
Method Article
Transcripción Iniciar asignación de sitios usando portadora de entrada súper baja-CAGE
En este artículo
Resumen
Cap Analysis of Gene Expression (CAGE) es un método para la cartografía cuantitativa de los extremos de mRNA 5 para capturar sitios de inicio de transcripción de ARN polimerasa II con una resolución de un solo nucleótido. Este trabajo describe un protocolo de baja entrada (SLIC-CAGE) para la generación de bibliotecas de alta calidad utilizando nanogramos-cantidades de ARN total.
Resumen
El análisis de la tapa de la expresión génica (CAGE) es un método utilizado para la detección de resolución de un solo nucleótido de los sitios de inicio de transcripción de ARN polimerasa II (TSS). La detección precisa de TSS mejora la identificación y el descubrimiento de los promotores principales. Además, los potenciadores activos se pueden detectar a través de firmas de iniciación de transcripción bidireccional. Aquí se describe un protocolo para realizar la portadora de entrada súper baja-CAGE (SLIC-CAGE). Esta adaptación SLIC del protocolo CAGE minimiza las pérdidas de ARN aumentando artificialmente la cantidad de ARN mediante el uso de una mezcla de portadoras de ARN transcrita in vitro que se añade a la muestra de interés, permitiendo así la preparación de la biblioteca a partir de nanogramos-cantidades de total ARN (es decir, miles de células). El portador imita la distribución esperada de la longitud del fragmento de la biblioteca de ADN, eliminando así los sesgos que podrían ser causados por la abundancia de un portador homogéneo. En las últimas etapas del protocolo, el portador se elimina a través de la degradación con endonucleases homing y la biblioteca de destino se amplifica. La biblioteca de muestras objetivo está protegida contra la degradación, ya que los sitios de reconocimiento de endonucleasa homing son largos (entre 18 y 27 bp), lo que hace que la probabilidad de su existencia en los genomas eucariotas sea muy baja. El resultado final es una biblioteca de ADN lista para la secuenciación de próxima generación. Todos los pasos en el protocolo, hasta la secuenciación, se pueden completar dentro de 6 días. La preparación del portaaviones requiere un día de trabajo completo; sin embargo, se puede preparar en grandes cantidades y mantenerse congelado a -80 oC. Una vez secuenciadas, las lecturas se pueden procesar para obtener TSS de resolución de nucleótidos de un solo nucleótido para todo el genoma. Una vez agregados a los promotores, los datos también se pueden utilizar para la generación de perfiles de expresiones centrada en 5'.
Introducción
El análisis de la tapa de la expresión génica (CAGE) es un método utilizado para el mapeo de resolución de un solo nucleótido en todo el genoma de los sitios de inicio de transcripción de ARN polimerasa II (TSSs)1. Su naturaleza cuantitativa también permite la generación de perfiles de expresión centrada de 5'-end. Las regiones que rodean los TSS (alrededor de 40 bp aguas arriba y aguas abajo) son promotores principales y representan la ubicación física donde el ARN polimerasa II y los factores de transcripción general se unen (revisado previamente2,3). La información sobre las ubicaciones exactas de los TSS se puede utilizar para el descubrimiento del promotor principal y para supervisar la dinámica del promotor. Además, a medida que los potenciadores activos exhiben firmas de transcripción bidireccional,los datos CAGE también se pueden utilizar para el descubrimiento de potenciadores y la supervisión de la dinámica del potenciador 4. La metodología CAGE ha aumentado recientemente en popularidad debido a su amplia aplicacióny uso en proyectos de investigación de alto perfil como ENCODE 5, modENCODE6y proyectos FANTOM7. Además, la información del SST también está demostrando ser importante para distinguir el tejido sanoy enfermo, ya que los TSS específicos de la enfermedad pueden utilizarse con fines de diagnóstico 8.
A pesar de que hay varios métodos para la asignación de TSS disponibles (CAGE, RAMPAGE, STRT, nanoCAGE, nanoCAGE-XL, oligo-capping), nosotros y otros hemos demostrado recientemente que CAGE es el método más imparcial para capturar verdaderos TSS con el menor número de falsos positivos9 , 10. El protocolo CAGE reciente, nAnT-iCAGE11, es el protocolo más imparcial para la generación de perfiles TSS, ya que evita cortar los fragmentos a etiquetas cortas utilizando enzimas de restricción y no utiliza amplificación de PCR. Una limitación del protocolo nAnT-iCAGE es el requisito de una gran cantidad de material de partida (por ejemplo, 5 g de ARN total para cada muestra). Para responder a preguntas específicas y biológicamente relevantes, a menudo es imposible obtener cantidades tan altas de material de partida (por ejemplo, para células clasificadas con FACS o etapas embrionarias tempranas). Por último, si nAnT-iCAGE tiene éxito, solo 1-2 ng de material de la biblioteca de ADN está disponible en cada muestra, lo que limita la profundidad de secuenciación alcanzable.
Para permitir el perfilado TSS utilizando sólo nanogramos de ARN total, recientemente hemos desarrollado Super-low Input Carrier-CAGE10 (SLIC-CAGE, Figura 1). SLIC-CAGE requiere sólo 10 ng de ARN total para obtener bibliotecas de alta complejidad. Nuestro protocolo se basa en el portador de ARN sintético cuidadosamente diseñado añadido al ARN de interés para lograr un total de 5 g de material de ARN. El portador sintético imita la biblioteca de ADN objetivo en la distribución de la longitud para evitar posibles sesgos que podrían ser causados por moléculas homogéneas en exceso. La secuencia del portador se basa en la secuencia del gen Escherichia coli leucyl-tRNA sintetasa (Tabla 1) por dos razones. En primer lugar, cualquier resto del portador en la biblioteca final, incluso si se secuencia, no se asignará a un genoma eucariota. En segundo lugar, como E. coli es una especie mesofílica, sus genes de limpieza están optimizados para el rango de temperatura adecuado para SLIC-CAGE. La secuencia portadora también está integrada con sitios de reconocimiento de endonucleasa de homing para permitir la degradación específica del ADN derivado de las moléculas de ARN portadora. La biblioteca de destino derivada de la muestra permanece intacta, ya que los sitios de reconocimiento de endonucleasa de homing son largos (I-CeuI a 27 bp; I-SceI a 18 bp) y estadísticamente poco probable que se encuentre en los genomas eucariotas. Después de la degradación específica del portador y la eliminación de fragmentos por exclusión de tamaño, la biblioteca de destino se amplifica y está lista para la secuenciación de próxima generación. Dependiendo de la cantidad de ARN inicial (1-100 ng), se espera que se requieran entre 13-18 ciclos de amplificación de PCR. La cantidad final de ADN por cada muestra oscila entre 5-50 ng, produciendo suficiente material para una secuenciación muy profunda. Cuando se utiliza sólo 1-2 ng de ARN total, se pueden detectar Verdaderos SST; sin embargo, se espera que las bibliotecas sean de menor complejidad. Por último, como SLIC-CAGE se basa en el protocolo nAnT-iCAGE11, permite la multiplexación de hasta ocho muestras antes de la secuenciación.
Protocolo
1. Preparación del Transportista
-
Preparación de plantillas de ADN para transcripción in vitro
- Preparar la mezcla de PCR para cada plantilla de PCR combinando 41 l de agua, 20 l de búfer de HF de 5x, 8 l de dNtPs de 2,5 mM, 10 ml de imprimación delantera única de 10 m (PCR_GN5_f1, Tabla2; imprimaciones se disuelven y se diluyen en agua) , 10 l de plásmido de plantilla de 2 ng/L que contenga el gen portador sintético y 1 l de phusion polimerasa. Mezclar la mezcla de PCR mediante pipeteo. Se puede preparar una mezcla maestra para las 10 plantillas a la vez (prepárese para 11 reacciones).
- Añadir 90 l de la mezcla de PCR a 10 sL de cada imprimación inversa de 10 m (PCR_N6_r1-r10, Tabla 2). Mezclar por pipeteo.
- PCR amplificar las plantillas utilizando el siguiente programa: 98 oC para 60 s, (98 oC para 10 s, 50 oC para 30 s, 72 oC para 30 s) 35 ciclos, 72 oC para 10 min, mantener a 4 oC.
-
Purificación en gel de plantillas de ADN amplificadas por PCR
- Preparar un gel de agarosa al 1% (se recomienda agarosa de baja fusión).
- Para disminuir el volumen, concentre las mezclas de reacción de PCR de 100 a 20 l de volumen total utilizando el concentrador de vacío a una temperatura baja-media (30-40 oC).
- Añadir 6 l del tinte de carga 6x, mezclar bien y cargar en el gel. Ejecute la electroforesis durante 30 minutos en 1 búfer TAE a la tensión adecuada para el tanque de electroforesis utilizado (5-10 V/cm). En paralelo, ejecute una escalera de ADN de 100 bp o 1.000 bp.
- Usando un bisturí limpio, exbose las rodajas de gel que contienen el producto PCR objetivo. Evite el exceso de gel de agarosa. Purificar los productos PCR utilizando un kit de extracción de gel (de acuerdo con las instrucciones del fabricante).
NOTA: Las proporciones A260/A230 de ADN aislado de geles de agarosa son típicamente bajas (0,1–0,3). El producto de destino esperado y los productos secundarios se muestran en la Figura 2A. Los rendimientos esperados a partir de reacciones de PCR de 100 ml son de 1,2 a 3 g. Las reacciones se pueden escalar para obtener un mayor rendimiento.
-
Transcripción in vitro de moléculas portadoras
- Transcribir el ARN portador in vitro utilizando la polimerasa de ARN T7 de acuerdo con las instrucciones del fabricante. Configurar reacciones de 10 a 20 l (el kit recomendado está en tabla de materiales).
- Purifique el ARN transcrito in vitro utilizando un kit de purificación de ARN. Configure la digestión del ADN en solución utilizando DNase I siguiendo las instrucciones estándar del fabricante y eluda el ARN en 50 ml de agua. Para aumentar el rendimiento de elución, deje el agua en la columna durante 5 minutos antes de la centrifugación.
NOTA: Tenga cuidado de no exceder la capacidad máxima de encuadernación de las columnas (en el kit mencionado en la Tabla de materiales,la capacidad es de hasta 100 g). El rendimiento esperado de las plantillas de PCR de 1 a 10 (1 kbp a 200 bp de longitud) es de 25 a 50 g a partir de reacciones de transcripción in vitro de 10 l. Las reacciones se pueden escalar para obtener un mayor stock de moléculas portadoras.
-
Tapado de moléculas de ARN portadoras transcritas in vitro
- Preparar la mezcla de tapón combinando 2 l de tampón de taponamiento de 10x, 1 l de 10 mM GTP, 1 ml de SAM de 2 mM (recién diluido) y 1 l de enzima de tapado de Vaccinia por ARN portador.
- Mezclar hasta 10 g de cada molécula portadora en 15 ml de volumen total y desnaturalidad durante 10 min a 65 oC. Colocar sobre hielo inmediatamente para evitar la formación de la estructura secundaria.
- Mezclar el ARN portador desnaturalizado con 5 l de la mezcla de taponamiento e incubar durante 1 h a 37 oC.
- Purificar moléculas de ARN tapadas utilizando un kit de purificación de ARN — siga el protocolo de limpieza del fabricante. Elute el ARN en 30 l de agua. Para aumentar el rendimiento de elución, deje el agua en la columna durante 5 minutos antes de la centrifugación.
NOTA: Mida la concentración utilizando el espectrofotómetro de microvolúmenes. La relación A260/A280 esperada es >2 y A260/A230 es >2. Tenga en cuenta que para algunas muestras de ARN A260/A230 puede estar entre 1.3–2. El rendimiento esperado cuando se utilizan 10 g de ARN sin tapar es de 9-10 g de ARN tapado.
- Preparar la mezcla del portador con tapa y sin tapar combinando las cantidades descritas en el Cuadro 3. Mezcle bien moviendo el tubo y mida la concentración utilizando el espectrofotómetro de microvolúmenes.
NOTA: Si se requiere una mayor concentración del portador para encajar en la reacción de transcripción inversa (ver más abajo), la mezcla portadora se puede concentrar utilizando el concentrador de vacío a baja temperatura media (30-35 oC) hasta alcanzar la concentración final deseada. Los pasos 2–14 se modifican del protocolo estándar nAnT-iCAGE reportado por Murata et al.
2. Transcripción inversa
- Combinar 1 l de la imprimación RT (2,5 mM TCT-N6 disuelto en agua, para secuencia ver Tabla Suplementaria 1), 10 ng de ARN total de interés y 4.990 ng de mezcla portadora (Tabla3) en 10 ml de volumen total en una placa PCR de baja unión. Mezclar moviendo el tubo.
NOTA: Si el ARN de la muestra está demasiado diluido para la transcripción inversa (véase más adelante), combínelo con la cantidad apropiada del portador, concéntreconel a través del concentrador de vacío a un volumen total de 9 l y agregue 1 l de la imprimación RT. La adición del conde portador, para llegar a 5 g de ARN en total evita la pérdida de la muestra. - Calentar la mezcla del paso 2.1 a 65oC durante 5 min y colocarla sobre hielo inmediatamente para evitar la renaturalización.
-
Preparación de la mezcla de transcripción inversa (RT).
- Para cada muestra, combine 6,1 l de agua (libres de RNase y DNase), 7,6 ml de búfer de primera cadena de 5x, 1,9 l de TDT de 0,1 M, 1 l de dNTPs de 10 mM, 7,6 ml de mezcla de trehalosa/sorbitol (véase la receta en Murata et al.11) y 3,8 l de la transcriptasa inversa recomendada (véase Tabla de Materiales). Mezclar bien moviendo el tubo.
- Añadir 28 l de la mezcla RT en el tubo de PCR con 10 s de ARN, portador y la imprimación RT (volumen total 38 l). Mezcle bien pipeteando.
NOTA: La mezcla es altamente viscosa debido a la trehalosa/sorbitol. Mezclar hasta que sea visiblemente homogéneo. - Incubar en un ciclor térmico utilizando el siguiente programa: 25 oC durante 30 s, 50 oC durante 60 min, y mantener a 4 oC.
-
Purificación de híbridos cDNA:RNA usando perlas magnéticas SPRI
- Añadir 68,4 l de las perlas SPRI recomendadas libres de ARSa y DNase (ver Tabla de Materiales)a 38 l de la mezcla RT (perlas a la relación de muestra 1.8:1). Mezclar bien pipeteando e incubar durante 5 min a temperatura ambiente (RT).
- Separe las perlas en un soporte magnético durante 5 min. Deseche el sobrenadante y lave las perlas dos veces con 200 s de etanol 70% (recién preparado).
NOTA: El etanol se añade a las perlas sin mezclar y mientras el tubo está en el soporte magnético. El etanol añadido se retira inmediatamente. Se debe tener cuidado de no perder ninguna perla durante los lavados, ya que puede conducir a la pérdida de la muestra. - Mientras el tubo todavía está en el soporte magnético, retire todos los rastros de etanol. Las gotas de etanol se pueden retirar y empujar fuera del tubo con una pipeta P10. No deje que las cuentas se sequen.
- Añadir 42 s de agua precalentada a 37 oC a las perlas y eluir la muestra pipeteando hacia arriba y hacia abajo 60x.
NOTA: Tenga cuidado de no causar espuma por pipeteo, ya que puede causar la pérdida de cuentas (es decir, muestra encuadernada) en la espuma. - Incubar a 37oC durante 5 min sin tapa para permitir la evaporación de trazas de etanol.
- Separe las perlas en un soporte magnético durante 5 minutos y transfiera el sobrenadante a una placa nueva.
NOTA: Trate de recuperar todo el sobrenadante para evitar la pérdida de la muestra evitando el arrastre de cuentas. Utilice la pipeta P10 para obtener las últimas gotas de muestra.
3. Oxidación
- Añadir 2 ml de NaOAc de 1 M (pH 4.5) a la reacción RT purificada. Mezclar con pipeteo, añadir 2 sl de 250 mM NaIO4 y mezclar de nuevo.
- Incubar sobre hielo durante 45 min. Cubra la placa con papel de aluminio para evitar la luz.
- Añadir 16 l de Tris-HCl (pH 8.5) a la mezcla de oxidación para neutralizar el pH.
- Purificar híbridos oxidados cDNA:RNA usando perlas magnéticas SPRI. Añadir 108 l de perlas SPRI a 60 l de la mezcla de oxidación (1,8:1 perlas a la relación de la muestra). Repita la purificación como se describe en los pasos 2.6.1–2.6.6. Eluir con 42 oC de agua precalentada a 37oC.
NOTA: Recién preparar 250 mM NaIO4 añadiendo 18,7 ml de agua por 1 mg de NaIO4. NaIO4 es sensible a la luz; por lo tanto, mantenga la solución en un tubo cubierto con papel de aluminio o en un tubo resistente a la luz.
4. Biotinylación
- Añadir 4 ml de NaOAc de 1 M (pH 6.0) en el tubo que contiene la muestra oxidada purificada y mezclar mediante pipeteo.
- Añadir 4 sl de solución de biotina de 10 mM, mezclar mediante pipeteo e incubar durante 2 h a 23 oC en un ciclor térmico para evitar la luz.
NOTA: Preparar la solución de biotina mezclando 50 mg de biotina con 13,5 ml de DMSO. Hacer alícuotas de un solo uso y congelar a -80 oC. - Purificar híbridos de ADNc:ARN biotinilado utilizando perlas magnéticas SPRI. Añadir 12 s de 2-propanol y mezclar mediante pipeteo. Añadir 108 l de perlas SPRI (1,8:1 perlas a la relación de la muestra) y repetir la purificación como se describe en los pasos 2.6.1–2.6.6. Eluir con 42 oC de agua precalentada a 37oC.
NOTA: El protocolo se puede pausar aquí, y las muestras se congelan a -80 oC.
5. RNase I Ddigestion
- Preparar la mezcla de RNase I mezclando 4,5 ml de 10x de tampón RNase I con 0,5 ml de RNase I (10 U/L) por cada muestra. Mezclar por pipeteo.
- Añadir 5 ml de la mezcla a cada muestra purificada (45 ml en total). Mezclar mediante pipeteo e incubar durante 30 min a 37oC.
6. Preparación de cuentas de streptavidina
- Para cada muestra, mezcle 30 ml de la perla de estreptavidina con 0,38 ml de ARNm de 20 mg/ml. Incubar en hielo durante 30 minutos y mezclar cada 5 minutos moviendo el tubo.
NOTA: Resuspenda las perlas de estreptavidina se hundan bien antes de pipetear moviendo la botella. La solución de ARNm debe prepararse de acuerdo con Murata et al. - Separe las perlas en el soporte magnético durante 2-3 min. Retire el sobrenadante.
- Lavar las perlas resuponiendo en 15 sde tampón A. Separe las perlas en el soporte magnético durante 2-3 min y retire el sobrenadante. Repita el lavado y retire el sobrenadante.
- Resuspenda las perlas en 105 ml de tampón A y agregue 0,19 l de 20 mg/ml de ARN. Mezcle bien pipeteando.
NOTA: Las perlas deben prepararse frescas antes de su uso. Comienza la preparación de las perlas durante la digestión de RNase I. Para múltiples muestras, prepare las perlas juntas en un solo tubo.
7. Captura de tapa
-
Encuadernación de muestras
- Añadir 105 l de perlas de estreptavidina preparadas a 45 l de la muestra tratada con RNase I. Mezcle bien pipeteando e incubar a 37 oC durante 30 min. Mezcle pipeteando cada 10 minutos.
- Separe las perlas en el soporte magnético durante 2-3 min. Retire el sobrenadante.
-
Perlas de lavado
- Añadir 150 s de tampón de lavado A y resuspender las perlas mediante pipeteo. Separe las perlas en el soporte magnético durante 2-3 min y retire el sobrenadante.
- Añadir 150 l del tampón de lavado B y resuspender las perlas mediante pipeteo. Separe las perlas en el soporte magnético durante 2-3 min y retire el sobrenadante.
- Añadir 150 l del tampón de lavado C y resuspender las perlas mediante pipeteo. Separe las perlas en el soporte magnético durante 2-3 min y retire el sobrenadante.
NOTA: Los búferes B y C deben precalentarse a 37 oC. Las recetas para los tampones de lavado A, B y C son como se describe en Murata et al.
-
liberación de adNc
- Preparar 1 tampón RNase I mezclando 58,5 ml de agua con 6,5 l de tampón RNase I de 10x.
- Resuspenda las perlas en 35 l de 1 x tampón RNase I. Incubar a 95oC durante 5 min y transferir directamente sobre hielo durante 2 min para evitar la reasociación del ADNc. Sostenga las tapas durante la transferencia al hielo, ya que pueden estallar debido a la acumulación de presión.
- Separe las perlas durante 2-3 minutos en un soporte magnético y transfiera el sobrenadante a una placa nueva.
- Resuspenda las perlas en 30 l de 1 x tampón RNase I. Separe las perlas en el soporte magnético durante 2-3 min y transfiera el sobrenadante al sobrenadante previamente recolectado (el volumen total de ADNc eludado debe ser de unos 65 l).
8. Eliminación de ARN por RNase H y RNase I Digestion
- Por muestra, combinar 2,4 l de agua, 0,5 l de tampón de 10 x RNase I, 0,1 l de RNase H y 2 l de RNase I.
- Añadir 5 s de la mezcla a los 65 s de la muestra de ADNc liberada y mezclar mediante pipeteo. Incubar a 37oC durante 15 min y mantener a 4oC.
- Purifica el ADNa de la mezcla de digestión RNase usando perlas magnéticas SPRI. Añadir 126 sl de perlas SPRI a 70 l de reacción de degradación y mezclar mediante pipeteo. Siga los pasos de purificación descritos para la purificación de perlas SPRI en 2.6.1–2.6.6. Eluir con 42 ml de agua precalentada a 37 oC como se describe.
- Preparar RNase Mix combinando 4,5 l de tampón RNase I de 10x y 0,5 l de RNase I.
- Añadir 5 ml de la mezcla de RNase a los 40 s de la muestra de ADNc purificado. Mezclar con pipeteo e incubar a 37oC durante 30 min. Mantener a 4oC.
- Purificar la muestra usando perlas magnéticas SPRI. Añadir 81 l de perlas SPRI a 45 l de reacción de degradación y mezclar mediante pipeteo. Siga los pasos de purificación descritos para la purificación de perlas SPRI en 2.6.1–2.6.6. Eluir con 42 ml de agua como se describe.
9. Ligadura de 5' Linker
- Concentrar la muestra de ADNc purificada a 4 ml utilizando el concentrador de vacío. Mantener la temperatura a 30–35 oC. Pruebe el volumen con una pipeta. Si la muestra se ha secado a la integridad, disolver añadiendo 4 s de agua.
NOTA: Es mejor evitar el secado a la integridad para evitar la pérdida de la muestra. - Incubar la muestra concentrada a 95oC durante 5 min y colocar inmediatamente sobre hielo durante 2 min para evitar la renaturalización. Sujete las tapas mientras transfiere los tubos, ya que las tapas pueden estallar debido a la acumulación de presión.
- Incubar 4 s del vinculador de 2,5 m y 5' a 55 oC durante 5 min y colocar inmediatamente sobre hielo durante 2 min para evitar la renaturalización.
- Mezclar 4 sl del vinculador de 2,5 m y 5' con 4 s de la muestra.
NOTA: El vinculador de 5' debe prepararse de acuerdo con el Cuadro Suplementario 2, el Cuadro Complementario 3, el Cuadro Suplementario 4y el Cuadro Suplementario 5. Diluir el vinculador de 10 M 5' a una concentración de 2,5 M utilizando 100 mM de NaCl antes de su uso. - Añadir 16 l de la premezcla de ligadura (ver Tabla de Materiales)al vinculador mixto de 5' y la muestra y mezclar bien pipeteando. Incubar a 16oC durante 16 h.
- Purificar la mezcla de ligadura usando perlas magnéticas SPRI. Agregue 43,2 l de perlas SPRI y siga los pasos 2.6.1–2.6.6. Eluir como se describe utilizando 42 s de agua precalentada a 37 oC.
- Repita la purificación realizada en el paso 9.6 añadiendo 72 sdes SPRI al sobrenadante transferido (1.8:1 perlas a la relación de la muestra).
NOTA: Los enlaces de 5' contienen códigos de barras que permiten la agrupación de hasta ocho muestras antes de la secuenciación (ocho códigos de barras de trinucleótidos están disponibles, como se describe en Murata et al.11 y Supplementary Table 1).
10. Ligadura de 3' Linker
- Concentrar la muestra purificada a 4 l utilizando el concentrador de vacío como se describe en el paso 9.1.
- Incubar la muestra concentrada a 95oC durante 5 min y colocar inmediatamente sobre hielo durante 2 min para evitar la renaturalización. Sujete las tapas mientras transfiere los tubos, ya que las tapas pueden estallar debido a la acumulación de presión.
- Incubar 4 s del vinculador de 2,5 m 3' a 65 oC durante 5 min y colocar inmediatamente sobre hielo durante 2 min para evitar la renaturalización.
- Añadir 4 sl del vinculador de 2,5 m 3' a los 4 s de la muestra concentrada.
- Añadir 16 l de la premezcla de ligadura y mezclar bien pipeteando. Incubar a 16oC durante 16 h.
- Purificar la mezcla de ligadura usando perlas magnéticas SPRI. Agregue 43,2 l de perlas SPRI y siga los pasos 2.6.1–2.6.6. Eluir como se describe utilizando 42 s de agua precalentada a 37 oC.
NOTA: El vinculador de 3' debe prepararse de acuerdo con las Tablas Suplementarias 6 y el Cuadro Suplementario 7. Diluir el vinculador de 10 M 3' a una concentración de 2,5 M utilizando 100 mM de NaCl.
11. Defosforilación
- Preparar la mezcla SAP combinando 4 l de agua, 5 l de 10 x tampón SAP y 1 l de enzima SAP.
- Añadir 10 l de mezcla SAP a la muestra ligada purificada (volumen total 50 l) e incubar en el termociclador utilizando el siguiente programa: 37 oC durante 30 min, 65 oC durante 15 min, y mantener a 4 oC.
12. Degradación de la hebra superior del vinculador de 3' usando enzima de escisión específica de Uracil
- Añadir 2 l de enzima de escisión específica de uracilo (ver Tabla de Materiales)a la muestra defosforilada, mezclar por pipeteo e incubar en el termocicleador utilizando el siguiente programa: 37 oC durante 30 min, 95 oC durante 5 min, y colocar inmediatamente en hielo durante 2 min para evitar el recocido de la hebra superior fragmentada.
- Purificar la mezcla de reacción añadiendo 93,6 l de perlas magnéticas SPRI a la mezcla de 52 l y mezclar bien mediante pipeteo. Repita los pasos de purificación 2.6.1–2.6.6. Elute con 42 ml de agua precalentada a 37 oC como se describe.
13. Síntesis de segundo hilo
- Preparar la segunda mezcla de síntesis de hebras (los volúmenes se expresan por muestra) combinando 5 l de tampón de reacción de polimerasa de ADN 10x, 2 ml de agua, 1 l de dNPS de 10 mM, 1 l de 50 m de segundo hilo de imprimación (la secuencia está en la Tabla Suplementaria 1) y 1 l de D NA exonucleasa-deficiente de la polimerasa (ver polimerasa recomendada en la Tabla de Materiales).
- Añadir 10 l de la mezcla a la muestra purificada y mezclar bien mediante pipeteo (el volumen total es de 50 l). Incubar en el ciclor térmico utilizando el siguiente programa: 95 oC durante 5 min, 55 oC durante 5 min, 72 oC durante 30 min, y mantener a 4 oC.
14. Degradación de La primera de síntesis de segundo hilo utilizando exonucleasa I
- Añadir 1 l de Exonucleasa I a la segunda mezcla de síntesis de hebras. Mezcle bien pipeteando e incubar a 37oC durante 30 min seguido de sostenerlo a 4oC.
- Purificar el ADN de doble trenzado añadiendo 91,8 l de perlas magnéticas SPRI a 51 l de la muestra tratada con Exonucleasa I. Repita los pasos de purificación descritos en 2.6.1–2.6.6. y eluir con 42 s de agua precalentada a 37 oC como se describe.
- Concentrar la muestra utilizando el concentrador de vacío a 15 ml como se describe en el paso 9.1.
15. Control de calidad y cantidad
- Utilice 1 l de las muestras concentradas y ejecute un chip de ADN de alta sensibilidad en un analizador de calidad de ADN. El perfil/cantidad esperado se presenta en la Figura3.
16. Primera Ronda de Degradación de los Transportistas
- Preparar la mezcla de degradación combinando 2 ml de agua, 2 l de tampón de enzimas de restricción de 10x, 1 l de I-SceI y 1 l de I-CeuI.
- Añadir 6 l de la mezcla de degradación a 14 l de la muestra concentrada y mezclar mediante pipeteo. Incubar a 37oC durante 3 h seguida de 20 min de desactivación a 65oC y mantener a 4oC.
- Purificar la mezcla de degradación utilizando perlas magnéticas SPRI. Añadir 5 l de agua para aumentar el volumen de la mezcla de degradación y añadir 45 l de perlas SPRI (1,8:1 perlas a la relación de la muestra). Repita la purificación como se describe en los pasos 2.6.1–2.6.6. y eluir con 42 oC de agua precalentada a 37oC.
- Concentrar la muestra eluida de 42 a 20 ml del volumen total como se describe en el paso 9.1.
17. Control del nivel de degradación y determinación del número de ciclos de amplificación de PCR
- Prepare la mezcla qPCR para amplificar bibliotecas enteras (mezcla de adaptadores). Combinar 3,8 l de agua, 5 l de premezcla qPCR (2x), 0,1 l de imprimación adaptor_f1 de 10 m (5'-AATGATACGGCGCGACCACCGA-3') y 0,1 l de imprimación adaptor_r1 de 10 m (5'-CAAGCAGAAGACGGCATACGA-3') para cada muestra (ver Tabla de Materiales para qPCR recomendado).
- Combine la mezcla del adaptador qPCR de 9 l con 1 l de muestra del paso 16.4 y mezcle bien pipeteando.
- Preparar la mezcla qPCR para amplificar el ADN derivado del portador (mezcla portadora). Combinar 3,8 l de agua, 5 l de premezcla qPCR (2x), 0,1 l de imprimación carrier_f1 de 10 m (5'-GCGGCAGCGTTCGCTATAAC-3') y 0,1 l de imprimación adaptor_r1 de 10 m para cada muestra
- Combine 9 l de mezcla portadora qPCR con 1 l de la muestra del paso 16.4 y mezcle bien pipeteando.
- Ajuste del programa qPCR: 95 oC durante 3 min (95 oC para 20 s, 60 oC para 20 s, 72 oC durante 2 min) repetidos 40x, seguidos de una curva de desnaturalización específica del instrumento (65-95 oC) y mantener a 4 oC.
NOTA: Prepare un control negativo reemplazando la muestra por agua.
18. Amplificación pcR de la biblioteca de destino
- Preparar la mezcla de amplificación de PCR combinando 6 ml de agua, 0,5 l de imprimación adaptor_f1 de 10 m, imprimación de 10 ml de adaptador_r1 de 10 m y 25 ml de premezcla de PCR (2x). Mezclar mediante pipeteo (consulte la Tabla de materiales para la premezcla de PCR recomendada).
- Agregue 32 s de la mezcla de PCR a 18 l de la muestra del paso 16.4. Mezcle bien pipeteando.
- Ajuste de la amplificación de PCR: 95 oC durante 3 min, (98 oC para 20 s, 60 oC durante 15 s, 72 oC durante 2 min) 12-18 ciclos, 72 oC durante 2 min y mantener a 4 oC.
NOTA: El número exacto de ciclos de PCR se determina por los resultados qPCR y corresponde al valor Ct obtenido con la mezcla de imprimación del adaptador (el número de ciclos de PCR es igual al valor Ct). - Purificar la muestra amplificada añadiendo 90 l de perlas magnéticas SPRI a 50 l de la muestra amplificada y mezclar bien mediante pipeteo. Repita los pasos de purificación descritos en los pasos 2.6.1–2.6.6. y eluir la muestra con 42 ml de agua como se describe.
19. Segunda Ronda de Degradación de los Transportistas
- Repita los pasos 16.1–16.3.
- Purificar la mezcla de degradación utilizando perlas magnéticas SPRI. Añadir 10 l de agua a la muestra para aumentar el volumen y mezclar con 30 l de perlas SPRI (1:1 perlas a la relación de la muestra). Repita la purificación como se describe en los pasos 2.6.1–2.6.6. y elugar con 42 oL de agua precalentada a 37 oC como se describe.
- Concentrar la muestra eluida de 42 a 30 ml de volumen total.
20. Selección del tamaño de la biblioteca
- Mezclar 24 l de perlas magnéticas SPRI con 30 s de la muestra del paso 19.3. (0,8:1 perlas a la relación de muestra). Repita los pasos de purificación como se describe en los pasos 2.6.1–2.6.6. y eluye la muestra en 42 ml de agua como se describe.
- Concentrar la muestra a aproximadamente 14 l como se describe en el paso 9.1.
21. Control de calidad
-
Evaluación de la distribución del tamaño
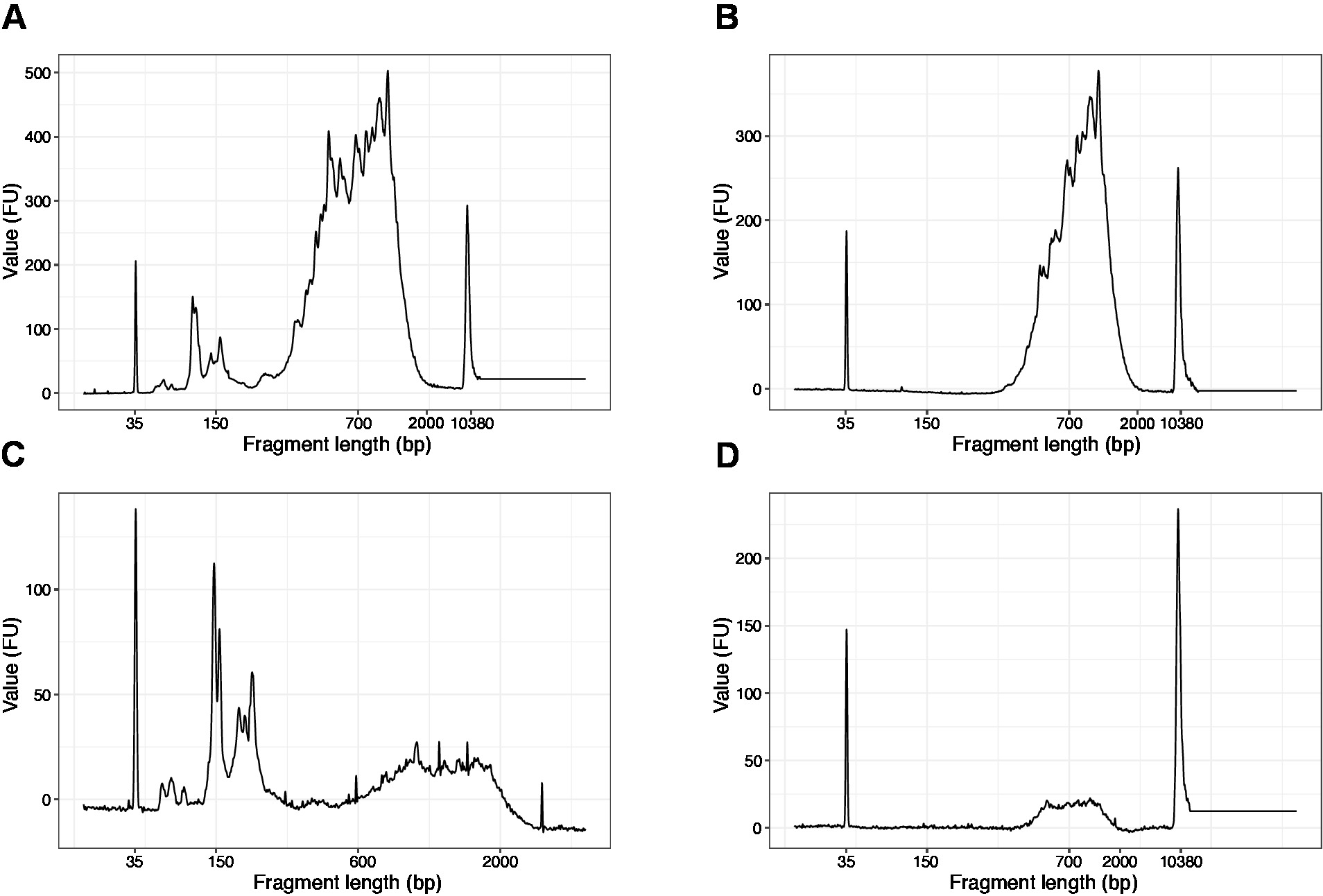
- Ejecutar 1 l de la muestra en el chip de ADN de alta sensibilidad. Los resultados esperados se presentan en la Figura4.
NOTA: Si los fragmentos inferiores a 200 bp son visibles (véase el ejemplo de la Figura 4A, C), la selección de tamaño (pasos 20.1–20.2) debe repetirse hasta que se eliminen los fragmentos cortos (Figura 4B,D). Por lo general, una ronda adicional de selección de tamaño es suficiente. Si la cantidad de fragmentos cortos es grave (como en la Figura 4C),la relación perlas-muestra debe reducirse a 0,6:1.
- Ejecutar 1 l de la muestra en el chip de ADN de alta sensibilidad. Los resultados esperados se presentan en la Figura4.
-
Control de calidad de degradación del portador
- Repita los pasos 17.1–17.5.
NOTA: Dependiendo de la concentración de las bibliotecas estimadas en la ejecución del chip de ADN del SA (análisis de la región), las muestras deben diluirse antes de qPCR. Utilice 0,5 l de la muestra para evitar la pérdida de la muestra y diluir de 100 a 500x en agua (diluir a 1–20 pg/L de concentración final). La diferencia esperada entre los valores Ct obtenidos con el adaptador y la mezcla portadora es de 5-10.
- Repita los pasos 17.1–17.5.
-
Cuantificación de bibliotecas
- Preparar la dilución de trabajo de la norma de ADN lambda mezclando 20 ml de 100 mg/ml de ADN lambda estándar con 980 l de 1x TE (preparar mediante la dilución de 20x TE proporcionada en el kit de cuantificación de ADN). La dilución del ADN lambda se puede almacenar a -20 oC.
- Preparar las diluciones seriales estándar de ADN lambda mezclando el estándar lambda diluido y 1x TE de acuerdo con la Tabla Suplementaria 8.
NOTA: Para una mayor precisión, se recomienda añadir 100 l de búfer de TE 1x a todos los tubos y eliminar el volumen de TE 1x según sea necesario por volumen de la lambda diluida que se va a añadir. No utilice más de 1 l de la biblioteca; se recomienda el uso de 384 placas de pozo para esta medición.
Resultados
Este informe describe el protocolo SLIC-CAGE completo para obtener bibliotecas listas para secuenciación a partir de nanogramos de material de ARN total inicial (Figura1). Para obtener la mezcla de portadoras de ARN sintético, en primer lugar, las plantillas portadoras de PCR deben prepararse y purificarse en gel para eliminar los productos secundarios de PCR (Figura2A). Cada plantilla de PCR (diez en total) se produce utilizando unaimprimación directa común, pero una imprimación inversa diferente (Tabla 2), que conduce a diferentes longitudes de la plantilla de PCR para permitir la variabilidad del tamaño de los portadores de ARN sintéticos. Una vez purificadas, las plantillas de PCR se utilizan para la transcripción in vitro de las moléculas portadoras. Se espera un único producto portador de ARN si las plantillas están purificadas en gel (ver análisis de gel representativo en la Figura 2B). La preparación del portador se puede ampliar dependiendo de la necesidad, y cuando se prepara, se mezcla y se congela a -80 oC para su uso futuro.
Utilizando la cantidad mínima recomendada de ARN total de muestra (10 ng) combinado con 16-18 ciclos de amplificación de PCR, se pueden lograr bibliotecas SLIC-CAGE de alta complejidad. El número de ciclos de PCR necesarios para amplificar la biblioteca final depende en gran medida de la cantidad de ARN de entrada total utilizado (el número esperado de ciclos se presenta en el Cuadro 4).
Después de la primera ronda de degradación, en los resultados qPCR (paso 17), la diferencia esperada entre los valores Ct obtenidos mediante la imprimación adaptor_f1 o carrier_f1 es 1-2, con valores Ct obtenidos con adaptor_f1 inferior que con carrier_f1.
La distribución de las longitudes de los fragmentos en la biblioteca final es de entre 200-2.000 bp con el tamaño medio del fragmento de 700-900 bp (basado en el análisis de la región utilizando el software Bioanalyzer, Figura 4B,D). Los fragmentos más cortos, tal como se presentan en la Figura 4A,C, deben eliminarse mediante rondas adicionales de exclusión de tamaño (pasos 20-21). Estos fragmentos cortos son artefactos de amplificación de PCR y no la biblioteca de destino. Tenga en cuenta que los fragmentos más cortos se agrupan mejor en las celdas de flujo de secuenciación y pueden causar problemas de secuenciación.
La cantidad esperada de material de biblioteca obtenido por muestra está entre 5-50 ng. Las cantidades significativamente más bajas son indicativas de la pérdida de muestra durante el protocolo. Si la cantidad baja obtenida es suficiente para la secuenciación (se necesitan 2-3 ng de las bibliotecas agrupadas), las bibliotecas pueden ser de menor complejidad (véase más adelante).
Dependiendo de la máquina de secuenciación, es posible que sea necesario optimizar la cantidad de la biblioteca cargada en la celda de flujo. Utilizando un Illumina HiSeq 2500, la carga de bibliotecas SLIC-CAGE de 8-12 pM da en promedio 150-200 millones de lecturas, con >80% de las lecturas pasando la puntuación de calidad Q30 como umbral.
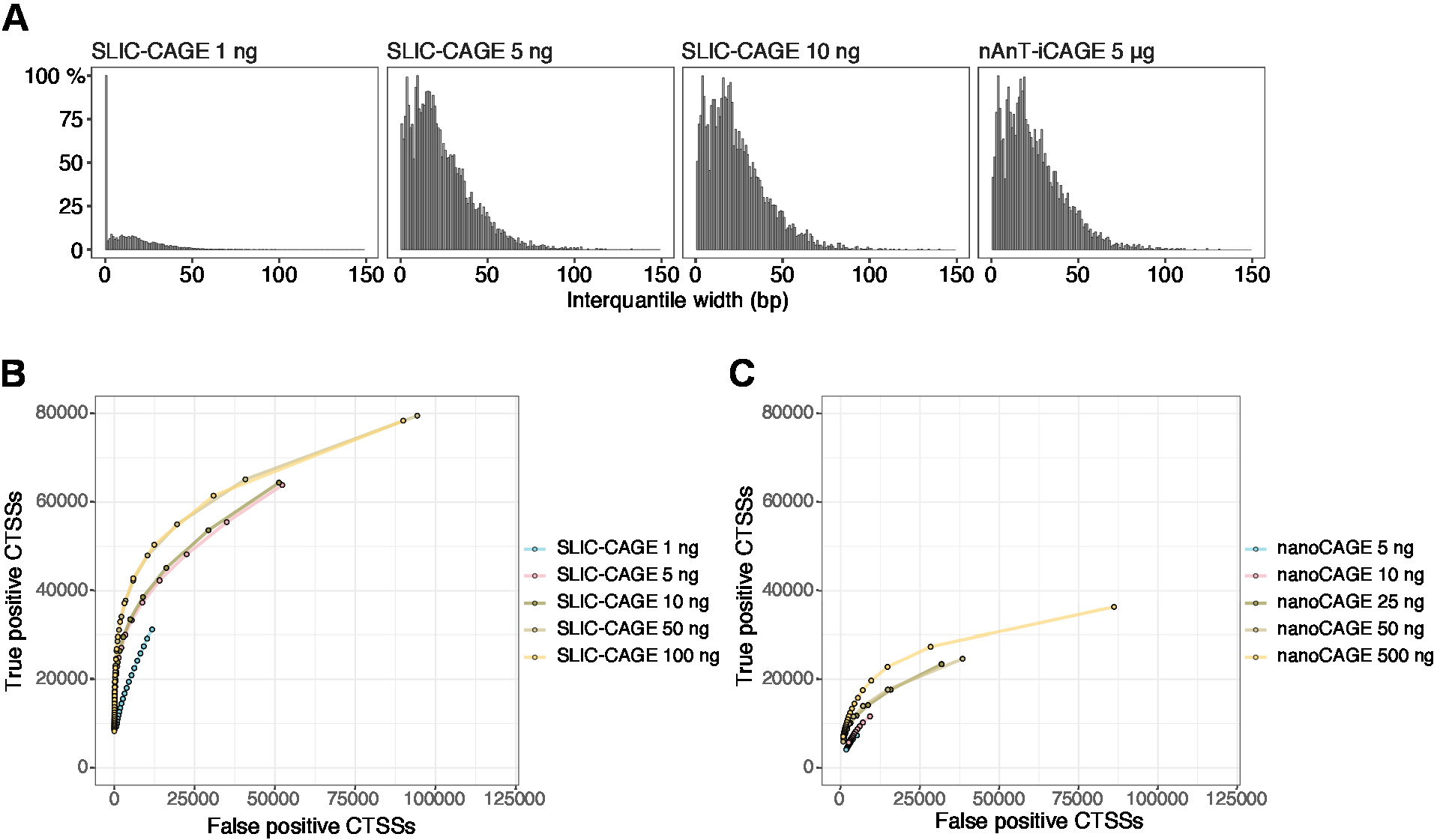
Las lecturas obtenidas se asignan al genoma de referencia [para lecturas de 50 bp, Bowtie212 se puede utilizar con parámetros predeterminados que permiten cero desajustes por secuencia de semillas (22 bp)]. Las eficiencias de mapeo esperadas dependen de la cantidad total de entrada de ARN y se presentan en el Cuadro5. Las lecturas de mapa único se pueden cargar en el entorno de computación gráfica y estadística de R13 y procesarse utilizando CAGEr (paquete Bioconductor14). La viñeta del paquete es fácil de seguir y explica el flujo de trabajo y el procesamiento de los datos asignados en detalle. Un fácil control visual de la complejidad de la biblioteca es la distribución del ancho del promotor, ya que las bibliotecas de baja complejidad tendrán promotores artificialmente estrechos (Figura5A,Biblioteca SLIC-CAGE derivada de 1 ng de ARN total, para más detalles ver publicación10). Sin embargo, incluso las bibliotecas SLIC-CAGE de baja complejidad permiten la identificación de verdaderos CTSS,con mayor precisión que los métodos alternativos para la asignación TSS de entrada baja/media (Figura 5B,C).

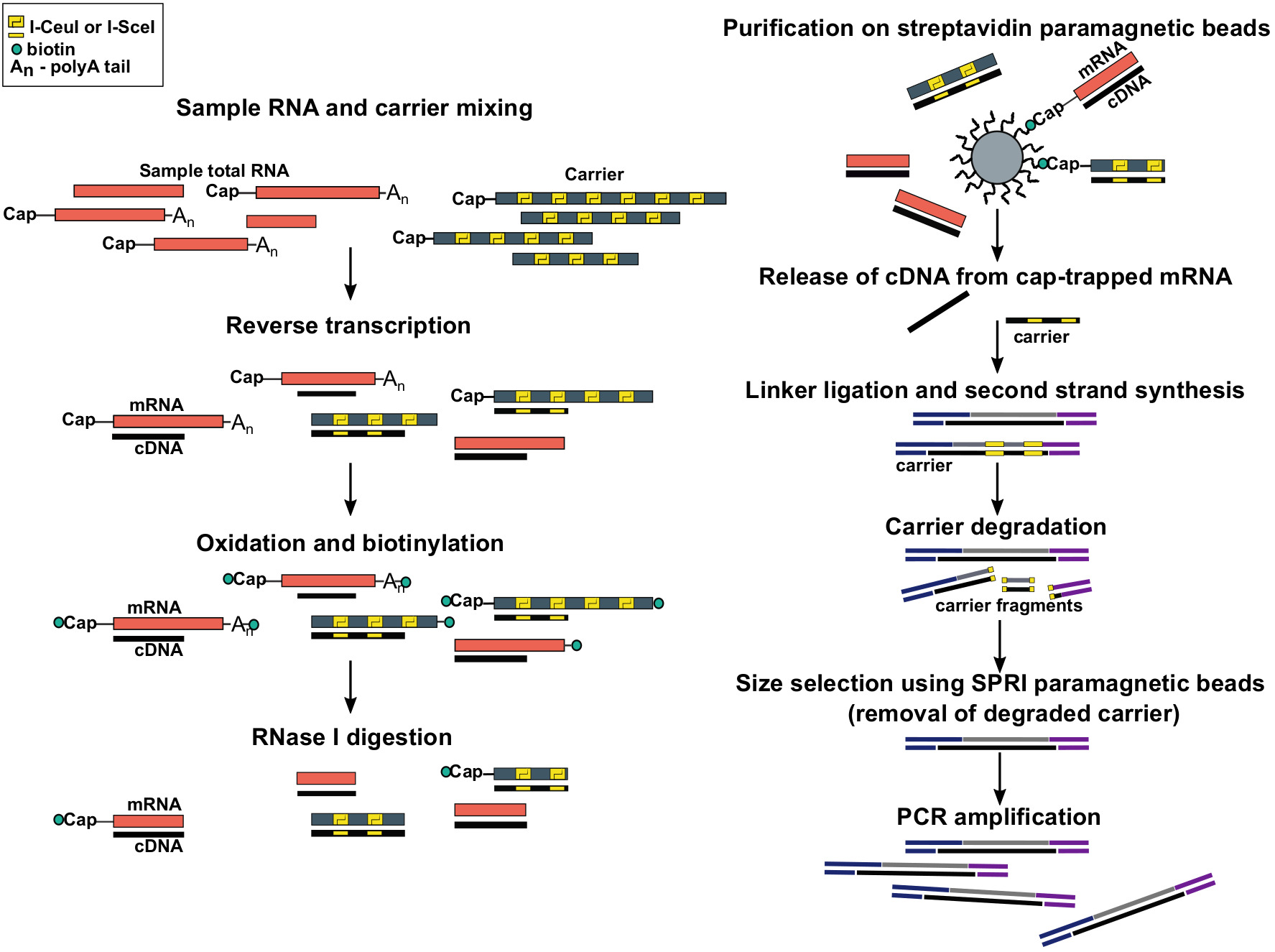
Figura 1: Pasos en el protocolo SLIC-CAGE. El ARN de muestra se mezcla con la mezcla portadora de ARN para lograr 5 g de material de ARN total. el ADNc se sintetiza a través de la transcripción inversa y la tapa se oxida mediante el periodato de sodio. La oxidación permite la unión de la biotina a la tapa utilizando hidrida de biotina. La biotina se une al extremo de 3o del ARNm, ya que también se oxida con periodoato de sodio. Para eliminar la biotina de los híbridos mRNA:cDNA con ADNc sintetizado incompletamente y de los extremos de 3o de ARNm, las muestras se tratan con RNase I. cDNA que alcanzó el extremo de 5o de ARNm es seleccionado por purificación de afinidad en cuentas magnéticas de streptavidina ( captura de la tapa). Después de la liberación de cDNA, se ligan los enlaces de 5 y 3o. Las moléculas de la biblioteca que se originan en el portador se degradan utilizando endonucleaseas I-SceI e I-CeuI y los fragmentos se eliminan utilizando perlas magnéticas SPRI. La biblioteca se amplifica entonces PCR. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Análisis en gel representativo de las plantillas de PCR portadoras y transcripciones in vitro portadoras. (A) Plantillas de PCR portadoras antes de la purificación de gel: el primer pozo contiene el marcador de 1 kbp, seguido de las plantillas de PCR portadoras 1, 1-10. (B) Transcripciones in vitro del transportista: el primer pozo contiene el marcador de 1 kbp, seguido de las transcripciones del portador 1-10. Las transcripciones portadoras fueron desnaturalizadas por calentamiento durante 5 minutos a 95 oC antes de la carga. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Calidad representativa del ADN (chip de ADN de alta sensibilidad) rastro de SLIC-CAGE antes de la primera ronda de degradación portadora. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Rastros representativos de calidad de ADN (chip de ADN de alta sensibilidad) de las bibliotecas SLIC-CAGE después de la amplificación de PCR. (A) Biblioteca SLIC-CAGE que requiere selección de tamaño adicional para la eliminación de fragmentos cortos. (B) Biblioteca SLIC-CAGE después de la selección del tamaño utilizando 0,6 x perlas SPRI a la relación de muestra. (C) Biblioteca SLIC-CAGE de menor cantidad de salida que requiere selección de tamaño para la eliminación de fragmentocorto. (D) Biblioteca SLIC-CAGE de menor cantidad de salida después de la selección de tamaño utilizando 0.6:1 SPRI perlas a la relación de muestra. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Validación de bibliotecas SLIC-CAGE. (A) Distribución de anchos interdirequisitos de clúster de etiquetas en bibliotecas SLIC-CAGE preparadas a partir de 1, 5 o 10 ng de ARN total de S. cerevisiae, y en la biblioteca nAnT-iCAGE preparada a partir de 5 g de ARN total de S. cerevisiae. Una gran cantidad de clústeres de etiquetas estrechas en la biblioteca SLIC-CAGE de 1 ng indica su baja complejidad. (B) curvas ROC para la identificación CTSS en bibliotecas S. cerevisiae SLIC-CAGE. Todos los CTSS de S. cerevisiae nAnT-iCAGE se utilizaron como un verdadero conjunto. (C) curvas ROC para la identificación CTSS en bibliotecas de NanoCAGE de S. cerevisiae. Todos los CTSS de S. cerevisiae nAnT-iCAGE se utilizaron como un verdadero conjunto. La comparación de las curvas ROC muestra que SLIC-CAGE supera fuertemente a nanoCAGE en la identificación de CTSS. Se utilizaron datos de ArrayExpress E-MTAB-6519. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Tabla 1: Secuencia del gen sintético portador. Los sitios de I-SceI son negritas y cursivas en púrpura, y los sitios de reconocimientos I-CeuI son verdes. Haga clic aquí para descargar este archivo.
| Portador | imprimación inversa 5'-3' | Longitud del producto PCR / bp | |
| 1 | PCR_N6_r1: NNNNNNCTACGTCGCAGACGAATT | 1034 | |
| 2 | PCR_N6_r2: NNNNNNTATCCAGATCGTTGAGCTGC | 966 | |
| 3 | PCR_N6_r3: NNNNNNCACTGCGGGATCTCTTTACG | 889 | |
| 4 | PCR_N6_r4: NNNNNNNGCCGTCGATAACTTTCGT | 821 | |
| 5 | PCR_N6_r5: NNNNNNAGTTGACCGCAGAGTCTTC | 744 | |
| 6 | PCR_N6_r6: NNNNNNNNGTGAAGAATTTCTGTTCCCA | 676 | |
| 7 | PCR_N6_r7: NNNNNNCTCGCGGCTCCAGTAACAC | 599 | |
| 8 | PCR_N6_r8: NNNNNNNTATACGCGATGTTGTCGTAC | 531 | |
| 9 | PCR_N6_r9: NNNNNNNACCGCCGCCTCTTCCGCAGG | 454 | |
| 10 | PCR_N6_r10: NNNNNNCAGGACGTTTTTGCCCAGCA | 386 | |
| * La imprimación delantera es la misma para todas las plantillas de portadora. Se subraya la secuencia del promotor T7. PCR_GN5_f1: TAATACGACTCACTATAGNNNNNNNNNCGTTCGCTA | |||
Tabla 2: Primers para amplificación de la plantilla portadora. La imprimación directa es la misma para todas las plantillas de portadora. Se subraya la secuencia del promotor T7. PCR_GN5_f1: TAATACGACTCACTATAGNNNNNCAGCGTTCGCTA. Utilizando imprimaciones inversas diferentes, se producen plantillas de PCR y, por lo tanto, ARN portadoras de diferente longitud.
| Portador | Longitud | sin tapar/g | tapado/g |
| 1 | 1034 | 3.96 | 0.45 |
| 2 | 966 | 8.36 | 0.95 |
| 3 | 889 | 4.4 | 0.5 |
| 4 | 821 | 6.6 | 0.75 |
| 5 | 744 | 4.4 | 0.5 |
| 6 | 676 | 3.08 | 0.35 |
| 7 | 599 | 4.4 | 0.5 |
| 8 | 531 | 3.96 | 0.45 |
| 9 | 454 | 2.64 | 0.3 |
| 10 | 386 | 2.2 | 0.25 |
Tabla 3: Mezcla portadora de ARN. En total 49 g de la mezcla portadora 0,3-1 kbp: sin tapar a 44 g, con un límite de 5 g.
| Entrada total de ARN /ng | Ciclos de PCR |
| 1 ng | 18 |
| 2 ng | 17 |
| 5 ng | 16 |
| 10 ng | 15-16 |
| 25 ng | 14-15 |
| 50 ng | 13-15 |
| 100 ng | 12-14 |
Cuadro 4: Número esperado de ciclos de PCR en dependencia de la entrada total de ARN de la muestra. El número aproximado de ciclos se basa en experimentos realizados con Saccharomyces cerevisiae, Drosophila melanogaster y ARN total de Mus musculus.
| Entrada total de ARN/ng | % mapa general | % de mapa único | % portador |
| 1 ng | 30 | 20-30 | 30 |
| 2 ng | 60 | 20-50 | 10 |
| 5 ng | 60-70 | 40-60 | 5-10 |
| 10 ng | 60-70 | 40-60 | 5-10 |
| 25 ng | 65-80 | 40-70 | 0-5 |
| 50 ng | 65-80 | 40-70 | 0-3 |
| 100 ng | 70-85 | 40-70 | 0-2 |
Tabla 5: Eficiencia cartográfico esperada y en dependencia de la cantidad total de entrada de ARN. Los números aproximados se presentan y se basan en experimentos realizados con Saccharomyces cerevisiae y MUS musculus ARN total.
Discusión
Para preparaciones exitosas de la biblioteca SLIC-CAGE, es fundamental utilizar puntas y tubos de baja unión para evitar la pérdida de muestras debido a la adsorción de muestras. En todos los pasos que implican la recuperación del sobrenadante, se recomienda recuperar todo el volumen de muestra. Como el protocolo tiene varios pasos, la pérdida continua de muestras dará lugar a bibliotecas sin éxito.
Si CAGE (nAnT-iCAGE) no se ha realizado de forma rutinaria, lo mejor es probar SLIC-CAGE con diferentes cantidades de entrada (10 ng, 20 ng, 50 ng, 100 ng, 200 ng) de la misma muestra total de ARN y compararlo con las bibliotecas nAnT-iCAGE que se preparan utilizando 5 g de ARN total. Si la biblioteca nAnT-iCAGE no tiene éxito (menos de 0,5-1 ng de la biblioteca de ADN obtenida por muestra), es poco probable que SLIC-CAGE funcione y que la pérdida de muestra sin batería deba minimizarse.
Un paso crítico para garantizar bibliotecas de alta calidad carentes de ARN degradado o ARNm sin acoto es el taponamiento de tapa descrito en la sección 7. Es muy importante que las perlas de estreptavidina se resuspendan a fondo en los tampones de lavado y que los tampones de lavado se retiren antes de continuar con el siguiente paso de lavado o la elución del ADNC.
Si los resultados del qPCR después de la primera ronda de degradación de portadora no muestran ninguna diferencia entre el uso de las imprimaciones adaptor_f1 y carrier_f1, se sigue recomendando continuar con el protocolo. Si después de la segunda ronda de degradación de portadora, la diferencia en los valores ct es inferior a cinco, se recomienda una tercera ronda de degradación de portadora. Nunca hemos encontrado una tercera ronda de degradación necesaria, y si se produce, se recomienda reemplazar las poblaciones de endonucleasa homing.
Se pueden añadir rondas adicionales de amplificación de PCR al protocolo si la cantidad final de la biblioteca obtenida no es suficiente para la secuenciación. La amplificación de PCR se puede ajustar con un número mínimo de ciclos de amplificación necesarios para producir suficiente material para la secuenciación, teniendo en cuenta la pérdida de muestra que no se puede evitar en la selección de tamaño. La purificación o selección de tamaño utilizando perlas magnéticas SPRI debe realizarse hasta que se eliminen todos los fragmentos pequeños (<200 bp) (si es necesario, utilice 0.6:1 perlas a la relación de muestra), y la biblioteca debe cuantificarse con Picogreen.
Las bibliotecas se pueden secuenciar en modo de extremo único o de extremo emparejado. Mediante la secuenciación de extremo emparejado, se puede obtener información sobre isoformas de transcripción. Además, como la transcripción inversa se realiza utilizando una imprimación aleatoria (TCT-N6, N6 siendo un hexamer aleatorio), la información del 3'-end secuenciado se puede utilizar como identificadores moleculares únicos (UMI) para contraer duplicados de PCR. Como se utiliza un número moderado de ciclos de amplificación de PCR (hasta 18), el uso de UMI se ha encontrado previamente innecesario.
Como el núcleo del protocolo se basa en nAnT-iCAGE11, SLIC-CAGE utiliza ocho códigos de barras. Por lo tanto, actualmente no se admite la multiplexación de más de ocho muestras. Además, tanto SLIC-CAGE como nAnT-iCAGE no son adecuados para capturar ARN inferiores a 200 bp, ya que los protocolos están diseñados para eliminar los enlaceres y artefactos PCR mediante la exclusión de tamaño con perlas AMPure XP.
SLIC-CAGE es el único método de resolución de un solo nucleótido de entrada baja imparcial para mapear los sitios de inicio de inicio de transcripción utilizando nanogramos de material de ARN total. Los métodos alternativos se basan en la actividad de conmutación de plantillas de la transcriptasa inversa al ARN tapado de código de barras en lugar de a la captura de tapa (por ejemplo, NanoCAGE15 y NanoPARE16). Debido a la conmutación de plantillas, estos métodos presentan sesgos específicos de la secuencia en la detección de TSS, lo que conduce a un mayor número de TSS falsos positivos y disminución del número de TSSs9,10.
Divulgaciones
Se ha llenado una patente para ARN/ADN portador degradable.
Agradecimientos
Este trabajo fue apoyado por la subvención Wellcome Trust (106954) otorgada a B. L. y Medical Research Council (MRC) Core Funding (MC-A652-5QA10). N. C. fue apoyado por EMBO Long-Term Fellowship (EMBO ALTF 1279-2016); E. P. contó con el apoyo del Consejo de Investigación Médica del Reino Unido; B. L. contó con el apoyo del Medical Research Council UK (MC UP 1102/1).
Materiales
| Name | Company | Catalog Number | Comments |
| 2-propanol, Bioultra, for molecular biology, ≥99.5% | Sigma-Aldrich | 59304-100ML-F | Used in RNAclean XP purification. |
| 3' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 3'linkers is described in the supplementary of this protocol. | ||
| 5' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 5'linkers is described in the supplementary of this protocol. | ||
| Agencourt AMPure XP, 60 mL | Beckman Coulter | A63881 | Purification of DNA |
| Agencourt RNAClean XP Kit | Beckman Coulter | A63987 | Purification of RNA and RNA:cDNA hybrids in CAGE steps. |
| Axygen 0.2 mL Polypropylene PCR Tube Strips and Domed Cap Strips | Axygen (available through Corning) | PCR-0208-CP-C | Or any 8-tube PCR strips (used only for water and mixes). |
| Axygen 1 x 8 strip domed PCR caps | Axygen (available through Corning) | PCR-02CP-C | Caps for PCR plates. |
| Axygen 1.5 mL Maxymum Recovery Snaplock Microcentrifuge Tube | Axygen (available through Corning) | MCT-150-L-C | Low-binding 1.5 mL tubes, used for enzyme mixes or sample concentration. |
| Axygen 96 well no skirt PCR microplate | Axygen (available through Corning) | PCR-96-C | Low-binding PCR plates - have to be used for all steps in the protocol. Note that plates should be cut to contain 2 x 8 wells for easier visibility of the samples |
| Bioanalyzer (or Tapestation): RNA nano and HS DNA kits | Agilent | To determine quality of RNA, efficient size selection and final quality of the library (Tapestation can also be used) | |
| Biotin (Long Arm) Hydrazide | Vector laboratories | SP-1100 | Biotinylation/tagging |
| Cutsmart buffer | NEB | Restriction enzyme buffer | |
| Deep Vent (exo-) DNA Polymerase | NEB | M0259S | Second strand synthesis |
| DNA Ligation Kit, Mighty Mix | Takara | 6023 | Used for 5' and 3'-linker ligation |
| dNTP mix (10 mM each) | ThermoFisher Scientific | 18427013 | dNTP mix for production of carrier templates (or any dNTPs suitable for PCR) |
| Dynabeads M-270 Streptavidin | Invitrogen | 65305 | Cap-trapping. Do not use other beads as these are optimised with the buffers used. |
| DynaMag-2 Magnet | ThermoFisher Scientific | 12321D | Magnetic stand for 1.5 mL tubes - used to prepare Streptavidin beads. |
| DynaMag-96 Side Skirted Magnet | ThermoFisher Scientific | 12027 | Magnetic stand for PCR plates (96 well-plates) - used with cut plates to contain 2 x 8 wells. |
| Ethanol, BioUltra, for molecular biology, ≥99.8% | Sigma-Aldrich | 51976-500ML-F | Used in AMPure washes. Any molecular biology suitable ethanol can be used. |
| Exonuclease I (E. coli) | NEB | M0293S | Leftover primer degradation |
| Gel Loading Dye, Purple (6x), no SDS | NEB | B7025S | agarose gel loading dye |
| HiScribe T7 High Yield RNA Synthesis Kit | New England Biolabs | E2040S | Kit for carrier in vitro transcription |
| Horizontal electrophoresis apparatus | purification of carrier DNA templates from agarose gels | ||
| I-Ceu | NEB | R0699S | Homing endonuclease used for carrier degradation. |
| I-SceI | NEB | R0694S | Homing endonuclease used for carrier degradation. |
| KAPA HiFi HS ReadyMix (2x) | Kapa Biosystems (Supplied by Roche) | KK2601 | PCR mix for target library amplification |
| KAPA SYBR FAST qPCR kit (Universal) 2x | Kapa Biosystems (Supplied by Roche) | KK4600 | qPCR mix to assess degradation efficiency and requiered number of PCR amplification cycles |
| Micropipettes and multichannel micropipettes (0.1-10 µL, 1-20 µL, 20-200 µL) | Gilson | Use of Gilson with the low-binding Sorenson tips is recommended. Other micropippetes might not be compatible. Different brand low-binding tips may not be of equal quality and may increase sample loss. | |
| Microplate reader | For Picogreen concentration measurement of the final library. Microplates are used to allow small volume measurement and reduce sample waste. | ||
| nuclease free water | ThermoFisher Scientific | AM9937 | Or any nuclease (DNase and RNase) free water |
| PCR thermal cycler | incubation steps and PCR amplficication | ||
| Phusion High-Fidelity DNA Polymerase | ThermoFisher Scientific | F530S | DNA polymerase for amplification of carrier templates (or any high fidelity polymerase) |
| QIAquick Gel Extraction Kit (50) | Qiagen | 28704 | Purification of carrier PCR templates from agarose gels. |
| qPCR machine | determining PCR amplification cyle number and degree of carrier degradation | ||
| Quant-iT PicoGreen dsDNA Reagent | ThermoFisher Scientific | P11495 | Used to measure final library concentration - recommended as, in our hands, it is more accurate and reproducible than Qubit. |
| Quick-Load Purple 100 bp DNA Ladder | NEB | N0551S | DNA ladder |
| Quick-Load Purple 1 kb Plus DNA Ladder | NEB | N0550S | DNA ladder |
| Ribonuclease H | Takara | 2150A | Digestion of RNA after cap-trapping. |
| RNase ONE Ribonuclease | Promega | M4261 | Degradation of single stranded RNA not protected by cDNA. |
| RNase-Free DNase Set | Qiagen | 79254 | Removal of carrier DNA templates after in vitro transcription. |
| RNeasy Mini Kit | Qiagen | 74104 | For cleanup of carrier RNA from in vitro transcription or capping |
| Sodium acetate, 1 M, aq.soln, pH 4.5 RNAse free | VWR | AAJ63669-AK | Or any nuclease (DNase and RNase) free solution |
| Sodium acetate, 1 M, aq.soln, pH 6.0 RNAse free | Or any nuclease (DNase and RNase) free solution | ||
| Sodium periodate | Sigma-Aldrich | 311448-100G | Oxidation of vicinal diols |
| Sorenson low binding aerosol barrier tips, MicroReach Guard, volume range 10 μL, Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719390-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 1,000 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719463-1000EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 20 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719412-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 200 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719447-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| SpeedVac Vacuum Concentrator | concentrating samples in various steps to lower volume | ||
| SuperScript III Reverse Transcriptase | ThermoFisher Scientific | 18080044 | Used for reverse transcription (1st CAGE step) |
| Trehalose/sorbitol solution | Preparation is described in Murata et al. 2014. | ||
| Tris-HCl, 1 M aq.soln, pH 8.5 | 1 M solution, DNase and RNase free | ||
| tRNA (20 mg/mL) | tRNA solution. Preparation is described in Murata et al. 2014. | ||
| UltraPure Low Melting Point Agarose | ThermoFisher Scientific | 16520050 | Or any suitable pure low-melt agarose. |
| USB Shrimp Alkaline Phosphatase (SAP) | Applied Biosystems (Provided by ThermoFisher Scientific) | 78390500UN | |
| USER Enzyme | NEB | M5505S | Degradation of 3'linker's upper strand, Uracil Specific Excision Reagent/Enzyme |
| Vaccinia Capping System | NEB | M2080S | Enzymatic kit for in vitro capping of carrier molecules |
| Wash buffer A | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer B | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer C | Cap trapping washes. Preparation is described in Murata et al. 2014. |
Referencias
- Shiraki, T., et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proceedings of the National Academy of Sciences of the United States of America. 100 (26), 15776-15781 (2003).
- Haberle, V., Lenhard, B. Promoter architectures and developmental gene regulation. Seminars in Cell and Developmental Biology. 57, 11-23 (2016).
- Haberle, V., Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nature Reviews Molecular Cell Biology. 19 (10), 621-637 (2018).
- Andersson, R., et al. An atlas of active enhancers across human cell types and tissues. Nature. 507 (7493), 455-461 (2014).
- Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Celniker, S. E., et al. Unlocking the secrets of the genome. Nature. 459 (7249), 927-930 (2009).
- Consortium, F., et al. A promoter-level mammalian expression atlas. Nature. 507 (7493), 462-470 (2014).
- Boyd, M., et al. Characterization of the enhancer and promoter landscape of inflammatory bowel disease from human colon biopsies. Nature Communications. 9 (1), 1661 (2018).
- Adiconis, X., et al. Comprehensive comparative analysis of 5'-end RNA-sequencing methods. Nature Methods. , (2018).
- Cvetesic, N., et al. SLIC-CAGE: high-resolution transcription start site mapping using nanogram-levels of total RNA. Genome Research. 28 (12), 1943-1956 (2018).
- Murata, M., et al. Detecting expressed genes using CAGE. Methods in Molecular Biology. 1164, 67-85 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9, 357 (2012).
- A language and environment for statistical computing. Available from: https://www.R-project.org/ (2017)
- Haberle, V., Forrest, A. R., Hayashizaki, Y., Carninci, P., Lenhard, B. CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic Acids Research. 43 (8), e51 (2015).
- Poulain, S., et al. NanoCAGE: A Method for the Analysis of Coding and Noncoding 5'-Capped Transcriptomes. Methods in Molecular Biology. 1543, 57-109 (2017).
- Schon, M. A., Kellner, M. J., Plotnikova, A., Hofmann, F., Nodine, M. D. NanoPARE: parallel analysis of RNA 5' ends from low-input RNA. Genome Research. 28 (12), 1931-1942 (2018).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados