
Method Article
Транскрипция Запуск сайта Картирование С использованием супер-низкий вход Перевозчик-CAGE
В этой статье
Резюме
Анализ экспрессии генов (CAGE) является методом для генома всей количественного картирования мРНК 5'ends для захвата РНК-полимераза II транскрипции начала сайтов с разрешением одного нуклеотида. Эта работа описывает протокол с низким уровнем ввода (SLIC-CAGE) для генерации высококачественных библиотек с использованием нанограммобщего объема РНК.
Аннотация
Анализ пределов экспрессии генов (CAGE) является методом, используемым для однонуклеотидного разрешения обнаружения РНК-полимераза II транскрипции сайтов (TSSs). Точное обнаружение TSS s улучшает идентификацию и обнаружение основных промоутеров. Кроме того, активные усилители могут быть обнаружены с помощью подписей двунаправленной инициации транскрипции. Описан очеркнутый здесь протокол для выполнения супер-низкого ввода перевозчика-CAGE (SLIC-CAGE). Эта SLIC адаптация протокола CAGE минимизирует потери РНК за счет искусственного увеличения количества РНК за счет использования транскрибированной РНК-микси инкрустированной РНК, которая добавляется в образец интереса, что позволяет библиотеке подготовку из нанограмм-суммы общего РНК (т.е. тысячи клеток). Перевозчик имитирует ожидаемое распределение длины фрагмента БИБЛИОТЕКи ДНК, тем самым устраняя предубеждения, которые могут быть вызваны обилием однородного носителя. На последних этапах протокола, перевозчик удаляется путем деградации с самонаводящимися эндонуклеазами и целевой библиотекой усиливается. Целевая библиотека образца защищена от деградации, так как места распознавания самонаводящихся эндонуклеазы длинные (между 18 и 27 б.п.), что делает вероятность их существования в эукариотических геномах очень низкой. Конечным результатом является библиотека ДНК, готовая к секвенированию следующего поколения. Все этапы протокола, вплоть до секвенирования, могут быть выполнены в течение 6 дней. Подготовка перевозчика требует полного рабочего дня; однако, он может быть подготовлен в больших количествах и храниться в замороженном состоянии при -80 градусах Цельсия. После секвенирования, считываний могут быть обработаны для получения генома всей однонуклеотидной резолюции TSSs. TSSs могут быть использованы для основного промотора или повышения открытия, обеспечивая понимание регуляции генов. После агрегирования в промоутеры данные также могут быть использованы для профилирования 5'centric выражения.
Введение
Кап-анализ экспрессии генов (CAGE) является методом, используемым для однонуклеотидного разрешения генома всей картографии РНК-полимераза II транскрипции начала сайтов (TSSs)1. Его количественный характер также позволяет 5'конец ориентированного выражения профилирования. Регионы, окружающие TSSs (около 40 bp вверх и вниз по течению) являются основными промоутеров и представляют физическое местоположение, где РНК-полимераза II и общие факторы транскрипции связывают (рассмотрено ранее2,3). Информация о точном местоположении TSSs может быть использована для обнаружения основных промоутеров и для мониторинга динамики промоутера. Кроме того, в качестве активных усилителей экспонат подписи двунаправленной транскрипции, CAGE данные также могут быть использованы для обнаружения усилителя и мониторинга динамики усилителя4. Методология CAGE в последнее время возросла в популярности благодаря ее широкому применению и использованию в громких исследовательских проектах, таких как ENCODE5,modENCODE6, и FANTOM проектов7. Кроме того, информация TSS также оказывается важным для различения здоровых и больных тканей, таккак специфические для болезней СтС могут быть использованы для диагностических целей 8.
Несмотря на то, что доступно несколько методов картирования TSS (CAGE, RAMPAGE, STRT, nanoCAGE, nanoCAGE-XL, oligo-capping), мы и другие недавно показали, что CAGE является самым беспристрастным методом захвата истинных TSS с наименьшим количеством ложных срабатываний9 , 10. Недавний протокол CAGE, nAnT-iCAGE11, является самым беспристрастным протоколом для профилирования TSS, так как он позволяет избежать разрезания фрагментов на короткие метки с использованием ферментов ограничения и не использует усиливание ПЦР. Ограничением протокола nAnT-iCAGE является требование к большому количеству исходного материала (например, 5 мкг общей РНК для каждого образца). Чтобы ответить на конкретные, биологически значимые вопросы, часто невозможно получить такое большое количество исходного материала (например, для сортированных FACS клеток или ранних эмбриональных стадиях). Наконец, если nAnT-iCAGE является успешным, только 1-2 нг материала библиотеки ДНК доступна из каждого образца, тем самым ограничивая достижимую глубину секвенирования.
Для того, чтобы TSS профилирования с использованием только нанограммы общей РНК, мы недавно разработали супер-низкий вход Carrier-CAGE10 (SLIC-CAGE, Рисунок 1). SLIC-CAGE требует только 10 нг от общей РНК для получения библиотек высокой сложности. Наш протокол опирается на тщательно разработанный синтетический РНК-носитель, добавленный в РНК, представляющий интерес для достижения в общей сложности 5 мкг МАТЕРИАЛА РНК. Синтетический носитель имитирует библиотеку ДНК-мишени в распределении длины, чтобы избежать потенциальных предубеждений, которые могут быть вызваны однородными молекулами в избытке. Последовательность носителя основана на последовательности гена синтеза Escherichia coli leucyl-tRNA(Таблица 1) по двум причинам. Во-первых, любые остатки носителя в окончательной библиотеке, даже если они секвенированы, не будут отображаться на эукариотический геном. Во-вторых, поскольку кишечная палочка является мезофильной вид, ее гены домашнего хозяйства оптимизированы для температурного диапазона, подходящего для SLIC-CAGE. Последовательность носителя также встроена с местами распознавания самонаводящихся эндонуклеазы, чтобы позволить специфическую деградацию ДНК, полученной из молекул РНК-носителя. Целевая библиотека, полученная из образца, остается нетронутой, так как места распознавания самонаводящихся эндонуклеаза являются длинными (I-CeuI - 27 bp; I-SceI 18 bp) и статистически вряд ли можно найти в эукариотических геномах. После специфической деградации носителя и удаления фрагментов по исключению размера, целевая библиотека усиливается пЦР и готова к секвенированию следующего поколения. В зависимости от стартовой суммы РНК (1-100 нг), ожидается, что потребуется 13-18 циклов усиления ПЦР. Окончательное количество ДНК на каждый образец колеблется между 5-50 нг, что дает достаточно материала для очень глубокого секвенирования. При использовании только 1-2 нг от общего ОБЪЕМА РНК, истинные TSSs могут быть обнаружены; однако ожидается, что библиотеки будут более сложными. Наконец, поскольку SLIC-CAGE основан на протоколе nAnT-iCAGE11,он позволяет мультиплексировать до восьми образцов до секвенирования.
протокол
1. Подготовка Перевозчика
-
Подготовка шаблонов ДНК для транскрипции in vitro
- Подготовьте смесь ПЦР для каждого шаблона ПЦР, объединив 41 кЛ воды, 20 л 5-х буфера HF, 8 л 2,5 мМ dNTPs, 10 зЛ уникального переднего грунта (PCR-GN5'f1, Таблица 2; праймеры растворяются и разбавляются в воде) , 10 л 2 нг/Л шаблон плазмида, содержащего синтетический ген носителя и 1 L Фузион полимераза. Смешайте смесь ПЦР путем пипетки. Мастер-микс для всех 10 шаблонов может быть подготовлен сразу (подготовьте 11 реакций).
- Добавляем 90 qL смеси ПЦР до 10 qL из каждого реверсивного грунта 10 мкм (PCR-N6-r10, Таблица 2). Смешайте путем пипетки.
- ПЦР усиливают шаблоны, используя следующую программу: 98 градусов по Цельсию на 60 с, (98 градусов по Цельсию на 10 с, 50 градусов по Цельсию для 30 с, 72 градуса по Цельсию) 35 циклов, 72 кв.к. в течение 10 мин, удерживайте при 4 градусах по Цельсию.
-
Гель очистки ПЦР-усиленных шаблонов ДНК
- Приготовьте 1% агарозный гель (рекомендуется низкоплавительная агароза).
- Чтобы уменьшить объем, сконцентрируйте смеси реакции ПЦР от 100 qL до 20 qL общего объема с помощью вакуумного концентратора при низкой средней температуре (30-40 градусов по Цельсию).
- Добавьте 6 кЛ 6x погрузочного красителя, хорошо перемешайте и загрузите на гель. Запуск электрофорез в течение 30 мин в 1x TAE буфера на напряжение подходит для используемого электрофореза танк (5-10 V/cm). Параллельно запустить 100 bp или 1000 bp ДНК лестнице.
- Используя чистый скальпель, вырезать гель ломтиками, содержащими целевой продукт ПЦР. Избегайте избыточного геля агарозы. Очистите продукты ПЦР с помощью комплекта для извлечения геля (в соответствии с инструкциями производителя).
ПРИМЕЧАНИЕ: Коэффициенты A260/A230 ДНК, выделенной из гелей агарозы, как правило, низки (0,1–0,3). Ожидаемый целевой продукт и побочные продукты показаны на рисунке 2A. Ожидаемые выходы от 100 ПЦР-реакций на 100 МЛ составляют 1,2-3 мкг. Реакции могут быть масштабированы, чтобы получить более высокую урожайность.
-
В пробирке транскрипция молекул носителя
- Транскрибный носитель РНК in vitro с использованием T7 РНК-полимераза в соответствии с инструкциями производителя. Настройка реакций на 10–20 л (рекомендуемый комплект находится в таблицематериалов).
- Очистите в пробирке транскрибированной РНК с помощью комплекта очистки РНК. Навяжните пищеварение ДНК в растворе с помощью DNase I в соответствии со стандартными инструкциями производителя, и увяните РНК в 50 зликат воды. Чтобы увеличить выход из elution, оставьте воду в колонке в течение 5 минут до центрифугации.
ПРИМЕЧАНИЕ: Будьте осторожны, чтобы не превысить максимальную связующую емкость столбцов (в комплекте, упомянутом в таблицематериалов, емкость до 100 мкг). Ожидаемый выход из шаблонов ПЦР 1-10 (1 кбит/с до 200 bp в длину) составляет 25-50 мкг от 10 кЛ реакций транскрипции. Реакции могут быть масштабированы, чтобы получить больший запас молекул носителя.
-
Урезание инкрушующих молекул РНК in vitro
- Приготовьте укупорки смесь, объединив 2 злицу 10x укупорки буфера, 1 л 10 мМ GTP, 1 л 2 мМ SAM (свеже разбавленный) и 1 Зл фермента Vaccinia capping на несущую РНК.
- Смешайте до 10 мкг каждой молекулы носителя в 15 qL от общего объема и денатурировать в течение 10 мин при 65 градусах Цельсия. Поместите на лед немедленно, чтобы предотвратить формирование вторичной структуры.
- Смешайте денатурированную РНК-носитель с 5 qL укупорки смеси и инкубировать в течение 1 ч при 37 градусов по Цельсию.
- Очистка ограниченных молекул РНК с помощью комплекта очистки РНК - следуйте протоколу очистки производителя. Elute РНК в 30 л воды. Чтобы увеличить выход из elution, оставьте воду в колонке в течение 5 минут до центрифугации.
ПРИМЕЧАНИЕ: Измеряйте концентрацию с помощью микротомного спектрофотометра. Ожидаемое соотношение A260/A280 составляет 2,2 и A260/A230, это йgt;2. Обратите внимание, что для некоторых образцов РНК A260/A230 может быть между 1,3-2. Ожидаемая урожайность при использовании 10 мкг необнаруженной РНК составляет 9-10 мкг ограниченной РНК.
- Подготовьте смесь ограниченного и неосвоенный перевозчика, объединив суммы, описанные в таблице 3. Хорошо перемешать, щелкнув трубку и измерить концентрацию с помощью микротома спектрофотометра.
ПРИМЕЧАНИЕ: Если требуется более высокая концентрация носителя, чтобы поместиться в обратную реакцию транскрипции (см. ниже), переносица может быть сконцентрирована с помощью вакуумного концентратора при низкой средней температуре (30-35 градусов по Цельсию) до достижения желаемой конечной концентрации. Шаги 2–14 изменены из стандартного протокола nAnT-iCAGE, о которых сообщает Murata et al.11
2. Обратная транскрипция
- Комбинат 1 зЛ RT грунтовки (2,5 мм TCT-N6 растворяется в воде, для последовательности см Дополнительнаятаблица 1 ), 10 нг общей РНК интереса и 4990 нг перевозчика смеси (Таблица 3) в 10 л общего объема в низкосвязывающей пластины ПЦР. Смешайте, щелкнув трубку.
ПРИМЕЧАНИЕ: Если РНК образца слишком разбавлена для обратной транскрипции (см. ниже), совместите ее с соответствующим количеством носителя, сконцентрируйтесь с помощью вакуумного концентратора до 9 Зл тв общем объеме и добавьте 1 злицу rt primer. Добавление графа носителя, чтобы достичь 5 мкг РНК в общей сложности предотвращает потерю образца. - Нагрейте смесь со ступени 2.1 при температуре 65 градусов по Цельсию в течение 5 минут, и поместите на лед немедленно, чтобы предотвратить ренатурацию.
-
Подготовка обратной транскрипции (RT) микс.
- Для каждого образца объединить 6,1 л воды (RNase- и DNase-бесплатно), 7,6 л 5x первого стрима буфера, 1,9 л 0,1 М ДТТ, 1 л 10 мМ dNTPs, 7,6л трегалозы / сорбитол смеси (см. рецепт в Мурата и др. 6 "Таблицаматериалов). Хорошо перемешать, щелкнув трубку.
- Добавьте 28 зл и рвете RT в трубку ПЦР с 10 Зл РНК, носителем и rt primer (общий объем 38 л). Хорошо перемешать путем пипетки.
ПРИМЕЧАНИЕ: Смесь очень вязкая из-за трефалозы/сорбитола. Смешайте до заметно однородной. - Инкубируйте в тепловом циклическом цикле по следующей программе: 25 градусов по Цельсию на 30 с, 50 градусов по Цельсию в течение 60 минут и удерживайте при 4 градусах Цельсия.
-
Очистка гибридов кДНК:РНК с использованием магнитных бусин SPRI
- Добавьте 68,4 л рекомендуемых шариков SPRI и DNase (см. Таблицаматериалов) к 38 злу смеси RT (бисер к выборке соотношение 1.8:1). Хорошо перемешать путем пипетки и инкубировать в течение 5 минут при комнатной температуре (RT).
- Отделить бусы на магнитной подставке в течение 5 мин. Отбросьте супернатант и дважды промойте бисер 200 юл 70% этанола (свежеприготовленный).
ПРИМЕЧАНИЕ: Этанол добавляется в бисер без смешивания и в то время как трубка находится на магнитной подставке. Добавленный этанол немедленно удаляется. Следует позаботиться о том, чтобы не потерять ни бисера во время стиха, так как это может привести к потере образца. - В то время как трубка все еще находится на магнитной подставке, удалить все следы этанола. Капли этанола можно удалить и вытолкнуть из трубки с помощью пипетки P10. Не дайте бисеру высохнуть.
- Добавьте 42 qL воды, разогретой при 37 градусах По Цельсию, и удалите образец, прокладывая вверх и вниз в 60раз.
ПРИМЕЧАНИЕ: Будьте осторожны, чтобы не вызвать вспенивание путем пипетки, как это может привести к потере бисера (т.е., связанный образец) в пене. - Инкубировать при 37 градусах По области в течение 5 мин без крышки, чтобы испарить следовые количества этанола.
- Разделите бусины на магнитной подставке на 5 мин и перенесите супернатант на новую пластину.
ПРИМЕЧАНИЕ: Попробуйте получить все супернатант, чтобы предотвратить потерю образца, избегая при этом переноса бисера. Используйте пипетку P10, чтобы получить последний образец капель.
3. Окисление
- Добавьте 2 злиц из 1 M NaOAc (pH 4.5) в очищенную реакцию RT. Смешайте путем пипетки, добавьте 2 qL 250 mM NaIO4 и снова перемешайте.
- Инкубировать на льду в течение 45 мин. Накройте тарелку алюминиевой фольгой, чтобы избежать света.
- Добавьте 16 кЛ Tris-HCl (pH 8.5) в смесь окисления, чтобы нейтрализовать рН.
- Очистка окисленных гибридов cDNA:RNA с использованием магнитных бусин SPRI. Добавьте 108 л бусин SPRI к 60 злу смеси окисления (1,8:1 шарики к выборке). Повторите очистку, описанную в шагах 2.6.1-2.6.6. Выщелаживать, используя 42 л воды, разогретой до 37 градусов по Цельсию.
ПРИМЕЧАНИЕ: Свежеприготовленный 250 мМ NaIO 4, добавив 18,7 л воды на 1 мг NaIO4. NaIO4 светочувствительный; поэтому держите раствор в трубке, покрытой алюминиевой фольгой, или в светостойкой трубке.
4. Биотинилаация
- Добавьте 4 злиц из 1 M NaOAc (pH 6.0) в трубку, содержащую очищенный окисленный образец, и смешайте путем трубки.
- Добавьте 4 хл из 10 мМ биотин раствор, смешать путем pipetting и инкубировать в течение 2 ч при 23 градусов по Цельсию в тепловой цикл, чтобы избежать света.
ПРИМЕЧАНИЕ: Подготовьте раствор биотина, смешав 50 мг биотина с 13,5 мл ДМСО. Сделать одноразовые aliquots и заморозить при -80 градусов по Цельсию. - Очистка биотинилапопромоваемых гибридов cDNA:RNA с использованием магнитных бусин SPRI. Добавить 12 л 2-пропанола и перемешать путем пипетки. Добавьте 108 л бусин SPRI (1,8:1 шарика к соотношению выборки) и повторите очищение, как описано в шагах 2.6.1-2.6.6. Выщелаживать, используя 42 л воды, разогретой при 37 градусах Цельсия.
ПРИМЕЧАНИЕ: Протокол можно приложить здесь, а образцы заморожены при -80 градусов по Цельсию.
5. RNase I Ddigestion
- Подготовьте RNase I mix, смешивая 4,5 зл и 10x RNase I буфера с 0,5 л RNase I (10 U / QL) на каждый образец. Смешайте путем пипетки.
- Добавьте в каждый очищенный образец 5 qL смеси (всего 45 л). Смешайте путем пипетки и инкубировать в течение 30 минут при 37 градусах Цельсия.
6. Подготовка стрептавидина бисера
- Для каждого образца смешайте 30 л стрептавидиновых бусин оксера с 0,38 л 20 мг/мл tRNA. Инкубировать на льду в течение 30 минут и перемешать каждые 5 минут, щелкнув трубку.
ПРИМЕЧАНИЕ: Resuspend стрептавидин бусы суспензии задолго до pipetting, щелкая бутылку. Решение tRNA должно быть подготовлено в соответствии с Мурата и др. - Отделить бусы на магнитной подставке в течение 2-3 мин. Удалите супернатант.
- Вымойте бусины, повторно приостановив в 15 зл буфера A. Разделите бусы на магнитной подставке в течение 2-3 мин и удалите супернатант. Повторите мыть и удалить супернатант.
- Приостановите действие бисера в 105 л буфера А и добавьте 0,19 л 20 мг/мл tRNA. Хорошо перемешать путем пипетки.
ПРИМЕЧАНИЕ: Бусы должны быть приготовлены свежие перед использованием. Начните подготовку бисера во время RNase I пищеварения. Для нескольких образцов подготовить бисер вместе в одной трубке.
7. Кап-ловушки
-
Связывание образцов
- Добавьте 105 л подготовленных бусин стрептавидина к 45 злу образца RNase I-обработанный. Хорошо перемешать путем pipetting и инкубировать при 37 кв. М. В течение 30 мин. Микс путем pipetting каждые 10 минут.
- Отделить бусы на магнитной подставке в течение 2-3 мин. Удалите супернатант.
-
Стиральная бисер
- Добавьте 150 л буфера для мытья А и переприостановите бисер путем пайпетирования. Разделите бусины на магнитной подставке в течение 2-3 мин и удалите супернатант.
- Добавьте 150 л буфера для мытья B и переприостановите бисер путем пайпетирования. Разделите бусины на магнитной подставке в течение 2-3 мин и удалите супернатант.
- Добавьте 150 л из буфера мытья C и resuspend бисера путем pipetting. Разделите бусины на магнитной подставке в течение 2-3 мин и удалите супернатант.
ПРИМЕЧАНИЕ: Буферы B и C должны быть разогреты до 37 градусов по Цельсию. Рецепты буферов для мытья A, B и C описаны в Мурата и др.
-
кДНК релиз
- Подготовка 1x RNase I буфера путем смешивания 58,5 л воды с 6,5 л 10x RNase I буфера.
- Приостановите действие бисера в 35 кл из 1x буфера RNase I. Инкубировать при 95 градусах по Цельсию в течение 5 мин и передавать прямо на льду в течение 2 мин, чтобы предотвратить реассоциацию кДНК. Держите крышки во время передачи на лед, как они могут всплывающее из-за давления наращивать.
- Разделите бусины на 2-3 мин на магнитной подставке и перенесите супернатант на новую пластину.
- Приостановите действие бисера в 30 кл из 1x буфера RNase I. Разделите бусины на магнитной подставке на 2-3 мин и перенесите супернатант на ранее собранный супернатант (общий объем электора кДНК должен составлять около 65 л.с.).
8. Удаление РНК RNase H и RNase I Digestion
- В образец, объединить 2,4 л воды, 0,5 л 10x RNase I буфера, 0,1 л RNase H, и 2 Зл RNase I.
- Добавьте 5 кЛ смеси в 65 л высвобожденого образца кДНК и смешайте путем пипетки. Инкубировать при 37 градусах по Цельсию в течение 15 мин и удерживайте при 4 градусах По Цельсию.
- Очистите cDNA из смеси пищеварения RNase с использованием магнитных бусин SPRI. Добавьте 126 зЛ бусин SPRI к 70 л реакции деградации и смешайте путем пипетки. Следуйте шагам очищения, описанным для очищения бисера SPRI в 2.6.1-2.6.6. Elute использованием 42 л воды, разогретой при 37 градусах По Цельсию, как описано.
- Подготовка RNase я смешивать, сочетая 4,5 зл и 10x RNase I буфера и 0,5 зл и L RNase I.
- Добавьте 5 qL смеси RNase к 40 ЗЛ очищенного образца кДНК. Смешайте путем пипетки и инкубировать при 37 градусах по Цельсию в течение 30 мин. Держите при 4 градусах Цельсия.
- Очистите образец с помощью магнитных бусин SPRI. Добавьте 81 кЛ бусин SPRI к 45 л реакции деградации и смешайте путем пипетки. Следуйте шагам очищения, описанным для очищения бисера SPRI в 2.6.1-2.6.6. Elute использованием 42 л воды, как описано.
9. Лигация 5' Линкер
- Сосредоточьтесь на очищенной образце кДНК до 4 л с помощью вакуумного концентратора. Держите температуру на уровне 30-35 градусов по Цельсию. Проверьте громкость с помощью пипетки. Если образец высох до полноты, растворите, добавив 4 л воды.
ПРИМЕЧАНИЕ: Лучше избегать высыхания до полноты, чтобы предотвратить потерю образца. - Инкубировать концентрированный образец при 95 градусах по Цельсию в течение 5 мин и немедленно поставить на лед в течение 2 мин, чтобы предотвратить ренатурацию. Держите крышки при передаче труб, как крышки могут всплывающие из-за давления наращивания.
- Инкубировать 4 зл и 2,5 мкм 5' связующее при 55 градусах по Цельсию в течение 5 мин и немедленно поместите на лед в течение 2 мин, чтобы предотвратить ренатурацию.
- Смешайте 4 злиц из 2,5 мкм 5' связующим звеном с 4 зл и в образце.
ПРИМЕЧАНИЕ: 5' связующее должно быть подготовлено в соответствии с дополнительной таблицей 2, Дополнительная таблица 3, Дополнительная таблица 4, и Дополнительная таблица 5. Разбавить связующее соединение 10 км 5 'до концентрации 2,5 мкм с помощью 100 мМ NaCl до использования. - Добавьте 16 юл перевязочного примикса (см. ТаблицаМатериалов) к смешанному 5' связному и образцу и хорошо перемешайте путем пипетки. Инкубировать при 16 градусах по Цельсию на 16 ч.
- Очистите перевязочение смесь с помощью магнитных бусин SPRI. Добавьте 43,2 л бусин SPRI и выполните шаги 2.6.1-2.6.6. Elute, как описано с помощью 42 л воды, разогретой при 37 градусах Цельсия.
- Повторите очищение, выполненное в шаге 9.6, добавив 72 зЛ бусинs SPRI к переданному супернатанту (1,8:1 бусинки к соотношению выборки).
ПРИМЕЧАНИЕ: 5'связующие содержат штрих-коды, что позволяет объединить до восьми образцов до секвенирования (восемь тринуклеотид штрих-кодов доступны, как описано в Мурата и др.11 и Дополнительная таблица 1).
10. Лигация 3' Линкер
- Сосредоточьтесь на очищенном образце до 4 л с помощью вакуумного концентратора, как описано в шаге 9.1.
- Инкубировать концентрированный образец при 95 градусах по Цельсию в течение 5 мин и немедленно поставить на лед в течение 2 мин, чтобы предотвратить ренатурацию. Держите крышки при передаче труб, как крышки могут выскочить из-за давления наращивания.
- Инкубировать 4 зл и 2,5 мкм 3' связующее при 65 кв кв 5 минут и немедленно поместите на лед в течение 2 мин, чтобы предотвратить ренатурацию.
- Добавьте 4 qL из 2,5 qM 3' связующим звеном к 4 злу концентрированного образца.
- Добавьте 16 кл.л. перевязки премиксит и хорошо перемешать путем пипетки. Инкубировать при 16 градусах по Цельсию на 16 ч.
- Очистите перевязочение смесь с помощью магнитных бусин SPRI. Добавьте 43,2 л бусин SPRI и выполните шаги 2.6.1-2.6.6. Elute, как описано с использованием 42 л воды, разогретой до 37 градусов по Цельсию.
ПРИМЕЧАНИЕ: 3' связующее должно быть подготовлено в соответствии с дополнительными таблицами 6 и дополнительной таблицей 7. Разбавить связующее звено 10 мкм 3' до концентрации 2,5 мкм с помощью 100 мМ NaCl.
11. Дефосфорилирование
- Подготовьте смесь SAP, объединив 4 л воды, 5 л 10-х буфера SAP и 1 зл и фермента SAP.
- Добавьте 10 кЛ смеси SAP в очищенный образец (общий объем 50 л) и инкубируйте сьо, используя следующую программу: 37 градусов по Цельсию в течение 30 мин, 65 градусов по Цельсию в течение 15 мин и удерживайте при 4 градусах Цельсия.
12. Деградация 3' Linker Верхней Strand использованием Uracil Специфический иссечение фермента
- Добавьте 2 зЛ урацила специфического иссеционного фермента (см. ТаблицаМатериалов) в дефосфорилированный образец, смешайте путем трубача и инкубируйте в термоцикле, используя следующую программу: 37 градусов по Цельсию в течение 30 мин, 95 градусов по Цельсию в течение 5 мин, и немедленно поместите на лед в течение 2 мин. предотвратить reannealing фрагментированной верхней нити.
- Очистите реакционную смесь, добавив 93,6 л магнитных бусин SPRI в смесь 52 л и хорошо перемешайте путем пипетки. Повторите шаги очистки 2.6.1-2.6.6. Elute с 42 qL воды, разогретой при 37 градусах По Цельсию, как описано.
13. Второй синтез стрядей
- Подготовьте смесь синтеза второй нити (объемы выражаются в образце) путем объединения 5 зл и 10x буфера реакции полимеразы ДНК, 2 злитромы воды, 1 кЛ 10 мМ dNTPs, 1 кЛ 50 мкм nAnT-iCAGE вторая нить праймера (последовательность находится в дополнительной таблице 1) и 1 l D NA экзонуклеиз-дефицит полимераза (см. рекомендуемую полимеразу в таблицематериалов).
- Добавьте 10 кл/л смеси в очищенный образец и хорошо перемешайте путем пипетки (общий объем составляет 50 л). Инкубировать в тепловом циклическом с использованием следующей программы: 95 градусов по Цельсию в течение 5 мин, 55 градусов по Цельсию в течение 5 мин, 72 градусов по Цельсию в течение 30 мин, и удерживайте при 4 градусах По Цельсию.
14. Деградация второй Strand синтез айтор ы использованием Exonuclease I
- Добавьте 1 зл Exonuclease I ко второй смеси синтеза нити. Хорошо перемешать путем pipetting и инкубировать при 37 кв. c в течение 30 минут, а затем проведение на 4 градуса по Цельсию.
- Очистите двойную ДНК, добавив 91,8 л магнитных бусин SPRI к 51 ЗЛ образца Exonuclease I-обработанный. Повторите шаги очистки, описанные в 2.6.1-2.6.6. и elute с 42 qL воды, разогретой до 37 градусов по Цельсию, как описано.
- Сосредоточьтесь на образце с помощью вакуумного концентратора до 15 зл, как описано в шаге 9.1.
15. Контроль качества и количества
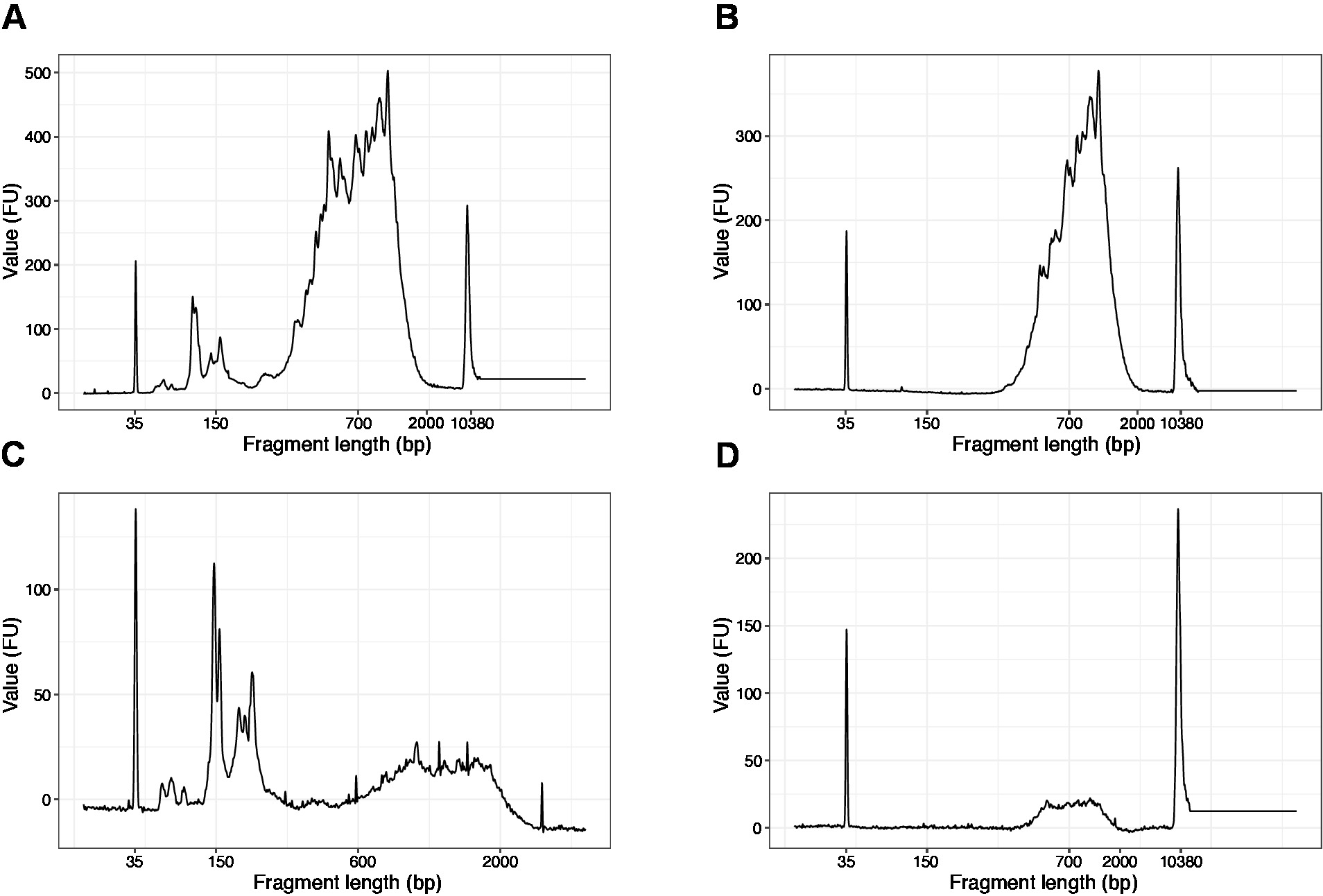
- Используйте 1 Зл из концентрированных образцов и запустить чип ДНК высокой чувствительности на анализаторе качества ДНК. Ожидаемый профиль/количество представлен на рисунке 3.
16. Первый раунд деградации перевозчиков
- Подготовьте смесь деградации, объединив 2 зл и воды, 2 зл 10-кратного буфера фермента ограничения, 1 зл I-SceI и 1 Зл I-CeuI.
- Добавьте 6 qL смеси деградации к 14 qL концентрированного образца и смешайте путем пипеттинг. Инкубировать при 37 градусах по Цельсию в течение 3 ч, затем 20 мин деактивации при 65 градусах по Цельсию и удерживать при 4 градусах Цельсия.
- Очистите смесь деградации с помощью магнитных бусин SPRI. Добавьте 5 qL воды, чтобы увеличить объем смеси деградации и добавить 45 л бусин SPRI (1,8:1 шарики к выборке). Повторите очищение, как описано в шагах 2.6.1-2.6.6. и elute с 42 qL воды, предварительно разогретой до 37 градусов по Цельсию.
- Концентрируйте элетированный образец от 42 qL до 20 qL от общего объема, как описано в шаге 9.1.
17. Контроль уровня деградации и определение количества циклов усиления ПЦР
- Подготовьте смесь qPCR для усиления целых библиотек (адапторная смесь). Комбинат 3,8 л воды, 5 qPCR премикс (2x), 0,1 л из 10 мкм адаптера-ф1 праймера (5'-AATGATACGGCGACCACCGA-3'), и 0,1 л из 10 мкм адаптера 1 праймера (5'-CAAGGAGAAGACACGA-3') для каждого образца (см. Таблица материалов для рекомендуемого кР).
- Смешайте 9 qPCR адаптерную смесь с 1 qL образца со ступени 16.4 и хорошо перемешайте путем пипетки.
- Подготовка qPCR смесь для усиления ДНК, полученных из перевозчика (перевозчик смесь). Объедините 3,8 л воды, 5 qPcR премикс (2x), 0,1 л из 10 км перевозчика no-f1 праймер (5'-GCGGGCGTCGCTATAAC-3'), и 0,1 л из 10 мкм адаптера
- Объедините 9 qPCR несущую смесь с 1 qL образца со ступени 16.4 и хорошо перемешайте путем пипетки.
- Набор программы qPCR: 95 градусов по Цельсию в течение 3 мин (95 градусов по Цельсию на 20 с, 60 градусов по Цельсию на 20 с, 72 градуса по Цельсию на 2 мин) повторяется 40x, а затем инструмент-специфическая кривая денатурации (65-95 градусов по Цельсию), и удерживайте при 4 c.
ПРИМЕЧАНИЕ: Подготовьте отрицательный контроль, заменив образец водой.
18. Усиление ПЦР Целевой библиотеки
- Подготовьте смесь усиления ПЦР, объединив 6 л воды, 0,5 л из 10 адаптера мМ-ф1 праймера, 0,5 л из 10 км адаптера-р1 и 25 л пЦР-премикса (2x). Смешайте путем пипетки (см. Таблицу материалов для рекомендуемого premix ПЦР).
- Добавьте 32 зЛ смеси ПЦР к 18 злу образца со ступени 16.4. Тщательно перемешайте путем пипетки.
- Установите усиливание ПЦР: 95 градусов по Цельсию в течение 3 мин, (98 градусов по Цельсию на 20 с, 60 градусов по Цельсию на 15 с, 72 градуса по Цельсию на 2 мин, 12-18 циклов, 72 градусов по Цельсию в течение 2 мин и удерживайте при 4 градусах по Цельсию.
ПРИМЕЧАНИЕ: Точное количество циклов ПЦР определяется результатами qPCR и соответствует значению Ct, полученному с помощью адаптера primer mix (количество циклов ПЦР равно значению Ct). - Очистите усиленный образец, добавив 90 л магнитных бусин SPRI до 50 злител к усиленному образцу и тщательно перемешайте путем пипетки. Повторите шаги очищения, описанные в шагах 2.6.1-2.6.6. и увядить образец, используя 42 л воды, как описано.
19. Второй раунд деградации перевозчиков
- Повторите шаги 16.1-16.3.
- Очистите смесь деградации с помощью магнитных бусин SPRI. Добавьте в образец 10 л воды, чтобы увеличить объем и смешать с 30 юл бусинs SPRI (1:1 шарики к выборке). Повторите очищение, как описано в шагах 2.6.1-2.6.6. и elute с 42 qL воды, разогретой при 37 градусах по Цельсию, как описано.
- Концентрируйте элетированный образец от 42 qL до 30 qL от общего объема.
20. Выбор размера библиотеки
- Смешайте 24 зл магнитных бусины SPRI с 30 зл и l образца со ступени 19.3. (0,8:1 бисера к выборке соотношение). Повторите шаги очищения, описанные в шагах 2.6.1-2.6.6. и увядить образец в 42 л воды, как описано.
- Сосредоточьтесь на выборке примерно до 14 qL, как описано в шаге 9.1.
21. Контроль качества
-
Оценка распределения размера
- Выполнить 1 зл и выл образца на чипе ДНК с высокой чувствительностью. Ожидаемые результаты представлены на рисунке 4.
ПРИМЕЧАНИЕ: Если видны фрагменты, превышающие 200 б.п.(см. пример на рисунке 4A,C), выбор размера (шаги 20.1-20.2) следует повторять до тех пор, пока не будут удалены короткие фрагменты (рисунок4B,D). Обычно достаточно одного дополнительного раунда выбора размера. Если количество коротких фрагментов является серьезным (как на рисунке 4C),соотношение бисера к выборке должно быть уменьшено до 0,6:1.
- Выполнить 1 зл и выл образца на чипе ДНК с высокой чувствительностью. Ожидаемые результаты представлены на рисунке 4.
-
Контроль качества перевозчика деградации
- Повторите шаги 17.1-17.5.
ПРИМЕЧАНИЕ: В зависимости от концентрации библиотек, оцениваемых в беге чипов ДНК HS (анализ региона), образцы должны быть разбавлены до qPCR. Используйте 0,5 л образца, чтобы избежать потери образца и разбавить 100-500x в воде (разбавить до 1-20 пг/ Л конечной концентрации). Ожидаемая разница между значениями Ct, полученными с помощью адаптера и переносного сочетания, составляет 5-10.
- Повторите шаги 17.1-17.5.
-
Количественная оценка библиотеки
- Подготовьте рабочий разбавление стандарта ДНК лямбда, смешивая 20 лк/л 100 мг/мл стандарта ДНК лямбда с 980 л.с. 1x TE (подготовьте путем разбавления 20x TE, предусмотренного в комплекте количественной оценки ДНК). Разбавление ДНК лямбды может храниться при -20 градусах Цельсия.
- Подготовка lambda ДНК стандартных серийных разбавлений путем смешивания разбавленной стандартля лямбда и 1x TE в соответствии с дополнительной таблицей 8.
ПРИМЕЧАНИЕ: Для более высокой точности рекомендуется добавить 100 юл 1x TE буфера для всех труб и удалить 1x TE объем по мере необходимости в объеме разбавленной лямбды, которые будут добавлены. Не используйте более 1 зл библиотеки; для этого измерения рекомендуется использовать 384 пластины скважин.
Результаты
В этом отчете описывается полный протокол SLIC-CAGE для получения библиотек, готовых к секвенированию, из нанограмм стартового общего материала РНК(рисунок 1). Для получения синтетической смеси носителя РНК, во-первых, шаблоны носителя ПЦР должны быть подготовлены и гель-очищены для устранения побочных продуктов ПЦР(рисунок 2A). Каждый шаблон ПЦР (всего десять) производится с помощью общего форварда, но другой реверсивной грунтовки(таблица2), что приводит к разной длине шаблона ПЦР, чтобы обеспечить изменчивость размера синтетических носителей РНК. После очистки, ПЦР шаблоны используются для экстракорпорной транскрипции молекул носителя. Один продукт рнк-носителя, как ожидается, если шаблоны гель-очищены (см. репрезентативный гель-анализ на рисунке 2B). Подготовка носителя может быть высококлассной в зависимости от необходимости, а при приготовлении, смешанной и замороженной при -80 градусов по Цельсию для будущего использования.
Используя рекомендуемое минимальное количество общей РНК выборки (10 нг) в сочетании с 16-18 циклами усиления ПЦР, можно достичь высокой сложности библиотек SLIC-CAGE. Количество циклов ПЦР, необходимых для усиления конечной библиотеки, сильно зависит от общего объема используемой РНК(ожидаемое количество циклов представлено в таблице4).
После первого раунда деградации, в результатах qPCR (шаг 17), ожидаемая разница между значениями Ct, полученными с помощью адаптера-f1 или проборщика carrier-f1, составляет 1-2, при этом значения Ct, полученные с адаптером,f1 ниже, чем с несущей,f1.
Распределение длинфрагментов в конечной библиотеке составляет от 200-2000 б.п. со средним размером фрагмента 700-900 bp (на основе анализа региона с использованием программного обеспечения Bioanalyzer, Рисунок 4B,D). Более короткие фрагменты, как утихло на рисунке 4A,C, должны быть удалены дополнительными раундами исключения размера (шаги 20-21). Эти короткие фрагменты являются артефактами усиления ПЦР, а не целевой библиотекой. Обратите внимание, что более короткие фрагменты лучше группируются в ячейках секвенирования потока и могут вызвать проблемы с секвенированием.
Ожидаемое количество библиотечного материала, полученного в выборке, составляет от 5 до 50 нг. Значительно меньшие суммы свидетельствуют о потере выборки во время протокола. Если полученного низкого количества достаточно для секвенирования (необходимо 2-3 нг объединенных библиотек), библиотеки могут быть более низкой сложности (см. ниже).
В зависимости от машины секвенирования, количество библиотеки, загруженной на ячейку потока, может потребоваться оптимизировать. Используя Illumina HiSeq 2500, загрузка 8-12 pM SLIC-CAGE библиотек дает в среднем 150-200 миллионов считывает, с йgt;80% считывает прохождение качества оценка 30 в качестве порога.
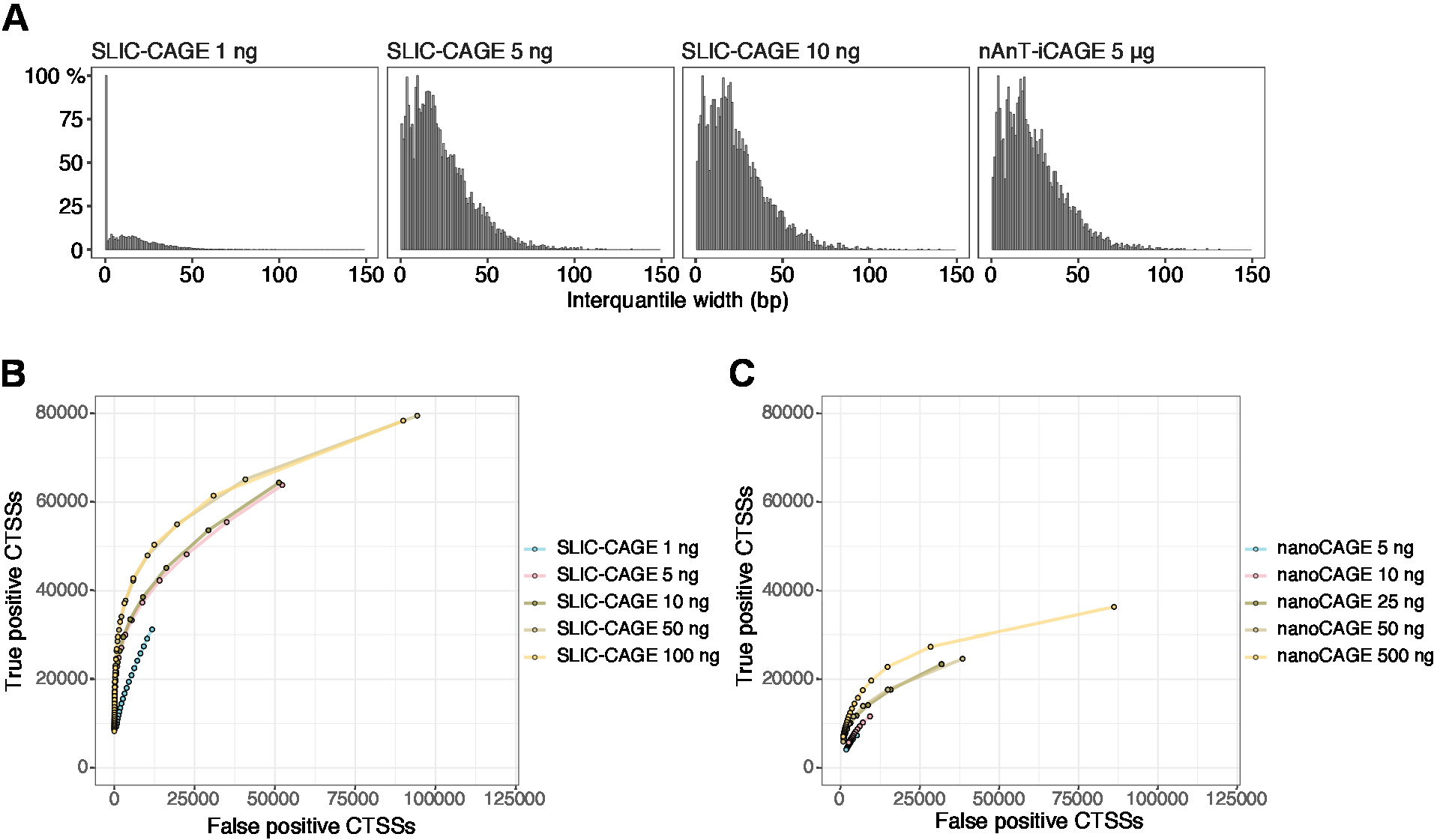
Полученные считывани затем отображаются на эталонном геноме (для 50 bp читает, Bowtie212 может быть использован с параметрами по умолчанию, которые позволяют нулевой несоответствия на последовательность семян (22 bp)». Ожидаемая эффективность картирования зависит от общего объема ввода РНК и представлена в таблице 5. Уникально отображенные считывания могут быть загружены в R графических и статистических вычислительных средах13 и обработаны с помощью CAGEr (Биопроводниковый пакет14). Пакетная виньетка легко следовать и подробно объясняет рабочий процесс и обработку отображенных данных. Легкий визуальный контроль сложности библиотеки является распределение ширины промотора, так как библиотеки с низкой сложностью будут иметь искусственно узкие промоутеры(Рисунок 5A, SLIC-CAGE библиотека, полученная от 1 нг от общей РНК, для деталей см. публикация10). Тем не менее, даже низкосложные библиотеки SLIC-CAGE позволяют идентифицировать истинные CTSS, с большей точностью, чем альтернативные методы для картирования tSS с низким/средним ввоза(рисунок 5B, C).

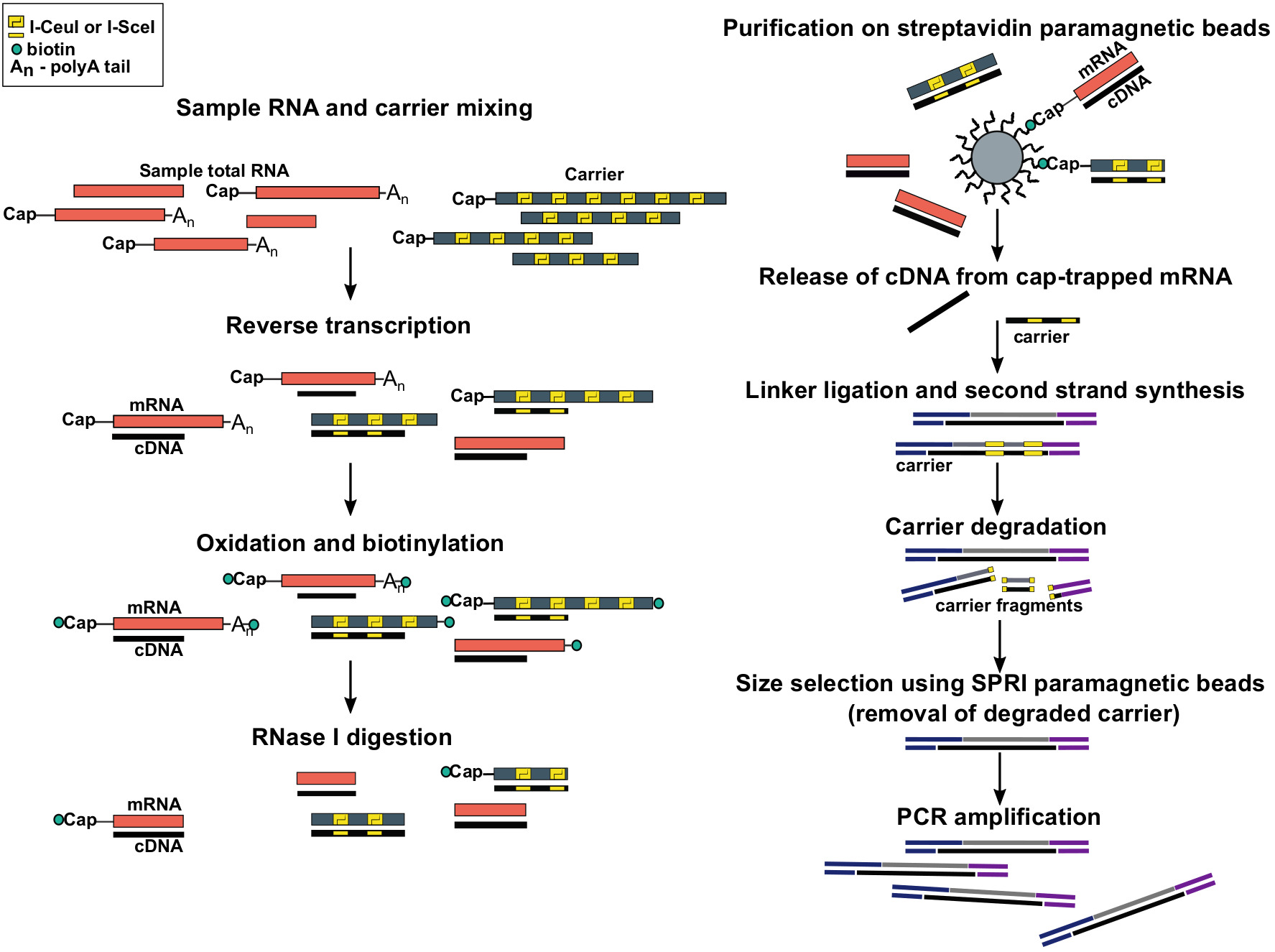
Рисунок 1: Шаги в протоколе SLIC-CAGE. Образец РНК смешивается с рнкрадной смесью РНК для достижения 5 мкг общего материала РНК. cDNA синтезируется через обратную транскрипцию и крышка окисляется с помощью периода натрия. Окисление позволяет прикреплять биотин к крышке с помощью биотина гидразида. Биотин привязывается к концу мРНК 3 ", так как он также окисляется с помощью периода натрия. Для устранения биотина из мРНК:cDNA гибриды с неполностью синтезированных кДНК и из 3 "концы мРНК, образцы обрабатываются с RNase I. кДНК, которые достигли 5 " конец мРНК затем выбирается сродство очищение на стуставидидин магнитных бусин ( крышка-ловушки). После выпуска cDNA, 5 "и 3"-linkers ligated. Молекулы библиотеки, которые происходят от носителя деградируют с помощью I-SceI и I-CeuI самонаводящихся эндонуклеазии и фрагменты удаляются с помощью магнитных бусинS SPRI. Затем библиотека усиливается PCR. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}

Рисунок 2: Представитель гель-анализ шаблонов ПЦР перевозчика и носителей в пробирке стенограммы. (A) Шаблоны ПЦР-носителя до очистки геля: первый колодец содержит маркер 1 кбитт, а затем шаблоны ПЦР-1, 1-10. (B) Carrier in vitro транскрипты: первый колодец содержит 1 кбитт маркер, а затем перевозчик стенограммы 1-10. Транскрипты перевозчика были денатурированы при нагревании в течение 5 мин при 95 градусах Цельсия до погрузки. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}

Рисунок 3: Представитель качества ДНК (высокая чувствительность ДНК чип) след SLIC-CAGE до первого раунда деградации носителя. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}

Рисунок 4: Репрезентативное качество ДНК (высокой чувствительности ДНК чипа) следы библиотек SLIC-CAGE после усиления ПЦР. (A) Библиотека SLIC-CAGE, которая требует дополнительного выбора размера для удаления коротких фрагментов. (B) SLIC-CAGE библиотека после выбора размера с использованием 0,6x SPRI бусы к выборке соотношение. (C) SLIC-CAGE библиотека с более низким объемом вывода, что требует выбора размера для удаления короткого фрагмента. (D) SLIC-CAGE библиотека более низкого объема вывода после выбора размера с использованием 0.6:1 SPRI бусы к выборке соотношение. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}

Рисунок 5: Проверка библиотек SLIC-CAGE. (A) Распределение кластера тегов интерквантилной ширины в библиотеках SLIC-CAGE, подготовленных из 1, 5 или 10 нг общей РНК S. cerevisiae, и в библиотеке nAnT-iCAGE, подготовленной из 5 мкг S. cerevisiae общей РНК. Большое количество узких кластеров тегов в библиотеке 1 нг SLIC-CAGE указывает на его низкую сложность. (B) КРИвые ROC для идентификации CTSS в библиотеках S. cerevisiae SLIC-CAGE. Все S. cerevisiae nAnT-iCAGE CTSSs были использованы в качестве истинного набора. (C) КРИвые ROC для идентификации CTSS в библиотеках S. cerevisiae nanoCAGE. Все S. cerevisiae nAnT-iCAGE CTSSs были использованы в качестве истинного набора. Сравнение кривых ROC показывает, что SLIC-CAGE сильно превосходит nanoCAGE в идентификации CTSS. Использовались данные ArrayExpress E-MTAB-6519. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
{kind=link}
Таблица 1: Последовательность синтетического гена носителя. I-SceI сайты смелые и курсивом в фиолетовый, и I-CeuI признания сайты зеленый. Пожалуйста, нажмите здесь, чтобы загрузить этот файл.
| Перевозчика | обратный грунт 5'-3' | PcR длина продукта / bp | |
| 1 | ПЦР-Н6-Р1: ННННКТАКГТКГКГКАГАГАГАГАТТ | 1034 год | |
| 2 | ПЦР-Н6ЗР2: NNNNNNTATCCAGATCGTGAGCTGC | 966 г. | |
| 3 | ПЦР-Н6-р3: ННННКАКТГГГГКТТТАКСГ | 889 год | |
| 4 | ПЦР-Н6ЗР4: НННННКГГГГГГТАТААКТГТГТГГГТГГТГГТГТГГТГТГТГТ | 821 год | |
| 5 | ПЦРЗН-Н6-р5: НННННАГТГАТАККККГАГКККККТТТЦТТЦТТТЦТТЦТТЦТТЦТТЦТЦТЦТЦТТЦТТЦ | 744 г. | |
| 6 | ПЦР-Н6ЗР6: NNNNNNGTGAAGAATTCTGTTCTTCTTCCCA | 676 г. | |
| 7 (г. | ПЦР-Н6-р7: NNNNNNCTCGCGGCTCTAGTCATCATAAC | 599 г. | |
| 8 | ПЦР-Н6ЗР8: NNNNNNTATACGCGATGTGTGTCGTACTAC | 531 | |
| 9 До 9 | ПЦР-Н6-р9: NNNNNNNNACCGCCGCCTCCGCAGG | 454 г. | |
| 10 Лет | ПЦРЗН-Н6-р10: NNNNCAGGACGTTTTGCCCAGCA | 386 г. | |
| Передняя грунтовка одинакова для всех шаблонов перевозчика. Подчеркнуто последовательность промоутера T7. ПЦРЗН5-ф1: ТААТАКАКТАКАКТАТАГННННКГГТКГКТА | |||
Таблица 2: Праймеры для усиления шаблона носителя. Передняя грунтовка одинакова для всех шаблонов несущих. Подчеркнуто последовательность промоутера T7. ПЦРЗН5-ф1: ТаАТАГКАКТАКАКТАТАГНННКГГГТКГКТКТА. С использованием различных обратных грунтовок, pcR шаблоны и, следовательно, РНК перевозчика различной длины производятся.
| Перевозчика | Длина | uncapped/мкг | ограничен/мкг |
| 1 | 1034 год | 3,96 | 0,45 |
| 2 | 966 г. | 8.36 Для | 0,95 |
| 3 | 889 год | 4.4 | 0,5 |
| 4 | 821 год | 6.6 | 0,75 |
| 5 | 744 г. | 4.4 | 0,5 |
| 6 | 676 г. | 3.08 | 0,35 |
| 7 (г. | 599 г. | 4.4 | 0,5 |
| 8 | 531 | 3,96 | 0,45 |
| 9 До 9 | 454 г. | 2.64 | 0,3 |
| 10 Лет | 386 г. | 2.2 | 0,25 |
Таблица 3: RNA несущая смесь. В общей сложности 49 мкг переносной смеси 0,3-1 кбитт: необнаруженный й 44 мкг, ограниченный 5 мкг.
| Общий объем ввода РНК /нг | Циклы ПЦР |
| 1 нг | 18 лет |
| 2 нг | 17 Лет |
| 5 нг | 16 Год |
| 10 нг | 15-16 |
| 25 нг | 14-15 |
| 50 нг | 13-15 |
| 100 нг | 12-14 |
Таблица 4: Ожидаемое количество циклов ПЦР в зависимости от общего объема РНК-ввода. Приблизительное количество циклов основано на экспериментах, проводимых с использованием Saccharomyces cerevisiae, Drosophila melanogaster и Mus musculus total RNA.
| Общий вход РНК/нг | % общий отображены | % однозначно отображено | % перевозчика |
| 1 нг | 30 год | 20-30 | 30 год |
| 2 нг | 60 лет | 20-50 | 10 Лет |
| 5 нг | 60-70 | 40-60 | 5-10 |
| 10 нг | 60-70 | 40-60 | 5-10 |
| 25 нг | 65-80 | 40-70 | 0-5 |
| 50 нг | 65-80 | 40-70 | 0-3 |
| 100 нг | 70-85 | 40-70 | 0-2 |
Таблица 5: Ожидаемая эффективность картирования и зависимость от общего объема ввода РНК. Приблизительные цифры представлены и основаны на экспериментах, проведенных с использованием Saccharomyces cerevisiae и Mus musculus общей РНК.
Обсуждение
Для успешной подготовки библиотеки SLIC-CAGE крайне важно использовать низкосвязные советы и трубки для предотвращения потери образца из-за адсорбции образцов. Во всех шагах, связанных с извлечением супернатанта, рекомендуется восстановить весь объем образца. Поскольку протокол состоит из нескольких этапов, непрерывная потеря выборки приведет к неудачным библиотекам.
Если CAGE (nAnT-iCAGE) не выполняется в обычном режиме, лучше всего протестировать SLIC-CAGE с различными объемами ввода (10 нг, 20 нг, 50 нг, 100 нг, 200 нг) того же общего образца РНК и сравнить с nAnT-iCAGE библиотек, которые готовятся с использованием 5 мкг общей РНК. Если библиотека nAnT-iCAGE не работает (менее 0,5-1 нг из библиотеки ДНК, полученной в образце), SLIC-CAGE вряд ли сработает, и потеря образца должна быть сведена к минимуму.
Важным шагом для обеспечения высококачественных библиотек, лишенных необнаруженной деградированных РНК или РНК, является ловушка крышки, описанная в разделе 7. Очень важно, чтобы стрептавидин бисер тщательно перенапрягается в буферах стирки и чтобы буферы для мытья удаляются до продолжения следующего шага стирки или утилиза кДНК.
Если результаты qPCR после первого раунда деградации носителя не показывают никакой разницы между использованием адаптера и праймеров carrier-f1, продолжение протокола по-прежнему рекомендуется. Если после второго раунда деградации носителей разница в значениях Ct составляет менее пяти, рекомендуется третий раунд деградации носителей. Мы никогда не находили третий раунд деградации необходимо, и если это происходит, рекомендуется заменить самонаводящихся эндонуклеазе запасов.
Дополнительные раунды усиления ПЦР могут быть добавлены в протокол, если окончательная сумма полученной библиотеки недостаточна для секвенирования. Затем усиление ПЦР может быть установлено с минимальным количеством циклов усиления, необходимых для получения достаточного количества материала для секвенирования, с учетом потерь выборки, которых нельзя избежать при выборе размера. Очистка или выбор размера с использованием магнитных бусин SPRI должны быть выполнены до тех пор, пока все небольшие (злт;200 bp) фрагменты удаляются (при необходимости, используйте 0,6:1 шарики для соотношения образца), и библиотека должна быть количественно с помощью Picogreen.
Библиотеки могут быть секвенированы в режиме одноконца или парного конца. С помощью сопряженного секвенирования можно получить информацию о изоформах транскрипта. Кроме того, по мере того, как обратная транскрипция выполняется с помощью случайного грунтовки (TCT-N6,N6, являющихся случайным гексеумером), информация из секвенированных 3'end может быть использована в качестве уникальных молекулярных идентификаторов (UMI) для сброса дубликатов ПЦР. Поскольку используется умеренное число циклов усиления ПЦР (до 18), использование UMIs ранее было признано ненужным.
Поскольку ядро протокола опирается на nAnT-iCAGE11,SLIC-CAGE использует восемь штрих-кодов. Таким образом, мультиплексирование более восьми образцов в настоящее время не поддерживается. Кроме того, как SLIC-CAGE, так и nAnT-iCAGE не подходят для захвата РНК короче 200 б.п., так как протоколы предназначены для удаления связующих и PCR артефактов путем исключения размера с amPure XP шариками.
SLIC-CAGE является единственным беспристрастным методом однонуклеотидного разрешения с низким уровнем ввода для картирования участков начала транскрипции с использованием нанограммов общего материала РНК. Альтернативные методы опираются на активность переключения шаблонов обратной транскриптазы на штрих-код, ограниченный РНК вместо крышки захвата (например, NanoCAGE15 и NanoPARE16). Из-за переключения шаблонов, эти методы демонстрируют последовательность конкретных предубеждений в обнаружении TSSs, что приводит к увеличению числа ложноположительных TSSs и снижение числа истинных TSSs9,10.
Раскрытие информации
Был заполнен патент на разлагаемую РНК/ДНК.
Благодарности
Эта работа была поддержана грантом Wellcome Trust (106954), присужденным B. L. и Совету медицинских исследований (MRC) Core Funding (MC-A652-5'A10). Н.К. был поддержан долгосрочным стипендией EMBO (EMBO ALTF 1279-2016); E. P. была поддержана Советом медицинских исследований Великобритании; B. L. была поддержана Советом медицинских исследований Великобритании (MC UP 1102/1).
Материалы
| Name | Company | Catalog Number | Comments |
| 2-propanol, Bioultra, for molecular biology, ≥99.5% | Sigma-Aldrich | 59304-100ML-F | Used in RNAclean XP purification. |
| 3' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 3'linkers is described in the supplementary of this protocol. | ||
| 5' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 5'linkers is described in the supplementary of this protocol. | ||
| Agencourt AMPure XP, 60 mL | Beckman Coulter | A63881 | Purification of DNA |
| Agencourt RNAClean XP Kit | Beckman Coulter | A63987 | Purification of RNA and RNA:cDNA hybrids in CAGE steps. |
| Axygen 0.2 mL Polypropylene PCR Tube Strips and Domed Cap Strips | Axygen (available through Corning) | PCR-0208-CP-C | Or any 8-tube PCR strips (used only for water and mixes). |
| Axygen 1 x 8 strip domed PCR caps | Axygen (available through Corning) | PCR-02CP-C | Caps for PCR plates. |
| Axygen 1.5 mL Maxymum Recovery Snaplock Microcentrifuge Tube | Axygen (available through Corning) | MCT-150-L-C | Low-binding 1.5 mL tubes, used for enzyme mixes or sample concentration. |
| Axygen 96 well no skirt PCR microplate | Axygen (available through Corning) | PCR-96-C | Low-binding PCR plates - have to be used for all steps in the protocol. Note that plates should be cut to contain 2 x 8 wells for easier visibility of the samples |
| Bioanalyzer (or Tapestation): RNA nano and HS DNA kits | Agilent | To determine quality of RNA, efficient size selection and final quality of the library (Tapestation can also be used) | |
| Biotin (Long Arm) Hydrazide | Vector laboratories | SP-1100 | Biotinylation/tagging |
| Cutsmart buffer | NEB | Restriction enzyme buffer | |
| Deep Vent (exo-) DNA Polymerase | NEB | M0259S | Second strand synthesis |
| DNA Ligation Kit, Mighty Mix | Takara | 6023 | Used for 5' and 3'-linker ligation |
| dNTP mix (10 mM each) | ThermoFisher Scientific | 18427013 | dNTP mix for production of carrier templates (or any dNTPs suitable for PCR) |
| Dynabeads M-270 Streptavidin | Invitrogen | 65305 | Cap-trapping. Do not use other beads as these are optimised with the buffers used. |
| DynaMag-2 Magnet | ThermoFisher Scientific | 12321D | Magnetic stand for 1.5 mL tubes - used to prepare Streptavidin beads. |
| DynaMag-96 Side Skirted Magnet | ThermoFisher Scientific | 12027 | Magnetic stand for PCR plates (96 well-plates) - used with cut plates to contain 2 x 8 wells. |
| Ethanol, BioUltra, for molecular biology, ≥99.8% | Sigma-Aldrich | 51976-500ML-F | Used in AMPure washes. Any molecular biology suitable ethanol can be used. |
| Exonuclease I (E. coli) | NEB | M0293S | Leftover primer degradation |
| Gel Loading Dye, Purple (6x), no SDS | NEB | B7025S | agarose gel loading dye |
| HiScribe T7 High Yield RNA Synthesis Kit | New England Biolabs | E2040S | Kit for carrier in vitro transcription |
| Horizontal electrophoresis apparatus | purification of carrier DNA templates from agarose gels | ||
| I-Ceu | NEB | R0699S | Homing endonuclease used for carrier degradation. |
| I-SceI | NEB | R0694S | Homing endonuclease used for carrier degradation. |
| KAPA HiFi HS ReadyMix (2x) | Kapa Biosystems (Supplied by Roche) | KK2601 | PCR mix for target library amplification |
| KAPA SYBR FAST qPCR kit (Universal) 2x | Kapa Biosystems (Supplied by Roche) | KK4600 | qPCR mix to assess degradation efficiency and requiered number of PCR amplification cycles |
| Micropipettes and multichannel micropipettes (0.1-10 µL, 1-20 µL, 20-200 µL) | Gilson | Use of Gilson with the low-binding Sorenson tips is recommended. Other micropippetes might not be compatible. Different brand low-binding tips may not be of equal quality and may increase sample loss. | |
| Microplate reader | For Picogreen concentration measurement of the final library. Microplates are used to allow small volume measurement and reduce sample waste. | ||
| nuclease free water | ThermoFisher Scientific | AM9937 | Or any nuclease (DNase and RNase) free water |
| PCR thermal cycler | incubation steps and PCR amplficication | ||
| Phusion High-Fidelity DNA Polymerase | ThermoFisher Scientific | F530S | DNA polymerase for amplification of carrier templates (or any high fidelity polymerase) |
| QIAquick Gel Extraction Kit (50) | Qiagen | 28704 | Purification of carrier PCR templates from agarose gels. |
| qPCR machine | determining PCR amplification cyle number and degree of carrier degradation | ||
| Quant-iT PicoGreen dsDNA Reagent | ThermoFisher Scientific | P11495 | Used to measure final library concentration - recommended as, in our hands, it is more accurate and reproducible than Qubit. |
| Quick-Load Purple 100 bp DNA Ladder | NEB | N0551S | DNA ladder |
| Quick-Load Purple 1 kb Plus DNA Ladder | NEB | N0550S | DNA ladder |
| Ribonuclease H | Takara | 2150A | Digestion of RNA after cap-trapping. |
| RNase ONE Ribonuclease | Promega | M4261 | Degradation of single stranded RNA not protected by cDNA. |
| RNase-Free DNase Set | Qiagen | 79254 | Removal of carrier DNA templates after in vitro transcription. |
| RNeasy Mini Kit | Qiagen | 74104 | For cleanup of carrier RNA from in vitro transcription or capping |
| Sodium acetate, 1 M, aq.soln, pH 4.5 RNAse free | VWR | AAJ63669-AK | Or any nuclease (DNase and RNase) free solution |
| Sodium acetate, 1 M, aq.soln, pH 6.0 RNAse free | Or any nuclease (DNase and RNase) free solution | ||
| Sodium periodate | Sigma-Aldrich | 311448-100G | Oxidation of vicinal diols |
| Sorenson low binding aerosol barrier tips, MicroReach Guard, volume range 10 μL, Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719390-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 1,000 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719463-1000EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 20 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719412-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 200 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719447-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| SpeedVac Vacuum Concentrator | concentrating samples in various steps to lower volume | ||
| SuperScript III Reverse Transcriptase | ThermoFisher Scientific | 18080044 | Used for reverse transcription (1st CAGE step) |
| Trehalose/sorbitol solution | Preparation is described in Murata et al. 2014. | ||
| Tris-HCl, 1 M aq.soln, pH 8.5 | 1 M solution, DNase and RNase free | ||
| tRNA (20 mg/mL) | tRNA solution. Preparation is described in Murata et al. 2014. | ||
| UltraPure Low Melting Point Agarose | ThermoFisher Scientific | 16520050 | Or any suitable pure low-melt agarose. |
| USB Shrimp Alkaline Phosphatase (SAP) | Applied Biosystems (Provided by ThermoFisher Scientific) | 78390500UN | |
| USER Enzyme | NEB | M5505S | Degradation of 3'linker's upper strand, Uracil Specific Excision Reagent/Enzyme |
| Vaccinia Capping System | NEB | M2080S | Enzymatic kit for in vitro capping of carrier molecules |
| Wash buffer A | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer B | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer C | Cap trapping washes. Preparation is described in Murata et al. 2014. |
Ссылки
- Shiraki, T., et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proceedings of the National Academy of Sciences of the United States of America. 100 (26), 15776-15781 (2003).
- Haberle, V., Lenhard, B. Promoter architectures and developmental gene regulation. Seminars in Cell and Developmental Biology. 57, 11-23 (2016).
- Haberle, V., Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nature Reviews Molecular Cell Biology. 19 (10), 621-637 (2018).
- Andersson, R., et al. An atlas of active enhancers across human cell types and tissues. Nature. 507 (7493), 455-461 (2014).
- Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Celniker, S. E., et al. Unlocking the secrets of the genome. Nature. 459 (7249), 927-930 (2009).
- Consortium, F., et al. A promoter-level mammalian expression atlas. Nature. 507 (7493), 462-470 (2014).
- Boyd, M., et al. Characterization of the enhancer and promoter landscape of inflammatory bowel disease from human colon biopsies. Nature Communications. 9 (1), 1661(2018).
- Adiconis, X., et al. Comprehensive comparative analysis of 5'-end RNA-sequencing methods. Nature Methods. , (2018).
- Cvetesic, N., et al. SLIC-CAGE: high-resolution transcription start site mapping using nanogram-levels of total RNA. Genome Research. 28 (12), 1943-1956 (2018).
- Murata, M., et al. Detecting expressed genes using CAGE. Methods in Molecular Biology. 1164, 67-85 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9, 357(2012).
- R Core Team. A language and environment for statistical computing. , Available from: https://www.R-project.org/ (2017).
- Haberle, V., Forrest, A. R., Hayashizaki, Y., Carninci, P., Lenhard, B. CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic Acids Research. 43 (8), e51(2015).
- Poulain, S., et al. NanoCAGE: A Method for the Analysis of Coding and Noncoding 5'-Capped Transcriptomes. Methods in Molecular Biology. 1543, 57-109 (2017).
- Schon, M. A., Kellner, M. J., Plotnikova, A., Hofmann, F., Nodine, M. D. NanoPARE: parallel analysis of RNA 5' ends from low-input RNA. Genome Research. 28 (12), 1931-1942 (2018).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены