Gene Dosage Effects Across Multiple Tissues in a Mouse Model of Down Syndrome. Genome Res. 14 (7), 1258-1267 (2004).
- Torres, E. M. Effects of Aneuploidy on Cellular Physiology and Cell Division in Haploid Yeast. Science. 317 (5840), 916-924 (2007).
- Itsara, A. Population Analysis of Large Copy Number Variants and Hotspots of Human Genetic Disease. Am. J. Human Gen. 84 (2), 148-161 (2009).
- Sudmant, P. H. Global diversity, population stratification, and selection of human copy-number variation. Science. 349 (6253), aab3761 (2015).
- Navin, N. Inferring tumor progression from genomic heterogeneity. Genome Res. 20 (1), 68-80 (2010).
- Torres, L., Ribeiro, F. R., Pandis, N., Andersen, J. A., Heim, S., Teixeira, M. R. Intratumor genomic heterogeneity in breast cancer with clonal divergence between primary carcinomas and lymph node metastases. Breast cancer res and treat. 102 (2), 143-155 (2007).
- Yates, L. R. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 21 (7), 751-759 (2015).
- Forsberg, L. A. Age-related somatic structural changes in the nuclear genome of human blood cells. Am. J. Human Gen. 90 (2), 217-228 (2012).
- Laurie, C. C. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Gen. 44 (6), 642-650 (2012).
- Jacobs, K. B. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Gen. 44 (6), 651-658 (2012).
- Knouse, K. A., Wu, J., Whittaker, C. A., Amon, A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. 111 (37), 13409-13414 (2014).
- Knouse, K. A., Wu, J., Amon, A. Assessment of megabase-scale somatic copy number variation using single-cell sequencing. Genome Res. 26 (3), 376-384 (2016).
- . KAPA Library Quantification Kit Available from: https://www.kapabiosystems.com/product-applications/products/next-generation-sequencing-2/library-quantification/ (2016)
- . DNAcopy Available from: https://bioconductor.org/packages/release/bioc/html/DNAcopy.html (2016)
- Lichti, U., Anders, J., Yuspa, S. H. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat. Protoc. 3 (5), 799-810 (2008).
- Brewer, G. J., Torricelli, J. R. Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2 (6), 1490-1498 (2007).
- Navin, N. Tumour evolution inferred by single-cell sequencing. Nature. 472 (7341), 90-94 (2011).
- Szulwach, K. E., Chen, P. Single-Cell Genetic Analysis Using Automated Microfluidics to Resolve Somatic Mosaicism. PLoS ONE. 10 (8), e0135007 (2015).
- Zong, C., Lu, S., Chapman, A. R., Xie, X. S. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science. 338 (6114), 1622-1626 (2012).
- Wang, Y. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 512 (7513), 155-160 (2014).
- Zhang, C. Z. Chromothripsis from DNA damage in micronuclei. Nature. 522 (7555), 179-184 (2015).
- Macaulay, I. C., Voet, T. Single Cell Genomics: Advances and Future Perspectives. PLoS Genetics. 10 (1), e1004126 (2014).
- Gawad, C., Koh, W., Quake, S. R. Single-cell genome sequencing: current state of the science. Nat. Rev. Gen. , 1-14 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.